Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(5):1270-1285. doi:10.7150/thno.22536 This issue Cite
Research Paper
GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade
Mingo Ming-Ho Yung1, Hermit Wai-Man Tang1, Patty Chun-Hui Cai1, Thomas Ho-Yin Leung1, Siew-Fei Ngu1, Karen Kar-Loen Chan1, Dakang Xu2, Huijuan Yang3,4, Hextan Yuen-Sheung Ngan1, David Wai Chan1 ![]()
1. Department of Obstetrics and Gynaecology, LKS Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, P. R. China.
2. Faculty of Medical Laboratory Science, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200030, People's Republic of China.
3. Department of Gynecological Oncology, Fudan University Shanghai Cancer Center, Fudan University, Shanghai, 200032, People's Republic of China.
4. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200032, People's Republic of China.
Received 2017-8-25; Accepted 2017-11-20; Published 2018-2-2
Citation:
Yung MMH, Tang HWM, Cai PCH, Leung THY, Ngu SF, Chan KKL, Xu D, Yang H, Ngan HYS, Chan DW. GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics 2018; 8(5):1270-1285. doi:10.7150/thno.22536. https://www.thno.org/v08p1270.htm
Other stylesAbstract

Intraperitoneal metastasis is a common occurrence and is usually involved in the poor prognosis of ovarian cancer. Its specific metastatic pattern implies that certain indispensable microenvironmental factors secreted in the peritoneal cavity can direct metastatic ovarian cancer cells to permissive niches for secondary lesion formation. However, the underlying molecular mechanisms are ill defined. Herein, we report that GRO-α and IL-8 are predominately upregulated in culture media derived from either normal or cancerous omenta and are associated with increased ovarian cancer aggressiveness.
Methods: OCM was established from culture medium of fresh human omental tissues. Primary and metastatic ovarian cancer cell lines were generated from human tumor tissues and verified by specific antibodies. The functional roles of GRO-α, IL-8, and their specific receptor CXCR2 were examined by neutralizing antibodies, shRNA gene knockdown, CRISPR/Cas9 gene knockout and pharmaceutical CXCR2 inhibitor SB225002. The oncogenic properties of ovarian cancer cells were examined by in vitro and in vivo mouse models.
Results: Both GRO-α and IL-8 can activate TAK1/NFκB signaling via the CXCR2 receptor. Intriguingly, TAK1/NFκB signaling activity was higher in metastatic ovarian cancer cells; this higher activity makes them more susceptible to OCM-induced tumor aggressiveness. Treatment of ovarian cancer cells with GRO-α and IL-8 neutralizing antibodies or ablation of CXCR2 by shRNA gene knockdown, CRISPR/Cas9 gene knockout, or CXCR2 inhibitor SB225002 treatment significantly attenuated TAK1/NFκB signaling and decreased in vitro and in vivo oncogenic and metastatic potential, suggesting CXCR2 plays a key role in the GRO-α and IL-8-governed metastatic spreading of ovarian cancer cells in the intraperitoneal cavity.
Conclusion: This study highlights the significance of GRO-α and IL-8 as the key chemokines in the peritoneal tumor microenvironment and suggests the utility of targeting their receptor CXCR2 as a potential target-based therapy for peritoneal metastases of ovarian cancer.
Keywords: chemokines, IL-8, GRO-α, CXCR2, NFκB signaling, ovarian cancer
Introduction
Unlike other human cancers, transcoelomic metastasis is the most common route of cancer spread in ovarian cancer [1]. Clinical evidence indicates that ovarian cancer with peritoneal metastases is usually accompanied with poor prognosis [2, 3]. Within the peritoneal cavity, omentum is one of the most preferential tissues for metastatic ovarian cancer seeding. However, this tissue is rarely studied, and the underlying molecular mechanism for the interaction between the omental microenvironment and metastatic ovarian cancer cells in tumor colonization remains obscure. Emerging evidence has suggested that chemokines play an active role in reprogramming the tumor microenvironment and promoting cancer cell dissemination. For example, omental adipocytes secrete numerous growth factors and hormones, such as IL-6, IL-8, MCP-1, TIMP-1 and adiponectin, to promote tumor growth and cancer cell aggressiveness in various human cancers [4, 5], suggesting the importance of chemokine-induced oncogenic alteration of the tumor microenvironment in promoting omental metastasis.
In addition to the effects of the tumor microenvironment, malignant cells intrinsically alter their behaviors and qualities to adapt to microenvironmental reprogramming during tumor development and progression. It is hypothesized that metastatic cancer cells escaping from primary tumor sites acquire genetic and epigenetic alterations to resist stresses and traverse boundaries [6, 7]. This hypothesis implies that metastatic cancer cells are considered more aggressive than primary tumor cells due to their gain of tumorigenic capacities [6, 7]. Numerous studies have documented that nuclear factor-kappa B (NFκB) deregulation is favorably associated with cancer progression via its promotion of cancer cell proliferation and metastatic capacity [8, 9]. Indeed, our previous report showed that aberrant phosphorylation of transforming growth factor (TGF)-β-activating kinase 1 (TAK1) at serine 412 is critically involved in the activation of NFκB signaling and is associated with ovarian cancer cell aggressiveness [10]. These findings suggest that TAK1/NFκB signaling plays a vital role in ovarian cancer metastasis.
In this study, we established omentum conditioned medium (OCM) as a model to study the interplay between metastatic ovarian cancer cells and omental tumor microenvironment during cancer cell colonization. We identified interleukin 8 (IL-8/CXCL8) and growth related oncogene-alpha (GRO-α/CXCL1) are the key chemokines in normal or cancerous OCMs stimulate the metastatic properties of ovarian cancer cells through activation of TAK1/NFκB signaling cascade via CXCR2. Our research provides a scientific basis for the importance of chemokines in the tumor microenvironment for the metastatic progression of ovarian cancer. Their related receptors and signaling may be potential therapeutic targets for impeding ovarian cancer metastasis.
Materials and Methods
Cell lines and human clinical samples
The human ovarian high-grade serous cancer cell line OVCA433 (advanced serous papillary cystadenocarcinoma) was kindly provided by Professor G. S. W. Tsao, Department of Anatomy, The University of Hong Kong, and ES-2 (verified as high grade serous subtype [11]). HEK293 cells were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). The human ovarian adenocarcinoma cell lines OVSAHO (JCRB1046) and OVKATE (JCRB1044) (Stage 3c adenocarcinoma) were purchased from Japanese Collection of Research Bioresources Cell Bank (JCRB Cell Bank, Japan). Another two high grade ovarian adenocarcinoma cell lines, SKOV3 (ATCC) and A2780cp (cisplatin cell line derived from A2780), were kindly provided by Prof. Benjamin Tsang, Department of Obstetrics and Gynaecology, University of Ottawa. The immortalized human ovarian surface epithelium (HOSE) cell line HOSE 96-9-18 was kindly provided by Prof. George Tsao, Department of Anatomy, University of Hong Kong. The cells were maintained in Dulbecco's modified Eagle's medium or MCDB 105 and medium 199 (1:1) (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% (v/v) fetal bovine serum (Invitrogen, Gibco, Gaithersburg, MD, USA) and 100 U/mL penicillin/streptomycin (Invitrogen Life Technologies, Carlsbad, CA, USA) in an incubator at 37°C with a humidified atmosphere of 5% CO2 and 95% air. All cell lines were authenticated by in-house Short Tandem Repeat (STR) DNA profiling and mycoplasma tests. Primary ovarian surface epithelium (OSE) cancer cells were prepared from fresh tumors collected from Queen Mary Hospital, The University of Hong Kong. The origins of all primary OSE cells were verified by immunofluorescence analysis using pan-cytokeratin (pan-CK) (1:1000; Dako, Denmark), cytokeratin 7 (CK7) (1:1000; Dako), and vimentin (Vim) (1:1000; Dako) staining as well as by flow cytometric analysis (BD FACSAria SORP) using APC-CD45 (BioLegend, San Diego, CA, USA) or CA125 (Abcam, UK) antibodies to analyze the percentages of CA125+CD45- cells in primary OSE cancer cells. All primary OSE cancer cells were maintained in MCDB 105 and medium 199 (1:1). The OCM was prepared using fresh human omental tissues collected from surgeries performed at Queen Mary Hospital, University of Hong Kong. After each sample was washed and dissected into small pieces, approximately 3 g of each omental sample was incubated in 45 mL MCDB 105 and medium 199 (1:1) for 24 h. After repeated filtering and centrifugation, the supernatant was stored at 4°C for future use.
Reagents
The TAK1 inhibitor, (5z)-7-oxozeaenol, was purchased from Sigma (Sigma Chemical Co., St. Louis, MO, USA). The CXCR2 chemokine receptor antagonist SB225002 was purchased from Selleck (Selleck Chemical, Houston, USA), and all recombinant human chemokines (CXCL7, GRO-α and IL-8) were obtained from Novus (NOVUS, Littleton, CO).
RNA extraction and real-time quantitative reverse transcription PCR (QPCR) analysis
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized using a reverse transcription reagent kit (Applied Biosystems, Foster City). The expression levels of GRO-α and IL-8 were evaluated by QPCR in an ABI PRISM 7500 system (Applied Biosystems) using Taqman gene expression assays: human GRO-α (Assay ID: Hs00236937_m1) and human IL-8 (Assay ID: APTZ9ZR). Human GAPDH (Assay ID: Hs02786624_g1) was used as an internal control.
Western blot analysis
Proteins lysates were isolated from cells using cell lysis buffer (Cell Signaling Technology) containing protease inhibitor cocktail (Roche) and phenylmethylsulfonyl fluoride (Sigma Chemical Co.). Samples were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blotted with 5% skimmed milk and subsequently probed overnight at 4°C with primary antibodies specific for anti-p-TAK1 (Ser412), anti-TAK1, anti-p-IKKα/β (Ser180/181), anti-IKKα/β, anti-p-IκBα, anti-IκBα (Cell Signaling, Beverly, MA, USA) and anti-β-actin (Sigma-Aldrich, St. Louis, MO, USA) and then incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibody (Amersham, Uppsala, Sweden). Immunodetection was performed with enhanced chemiluminescence reagent solution (AmershamTM ECLTM) and visualized by X-ray film.
