Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Acknowledgements
Abbreviations
Supplementary Material
References
Introduction
Results
Discussion
Materials and Methods
Acknowledgements
Abbreviations
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(5):1389-1398. doi:10.7150/thno.20706 This issue Cite
Research Paper
Longitudinal Multiplexed Measurement of Quantitative Proteomic Signatures in Mouse Lymphoma Models Using Magneto-Nanosensors
Jung-Rok Lee1, Iris Appelmann2,3, Cornelius Miething2,4, Tyler O. Shultz5, Daniel Ruderman6, Dokyoon Kim5, Parag Mallick7, Scott W. Lowe2, Shan X. Wang5,7,8 ![]()
1. Division of Mechanical and Biomedical Engineering, Ewha Womans University, Seoul, South Korea
2. Cancer Biology and Genetics Program, Memorial Sloan Kettering Cancer Center, New York, New York, USA
3. Department of Hematology, Oncology, Hemostaseology and Stem Cell Transplantation, University Hospital RWTH Aachen, Aachen, Germany
4. Department of Internal Medicine, Medical Center - University of Freiburg, Freiburg, Germany
5. Department of Materials Science and Engineering, Stanford University, Stanford, California, USA
6. Ellison Institute of Transformative Medicine of USC, USC Keck School of Medicine, Los Angeles, California, USA
7. Department of Medicine, Department of Radiology, Stanford University, Stanford, California, USA
8. Department of Electrical Engineering, Stanford University, Stanford, California, USA
Received 2017-4-24; Accepted 2017-12-12; Published 2018-2-3
Citation:
Lee JR, Appelmann I, Miething C, Shultz TO, Ruderman D, Kim D, Mallick P, Lowe SW, Wang SX. Longitudinal Multiplexed Measurement of Quantitative Proteomic Signatures in Mouse Lymphoma Models Using Magneto-Nanosensors. Theranostics 2018; 8(5):1389-1398. doi:10.7150/thno.20706. https://www.thno.org/v08p1389.htm
Other stylesAbstract

Cancer proteomics is the manifestation of relevant biological processes in cancer development. Thus, it reflects the activities of tumor cells, host-tumor interactions, and systemic responses to cancer therapy. To understand the causal effects of tumorigenesis or therapeutic intervention, longitudinal studies are greatly needed. However, most of the conventional mouse experiments are unlikely to accommodate frequent collection of serum samples with a large enough volume for multiple protein assays towards single-object analysis. Here, we present a technique based on magneto-nanosensors to longitudinally monitor the protein profiles in individual mice of lymphoma models using a small volume of a sample for multiplex assays.
Methods: Drug-sensitive and -resistant cancer cell lines were used to develop the mouse models that render different outcomes upon the drug treatment. Two groups of mice were inoculated with each cell line, and treated with either cyclophosphamide or vehicle solution. Serum samples taken longitudinally from each mouse in the groups were measured with 6-plex magneto-nanosensor cytokine assays. To find the origin of IL-6, experiments were performed using IL-6 knock-out mice.
Results: The differences in serum IL-6 and GCSF levels between the drug-treated and untreated groups were revealed by the magneto-nanosensor measurement on individual mice. Using the multiplex assays and mouse models, we found that IL-6 is secreted by the host in the presence of tumor cells upon the drug treatment.
Conclusion: The multiplex magneto-nanosensor assays enable longitudinal proteomic studies on mouse tumor models to understand tumor development and therapy mechanisms more precisely within a single biological object.
Keywords: Proteomic signature, magneto-nanosensors, longitudinal study, therapeutic intervention, mouse lymphoma, IL-6
Introduction
Cancer proteomics has been greatly investigated to elucidate the mechanism of tumorigenesis and to find early cancer detection biomarkers [1, 2]. Serum proteins or cytokines have been predominantly studied as early cancer detection biomarkers [1, 3, 4], because they can be easily collected via minimally invasive methods and sera contain much more cancer-associated proteins directly released from tumor masses than any other forms of samples such as saliva and urine. In addition, serum protein biomarkers have been used to predict the host's response to a cancer therapy [5], tumor progression and regression [6], and cancer recurrence [7]. Furthermore, proteomic signatures can reveal the activity of a host's immune system during the development of tumors or upon a drug treatment such as cancer immunotherapy [8].
To understand the mechanisms in immune responses and drug actions, researchers have mainly used mouse models because they can be performed in a more controlled manner, and experiments on humans are heavily restricted due to regulations. Despite differences between the species, mice are still a good candidate model to represent humans, because they have genetic, biological, and behavioral similarities to humans. However, the problem with experiments using mice is that the volume of blood in a mouse is over 1000 times less than that of humans, which limits the volume drawn at each time and the frequency of blood collection. These two factors of sample collection are practically important to experimental design and performing longitudinal experiments with a single object, especially when the measurements of multiple proteins are required. Due to the heterogeneity among mice, if blood or serum samples from different mice are pooled together, unique proteomic profiles rendered in a single mouse can be averaged out [9]. Therefore, only common trends among the mice will be captured in the measurements, or unfortunately the opposite trends could be cancelled out if they are expressed separately by two groups of mice in the pool.
As we witnessed, single cell analysis has brought a new perspective in bioscience and medicine [10, 11]. Similarly, a longitudinal study on a single mouse can bring a new insight into the protein profiles in a single object correlated with symptoms or outputs from the object during the course of tumor development [12]. More importantly, longitudinal monitoring can reveal temporal changes in cytokine levels induced by host-tumor interactions, which cannot even be recognized by conventional end-point measurements. To obtain such data, many approaches have been utilized to overcome the restrictions of sample collection in longitudinal studies. One method is microarrays [13, 14], which are high-throughput, but are not sensitive enough to measure low abundant proteins. Another approach is bead-based assays [15]. Bead-based assays are highly multiplexed and sensitive, but the process of the measurement is relatively slow because bead-counting is a serial process, and the costs of reagents are fairly high. Thus, a new tool is greatly needed to perform longitudinal studies with mouse models more effectively with a higher temporal resolution and the capability of sensitive multiplexing.
