Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(5):1243-1255. doi:10.7150/thno.22856 This issue Cite
Research Paper
Serum exosomes can restore cellular function in vitro and be used for diagnosis in dysferlinopathy
Xue Dong1*, Xianjun Gao1*, Yi Dai2*, Ning Ran1, HaiFang Yin1 ![]()
1. Department of Cell Biology, Tianjin Medical University, Qixiangtai Road, Heping District, Tianjin, 300070, China
2. Department of Neurology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Shuaifuyuan Street, Dongcheng District, Beijing, 100730, China
*These authors contribute equally to the work.
Received 2017-9-18; Accepted 2017-11-17; Published 2018-2-2
Citation:
Dong X, Gao X, Dai Y, Ran N, Yin H. Serum exosomes can restore cellular function in vitro and be used for diagnosis in dysferlinopathy. Theranostics 2018; 8(5):1243-1255. doi:10.7150/thno.22856. https://www.thno.org/v08p1243.htm
Other stylesAbstract

Purpose: It is challenging to deliver the full-length dysferlin gene or protein to restore cellular functions of dysferlin-deficient (DYSF-/-) myofibres in dysferlinopathy, a disease caused by the absence of dysferlin, which is currently without effective treatment. Exosomes, efficient membranous nanoscale carriers of biological cargoes, could be useful.
Experimental design: Myotube- and human serum-derived exosomes were investigated for their capabilities of restoring dysferlin protein and cellular functions in murine and human DYSF-/- cells. Moreover, dysferlinopathic patient serum- and urine-derived exosomes were assessed for their abilities as diagnostic tools for dysferlinopathy.
Results: Here we show that exosomes from dysferlin-expressing myotubes carry abundant dysferlin and enable transfer of full-length dysferlin protein to DYSF-/- myotubes. Exogenous dysferlin correctly localizes on DYSF-/- myotube membranes, enabling membrane resealing in response to injury. Human serum exosomes also carry dysferlin protein and improve membrane repair capabilities of human DYSF-/- myotubes irrespective of mutations. Lack of dysferlin in dysferlinopathic patient serum and urine exosomes enables differentiation between healthy controls and dysferlinopathic patients.
Conclusions: Our findings provide evidence that exosomes are efficient carriers of dysferlin and can be employed for the treatment and non-invasive diagnosis of dysferlinopathy.
Keywords: Dysferlinopathy, exosome, therapeutics, diagnostics
Introduction
Muscular dystrophies are heterogeneous, hereditary neuromuscular disorders and are associated with frame-disrupting or missense mutations in several genes involved in muscular structure and function [1]. Mutations in the dysferlin gene, encoding a 230 kDa transmembrane protein implicated in membrane fusion and repair [2], are linked to clinically distinct muscle diseases, i.e., limb-girdle muscular dystrophy type 2B (LGMD2B), Miyoshi myopathy (MM) and distal myopathy with anterior tibial (DMAT)[3, 4],which are also collectively referred to as dysferlinopathy, and are characterized by wasting and weakness of skeletal muscle. Dysferlinopathy generally manifests after puberty with variable severity in different muscles [5]. Currently no therapy is available for dysferlinopathic patients [6]. Transfecting functional truncated variants of dysferlin genes in animal models has proven successful at restoring membrane repair, yet has failed to arrest progressive muscle wasting. This suggests that the full-length dysferlin protein maybe required to reverse disease progression [7][8].However, the large size of the dysferlin gene represents a challenge for gene transfer approaches, since most viral vectors used in gene therapy have a limited cargo capacity. Although adeno-associated virus (AAV) vector systems have been developed to deliver full-length or nano-sized dysferlin genes into dysferlin-deficient mice and non-human primates with encouraging results [9, 10], AAV can trigger immune response that limits its therapeutic efficacy upon repeat treatment. Recent studies have made some progress in correcting the deficiency by inducing stable expression of full-length dysferlin integrated in dysferlin-deficient myoblasts using sleeping beauty transposons. Transfected myoblasts grafted in immune-deficient mice exhibited normal regenerative capabilities in situ. Although such ex vivo therapeutic approaches appear attractive, they require substantial development prior to clinical applications [11].