Immunohistochemical (IHC) staining
Tumors and omental tissues were washed with PBS and cut into 1.5 cm × 1.5 cm × 5 mm sections, followed by overnight fixation in a 4% paraformaldehyde fixative solution (0.4 g paraformaldehyde and 5×10-6 M NaOH in 10 mL PBS) at 4°C. IHC was carried out using anti-GRO-α and anti-IL-8 (Abcam) antibodies, and all IHC slides were analyzed by the Aperio ScanScope System (Department of Pathology, University of Hong Kong).
Luciferase reporter assay
NFκB luciferase reporter assay was performed using a NFκB luciferase reporter, Renilla luciferase plasmids and the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA) as described previously [10].
Cell proliferation and focus formation assays
Cell proliferation was examined by XTT cell proliferation kit (Roche, Basel, Switzerland). For focus formation assays, approximately 1000 cells were cultured in each well of a six-well plate and incubated with different treatments. After incubation at 37°C in an incubator with a humidified atmosphere of 5% CO2 and 95% air for two weeks, colonies were stained with crystal violet and counted.
Soft agar assay
Soft agar assays were used to determine the anchorage-independent growth ability of cancer cells. Approximately 2500 cancer cells were embedded in 0.2% agarose-medium and laid on the top of a supporting layer of 1% agarose-medium (without FBS) in each well of a six-well plate. 1 mL culture medium was added to each well to avoid dryness. After three to four weeks, viable colonies containing more than 20 cells were counted and photographed under a microscope (Nikon ECLIPSE Ti-S) with 4X and 200X magnification.
Matrigel cell migration and invasion assays
According to the manufacturer's (Corning, NY, USA) instructions, a cell suspension containing 5 × 104 cells in serum-free medium was added to each insert. The medium (500 μL) containing 1% fetal bovine serum OCM or chemokines was added to the lower chamber as a chemoattractant. After incubation, the migrated/invaded cells were stained and counted by microscopy.
Ex vivo colonization assay
The protocol for the ex vivo culture of the omentum was modified from Khan SM et al. In brief, omenta were resected from mice and washed with PBS. Each specimen was divided into two pieces and cultured in DMEM/F12 with 10% FBS. Approximately 1 × 105 stable GFP-labeled CXCR2 knockdown cells or scramble control cells were seeded on the respective cultured omentum and incubated in an incubator at 37°C with a humidified atmosphere of 5% CO2 and 95% air for 2 weeks to allow cancer cell colonization. The medium was changed every 4 days, and the number of colonies were counted under a ZOETM Fluorescence Cell Imager (Bio-Rad).
In vivo tumorigenicity assay
To study the effect of CXCR2 on tumor growth in vivo, 2 × 106 ovarian cancer CXCR2-/- SKOV3 or parental control cells were implanted intraperitoneally (i.p.) into four-week-old female CB17/Icr-Prkdcscid (SCID) mice in groups of five. To study the effect of the CXCR2 inhibitor SB225002 on tumor growth in vivo, SKOV3 parental cells (2 × 106 per mouse) were intraperitoneally injected into four-week-old SCID mice. When palpable tumors formed at week two, the experimental group was treated once every 2 days by injection of SB225002 (2.5 mg/kg) in 200 μL carrier solution (10% DMSO). For the control group, only PBS was i.p. injected. After approximately 45 days, all mice were sacrificed, and the distribution and weight of tumor nodules were evaluated. The entire animal study was performed according to the guidelines approved by The Committee on the Use of Live Animals in Teaching and Research of The University of Hong Kong (CULATR number: 2560-11).
Data analysis
All experiments were repeated at least three independent times, unless otherwise stated. Values are represented as the mean ± SEM, and a two-tailed Student's t-test was used for comparisons. Fisher's exact test (for parametric data) and the Mann-Whitney test (for non-parametric data) were used, and P ≤ 0.05 was considered statistically significant.
Results
Metastatic ovarian cancer cells exhibit higher oncogenic induction in OCM
The omentum is considered a preferential site of ovarian cancer metastasis [5, 12, 13], and thus, it was of interest to determine whether the omental microenvironment specifically modulated ovarian tumor cells to promote metastatic cancer cell dissemination. To investigate the role of the tumor microenvironment in the aggressiveness of ovarian cancer cells, a good tumor cell model is needed that closely mimics clinical tumor development. Considering the limitations of commercial ovarian cancer cell lines, primary ovarian cancer cells obtained from the omentum or other intraperitoneal organs (metastatic) and ovaries (primary cancer cells) were used for this study. To this end, four primary cell lines were established from two patients' tumors: 3bU2S8 and 3bL8O17 were derived from the primary ovary tumor tissue and omental tumor tissue, respectively, of a 50-year-old patient who was diagnosed with ovarian clear cell carcinoma with the International Federation of Gynecology and Obstetrics (FIGO) Stage 4, Grade 3; Pod7 and Om8 were derived from tumor tissue of the pouch of Douglas (POD) and the cancerous omental tissue, respectively, of a 52-year-old patient who was diagnosed with high-grade serous ovarian carcinoma with FIGO Stage 3b (Fig. 1A). These cells were verified as epithelial ovarian cancer (EOC) cells by immunofluorescent staining using pan cytokeratin (pan-CK), cytokeratin 7 (CK7) and vimentin (Vim) cell markers (Fig. 1B) and flow cytometric analysis for CA125 expression (data not shown).
To mimic the effects of the omental microenvironment, OCM was established according to our laboratory protocol. To investigate the differential responses of metastatic and primary tumor ovarian cancer cells, both abovementioned primary ovarian cancer cell lines, the human ovarian cancer cell lines OVCA433, ES-2, OVKATE and OVSAHO, and the immortalized ovarian epithelial cell line HOSE96 were used for OCM treatment. The results showed that OCM could enhance the proliferation of all the ovarian cancer cell lines. Pod7, Om8, OVCA433, ES-2, OVKATE and OVSAHO cells increased from ~20% to 50% compared with their controls, whereas there was no significant effect of OCM observed in HOSE96 cells (Fig. 1C). OCM also promoted cell growth in the primary clear cell carcinoma cell line 3bU2S8 but not in its corresponding omental metastatic tumor cell line 3bL8O17 (Fig. 1C). However, soft agar assays demonstrated that OCM treatment enhanced not only the size but also the number of colonies in 3bU2S8, 3bL8O17 and ES-2 cells by nearly 39-fold (Fig. 1D). Of note, the omental metastatic tumor cell line 3bL8O17 displayed an ~2-fold higher capacity for colony formation than its corresponding primary tumor cell line 3bU2S8 (Fig. 1D), indicating that OCM can promote the cell proliferation and anchorage independence of metastatic ovarian cancer cells. Moreover, transwell cell migration assays showed that the ~90% increase in cell migration in 3bU2S8, 3bL8O17 and ES-2 cells compared with untreated controls was caused by OCM treatment and that the motility of 3bL8O17 cells was even 1.5-fold faster than 3bU2S8 cells cultured in OCM (Fig. 1E). Transwell invasion assays indicated that the number of cells that invaded through the Matrigel was increased from ~40% to 180% in 3bU2S8, 3bL8O17 and ES-2 cells treated with OCM and, more importantly, that 3bL8O17 cells showed a 1.5-fold higher invasion rate than 3bU2S8 cells upon OCM treatment (Fig. 1F), implying that the OCM augmented the cell growth, anchorage-independent cell growth, and cell migration and invasion capacities of ovarian cancer cells, particularly in highly metastatic ovarian cancer cells.
Figure 1
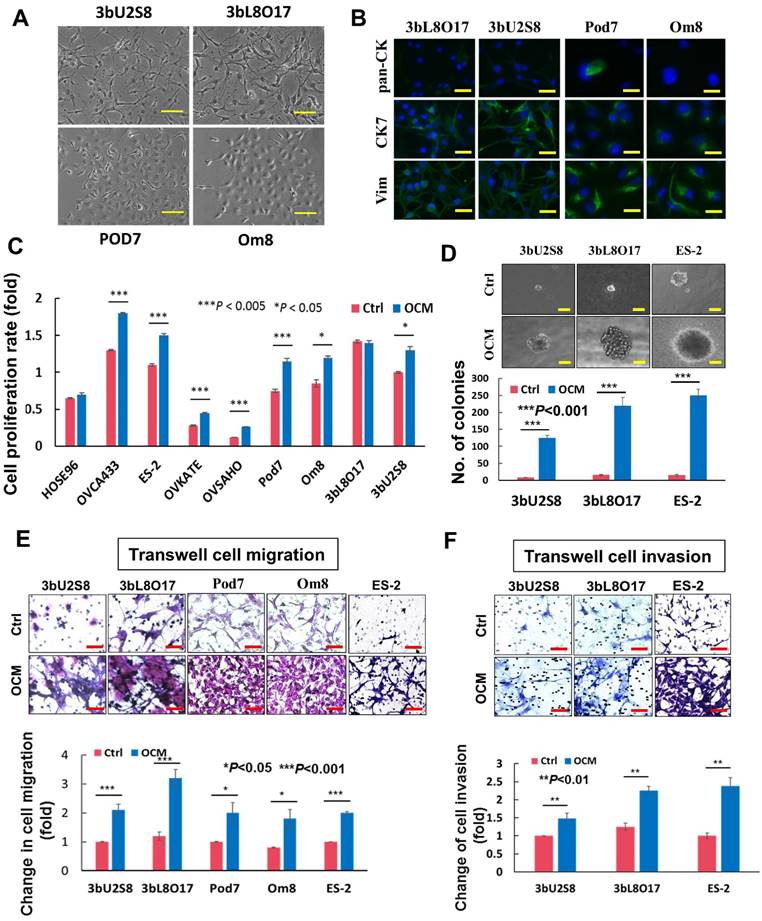
OCM exacerbates the aggressiveness of ovarian cancer cells. (A) Upper, photomicrographs of 3bU2S8 and 3bL8O17 cells respectively isolated from primary ovarian tumor tissues and omental tumor tissues of a 50-year-old patient with ovarian clear cell carcinoma. Lower, photomicrographs of Pod7 and Om8 cells correspondingly isolated from POD and cancerous omentum tissue of a 52-year-old patient with high-grade serous ovarian carcinoma. Representative images are presented from the above experiments at a magnification of 100×. Scale bar = 50 μm. (B) Immunofluorescent verification of the primary ovarian cancer cells as epithelial origin. 3bU2S8, 3bL8O17, Pod7 and Om8 cells seeded on glass cover slips were methanol-fixed and incubated with primary mouse monoclonal antibodies including anti-pan-CK (pan cytokeratin), anti-CK7 (cytokeratin 7) and anti-Vim (vimentin), followed by immunofluorescent-staining with Alexa Fluor 488-conjugated secondary rabbit anti-mouse antibody (green). Nuclei of cells were counterstained with DAPI (blue). Images were taken at a magnification of 100×. Scale bar = 25 μm. (C) XTT cell proliferation assay demonstrated that OCM promoted cell proliferation in all ovarian cancer cell lines employed including OVCA433, ES-2, OVKATE and OVSAHO and most primary tumor cells such as Pod7, Om8 and 3bU2S8 (*P < 0.05 and ***P < 0.005). (D) Soft-agar assay revealed that OCM enhanced anchorage independent growth ability in primary and omental metastatic tumor cells as well as representative ovarian cancer cell line ES-2 (***P < 0.001). Images were taken at a magnification of 100×. Scale bar = 7.5 μm. (E) Transwell migration assay showed that primary cultured tumor cells 3bU2S8, 3bL8O17, Pod7 and Om8 and cancer cell line ES-2 with OCM treatment (18 h) resulted in a significant increase in the number of cells penetrating through the membrane when compared with their respective controls with 1% FBS normal medium (*P < 0.05 and ***P < 0.001). Scale bar = 50 μm. (F) 3bU2S8, 3bL8O17 and ES-2 cells treated with OCM (48 h) displayed more invaded cells through the Matrigel as compared with their respective controls with 1% FBS normal medium (**P < 0.01). Numbers of migrated or invaded cells in three randomly chosen fields were counted for three independent experiments and the normalized numerical data are presented in bar charts with error bars. Scale bar = 50 μm.