We have developed magneto-nanosensors capable of monitoring multiple proteins at femtomolar sensitivities with a large dynamic range [16, 17]. The magneto-nanosensors have been demonstrated for the detection of radiation biomarkers [18], autoantibodies [19, 20], and small molecules [21] as well as for monitoring protein-protein interactions [22, 23]. Here, we present a method using magneto-nanosensors with 6-plex cytokine assays to measure longitudinal protein profiles in response to cyclophosphamide treatment in mouse lymphoma models. High levels of IL-6 and GCSF in serum samples from the drug-treated mice were observed after the treatment, and it was found that IL-6 is produced by the host in the presence of tumor cells upon the treatment by using IL-6 knock-out (KO) mice. We believe that our technique enables researchers to longitudinally monitor protein profiles during the course of tumor development or therapeutic intervention, which allows for studies on mechanisms of tumorigenesis and drug action in a single object of interest.
Results
Development of multiplexed magneto-nanosensor cytokine assays
The magneto-nanosensor employs the effect of giant magnetoresistance (GMR) to detect magnetic nanoparticles (MNPs) in its proximity [24]. MNPs are surrogate labels for target proteins in a sandwich complex with a pair of antibodies. The MNPs used in this study have an average diameter of 46±13 nm with multiple cores of iron oxide embedded in a dextran polymer [23]. To measure multiple proteins in a sample of interest, an array of 8 × 8 magneto-nanosensors was fabricated on a 10 mm × 12 mm chip using photolithography techniques as shown in Figure 1A. Individual magneto-nanosensors on the chip were functionalized with different antibodies that specifically capture their target cytokines such as granulocyte colony-stimulating factor (GCSF), tumor necrosis factor alpha (TNF-α), eosinophil chemotactic protein (Eotaxin or CCL11), fms-related tyrosine kinase 3 ligand (FLT3LG), interleukin 6 (IL-6), and vascular endothelial growth factor (VEGF) (Figure 1B). In addition to the capture antibodies, some sensors were coated with bovine serum albumin (BSA) or biotinylated BSA to serve as negative and positive controls, respectively.
Figure 1
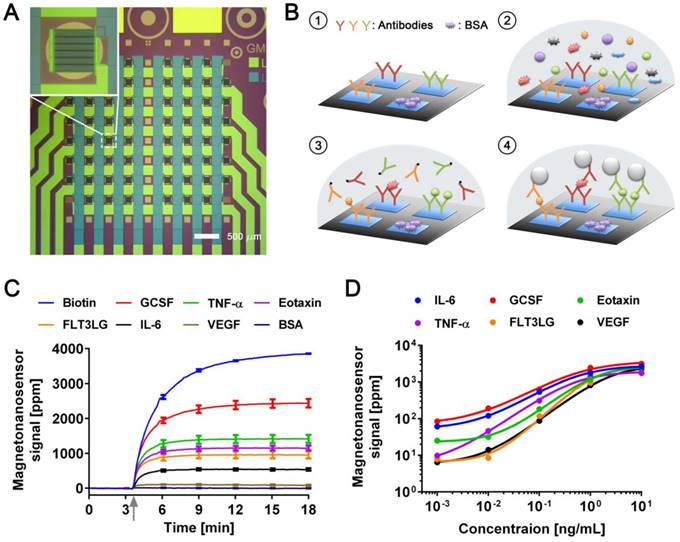
Development of multiplexed magneto-nanosensor cytokine assays. (A) Optical image of an 8 × 8 array of magneto-nanosensor chip. The chip consists of 64 individually-accessible magneto-nanosensors. Inset: optical image of an individual magneto-nanosensor. (B) Schematic of sandwich protein immunoassays using magneto-nanosensors. (1) Different capture antibodies that specifically capture their target proteins were immobilized on different magneto-nanosensors. Bovine serum albumin (BSA) and biotinylated BSA (Biotin, not shown here) were also immobilized on different sensors as negative and positive controls, respectively. (2) Serum containing target proteins was added to the magneto-nanosensor chip, and allowed to bind to the corresponding capture antibodies. (3) After washing unbound proteins, biotinylated detection antibodies were added to the chip, and allowed to bind to the bound proteins to form sandwich complexes. (4) After unbound detection antibodies were washed, the chip was loaded into a reader station, and streptavidin-coated MNPs were introduced. The binding signals of MNPs to the sandwich complexes were monitored and recorded by the reader station. (C) Real-time monitoring of binding signals of MNPs. The baseline signals were measured after the chip was loaded into the reader station. Then, MNPs were introduced to the chip as indicated by the gray arrow. The binding signals were monitored until they reached their plateaus. Six recombinant proteins (GCSF: 1 ng/mL, TNF-α: 1 ng/mL, Eotaxin: 1 ng/mL, FLT3LG: 1 ng/mL, IL-6: 0.1 ng/mL, and VEGF: 0.1 ng/mL) were used as a test sample. The error bars represent standard deviations of 4 identical magneto-nanosensors. (D) Titration curves of 6-plex protein assays measured by multiplexed magneto-nanosensors. Six recombinant proteins were mixed to be at indicated final concentrations (10 and 1 ng/mL, and 100, 10, and 1 pg/mL), and measured using multiplexed magneto-nanosensor protein assays. Each data point is the average signal of 4 identical magneto-nanosensors, and titration curves (solid lines) were calculated using four parameter logistic (4-PL) regression (IL-6: R2 = 0.9997, GCSF: R2 = 0.9892, Eotaxin: R2 = 0.9936, TNF-α: R2 = 0.9950, FLT3LG: R2 = 0.9524, and VEGF: R2 = 0.9969).