Exosomes are membranous nanovesicles with diameters of 50-150nm, which are secreted by various types of cells upon fusion of multi-vesicular bodies with the plasma membrane [12]. Exosomes are known to be intercellular messengers and can shuttle cargoes such as proteins, lipids, mRNA and microRNAs between cells [13]; thus, exosomes have been extensively used for diagnostic and therapeutic purposes [14]. Antigen-carrying tumor exosomes were shown to activate dendritic cells and elicited effective regression in hepatocellular carcinoma and other tumor models [15, 16]; while exosomes from neurons can shuttle endogenous proteins such as alpha-synuclein oligomers and amyloid-β (Aβ) in neurodegenerative diseases [17, 18]. Proteomic analysis of exosome-like vesicles released from C2C12 myoblasts and myotubes revealed protein involved in the regulation of myoblast fusion, membrane repair and regeneration [19]. Fusion of exosomes with a target cell membrane could also facilitate the exchange of membrane proteins between two cell types [20, 21]. Thus, exosomes could potentially deliver therapeutic proteins in dysferlinopathy.
Considering the unique features of exosomes, e.g., low immunogenicity and scalability, exosomes have been extensively employed to carry therapeutic moieties [22]. Here, we present evidence that exosomes derived from dysferlin-expressing muscle cells or serum are capable of delivering full-length dysferlin protein and may be useful for treating dysferlinopathy. The lack of dysferlin protein in urine exosomes derived from dysferlinopathic patients also presents non-invasive diagnostic opportunities for dysferlinopathy compared to the dysferlin Western blots from muscle biopsy that are currently necessary for confirmation of diagnosis.
Materials and Methods
Cell culture
Immortalized murine H2K, A/J, BL6 and dysferlin-deficient H2K cells were kindly provided by Professor Terry Partridge (Children's National Medical Center,Center for Genetic Medicine Research,Washington DC, US) and cultured as previously reported [23, 24, 25]. Briefly, cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% fetal calf serum (FBS), 2% chick embryo extract, 2% L-glutamine, 1% penicillin and streptomycin and 20U/mL mouse recombinant IFN-γ (Invitrogen, US) at 33°C in 10% CO2. Immortalized human myoblasts were kindly provided by Professor Jennifer Morgan (University College London, Institute of Child Health, London, UK) and were cultured in skeletal muscle cell growth medium (PromoCell, Germany) supplemented with 1.5% GlutaMAX (Gibco, US), 20% FBS and 1% penicillin and streptomycin. Myotubes were obtained from confluent H2K, dysferlin-deficient H2K or human myoblasts seeded in gelatin-coated 12-well plates after 3 days of serum deprivation at 37°C under a 5% CO2 atmosphere (DMEM with 5% horse serum (HS, Hyclone, US)). Murine C2C12 cells and NIH 3T3 cells were kept in house and grown at 37°C in 5% CO2 in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin.