OCM activates the TAK1/NFκB signal transduction arm in ovarian cancer cells
Our recent study found that distant metastases samples of ovarian cancer patients exhibit a higher level of transforming growth factor (TGF)-β-activating kinase 1 (TAK1) compared to the primary ovarian tumors [10]. Importantly, TAK1 is a serine/threonine kinase, and the increase in p-TAK1 (Ser412) could profoundly activate NFκB activity and increase its associated tumor aggressiveness [10, 14]. Consistent with our previous findings, western blot analysis revealed that the metastatic ovarian cancer cell line 3bL8O17 had stronger expression of TAK1 than 3bU2S8 cells (Fig. 2A). Further study of our cell lines and clinical samples showed that the levels of both TAK1 and p-TAK1 (Ser412) were significantly higher in metastatic cell lines such as Pod7 (metastatic cells from the POD, high-grade serous carcinoma, Stage 2b), Om8 (metastatic cancer cells from the omentum removed from the same patient as Pod7), and cancerous omental tissue (HGSC (O), high-grade serous carcinoma, Stage 3c and Grade 3) than those in the primary tumor tissue (HGSC (P), high-grade serous carcinoma, Stage 3c and Grade 3), the primary ovarian cancer cell line 3bU2S8 and another primary ovarian tumor tissue (EC, endometrioid adenocarcinoma, Stage 1c and Grade 2) (Fig. 2B). These findings indicate that the metastatic ovarian cancer cells intrinsically alter tumor progression to support their aggressiveness.
Figure 2
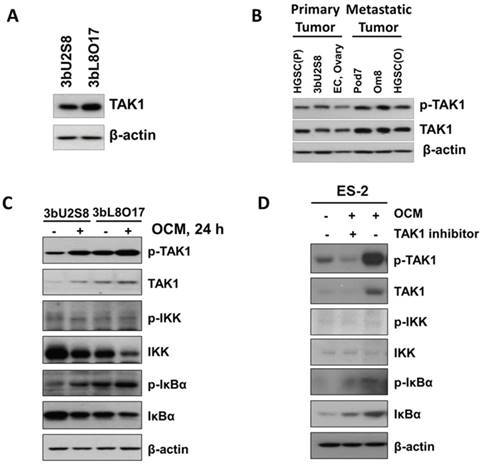
OCM induces TAK1/NFκB activation in ovarian cancer cells. (A) Higher expression level of TAK1 was observed in omental metastasized ovarian cancer cells, such as 3bL8O17, as compared with its primary ovarian cancer counterpart cells, such as 3bU2S8. (B) The expression levels of p-TAK1 (Ser412) and TAK1 in distant metastatic specimens such as omentum (Om8 and HGSC(O) cells) and POD (Pod7 cells) were relatively stronger than those in primary ovarian tumor tissues (3bU2S8, EC, and HGSC(P)). (C) Western blot analysis was performed to reveal the differential initiation of TAK1/NFκB signalling between primary (3bU2S8 cells) and metastatic (3bL8O17 cells) ovarian tumor cells in response to OCM treatment (24 h). (D) A TAK-1 inhibitor blocked OCM-mediated phosphorylation on TAK1/NFκB signalling in ES-2 cells. Treatment with (5z)-7-oxozeaenol (2.5 μM) reduced Ser412 phosphorylation and NFκB activity induced by OCM (24 h). Representative cropped blots are presented.
Next, we determined whether the activation of the TAK1/NFκB pathway in primary ovarian cancer cells such as 3bU2S8 and 3bL8O17 was induced by OCM. The results revealed that OCM treatment yielded a marked increase in the phosphorylation of TAK1 at Ser412 and the NFκB target IκBα at Ser32 compared with their respective untreated controls. Such OCM-mediated TAK1 activation was considerably more apparent in 3bL8O17 cells, as evidenced by higher expression levels of p-TAK1 (Ser412) and p-IκBα (Ser32) when compared with those in 3bU2S8 cells (Fig. 2C), suggesting that TAK1/NFκB activity was differentially regulated in metastatic cells and primary tumor cells upon OCM treatment. To further demonstrate that OCM enhances the growth of ovarian cancer cells through initiation of TAK1/NFκB activity, the TAK1 inhibitor (5z)-7-oxozeaenol was applied to suppress TAK1 activity. After treatment with (5z)-7-oxozeaenol (2.5 μM), not only was the expression of p-TAK1 (Ser412) markedly attenuated but also OCM-induced NFκB activity was significantly counteracted in ES-2 cells compared with the positive control treated with only OCM (Fig. 2D). These findings support the idea that increased TAK1 facilities the activation of the NFκB pathway in ovarian cancer, which in turn promotes the metastatic progression of ovarian cancer cells in OCM.
GRO-α and IL-8 are the predominant chemokines in OCM
Emerging evidence has suggested that chemokines are the dominant factors in the tumor microenvironment that promote metastatic progression [15-17]. To this end, to elucidate the molecular mechanisms driving ovarian cancer aggressiveness, we analyzed OCM established from either a normal (omental tissue obtained from a normal female) or cancerous omentum (omental tissue obtained from an ovarian cancer patient) using a human cytokine array. We found that the cancerous OCM had more chemokines expressed than the normal OCM (Fig. 3A). Among these chemokines, GRO-α, IL-8 and CXCL7 were commonly found in both types of OCM (Fig. 3A), while chemerin, IL-16 and CXCL4 solely existed in cancerous OCM (Fig. 3A). As OCM activated cancer cell metastasis by mediating NFκB activity, we therefore studied the interaction network of chemokines and NFκB signaling by bioinformatics online software at http://pathwaynet.princeton.edu (Fig. 3B). The pathway analysis identified ELR+ CXC chemokines GRO-α (CXCL1) and IL-8 as the only cytokines that exhibited a strong relationship with NFκB regulation (Fig. 3B). On the other hand, CXCL7, chemerin and other chemokines, could neither be found in the bioinformatics analysis nor activate TAK1/NFκB signaling when their recombinant proteins were used in ovarian cancer cells (Fig. S1). Therefore, we concentrated our attention on the expression levels and functional roles of IL-8 and GRO-α.
Given that both IL-8 and GRO-α are the predominant players in OCM that are responsible for activating TAK1/NFκB signaling and ovarian cancer aggressiveness, it is important to quantify their levels in OCM obtained from patients. Sixteen normal or cancerous omental samples were used to prepare OCM. Using human CXCL1/GRO-α and IL-8 ELISA assays, the average concentrations of GRO-α and IL-8 were 9.26 ng/mL and 75 ng/mL respectively in OCM (Fig. 3C). After normalizing the dry weights of the omental tissues, the concentrations of GRO-α in normal and cancerous OCM were calculated as 0.16 ± 0.06 μg/g and 0.24 ± 0.1 μg/g respectively (Fig. 3D). There were no significant changes in the concentration of GRO-α (P = 0.56) between normal and cancerous OCM (Fig. 3D). On the other hand, the concentrations of IL-8 were 0.64 ± 0.16 μg/g and 2.58 ± 0.66 μg/g in normal and cancerous OCM, respectively (Fig. 3D). IL-8 was at a 3-fold higher concentration in cancerous OCM when compared with that in normal OCM (Fig. 3D). These data indicate that both GRO-α and IL-8 are abundantly released from omental tissues, whereas IL-8 might be secreted not only by the omental tissues but also by cancer cells.