Then, a serum sample was added to the chip, and the target proteins in the sample bound to the capture antibodies on the sensors. The bound proteins were labeled with biotinylated antibodies (detection antibodies), which were further labeled with streptavidin-coated MNPs. To demonstrate the 6-plex assay with the magneto-nanosensors, a chip was incubated with a sample containing recombinant proteins of GCSF, TNF-α, Eotaxin, and FLT3LG all at 1 ng/mL and both IL-6 and VEGF at 0.1 ng/mL. The concentrations of the proteins were tailored to show various levels of magneto-nanosensor signals. Real-time signals were monitored after MNPs were added to the chip (Figure 1C). The positive control sensors always produce a maximum signal because they bind to MNPs directly without forming any sandwich structure with the antibodies and proteins. Due to the different number densities of the proteins in the sample and the binding affinities of the antibodies, it is difficult to precisely compare the concentrations of different proteins using the raw signals, but generally higher concentrations produce higher magneto-nanosensor signals because it is known that the magneto-nanosensor signals are proportional to the number of bound MNPs [23, 25]. More precise conversion factors to concentrations from measured signals for each assay are typically obtained with titration curves where signals are plotted versus varying concentrations of each target protein. However, before measuring the titration curves, we tested the cross-reactivity between the 6-plex protein assays by using the method described previously [26], because there are concerns about the cross-reactivity of antibodies used in immunoassays [27]. No significant cross-reactivity was observed across the assays (Figure S1). With these antibody pairs, titration curves were measured with proteins ranging from 1 pg/mL to 10 ng/mL (Figure 1D). The parameters of analyzed titration curves are summarized in Table S1. After demonstrating that the assays show clear dose-response with varying concentrations of proteins, we further tested the variations of the assays by measuring pre-aliquoted and frozen quality control (QC) samples with different reader stations on different days (Figure S2). The calculated coefficients of variation (CV) of the assays were less than 25% in all the cases that we tested, which is comparable to or better than the performance of conventional microarray techniques [28-30].
Figure 2
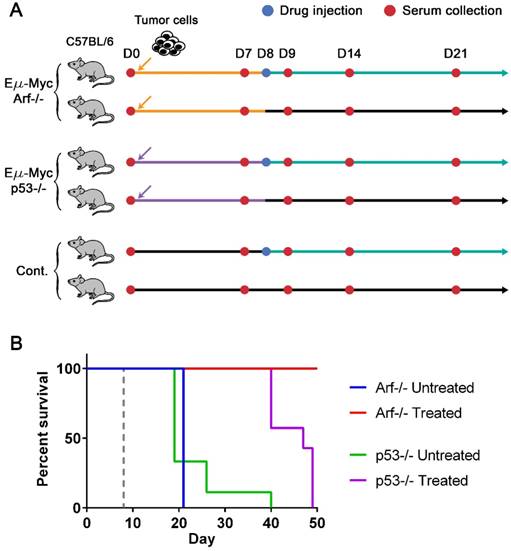
Lymphoma mouse models and survival curves. (A) Experimental design. Three groups of syngeneic immunocompetent mice (C57BL/6) were injected with either Eµ-Myc/Arf-/-, Eµ-Myc/p53-/-, or no tumor cells (control), respectively, after the initial bleeding. Serum samples were collected from each mouse every 7 days and on day 9 until mice died or became moribund. Two subgroups of each cell line group were treated with either drug (cyclophosphamide) or vehicle solution (sham injection) on day 8. (B) The Kaplan-Meier curves of four groups of mice (both untreated (n=10) and drug-treated (n=10) groups of Eµ-Myc/Arf-/- challenged mice, and both untreated (n=9) and drug-treated (n=7) groups of Eµ-Myc/p53-/- challenged mice). The mice were observed until day 50, and all Eµ-Myc/Arf-/- challenged mice in the drug-treated group survived until day 50. The dashed line indicates the drug treatment.
Mouse lymphoma models
To generate drug-sensitive/resistant mouse tumor models where protein profiles are monitored with magneto-nanosensors upon therapeutic intervention, we inoculated 3 × 105 Eµ-Myc/p53-/- or Eµ-Myc/Arf-/- mouse lymphoma cells into syngeneic and immunocompetent C57BL/6 mice (Figure 2A). The oncogene of c-Myc was coupled to the immunoglobulin heavy chain µ enhancer (Eµ) and was overexpressed in these cell lines, leading to the development of non-Hodgkin's lymphoma, which closely resembles human Burkitt's lymphoma [31, 32]. Arf (its product: p19Arf, p14Arf in humans) and p53 are tumor suppressor genes, and their mutations combined with Eµ-Myc are known to develop tumors much faster than Eµ-Myc only [33]. Interestingly, although Arf and p53 closely interplay via Mdm2 [32, 34], their mutations' outcomes in response to chemotherapy using cyclophosphamide are dramatically different: Eµ-Myc/Arf-/- (or Arf null) is sensitive to the drug (similar response as Eµ-Myc), but Eµ-Myc/p53-/- (or p53 null) shows primary resistance [33]. Among two subgroups of mice inoculated with Arf null cells, one subgroup was injected with cyclophosphamide 8 days after transplantation of the cells, and the other subgroup was treated with vehicle solution (Hank's balanced salt solution, HBSS) (Figure 2A). The drug treatment was scheduled on day 8 post-transplantation to maximize the effect of the drug, because a huge efflux of tumor cells into lymph nodes from spleen was observed on day 10 post-transplantation in similar mouse models [35]. Serum samples were collected from both subgroups before inoculation of the cells (D0) as well as 7 (i.e., pre-treatment), 9, 14, and 21 days after the cell injection (D7, D9, D14, and D21). The same drug treatment and serum collections were performed for the subgroups of the mice inoculated with p53 null cells and the control mice (no cells were injected as a negative control). The Kaplan-Meier curves of four groups (treated and untreated Arf null, and treated and untreated p53 null) clearly display their sensitivities to the drug (Figure 2B). All mice in the treated Arf null group (n=10) survived until 80 days after the cell injection when the experiment was terminated, while the treated p53 null group (n=7) all died before day 50. Both untreated Arf null (n=10) and p53 null (n=9) groups started to die around day 20, or become moribund and then sacrificed after the final bleeding. As expected, the treated (n=3) and untreated (n=3) groups of the control mice did not show any symptoms of the disease, and survived until the experiment was terminated.