To examine which tissue types secrete GRO-α and IL-8, normal omenta (n = 9), cancerous omenta (n = 6) and omentum-metastasized tumors (n = 2) were sectioned for IHC staining for GRO-α and IL-8. Among normal and cancerous omental sections, four tissue types, lymph node, milky spot, blood vessel and adipose tissue, preferentially expressed GRO-α and IL-8 (Fig. 3E). These chemokines were also found in tumor samples, and intriguingly, the expression of IL-8 in omentum-metastasized tumor cells was extraordinarily high (Fig. 3E), implying that the upregulated IL-8 in cancerous OCM potentially comes from ovarian metastatic cancer cells. Indeed, QPCR analysis revealed that co-treatment of IL-8 could induce a positive feedback loop to further elevate IL-8 but not GRO-α dose dependently in ES-2 and SKOV3 cells (Fig. 3F and Fig. S2). In contrast, blocking CXCR2/TAK1/NFκB signaling by CXCR2 inhibitor SB225002 or TAK1 inhibitor (5z)-7-oxozeaenol could impair IL-8 positive feedback response (Fig. 3F and Fig. S2). These data support the notion that a positive feedback loop of IL-8 secretion is bridging between tumor cells and cancerous omental tissues.
GRO-α and IL-8 are capable of enhancing cancer cell aggressiveness by activation of the TAK/NFκB cascade
We have shown that OCM can enhance the oncogenic properties of ovarian cancer cells. Because GRO-α and IL-8 are the predominant chemokines in OCM, their functional roles in ovarian cancer cells were further examined. The results of XTT cell proliferation assays showed that GRO-α (100 ng/mL) and IL-8 (25 ng/mL) resulted in the highest proliferation rate in OVCA433 cells on day 4 (Fig. 4A), while GRO-α (40 ng/mL) and IL-8 (50 ng/mL) induced the same cell proliferation rate in A2780cp cells (Fig. 4A). This finding indicates that GRO-α and IL-8 function similar to OCM in activating the growth of human ovarian cancer cells, but there is a differential response of ovarian cancer cells to GRO-α and IL-8 stimulation. In addition, Transwell cell migration assays revealed that there was an ~20% to 50% increase in ES-2 cell migration caused by IL-8 (50 ng/mL) or GRO-α (80 ng/mL) treatment compared with the untreated control (Fig. 4B). Furthermore, transwell invasion assays showed that the number of cells that invaded through the Matrigel also increased by at least 10% to 30% in ES-2 cells treated with IL-8 (50 ng/mL) or GRO-α (80 ng/mL). To further confirm the functional importance of IL-8 and GRO-α in OCM, transwell migration and invasion assays were additionally conducted in ES-2 cells through co-treatment of anti-GRO-α, anti-IL-8 or anti-CXCR2 antibody (2 μg/mL) with OCM. Compared with the untreated control, OCM could profoundly enhance the migration and invasion capabilities of ES-2 cells by 3.8-fold and 1.8-fold respectively. Conversely, the OCM-promoted cell migration was reduced to ~2.5-fold when ES-2 cells were co-treated with anti-GRO-α, anti-IL-8 or anti-CXCR2 antibody (Fig. 4C). Similarly, the OCM-promoted cell invasion in ES-2 cells could be inhibited by anti-CXCR2 and anti-GRO-α antibodies, but not by anti-IL-8 (Fig. 4D), suggesting that both IL-8 and GRO-α function separately in OCM to promote the tumorigenicity of ovarian cancer.
Figure 3
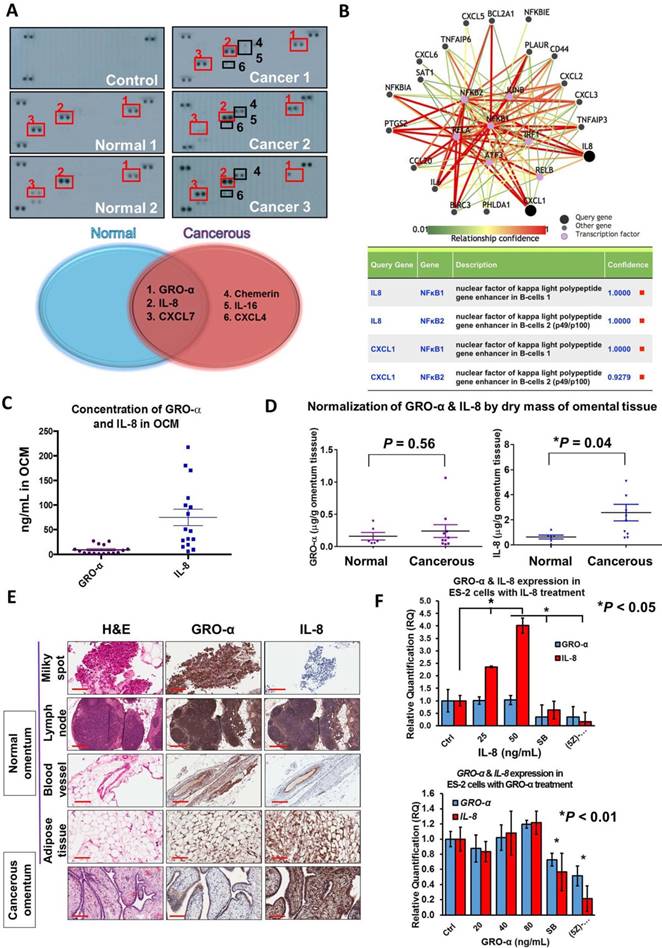
CXC chemokines GRO-α (CXCL1) and IL-8 (CXCL8) are frequently upregulated in OCM. (A) Human cytokine array analysis was carried out on normal OCMs (n = 2) and cancerous OCMs (n = 5). The control membrane represented the 1% FBS medium. Among all these chemokines examined, GRO-α, IL-8 and CXCL7 were highly expressed in both normal and cancerous OCMs, whereas chemerin, IL-16 and CXCL4 were only released in cancerous OCMs. (B) Pathway analysis of transcription factors that potentially interacted with GRO-α (CXCL1) and IL-8 by PathwayNet analysis program. The candidate transcription factors with the highest confidence including NFκB are shown in the diagram as purple coloured dots. (C) The concentrations of GRO-α and IL-8 in normal OCMs (n = 6) and cancerous OCMs (n = 10) were respectively measured by Human CXCL1/GRO-α DuoSet ELISA and Human IL-8 ELISA. The ranges and mean values of concentrations were calculated and plotted. (D) Expression levels of GRO-α and IL-8 were normalized by the corresponding dry mass of omentum tissue employed for preparing each OCM sample and grouped into normal or cancerous OCM. (E) IHC analysis was conducted against GRO-α and IL-8 (1: 500) in 15 omental specimens including milky spots, lymph nodes, blood vessels and adipose tissues as well as 2 omental metastatic ovarian tumors. Higher expression level of IL-8 was observed in ovarian tumor cells metastasized to omentum and representative tumor sample (Ov75, serous adenocarcinoma, Stage 4 and Grade 3 with omental metastasis) is presented. (F) QPCR analysis revealed that treatment with IL-8 (25 and 50 ng/mL) for 24 h induced a positive feedback loop in ES-2 cells in a dose dependent manner, whereas GRO-α (20, 40 and 80 ng/mL) treatment was unlikely to trigger a similar phenomenon. In addition, the IL-8-induced positive feedback was apparently retarded in ES-2 cells when co-treated with IL-8 (50 ng/mL) and SB225002 (5 μM) or (5z)-7-oxozeaenol (2.5 μM) for 24 h. Scale bar = 200 μm.
Figure 4
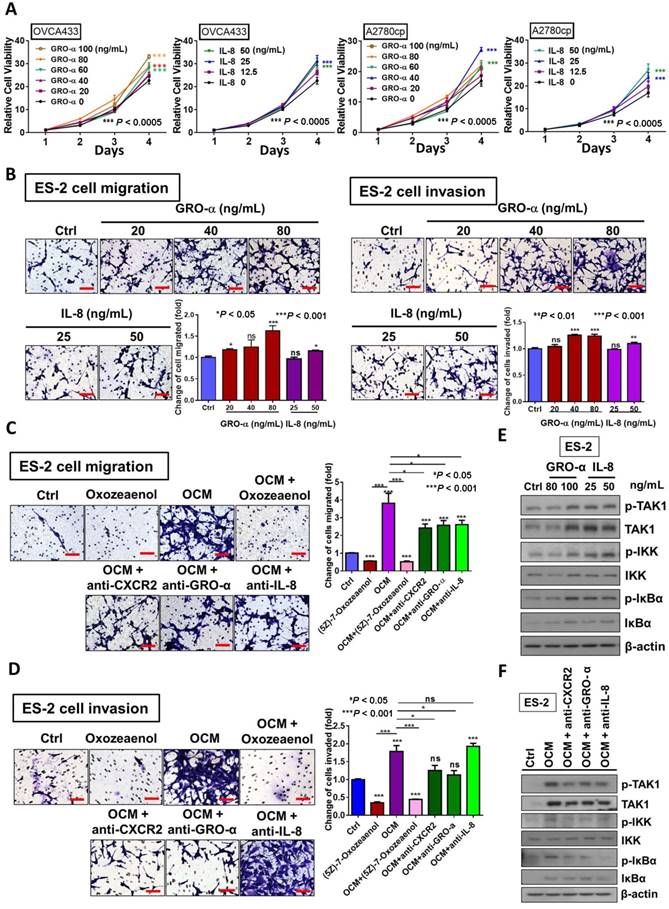
GRO-α and IL-8 are the decisive chemokines in exaggerating ovarian cancer cell aggressiveness via activation of TAK1/NFκB signaling. (A) XTT cell proliferation assay demonstrated that GRO-α and IL-8 promoted cell growth in OVCA433 and A2780cp cells as compared with the respective controls. Additionally, the growth stimulatory effect was most obvious in OVCA433 cells (*** P < 0.0005) treated with GRO-α (100 ng/mL) and IL-8 (25 ng/mL) over a time course of 4-days treatment, whereas GRO-α (40 ng/mL) and IL-8 (50 ng/mL) were required to produce a similar effect in A2780cp cells (*** P < 0.0005). (B) Transwell migration assay showed that ES-2 cells treated with IL-8 (50 ng/mL) and GRO-α (80 ng/mL) for 18 h exhibited a significant upsurge in the number of cells penetrating through the membrane as compared with untreated controls (*P < 0.05 and ***P < 0.001). In addition, ES-2 cells treated with same conditions of IL-8 and GRO-α showed more invaded cells through the Matrigel as compared with the control (**P < 0.01 and ***P < 0.001). Scale bar = 50 μm. (C) Transwell migration assay showed that OCM-mediated migration was significantly inhibited in ES-2 cells when co-treated with anti-GRO-α, anti-IL-8, anti-CXCR2 antibodies (2 μg/mL) (*P < 0.05) or (5z)-7-oxozeaenol (2.5 μM) (***P < 0.001) for18 h. Scale bar = 50 μm. (D) ES-2 cells co-treated with anti-GRO-α, anti-CXCR2 antibodies (2 μg/mL) (*P < 0.05) or (5z)-7-oxozeaenol (2.5 μM) (***P < 0.001) showed fewer invaded cells through the Matrigel as compared with the only OCM-treated cells. Numbers of migrated or invaded cells in three randomly chosen fields were counted for three independent experiments and the normalized numerical data are presented in bar charts with error bars. Scale bar = 50 μm. (E) Western blot analysis demonstrated that treatment with either GRO-α (80 and 100 ng/mL) or IL-8 (25 and 50 ng/mL) upregulated the TAK/NFκB activity in ES-2 cells as witnessed by the increase in the phosphorylation of TAK1 and its downstream targets IKKα/β and IκBα. (F) Co-treatment of antibodies (2 μg/mL) of CXCR2, GRO-α and IL-8 with OCM in ES-2 cells suppressed the promoting effect of OCM on TAK/NFκB signalling activity.
Considering our previous data that activation of the TAK1/NFκB signaling axis augments malignant cell migration and cell invasion, it was of paramount importance to determine whether blocking TAK1 activity could attenuate NFκB-mediated cancer cell migration and invasion in ovarian cancer. As expected, ES-2 cells exhibited a decreased capacity for cell migration and cell invasion upon co-treatment of TAK1 inhibitor (5z)-7-oxozeaenol (2.5 μM) with OCM (P < 0.0001) when compared with OCM only treatment (Fig. 4C and D). Mechanistic studies using a NFκB luciferase reporter assay in HEK293 cells revealed that both IL-8 and GRO-α markedly increased NFκB activity (Fig. S3A). Western blot analysis further confirmed that both chemokines could increase the phosphorylation of TAK1 at Ser412 and of its downstream targets IKKα/β at Ser180/181 and IκBα at Ser32 in ES-2 cells when compared with the untreated control (Fig. 4E). Conversely, the increased expression level of p-TAK1 (Ser412), p-IKKα/β (Ser180/181) and p-IκBα (Ser32) in ES-2 cells was remarkably suppressed by co-treatment of anti-GRO-α, anti-IL-8 or anti-CXCR2 antibody (2 μg/mL) with OCM (Fig. 4F). The inhibition of NFκB activity was also proven by a NFκB luciferase reporter assay in HEK293 cells (Fig. S3B). Taken together, our data indicate that GRO-α and IL-8 may work separately to govern ovarian cancer aggressiveness but that both are key factors in modulating the TAK1/NFκB cascade in ovarian cancer cells.
CXCR2 is required for IL-8, GRO-α and OCM to activate TAK1/NFκB signaling in ovarian cancer
Mounting evidence suggests that CXC chemokine receptor 2 (CXCR2) is the common receptor of IL-8 and GRO-α in human cancers, including in ovarian cancer cells [18, 19]. Aberrant upregulation of CXCR2 has been shown to promote tumor progression in ovarian cancers [19], indicating its importance in ovarian cancer peritoneal metastases. By analyzing the expression of CXCR2 in ovarian cancer using Kaplan-Meier Plotter (ovarian cancer) (http://kmplot.com/analysis/index.php?p=service&cancer=ovar), we found that the high expression of CXCR2 was significantly correlated with lower progression-free survival (PFS) (P = 0.049) and post-progression survival (PPS) (P = 0.015) but not overall survival (OS) (P = 0.21) (Fig. S4) in ovarian cancer. This finding indicates that the upregulated CXCR2 might affect ovarian cancer recurrence and metastasis, leading to poor PFS and PPS. However, there is no significance for GRO-α and IL-8 in patient survival (Fig. S4).
Figure 5
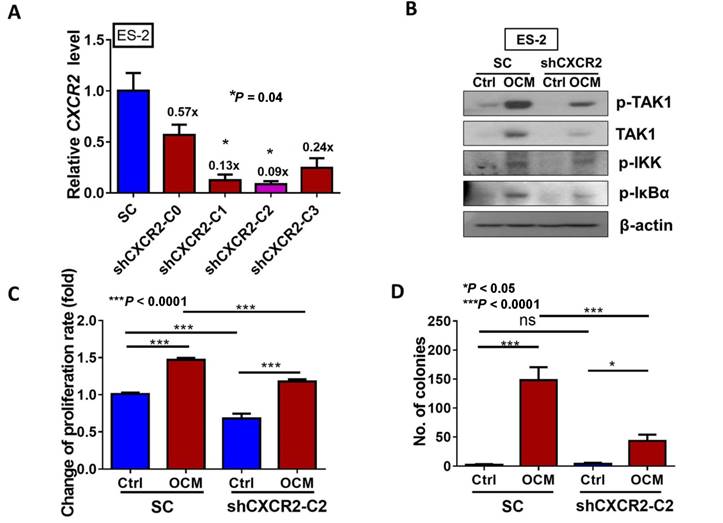
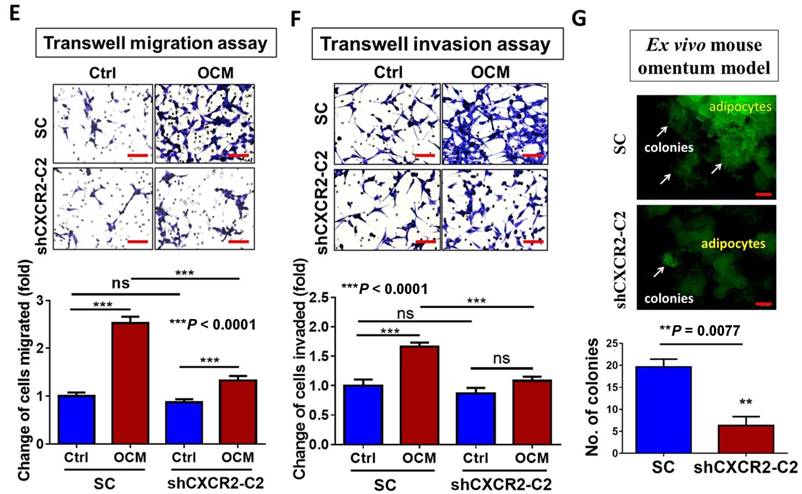
CXCR2 is required for GRO-α, IL-8 and OCM-mediated TAK/NFκB activation in ovarian cancer cells. (A) Quantitative PCR analysis was performed on ES-2 cells transfected with either shCXCR2 plasmid or scrambled control vector (SC). The selected stable clone, shCXCR2-C2, expressed the lowest CXCR2 level (*P = 0.04) among all four clones as compared with the SC. (B) Western blot analysis showed that OCM-induced TAK/NFκB activity in the scrambled control could be significantly suppressed upon depletion of CXCR2. 1% FBS normal medium was used in the control treatment. (C) XTT cell proliferation assay verified that OCM persuaded cell proliferation in ES-2 scrambled control cells (***P < 0.0001) when compared with treatment of 1% FBS normal medium, and such growth promotional effect was significantly attenuated upon knockdown of CXCR2 (***P < 0.0001). (D) Soft-agar assay revealed that OCM enhanced anchorage independent growth ability in ES-2 scrambled control cells (***P < 0.0001) as compared with treatment of 1% FBS normal medium for 20 days, whereas the OCM-initiated anchorage independence was decreased upon knockdown of CXCR2 (*P < 0.05). (E) Transwell migration assay showed that OCM enhanced cell migration in ES-2 scrambled control cells (***P < 0.0001) as compared with treatment of 1% FBS normal medium for 18 h, while the OCM-promoted cell migration capability was lessened upon silencing of CXCR2 (***P < 0.0001). Scale bar = 50 μm. (F) ES-2 scrambled control cells treated with OCM for 24 h displayed more invaded cells through the Matrigel as compared with the respective control with 1% FBS normal medium (***P < 0.0001), and such OCM-stimulated invasive capability was lowered in CXCR2-depleted ES-2 cells (***P < 0.0001). Numbers of migrated or invaded cells in three randomly chosen fields were counted for three independent experiments and the normalized numerical data are presented in bar charts with error bars. Scale bar = 50 μm. (G) An ex vivo mouse omentum model was developed to examine the cancer cell colonization ability upon CXCR2 knockdown. ES-2 shCXCR2-C2 cells and scrambled control cells were labeled with GFP using LV-CMV-RLuc-IRES-GFP lentiviral particles and selected by FASC sorting. The chosen GFP-labelled cells were co-cultured with omenta dissected from female mice in 10% FBS DMEM/F12 medium for 2 weeks. Fluorescent colonies were counted under a microscope and significant reduction in the number of colonies was observed in shCXCR2-C2 (* P= 0.0077) as compared with scrambled control. Representative images are presented from the above experiments at a magnification of 40× and arrows indicate the locations of tumor nodules. Scale bar = 100 μm.