Multiplexed cytokine measurements
To investigate the differences in protein profiles between the treated and untreated groups, 25 µL of serum at each sample collection point from each mouse were measured with the 6-plex magneto-nanosensor assays to construct protein profiles over the course of the experiment (Figure 3). The signals were converted into concentrations using the titration curves (Figure 1D) to estimate the actual concentrations of the proteins (Figure S3). For the measurement, the mice were randomly selected from Arf null (n=5 for treated and untreated, respectively) and p53 null (n=4 for treated and untreated, respectively) cohorts shown in Figure 2. Regarding the untreated p53 null cohort, however, we only include the mice that died before day 21 to ensure the same physiological outcomes among the 4 mice that we choose to measure. In addition, to examine the intermediate effect induced by the drug, serum samples were collected one day before and after the drug treatment (D7 and D9), which was possible due to the small sample consumption in magneto-nanosensor measurement. We clearly observed that the IL-6 levels in serum samples peaked on day 14 only in the treated subgroups of both Arf null and p53 null cohorts (Figure 3). Furthermore, the levels of GCSF were elevated rapidly just one day (D9) after the drug treatment only in the treated subgroups, and remained high until day 14 compared to the untreated subgroups. Comparable patterns of these proteins were not observed in control mice, implicating these findings as relevant to host-tumor interactions upon the treatment. To more carefully figure out which proteins display significant differences induced by the treatment, we performed statistical tests between the treated and untreated subgroups in each cell line cohort (Table 1). Prior to the treatment, both the treated and untreated groups were essentially identical as they were inoculated with the same number and type of tumor cells. As expected, they were not statistically different at the pre-treatment stage. Then, we again found that the differences in IL-6 and GCSF levels were statistically significant at the post-treatment stage. In addition, the statistical tests show that TNF-α and FLT3LG were highly expressed in the treated groups of Arf null and p53 null, respectively, after the treatment. These differences could be related to the final outputs of the treatment, and used to predict the prognosis.
Table 1
Statistical tests between drug-treated and untreated groups for each cytokine
| Cell type | Cytokine | p-value | |
|---|---|---|---|
| Pre-treatment | Post-treatment | ||
| Eµ-Myc/Arf-/- | IL-6 | 0.6956 | < 0.0001* |
| GCSF | 0.7370 | 0.0012* | |
| Eotaxin | 0.4920 | 0.4664 | |
| TNF-α | 0.6571 | 0.0001* | |
| FLT3LG | 0.2008 | 0.1508 | |
| VEGF | 0.8629 | 0.6108 | |
| Eµ-Myc/p53-/- | IL-6 | 0.6080 | 0.0047* |
| GCSF | 0.9560 | 0.0002* | |
| Eotaxin | 0.6378 | 0.0716 | |
| TNF-α | 0.3401 | 0.7766 | |
| FLT3LG | 0.6726 | 0.0007* | |
| VEGF | 0.3637 | 0.0606 | |
| Control | IL-6 | 0.8956 | 0.4223 |
| GCSF | 0.0206 | 0.1059 | |
| Eotaxin | 0.2628 | 0.2519 | |
| TNF-α | 0.8694 | 0.4604 | |
| FLT3LG | 0.0684 | 0.1199 | |
| VEGF | 0.0800 | 0.9713 | |
(*: FDR < 5%)
The origin of IL-6
While we observed an elevated level of IL-6 in the drug-responsive groups, it has been reported that affected lymph nodes in non-Hodgkin's diseases produce IL-6 and high serum IL-6 levels are correlated with worse prognosis [8, 36]. Thus, anti-IL-6 therapy has been under clinical trials for non-Hodgkin's diseases, especially Castleman disease [36]. However, IL-6 is known to be an important cytokine in immune response by enhancing T-cell and B-cell functions [37], and was demonstrated to have anti-tumor activity in mouse tumor models [38, 39]. To address this issue, we need to clarify whether IL-6 is produced by the host or tumor cells. The origin of IL-6 was investigated using IL-6 KO mice with the p53 null cell line (Figure 4A). The tumor cells were injected into two subgroups of IL-6 KO C57BL/6 mice (n=10 for both treated and untreated, respectively) using the same protocols described in Figure 2. The same drug was given to only one subgroup on day 8, and serum samples were collected every 7 days as well as the day after the drug treatment. The experiment was terminated on day 35, and we obtained similar survival curves that resemble those of the p53 null cohort using wild type mice (Figure 4B). Among 10 mice in each subgroup, 5 mice were randomly selected for IL-6 measurement (Figure 4C). The IL-6 levels in the treated group of IL-6 KO mice were as low as those in the untreated groups of IL-6 KO and wild type mice (Figure 4C). The elevated level of IL-6 in the treated wild type mice is statistically different from those in the IL-6 KO mice and untreated wild type mice. This indicates that IL-6 is generated by the host in the presence of tumor cells upon the drug treatment, not by the tumor cells themselves.