To further investigate the role of CXCR2 in OCM-mediated ovarian cancer cell aggressiveness, CXCR2 was stably knocked down in ES-2 cells by shRNAi. The expression of CXCR2 in four selected stable CXCR2 knockdown clones was verified by QPCR analysis (Fig. 5A). Western blot analysis showed that TAK1/NFκB signaling was reduced in CXCR2 knockdown clones (shCXCR2-C2) compared with the scrambled control, indicating that CXCR2 is involved in modulating the NFκB pathway under normal conditions (Fig. 5B). However, once ovarian cancer cells were cultured in OCM, the levels of p-TAK1, p-IKK and p-IκB were highly upregulated in the scrambled control, while CXCR2-depleted ovarian cancer cells (shCXCR2-C2) only showed no or minor effect(s) on the induction of p-TAK1, p-IKK and p-IκB (Fig. 5B). This finding suggests that CXCR2 is a crucial factor for mediating the roles of IL-8 and GRO-α in the activation of TAK1/NFκB signaling. Functionally, knockdown of CXCR2 significantly inhibited the cell proliferation rate by 33% (P < 0.0001) (Fig. 5C) but did not alter the capacities of ovarian cancer cells in anchorage-independent growth (P = 0.35) (Fig. 5D), migration (P = 0.20) (Fig. 5E), and invasion (P = 0.37) (Fig. 5F). In contrast, with OCM treatment, depletion of CXCR2 in ES-2 cells (shCXCR2-C2) remarkably reduced cell growth by 29% (P < 0.0001) (Fig. 5C), anchorage-independent growth by 70% (P<0.0001) (Fig. 5D), and cell migration by 120% (P < 0.0001) (Fig. 5E) compared with the respective scrambled controls. However, cell invasion capacity was not affected by OCM treatment in the CXCR2 knockdown clone (P = 0.09) (Fig. 5F). Moreover, we employed an ex vivo mouse omentum model to examine the role of CXCR2 in ovarian cancer cell colonization. With GFP-labeled cells, fluorescence microscopy revealed that knockdown of CXCR2 in ES-2 cells significantly reduced both the number and size of colonies by 70% on the surface of murine omenta (P = 0.0077) (Fig. 5G). Collectively, these data suggest that CXCR2 is vital for OCM-mediated cancer cell aggressiveness via the TAK1/NFκB signaling cascade.
Targeting of CXCR2 is capable of blocking IL-8/GRO-α-induced ovarian cancer aggressiveness
It is known that reducing the amount of chemokine receptors is not enough to block their amplification of downstream signaling completely [20]. Hence, to better understand the functional role of CXCR2 in mediating IL-8 and GRO-α-induced activation of TAK1/NFκB signaling, endogenous CXCR2 was completely silenced by CRISPR/Cas9-mediated gene knockout (Fig. 6A). Consistently, knockout of CXCR2 in ovarian cancer cell lines such as SKOV3 cells, strongly reduced the OCM-mediated activation of TAK1 and NFκB signaling activities (Fig. 6B). Similarly, SKOV3 CXCR2-/- cells showed increased suppressive effects on focus formation (55%; P < 0.05), although there was still a slight amount of cell growth (Fig. 6C). In addition, knockout of CXCR2 led to a 50% reduction in the cell migration and invasion capacities of SKOV3 cells cultured with GRO-α/IL-8, 1% FBS control medium or OCM (Fig. 6D, E and Fig. S5). Noticeably, knockout of CXCR2 showed a stronger inhibition effect than CXCR2 knockdown on the cell invasion capacity of SKOV3 cells (Fig. 6E), indicating that CXCR2 plays a more important role in ovarian cancer cell invasiveness. Because our ex vivo study showed that CXCR2 is vital for ovarian cancer cell colonization of the omentum, it was important to examine its effect on in vivo tumor dissemination in severe combined immunodeficiency (SCID) mice. To this end, SKOV3 CXCR2-/- and parental control cells were intraperitoneally injected into SCID mice. After 45 days, the mice were sacrificed. As expected, knockout of CXCR2 caused much less tumor seeding throughout the peritoneal cavity of the mice (Fig. 6F). There were 4-fold fewer tumor nodules developed with a 3.5-fold lower tumor weight in SCID mice grafted with SKOV3 CXCR2-/- cells as compared with SCID mice injected with SKOV3 parental control cells (Fig. 6F). These findings suggest that CXCR2 plays a key role in mediating TAK1/NFκB signaling and thus in its associated ovarian cancer aggressiveness.
Figure 6
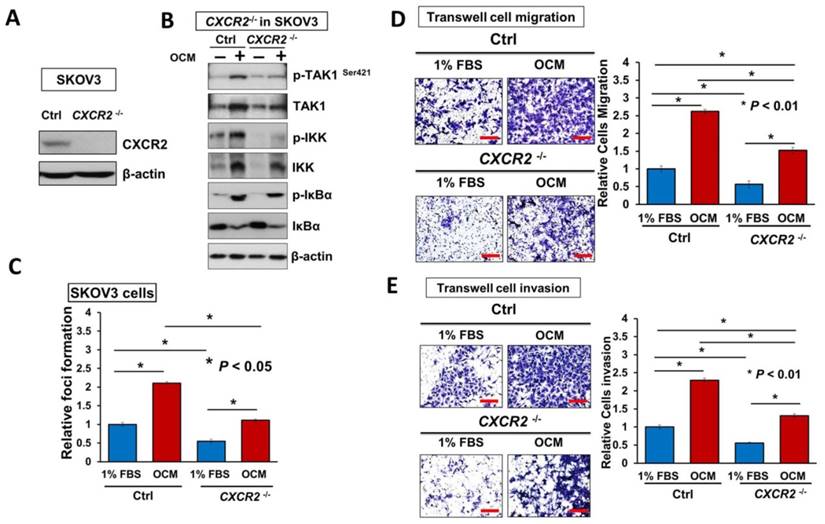
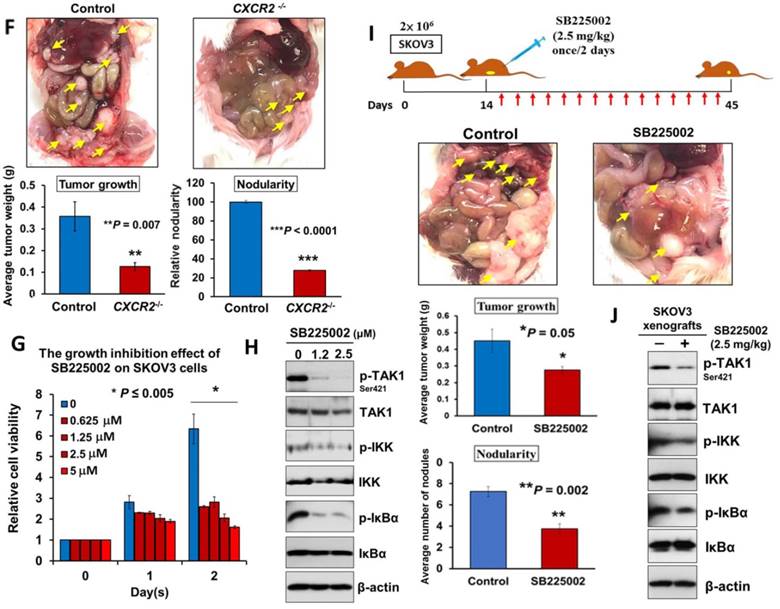
Directing therapeutic approaches against CXCR2 is a potential strategy in hindering GRO-α- and IL-8-modulated ovarian cancer aggressiveness. (A) Western blot analysis was conducted to check the knockout efficiency of CXCR2 on SKOV3 CXCR2-/- cells and it showed CXCR2 was thoroughly depleted by a CRISPR/Cas9 gene-editing approach. (B) Western blot analysis confirmed that SKOV3 CXCR2-/- cells exhibited a much lower activation of TAK1/NFκB signalling upon OCM treatment for 24 h when compared with the parental control. (C) Focus formation assay demonstrating a significant escalation of the number of colonies formed in SKOV3 parental cells treated with OCM, while apparent inhibition of colony formation occurred in SKOV3 CXCR2-/- cells treated with same condition. (D) Transwell migration assay showed that OCM enhanced cell migration in SKOV3 parental cells (*P < 0.01) as compared with treatment of 1% FBS normal medium for 18 h, while the OCM-promoted cell migration capability was lessened upon complete silencing of CXCR2 (*P < 0.01). Scale bar = 50 μm. (E) SKOV3 parental cells treated with OCM for 24 h displayed more invaded cells through the Matrigel as compared with the respective control with 1% FBS normal medium (*P < 0.01) and such OCM-stimulated invasive capability was lowered in SKOV3 CXCR2-/- cells (*P < 0.01). Numbers of migrated or invaded cells in three randomly chosen fields were counted for three independent experiments and the normalized numerical data are presented in bar charts with error bars. Scale bar = 50 μm. (F) Effects of CXCR2 knockout on ovarian tumor dissemination in a xenograft mouse tumor model. Ovarian cancer cell line with stable knockout of CXCR2 and parental control of SKOV3 cells were i.p. inoculated into 4-week-old female SCID mice. Average tumor burden and the relative number of tumor nodules formed by CXCR2 knockout were much lower than in the parental control, which exhibited tumor nodules throughout the peritoneal cavity as photographed on Day 45. The yellow arrows indicate the location of tumor nodules in the peritoneal cavity of the mouse. (G) Concentration-dependent inhibition of cell proliferation in SKOV3 cells (* P ≤ 0.005) treated with SB225002 (0.625, 1.25, 2.5 and 5 μM) for 2 days was revealed using XTT cell proliferation assay. (H) Western blot analysis confirmed that TAK1/NFκB signaling was inhibited dose-dependently in SKOV3 cells upon treatment with SB225002 (1.25 and 2.5 μM) as compared with untreated control. (I) Upper, a schematic presentation of the experimental set-up. Lower, effects of CXCR2 inhibitor on ovarian tumor dissemination in a xenograft mouse tumor model. SKOV3 cells were i.p. inoculated into 4-week-old female SCID mice. On Day 14 post-injection of tumor cells, treatment with SB225002 (2.5 mg/kg) was initiated and carried out every 2 days with a total of 15 injections. Average tumor burden and number of tumor nodules formed by CXCR2 inhibition were much lower than in the PBS-treated control, which exhibited tumor nodules throughout the peritoneal cavity as photographed on Day 45. The yellow arrows indicate the locations of tumor nodules in the peritoneal cavity of the mouse. (J) Western blot analysis verified that TAK1/NFκB signalling was inhibited in xenograft tumor tissues of the SB225002-treated group.
Given that depletion of CXCR2 could effectively impair IL-8 or GRO-α-mediated TAK1/NFκB signaling and ovarian cancer aggressiveness, it was of great importance to investigate any CXCR2 inhibitors that could be used as effective treatments for controlling the peritoneal metastases of ovarian cancer. Therefore, we investigated the efficiency and feasibility of a selective non-peptide CXCR2 antagonist, SB225002, to block the functions of CXCR2 in ovarian cancer cells. By XTT cell proliferation assay, we showed that SB225002 could significantly inhibit the cell viability of SKOV3 cells in a dose-dependent manner (Fig. 6G). Of note, a low dose of SB225002 (0.625 μM) was equivalent to the effects of CXCR2 knockout and resulted in ~58% reduction in the cell viability of SKOV3 cells for 2 days (Fig. 6C and G), indicating that SB225002 is a potent CXCR2 inhibitor. Correspondingly, western blot analysis revealed that SB225002 could strongly suppress TAK1 and NFκB activities, similar to that observed in SKOV3 CXCR2-/- cells (Fig. 6B and H). Moreover, to gain deeper insights into the functional effect of SB225002 on tumor seeding in the peritoneal cavity, SKOV3 cells were intraperitoneally xenografted into SCID mice. When palpable tumors formed on Day 14, SB225002 (2.5 mg/kg) in carrier solution was intraperitoneally administered into tumor-bearing mice once every 2 days with a total of 15 injections before all mice were sacrificed (Fig. 6I). Consistent with the findings regarding SKOV3 CXCR2-/- tumor growth in mice, we found that SB225002 could remarkably inhibit tumor seeding and tumor growth in the peritoneal cavity of SCID mice as evidenced by the ~45% reduction in tumor weight and the ~50% decrease in the number of tumor nodules formed (Fig. 6I). Western blot analysis also confirmed that tumor tissues from the SB225002-treated group exhibited reduction of TAK1 and NFκB signaling activities as compared with the control group (Fig. 6J). However, compared with SKOV3 CXCR2-/-, SB225002 treatment mainly inhibited metastatic spreading to the peritoneum instead of strongly reducing primary tumor growth (Fig. 6I). This different effect might be due to the limited diffusion of SB225002 into the inner portions of primary tumors. Nevertheless, these findings still indicate that SB225002 functions similar to the effects of CXCR2 knockout in ovarian cancer cells. Hence, SB225002 may be applied as an effective remedy to impair tumor dissemination in ovarian cancer.
Discussion
Clinical observations have shown that ovarian cancer accompanied with peritoneal metastases usually indicates a very poor prognosis [21]. Emerging evidence suggests that malignant ascites act as a reservoir, providing plenty of fatty acids, soluble growth factors and chemokines for use in remodeling shed ovarian cancer cells to more aggressive metastatic cancer cells [4, 22, 23]. In this study, we showed that OCM mimics the tumor microenvironment of the human omentum in promoting ovarian cancer cell aggressiveness and that omental-metastasized ovarian cancer cells per se behave more aggressively via activation of TAK1/NFκB signaling. Notably, we are the first to report that GRO-α and IL-8 released from normal and cancerous omenta are the key chemokines that activate TAK1/NFκB signaling and promote ovarian cancer seeding and growth in the peritoneal cavity via CXCR2. Therefore, targeting CXCR2 by genetic or pharmaceutical approaches could be promising therapeutic regimes for impeding ovarian cancer peritoneal metastases.
Previous studies have suggested that TGF-β signaling is initiated in cancer metastasis, indicating that this signaling is one of the potential therapeutic targets for combating ovarian cancer peritoneal metastases [24-26]. TGF-β-activated kinase 1 (TAK1) is a serine/threonine kinase and is frequently upregulated in ovarian cancer cells and clinical samples accompanied with high-grade and distant metastasized tumors [10]. The phosphorylation of TAK1 at serine 412 promotes cell proliferation, migration and invasion by activating NFκB signaling in ovarian cancer cells [10]. Coincidently, in the present study, higher TAK1/NFκB signaling was seen in metastatic ovarian cancer cells upon treatment with OCM, and the magnitude of enhanced signaling activity was proportional to the aggressiveness of the ovarian cancer cells. On the other hand, it is known that the aberrant activation of IKKβ/NFκB signaling triggers malignant cells to produce cytokines such as IL-6, IL-8 and GRO-α [27-29]. These chemokines are strongly associated with decreased cell mobility, angiogenesis and chemoresistance of human cancers [8, 29-32]. Likewise, our results using ELISA and IHC analyses illustrated that omentum-metastasized tumor cells intrinsically express an enriched array of chemokines, thereby implicating the potential of a positive feedback loop between these cytokines and TAK1/NFκB activity during metastatic progression in the peritoneal cavity.
Accumulating evidence has shown that the omentum is the preferential site for peritoneal metastasis of ovarian cancer cells [5, 12, 13]. Indeed, a wide spectrum of cells and tissues in the omentum have been reported to be crucial for this metastatic process [5, 33-35]. For instance, cancer-associated fibroblasts (CAFs) in the omentum interact with ovarian cancer cells to facilitate omental metastasis [35, 36]. Adipocytes near ovarian cancer cells are thought to provide CAFs with fatty acids and chemokines, supporting the growth and propagation of the tumors [4, 37]. Herein, our findings revealed that chemokines GRO-α and IL-8 are highly expressed in omental milky spots, lymph nodes, adipose tissue and blood vessels, which supports the idea that these tissue types of the omentum determine the dissemination of ovarian cancer cells. Moreover, GRO-α and IL-8 are detected in both the ascites and sera of ovarian cancer patients [38-42], wherein ascites intriguingly have a higher level of these chemokines than the respective serum samples from the same patients. This finding supports our data that GRO-α and IL-8 originate from both the omental tissues and cancer cells to attract ovarian cancer cell colonization.
Previous studies have documented that overexpression of G-protein-coupled receptors (GPCRs) such as CXCR2 is associated with poor prognosis and cancer metastasis in human cancers, including in epithelial ovarian cancer [43-45]. Kaplan-Meier analysis in this study consistently revealed that the high CXCR2 level was inversely correlated with progression-free and post-progression survival rates, suggesting that CXCR2 exerts substantial influences on the recurrence and metastasis of ovarian cancer. Notably, CXCR2 has also been reported to promote tumor progression in ovarian cancer by promoting angiogenesis, stimulating cell growth and inhibiting cell death through the EGFR/AKT/NFκB pathway [19, 43]. Apart from CXCR2, its relevant chemokines, such as IL-8, further enhance CXCR2-mediated cancer cell metastasis [46, 47]. The pro-angiogenic chemokine GRO-α, on the other hand, has been shown to mediate the activation of anti-apoptotic pathways in cancer cells by interacting with CXCR2 [48, 49]. However, GRO-α is also known to increase the oncogenic capacities of cancer invasion and migration [18]. Herein, we reported that IL-8 and GRO-α display functional effects similar to OCM in ovarian cancer cells, and in particular, GRO-α was highly expressed in normal OCM, suggesting that this is why shed ovarian cancer cells tend to preferentially metastasize to the omentum [2]. On the other hand, suppressing the functions of IL-8 and GRO-α in OCM via neutralizing antibodies attenuated OCM-induced cell proliferation and cell aggressiveness as well as TAK1/NFκB signaling activities, further highlighting the importance of these OCM chemokines in promoting ovarian cancer aggressiveness. In fact, CXCR2 plays a key role in mediating TAK1/NFκB signaling after stimulation by either IL-8 or GRO-α. Therefore, targeting CXCR2 could achieve useful therapeutic effects on ovarian cancer metastasis. As expected, our experiments inhibiting CXCR2 function by using neutralizing antibodies, shRNA-mediated gene knockdown or gene knockout strategies have clearly established the crucial role of CXCR2 in the activation of TAK1/NFκB signaling in the oncogenic capacities of ovarian cancer cells. Similarly, inhibition of CXCR2 by SB225002 effectively impeded CXCR2-mediated ovarian cancer metastasis in vivo has been reported in other human cancer studies [45, 50], suggesting that this type of CXCR2 antagonist could be developed as a potential therapeutic drug for preventing peritoneal metastases.
In summary, this study suggests that IL-8 and GRO-α in OCM and the omental tumor microenvironment are essential for promoting ovarian cancer aggressiveness through activation of the CXCR2/TAK1/NFκB signaling axis. Our findings underscore the relevance of the tumor microenvironment in facilitating ovarian cancer metastatic progression and aid in the exploration of CXCR2 as a potential therapeutic target for ovarian cancer.
Abbreviations
NFκB: nuclear factor-kappa B; TGF: transforming growth factor; TAK1: TGF-β-activating kinase 1; OCM: omentum conditioned medium; ELR+: glutamic acid-leucine-arginine; IL-8: interleukin 8; GRO-α: growth related oncogene-alpha; HOSE: immortalized human ovarian surface epithelium cells; IHC: immunohistochemical; FIGO: the International Federation of Gynecology and Obstetrics; POD: the pouch of Douglas; CXCR2: CXC chemokine receptor 2; PFS: progression-free survival; PPS: post-progression survival; OS: overall survival; i.p.: intraperitoneal; SCID: the severe combined immunodeficiency; CAFs : cancer-associated fibroblasts; GPCRs: G-protein-coupled receptors.
Acknowledgements
We thank Prof. Benjamin Tsang (Department of Obstetrics and Gynaecology, The University of Ottawa) and Prof. George Tsao (Department of Anatomy, The University of Hong Kong) for providing various human ovarian cancer and HOSE cell lines. This study was supported by HKSAR RGC General Research Fund (GRF) (HKU 761213M).