Figure 3
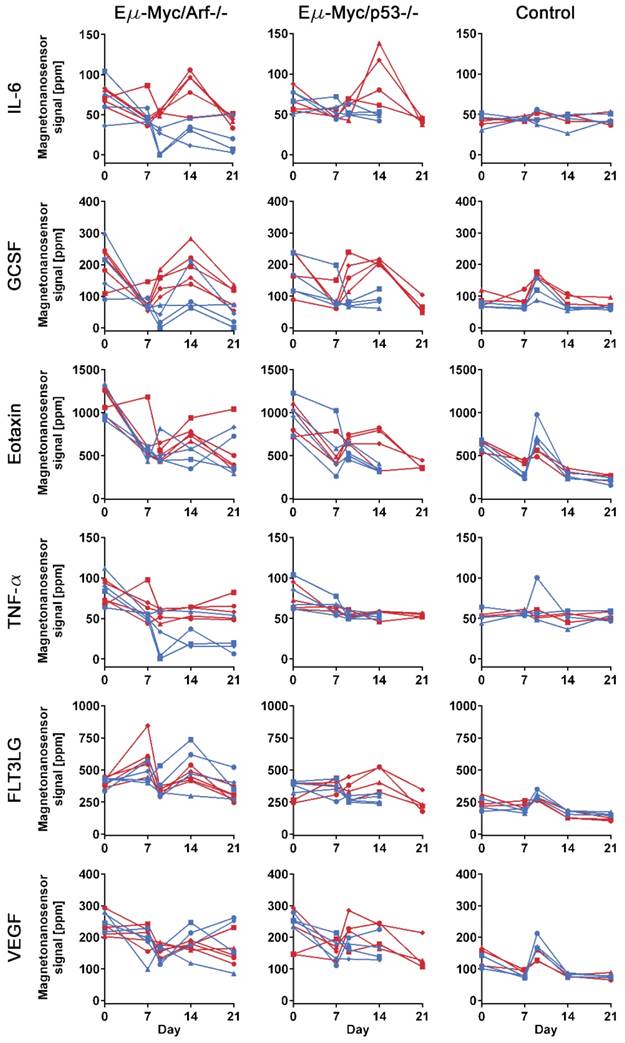
Protein profiles of lymphoma mouse models measured by multiplexed magneto-nanosensor assays. Protein signals from 6 different groups of mice. Red represents drug-treated groups, while blue represents untreated groups. Each row shows profiles of a protein (IL-6, GCSF, Eotaxin, TNF-α, FLT3LG, and VEGF), and y-axes were on the same scale for the same protein across different mouse groups. Each column represents the same group of mice challenged with the same type of cell line or control group (n=3 for drug-treated and untreated groups, respectively). The first column shows drug-treated (n=5) and untreated (n=5) groups of mice challenged with Eµ-Myc/Arf-/- lymphoma cells, while the second column displays drug-treated (n=4) and untreated (n=4) groups of Eµ-Myc/p53-/- lymphoma cells. Since the untreated Eµ-Myc/p53-/- groups died before day 21, the last data points are day 14. Each data point represents the average signal of 4 identical magneto-nanosensors.
Figure 4
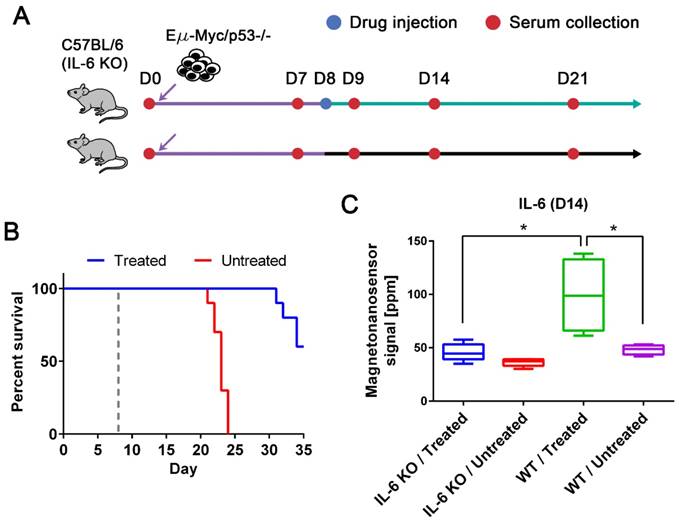
IL-6 KO mouse models and the origin of up-regulated IL-6. (A) Experimental design with IL-6 KO mice. Two groups of IL-6 KO mice were challenged with the Eµ-Myc/p53-/- cell line after the initial bleeding on day 0. Serum samples were taken every 7 days and on day 9. Only one of the groups was treated with the drug on day 8, and the other was injected with vehicle solution. (B) The Kaplan-Meier curves of untreated (n=10) and drug-treated (n=10) groups of IL-6 KO mice. The mice were observed until day 35, and the dashed gray line indicates the drug treatment. (C) IL-6 protein signals on day 14 from four groups (both drug-treated (n=5) and untreated (n=5) groups of IL-6 KO mice and both drug-treated (n=4) and untreated (n=4) groups of wild type mice challenged with Eµ-Myc/p53-/- lymphoma cells). Mann-Whitney tests (two-tailed) were performed to calculate p-values, and an asterisk indicates p < 0.05.
Discussion
We have demonstrated that longitudinal multiplexed protein assays can be performed using magneto-nanosensors for quantitative proteomic signature analysis on a single biological object. Small consumption of serum samples in magneto-nanosensors allowed frequent sample collection in a single mouse. In this study, undiluted samples were used but if diluted samples are acceptable based on the concentrations of proteins of interest, it would be possible to monitor protein profiles on an hourly basis or even with higher temporal resolution by taking smaller volumes of samples more often. Another approach to achieve this without losing any sensitivity is to integrate the magneto-nanosensors with microfluidic channels to compartmentalize the sensors [40, 41]. Furthermore, only 6-plex protein assays were demonstrated in this study as proof of concept, but since there are 64 individual sensors on a chip, theoretically 64 different proteins can be measured with a single chip. Based on the methods described in this study, the panel of proteins can be further expanded without increasing the volume of sample required. Thus, the number of proteins that can be measured with magneto-nanosensors will be the same as commercially available bead-based assays (~50-plex). The sensitivities of both assays are comparable, but the assay time of the magneto-nanosensors is shorter than the bead-based assays due to the parallel processes in magneto-nanosensors. Compared to the conventional microarray [42], the assay sensitivity of the magneto-nanosensors is better, as demonstrated in this study.
The magneto-nanosensor measurement revealed that IL-6 and GCSF were highly expressed after the treatment with cyclophosphamide. They peaked on day 14 compared to the untreated groups, but the levels of GCSF rapidly increased on the day after the treatment. Based on these observations, a possible scenario is that cyclophosphamide induced apoptosis of tumor cells or inflammation in the tumor microenvironment, which recruited immune cells. Then, the local immune cells such as macrophages produced GCSF, which further recruited granulocytes such as neutrophils. As the cellular immune response was initiated, the host cells generated IL-6 to activate T-cell and B-cell functions. Interestingly, the fully drug-responsive group (treated Arf null cohort) maintained statistically higher levels of TNF-α than the untreated group, while the partially responsive group (treated p53 null cohort; substantial extension in survival curve) produced more FLT3LG than its untreated counterpart. Since TNF-α is an indicator of apoptosis and a pro-inflammatory cytokine [43], this could reflect that the immune cells are actively eliminating tumor cells. It has also been reported that FLT3LG has anti-tumor activity in vivo, as was demonstrated with fibrosarcoma [44], but high levels of FLT3LG were not observed in the treated Arf null cohort. This might indicate that the p53 null cell line induces antagonists to prevent the production of FLT3LG as the tumor cells are growing and interacting with the host. Thus, the signature of these two cytokines could be a predictor of therapeutic outcomes.
Traditional cancer proteomics has been focused more on finding new biomarkers released from cancer cells or indicating the presence of tumor cells. However, proteomic signature of cytokines can represent host-tumor interactions including immune and drug responses, which help understand underlying mechanisms. With this technique, more precise and dense protein profile information in a single mouse can be obtained for any type of biological process or disease including cancer immunotherapy [43, 45], vaccination [46], autoimmune diseases [47], and cardiovascular diseases [48].
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center.
Lymphoma mouse models
Mice were housed at Memorial Sloan Kettering Cancer Center in a facility accredited by the American Association of Laboratory Animal Care. Procedures were performed in accordance with Institutional Animal Care and Use Committee and National Institutes of Health guidelines. Mice were gas anesthetized using isoflurane (2% in 100% oxygen, 1 L/min) during all procedures and kept at 37 °C. 3 × 105 cells of either Eµ-Myc/Arf-/- or Eµ-Myc/p53-/- lymphoma cells in 200 µL of HBSS were injected via the tail vein of 6-weeks-old female C57BL/6 mice. Mice injected with 200 µL of HBSS served as negative controls. For IL-6 KO C57BL/6 mice, 3 × 105 cells of only Eµ-Myc/p53-/- lymphoma cells in 200 µL of HBSS were injected. Each treated group in different cell lines or types of mice was injected with cyclophosphamide at 300 mg/kg at 8 days post-inoculation of tumor cells.
For serum collection, 50 µL of blood was collected by submandibular bleeding into a 1 mL Z-gel microtube (Sarstedt, Germany). 400 µL of saline was injected subcutaneously to replenish the fluids in the mice. The blood was allowed to clot at room temperature for 1 h and spun down at 1 × 104 × g for 10 min at 4 °C to remove the blood cells. The serum was transferred to 1.7 mL microcentrifuge tubes and stored at -80 °C until analyses.
Magneto-nanosensors
An 8 × 8 array of magneto-nanosensors was fabricated on a chip with a dimension of 10 mm × 12 mm, as described previously [22]. Briefly, serpentine structures of multiple stripes of spin valve stacks (seed layer/IrMn/CoFe/Ru/CoFe/Cu/CoFe) were patterned via photolithography, and connected through 300 nm thick Ta/Au/Ta conductive layers. Each sensor was connected in the manner of a grid network to electrical pads for external connection with a reader station. A 30 nm thick passivation layer was deposited on top of the sensors except on 4 sensors at the corners. On the rest of the chip surface, a 300 nm thick passivation layer was deposited to avoid chemical corrosion and electrical breakdown.
For immobilization of capture antibodies, the chip surface was cleaned twice using acetone, methanol, and isopropanol sequentially. Then, a reaction well with an inner diameter of a quarter inch was installed to accommodate samples and reagents during the assay. The chip was further cleaned using oxygen plasma for 3 min before the surface chemistry employed previously [22, 24]. Briefly, the chip was treated with 1% poly(allylamine hydrochloride) for 5 min at room temperature, and rinsed with distilled water (10977-023, Life Technologies). The chip was then cured at 120 °C on a hot plate for 1 h, followed by treatment with 2% poly(ethylene-alt-maleic anhydride) for 5 min. After the chip was rinsed with distilled water, a 1:1 mixture of N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) in distilled water was added to the well, and incubated with the chip for 1 h. The chip was then rinsed again, and fully dried. Each capture antibody (anti-IL-6: MAB406, anti-GCSF: MAB414, anti-Eotaxin: AF-420-NA, anti-TNF-α: AF-410-NA, anti-FLT3LG: AF427, and anti-VEGF: AF-493-NA from R&D Systems) was spotted on different magneto-nanosensors in quadruplicate using a non-contact arrayer (sciFLEXARRAYER S5, Scienion). BSA and biotinylated BSA were spotted on different magneto-nanosensors as positive and negative controls, respectively. The chip was placed in a humid chamber and incubated overnight at 4 °C.
Cytokine assays
The chip with immobilized capture antibodies was rinsed with rinsing buffer (Phosphate buffered saline (PBS) pH 7.4 with 0.1% BSA and 0.05% Tween20) and incubated with 1% BSA for 1 h at room temperature. After blocking with BSA, the chip was rinsed with rinsing buffer. 25 µL of an undiluted serum sample was then added to the chip and incubated for 1.5 h. For the titration curve measurements or development purpose, recombinant proteins were spiked into PBS and used at the indicated concentrations (IL-6: 406-ML, GCSF: 414-CS, Eotaxin: 420-ME, TNF-α: 410-MT, FLT3LG: 427-FL, and VEGF: 493-MV from R&D Systems). After rinsing the chip again, 50 µL of a cocktail of 6 different biotinylated detection antibodies (anti-IL-6: BAF406, anti-GCSF: BAF414, anti-Eotaxin: BAF420, anti-TNF-α: BAF410, anti-FLT3LG: BAF427, and anti-VEGF: BAF493 from R&D Systems), each of which was at a concentration of 0.5 µg/mL, were added to the chip and incubated for 1 h, allowing for formation of sandwich complexes around the target protein or cytokine. The chip was then rinsed to remove unbound detection antibodies, and inserted into the reader station equipped with a 3 in. Helmholz coil. The reader station calibrated each magneto-nanosensor and recorded baseline signals from individual magneto-nanosensors. After obtaining the baseline signals, streptavidin-coated MNPs (130-048-101, Miltenyi Biotec) were added to the chip, and the binding signals of MNPs to the sandwich complexes (more specifically binding between streptavidin on MNP and biotin on detection antibody) were recorded until the binding signals reached their plateaus. The plateau signals were taken as magneto-nanosensor signals. To avoid negative values, all magneto-nanosensor signals were shifted by 50 ppm during the post-processing.