Author Contributions
D.C., M.Y., H.T. and H.N. designed research; M.Y., H.T., P.C., and D.C. performed the experiments; D.C., T.L., D.X., S.N., K.C., H.Y. and H.N. contributed new reagents-analytic tools; D.C., M.Y., H.T., and H.N. analyzed and interpreted data; D.C. M.Y. and H.T wrote the manuscript. All authors were involved in editing the manuscript and had final approval of the submitted and published versions.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006;7:925-34
2. Joo WD, Schwartz PE, Rutherford TJ, Seong SJ, Ku J, Park H. et al. Microscopic Omental Metastasis in Clinical Stage I Endometrial Cancer: A Meta-analysis. Ann Surg Oncol. 2015;22:3695-700
3. Kapp KS, Kapp DS, Poschauko J, Stucklschweiger GF, Hackl A, Pickel H. et al. The prognostic significance of peritoneal seeding and size of postsurgical residual in patients with stage III epithelial ovarian cancer treated with surgery, chemotherapy, and high-dose radiotherapy. Gynecol Oncol. 1999;74:400-7
4. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498-503
5. Nowicka A, Marini FC, Solley TN, Elizondo PB, Zhang Y, Sharp HJ. et al. Human omental-derived adipose stem cells increase ovarian cancer proliferation, migration, and chemoresistance. PLoS One. 2013;8:e81859
6. Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13:497-510
7. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275-92
8. Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749-59
9. Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 2006;17:189-201
10. Cai PC, Shi L, Liu VW, Tang HW, Liu IJ, Leung TH. et al. Elevated TAK1 augments tumor growth and metastatic capacities of ovarian cancer cells through activation of NF-kappaB signaling. Oncotarget. 2014;5:7549-62
11. Kwok AL, Wong OG, Wong ES, Tsun OK, Chan KK, Cheung AN. Caution over use of ES2 as a model of ovarian clear cell carcinoma. J Clin Pathol. 2014;67:921-2
12. Drake LE, Macleod KF. Tumour suppressor gene function in carcinoma-associated fibroblasts: from tumour cells via EMT and back again? J Pathol. 2014;232:283-8
13. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423-37
14. Kobayashi Y, Mizoguchi T, Take I, Kurihara S, Udagawa N, Takahashi N. Prostaglandin E2 enhances osteoclastic differentiation of precursor cells through protein kinase A-dependent phosphorylation of TAK1. J Biol Chem. 2005;280:11395-403
15. Nasser MW, Elbaz M, Ahirwar DK, Ganju RK. Conditioning solid tumor microenvironment through inflammatory chemokines and S100 family proteins. Cancer lett. 2015;365:11-22
16. O'Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. Biochem J. 2008;409:635-49
17. Singh S, Sadanandam A, Singh RK. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev. 2007;26:453-67
18. Reiland J, Furcht LT, McCarthy JB. CXC-chemokines stimulate invasion and chemotaxis in prostate carcinoma cells through the CXCR2 receptor. Prostate. 1999;41:78-88
19. Yang G, Rosen DG, Liu G, Yang F, Guo X, Xiao X. et al. CXCR2 promotes ovarian cancer growth through dysregulated cell cycle, diminished apoptosis, and enhanced angiogenesis. Clin Cancer Res. 2010;16:3875-86
20. Boppana NB, Devarajan A, Gopal K, Barathan M, Bakar SA, Shankar EM. et al. Blockade of CXCR2 signalling: a potential therapeutic target for preventing neutrophil-mediated inflammatory diseases. Exp Biol Med. 2014;239:509-18
21. Halkia E, Spiliotis J, Sugarbaker P. Diagnosis and management of peritoneal metastases from ovarian cancer. Gastroenterol Res Pract. 2012;2012:541842
22. Weidle UH, Birzele F, Kollmorgen G, Rueger R. Mechanisms and Targets Involved in Dissemination of Ovarian Cancer. Cancer Genomics Proteomics. 2016;13:407-23
23. Yu S, Murph MM, Lu Y, Liu S, Hall HS, Liu J. et al. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. J Natl Cancer Inst. 2008;100:1630-42
24. Yamamura S, Matsumura N, Mandai M, Huang Z, Oura T, Baba T. et al. The activated transforming growth factor-beta signaling pathway in peritoneal metastases is a potential therapeutic target in ovarian cancer. Int J Cancer. 2012;130:20-8
25. Moon H, Ju HL, Chung SI, Cho KJ, Eun JW, Nam SW. et al. Transforming Growth Factor Beta Promotes Liver Tumorigenesis in Mice via Upregulation of Snail. Gastroenterology. 2017
26. Yang H, Fang F, Chang R, Yang L. MicroRNA-140-5p suppresses tumor growth and metastasis by targeting transforming growth factor beta receptor 1 and fibroblast growth factor 9 in hepatocellular carcinoma. Hepatology. 2013;58:205-17
27. Singha B, Gatla HR, Manna S, Chang TP, Sanacora S, Poltoratsky V. et al. Proteasome inhibition increases recruitment of IkappaB kinase beta (IKKbeta), S536P-p65, and transcription factor EGR1 to interleukin-8 (IL-8) promoter, resulting in increased IL-8 production in ovarian cancer cells. J Biol Chem. 2014;289:2687-700
28. Son DS, Parl AK, Rice VM, Khabele D. Keratinocyte chemoattractant (KC)/human growth-regulated oncogene (GRO) chemokines and pro-inflammatory chemokine networks in mouse and human ovarian epithelial cancer cells. Cancer Biol Ther. 2007;6:1302-12
29. Yang J, Wang Y, Gao Y, Shao J, Zhang XJ, Yao Z. Reciprocal regulation of 17beta-estradiol, interleukin-6 and interleukin-8 during growth and progression of epithelial ovarian cancer. Cytokine. 2009;46:382-91
30. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S. et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461-6
31. Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I. et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene. 2008;27:4712-23
32. Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene. 2001;20:4188-97
33. Chu Y, Tang H, Guo Y, Guo J, Huang B, Fang F. et al. Adipose-derived mesenchymal stem cells promote cell proliferation and invasion of epithelial ovarian cancer. Exp Cell Res. 2015
34. Klopp AH, Zhang Y, Solley T, Amaya-Manzanares F, Marini F, Andreeff M. et al. Omental adipose tissue-derived stromal cells promote vascularization and growth of endometrial tumors. Clin Cancer Res. 2012;18:771-82
35. Cai J, Tang H, Xu L, Wang X, Yang C, Ruan S. et al. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis. 2012;33:20-9
36. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J. et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515-33
37. Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. 2013;1831:1533-41
38. Matte I, Lane D, Laplante C, Rancourt C, Piche A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am J Cancer Res. 2012;2:566-80
39. Yang G, Rosen DG, Zhang Z, Bast RC Jr, Mills GB, Colacino JA. et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:16472-7
40. Lokshin AE, Winans M, Landsittel D, Marrangoni AM, Velikokhatnaya L, Modugno F. et al. Circulating IL-8 and anti-IL-8 autoantibody in patients with ovarian cancer. Gynecol Oncol. 2006;102:244-51
41. Lambeck AJ, Crijns AP, Leffers N, Sluiter WJ, ten Hoor KA, Braid M. et al. Serum cytokine profiling as a diagnostic and prognostic tool in ovarian cancer: a potential role for interleukin 7. Clin Cancer Res. 2007;13:2385-91
42. Lee Z, Swaby RF, Liang Y, Yu S, Liu S, Lu KH. et al. Lysophosphatidic acid is a major regulator of growth-regulated oncogene alpha in ovarian cancer. Cancer Res. 2006;66:2740-8
43. Dong YL, Kabir SM, Lee ES, Son DS. CXCR2-driven ovarian cancer progression involves upregulation of proinflammatory chemokines by potentiating NF-kappaB activation via EGFR-transactivated Akt signaling. PloS one. 2013;8:e83789
44. Zhao J, Ou B, Feng H, Wang P, Yin S, Zhu C. et al. Overexpression of CXCR2 predicts poor prognosis in patients with colorectal cancer. Oncotarget. 2017;8:28442-54
45. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L. et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29:832-45
46. Sun Q, Sun F, Wang B, Liu S, Niu W, Liu E. et al. Interleukin-8 promotes cell migration through integrin alphavbeta6 upregulation in colorectal cancer. Cancer Lett. 2014;354:245-53
47. Lee YS, Choi I, Ning Y, Kim NY, Khatchadourian V, Yang D. et al. Interleukin-8 and its receptor CXCR2 in the tumour microenvironment promote colon cancer growth, progression and metastasis. Br J Cancer. 2012;106:1833-41
48. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735-41
49. Loukinova E, Dong G, Enamorado-Ayalya I, Thomas GR, Chen Z, Schreiber H. et al. Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene. 2000;19:3477-86
50. Sueoka H, Hirano T, Uda Y, Iimuro Y, Yamanaka J, Fujimoto J. Blockage of CXCR2 suppresses tumor growth of intrahepatic cholangiocellular carcinoma. Surgery. 2014;155:640-9
Author contact
![]() Corresponding author: Dr. David W Chan, Department of Obstetrics and Gynaecology, L747 Laboratory Block, LKS Faculty of Medicine, 21 Sassoon Road, Pokfulam, Hong Kong. Phone: (852) 3917-9367; Fax: (852) 2816-1947. E-mail: dwchanhk.
Corresponding author: Dr. David W Chan, Department of Obstetrics and Gynaecology, L747 Laboratory Block, LKS Faculty of Medicine, 21 Sassoon Road, Pokfulam, Hong Kong. Phone: (852) 3917-9367; Fax: (852) 2816-1947. E-mail: dwchanhk.
Citation styles
APA
Yung, M.M.H., Tang, H.W.M., Cai, P.C.H., Leung, T.H.Y., Ngu, S.F., Chan, K.K.L., Xu, D., Yang, H., Ngan, H.Y.S., Chan, D.W. (2018). GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics, 8(5), 1270-1285. https://doi.org/10.7150/thno.22536.
ACS
Yung, M.M.H.; Tang, H.W.M.; Cai, P.C.H.; Leung, T.H.Y.; Ngu, S.F.; Chan, K.K.L.; Xu, D.; Yang, H.; Ngan, H.Y.S.; Chan, D.W. GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics 2018, 8 (5), 1270-1285. DOI: 10.7150/thno.22536.
NLM
Yung MMH, Tang HWM, Cai PCH, Leung THY, Ngu SF, Chan KKL, Xu D, Yang H, Ngan HYS, Chan DW. GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics 2018; 8(5):1270-1285. doi:10.7150/thno.22536. https://www.thno.org/v08p1270.htm
CSE
Yung MMH, Tang HWM, Cai PCH, Leung THY, Ngu SF, Chan KKL, Xu D, Yang H, Ngan HYS, Chan DW. 2018. GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics. 8(5):1270-1285.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.