The reader station applies both an AC magnetic field to modulate the magnetization of the magneto-nanosensors and an AC voltage to the sensors at different frequencies to employ the double modulation scheme [49, 50]. The amplified signals from the magneto-nanosensors were processed using Fast Fourier Transform (FFT), and corrected using temperature and magnetoresistance correction schemes described in detail in the previous study [51].
Statistical analysis
Multiplex magneto-nanosensor data were analyzed as mixed effects models using the MCMCglmm package (version 2.24) in the R statistical environment (version 3.3.2). Each analyte was modeled independently. The cell type (levels “p53 null”, “Arf null”, and “none”), treatment epoch (levels “pre-treatment” and “post-treatment”), and cyclophosphamide treated (“true”, “false”) were modeled as nested fixed effects. The mouse identifier was treated as a random effect. The day within each epoch was not considered as a factor. We employed 106 MCMCglmm iterations, and a burn-in of 10,000; computations with these settings proved to be highly reproducible. The reported significance is the p-value of the cyclophosphamide-treated effect. Using a hypothesis rejection criterion of false discovery rate (FDR) < 5%, cytokines with statistical significance were selected (Table 1).
Acknowledgements
We would like to thank Drs. D. Mitchell Magee and Joshua LaBaer at Arizona State University for their helpful discussions. This work was supported by Physical Science Oncology Center (U54CA143907), Center for Cancer Nanotechnology Excellence (U54CA151459 and U54CA199075), and the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2017R1C1B5076529). I.A. was supported by Dr. Mildred Scheel postdoctoral fellowship (project no. 109902) of the German Cancer Aid (Deutsche Krebshilfe).
Abbreviations
KO: knock-out; GMR: giant magnetoresistance; MNP: magnetic nanoparticle; BSA: bovine serum albumin; GCSF: granulocyte colony-stimulating factor; TNF: tumor necrosis factor; FLT3LG: fms-related tyrosine kinase 3 ligand; IL-6: interleukin 6; VEGF: vascular endothelial growth factor; QC: quality control; CV: coefficient of variation.
Supplementary Material
Supplementary figures and tables.
Competing Interests
S.X.W. has related patents or patent applications assigned to Stanford University and out-licensed for potential commercialization. S.X.W. has stock or stock options in MagArray, Inc., which has licensed relevant patents from Stanford University for commercialization of magneto-nanosensor chips.
References
1. Wulfkuhle JD, Liotta LA, Petricoin EF. Proteomic applications for the early detection of cancer. Nat Rev Cancer. 2003;3:267-75
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
3. Hori SS, Gambhir SS. Mathematical model identifies blood biomarker-based early cancer detection strategies and limitations. Sci Transl Med. 2011;3:109ra16
4. Borrebaeck CA. Precision diagnostics: moving towards protein biomarker signatures of clinical utility in cancer. Nat Rev Cancer. 2017;17:199-204
5. Hanash SM, Pitteri SJ, Faca VM. Mining the plasma proteome for cancer biomarkers. Nature. 2008;452:571-9
6. Lee MS, Kim JH, Lee JS, Yun SJ, Kim WJ, Ahn H. et al. Prognostic Significance of CREB-Binding Protein and CD81 Expression in Primary High Grade Non-Muscle Invasive Bladder Cancer: Identification of Novel Biomarkers for Bladder Cancer Using Antibody Microarray. Plos One. 2015;10:e0125405
7. Carlsson A, Wingren C, Kristensson M, Rose C, Ferno M, Olsson H. et al. Molecular serum portraits in patients with primary breast cancer predict the development of distant metastases. Proc Natl Acad Sci U S A. 2011;108:14252-7
8. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11-22
9. Zhang SD, Gant TW. Effect of pooling samples on the efficiency of comparative studies using microarrays. Bioinformatics. 2005;21:4378-83
10. Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17:175-88
11. Heath JR, Ribas A, Mischel PS. Single-cell analysis tools for drug discovery and development. Nat Rev Drug Discov. 2016;15:204-16
12. Lee JR, Chan CT, Ruderman D, Chuang HY, Gaster RS, Atallah M. et al. Longitudinal Monitoring of Antibody Responses against Tumor Cells Using Magneto-nanosensors with a Nanoliter of Blood. Nano Lett. 2017;17:6644-52
13. Fan R, Vermesh O, Srivastava A, Yen BK, Qin L, Ahmad H. et al. Integrated barcode chips for rapid, multiplexed analysis of proteins in microliter quantities of blood. Nat Biotechnol. 2008;26:1373-8
14. MacBeath G. Protein microarrays and proteomics. Nat Genet. 2002;32(Suppl):526-32
15. Vignali DA. Multiplexed particle-based flow cytometric assays. J Immunol Methods. 2000;243:243-55
16. Gaster RS, Hall DA, Nielsen CH, Osterfeld SJ, Yu H, Mach KE. et al. Matrix-insensitive protein assays push the limits of biosensors in medicine. Nat Med. 2009;15:1327-32
17. Osterfeld SJ, Yu H, Gaster RS, Caramuta S, Xu L, Han SJ. et al. Multiplex protein assays based on real-time magnetic nanotag sensing. Proc Natl Acad Sci U S A. 2008;105:20637-40
18. Kim D, Marchetti F, Chen Z, Zaric S, Wilson RJ, Hall DA. et al. Nanosensor dosimetry of mouse blood proteins after exposure to ionizing radiation. Sci Rep. 2013;3:2234
19. Lee JR, Haddon DJ, Wand HE, Price JV, Diep VK, Hall DA. et al. Multiplex giant magnetoresistive biosensor microarrays identify interferon-associated autoantibodies in systemic lupus erythematosus. Sci Rep. 2016;6:27623
20. Lee JR, Haddon DJ, Gupta N, Price JV, Credo GM, Diep VK. et al. High-Resolution Analysis of Antibodies to Post-Translational Modifications Using Peptide Nanosensor Microarrays. Acs Nano. 2016;10:10652-60
21. Lee JR, Choi J, Shultz TO, Wang SX. Small Molecule Detection in Saliva Facilitates Portable Tests of Marijuana Abuse. Anal Chem. 2016;88:7457-61
22. Lee JR, Bechstein DJ, Ooi CC, Patel A, Gaster RS, Ng E. et al. Magneto-nanosensor platform for probing low-affinity protein-protein interactions and identification of a low-affinity PD-L1/PD-L2 interaction. Nat Commun. 2016;7:12220
23. Gaster RS, Xu L, Han SJ, Wilson RJ, Hall DA, Osterfeld SJ. et al. Quantification of protein interactions and solution transport using high-density GMR sensor arrays. Nat Nanotechnol. 2011;6:314-20
24. Lee JR, Sato N, Bechstein DJ, Osterfeld SJ, Wang J, Gani AW. et al. Experimental and theoretical investigation of the precise transduction mechanism in giant magnetoresistive biosensors. Sci Rep. 2016;6:18692
25. Wang SX, Li G. Advances in giant magnetoresistance biosensors with magnetic nanoparticle tags: Review and outlook. IEEE T Magn. 2008;44:1687-702
26. Gaster RS, Hall DA, Wang SX. Autoassembly protein arrays for analyzing antibody cross-reactivity. Nano Lett. 2011;11:2579-83
27. Baker M. Reproducibility crisis: Blame it on the antibodies. Nature. 2015;521:274-6
28. Angenendt P, Glokler J, Murphy D, Lehrach H, Cahill DJ. Toward optimized antibody microarrays: a comparison of current microarray support materials. Anal Biochem. 2002;309:253-60
29. Shi LM, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC. et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151-61
30. Sato F, Tsuchiya S, Terasawa K, Tsujimoto G. Intra-platform repeatability and inter-platform comparability of microRNA microarray technology. Plos One. 2009;4:e5540
31. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S. et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533-8
32. Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658-69
33. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM. et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335-46
34. Sherr CJ, Weber JD. The ARF/p53 pathway. Current opinion in genetics & development. 2000;10:94-9
35. Ito K, Smith BR, Parashurama N, Yoon JK, Song SY, Miething C. et al. Unexpected dissemination patterns in lymphoma progression revealed by serial imaging within a murine lymph node. Cancer Res. 2012;72:6111-8
36. Kurzrock R, Voorhees PM, Casper C, Furman RR, Fayad L, Lonial S. et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res. 2013;19:3659-70
37. Akira S, Taga T, Kishimoto T. Interleukin-6 in biology and medicine. Advances in immunology. 1993;54:1-78
38. Mule JJ, Custer MC, Travis WD, Rosenberg SA. Cellular mechanisms of the antitumor activity of recombinant IL-6 in mice. J Immunol. 1992;148:2622-9
39. Mule JJ, McIntosh JK, Jablons DM, Rosenberg SA. Antitumor activity of recombinant interleukin 6 in mice. J Exp Med. 1990;171:629-36
40. Bechstein DJB, Lee JR, Ooi CC, Gani AW, Kim K, Wilson RJ. et al. High performance wash-free magnetic bioassays through microfluidically enhanced particle specificity. Sci Rep. 2015;5:11693
41. Bechstein DJ, Ng E, Lee JR, Cone SG, Gaster RS, Osterfeld SJ. et al. Microfluidic multiplexed partitioning enables flexible and effective utilization of magnetic sensor arrays. Lab Chip. 2015;15:4273-6
42. Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2:RESEARCH0004
43. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321-30
44. Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat Med. 1997;3:625-31
45. Lee S, Margolin K. Cytokines in cancer immunotherapy. Cancers. 2011;3:3856-93
46. Ahlers JD, Belyakov IM, Matsui S, Berzofsky JA. Mechanisms of cytokine synergy essential for vaccine protection against viral challenge. Int Immunol. 2001;13:897-908
47. O'Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat Rev Immunol. 2002;2:37-45
48. Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO 3rd, Criqui M. et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499-511
49. de Boer BM, Kahlman JA, Jansen TP, Duric H, Veen J. An integrated and sensitive detection platform for magneto-resistive biosensors. Biosens Bioelectron. 2007;22:2366-70
50. Han S-J, Yu H, Murmann B, Pourmand N, Wang SX. A High-Density Magnetoresistive Biosensor Array with Drift-Compensation Mechanism. Dig Tech Pap IEEE Int Solid State Circuits Conf. 2007:168-594
51. Hall DA, Gaster RS, Osterfeld SJ, Murmann B, Wang SX. GMR biosensor arrays: correction techniques for reproducibility and enhanced sensitivity. Biosens Bioelectron. 2010;25:2177-81
Author contact
![]() Corresponding author: sxwangedu (SXW)
Corresponding author: sxwangedu (SXW)
Citation styles
APA
Lee, J.R., Appelmann, I., Miething, C., Shultz, T.O., Ruderman, D., Kim, D., Mallick, P., Lowe, S.W., Wang, S.X. (2018). Longitudinal Multiplexed Measurement of Quantitative Proteomic Signatures in Mouse Lymphoma Models Using Magneto-Nanosensors. Theranostics, 8(5), 1389-1398. https://doi.org/10.7150/thno.20706.
ACS
Lee, J.R.; Appelmann, I.; Miething, C.; Shultz, T.O.; Ruderman, D.; Kim, D.; Mallick, P.; Lowe, S.W.; Wang, S.X. Longitudinal Multiplexed Measurement of Quantitative Proteomic Signatures in Mouse Lymphoma Models Using Magneto-Nanosensors. Theranostics 2018, 8 (5), 1389-1398. DOI: 10.7150/thno.20706.
NLM
Lee JR, Appelmann I, Miething C, Shultz TO, Ruderman D, Kim D, Mallick P, Lowe SW, Wang SX. Longitudinal Multiplexed Measurement of Quantitative Proteomic Signatures in Mouse Lymphoma Models Using Magneto-Nanosensors. Theranostics 2018; 8(5):1389-1398. doi:10.7150/thno.20706. https://www.thno.org/v08p1389.htm
CSE
Lee JR, Appelmann I, Miething C, Shultz TO, Ruderman D, Kim D, Mallick P, Lowe SW, Wang SX. 2018. Longitudinal Multiplexed Measurement of Quantitative Proteomic Signatures in Mouse Lymphoma Models Using Magneto-Nanosensors. Theranostics. 8(5):1389-1398.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.