Preparation and purification of exosomes
Cell culture medium was sequentially centrifuged at 1000 × g for 10 min, followed by 10,000×g for 30 min. The supernatant was collected and filtered with a 0.22µm filter (Millex, Germany), followed by ultracentrifugation at 100,000×g for 1 h to pellet exosomes. Exosome pellets were washed in a large volume of PBS and recovered by centrifugation at 100,000×g for 1 h. The total protein concentration of exosomes was quantified by the Bradford assay (Sangon Biotech, China). For human serum and urine exosome isolation, human serum and urine were obtained from healthy volunteers (provided by Tianjin Blood Center, Tianjin, China) and dysferlinopathic patients (provided by Peking Union Medical College Hospital, Beijing, China, approved by the hospital ethic committee, Permit No. JS-1317) and centrifuged at 3000×g, 6000×g and 10,000×g twice for 30 min each time to remove cell debris, followed by filtration with 0.22µm filter (Millex, Germany). The supernatant was transferred into a fresh tube and further pelleted by ultracentrifugation (Beckman Optimal-100 XP, Beckman Coulter, Germany) at 100,000×g for 1 h. Exosome pellets were washed in a large volume of PBS and recovered by ultracentrifugation at 100,000×g for 1 h. Exosomes were purified by sucrose density gradient centrifugation as previously reported [26]. Briefly, the gradients were centrifuged for 18 h at 100,000×g at 4°C. Six fractions from 0.65-1.65 M sucrose gradients were collected and ultracentrifuged at 100,000×g for 1 h at 4°C to pellet exosomes. The exosome pellets were dissolved in PBS and the total protein concentration of exosomes was quantified by the Bradford assay (Sangon Biotech, China).
Characterization of exosomes
The size distribution of exosomes was measured using a nanoparticle tracking and zeta potential distribution analyzer (Paricle Metrix-PMX, Germany) and the morphology of the exosomes was visualized using a high-resolution transmission electron microscope (TEM, Hitachi HT7700, Tokyo, Japan). Briefly, the re-suspended exosomes were mixed with an equal volume of 4% paraformaldehyde (PFA) and adsorbed onto a glow-discharged, carbon-coated formvar film, which was attached to a metal specimen grid. Excess solution was blotted off and the grid was immersed with a small drop (50 μL) of 1% glutaraldehyde for 5 min followed by washing with 100 μL still water 2 min each time for 8 times. Subsequently, the grid was transferred to 50 μL uranyl-oxalate solution (pH7.0) for 5 min and then 50 μL methyl cellulose-uranyl acetate (100 μL 4% uranyl acetate and 900 μL 2% methyl cellulose) for 10 min on ice. The excess solution was blotted off and the sample was dried and examined in the TEM.
Exosome uptake assay
To test the uptake of exosomes derived from H2K myotubes in dysferlin-deficient H2K myoblasts, exosomes (5 μg) labelled with membrane dye PKH67 (Sigma, US) were incubated with dysferlin-deficient H2K myoblasts cultured in differentiation medium containing 2% pre-centrifuged HS. Cells were observed and photographed using a conventional fluorescence microscope (Olympus FV1000, Olympus, Japan) at 0, 4, 8, 12, 24 and 48 h after incubation. For detection of the localization of exosomes in dysferlin-deficient H2K myotubes, 5 μg PKH67-labelled exosomes were incubated with dysferlin-deficient H2K myotubes for 48 h at 37°C in 5% CO2. Subsequently, cells were incubated with HBSS solution supplemented with the membrane dye FM4-64 (2.5 μM, Molecular Probes, Invitrogen, US) at 37°C for 5 min, followed by washing and visualization using a confocal fluorescence microscope (Olympus FV1000, Olympus, Japan).
Digestion of exosomes
To evaluate the location of dysferlin protein in exosomes, 25 μg exosomes derived from H2K myotubes were incubated with 0.025% trypsin (Sigma, US) and /or 0.1% saponin (Sigma, US) for 5 min at 37°C, followed by heating at 100°C for 3 min to terminate the digestion reaction, and were then analyzed by Western blot assay.
Protein extraction and western blotting
Exosome and cell pellets were lysed in lysis buffer (125 mM Tris-HCl, pH6.8, 10% sodium dodecyl sulfate (SDS), 2M urea, 20% glycerol and 5% 2-mercaptoethanol) and subjected to 10% SDS-polyacrylamide gel electrophoresis and gels were transferred to a PVDF membrane. Membranes were blocked in 5% skimmed milk and probed with primary antibodies including mouse monoclonal antibody to the C-terminus of dysferlin (1:200, NovoCastra, US), rabbit monoclonal antibodies to the N-terminus of dysferlin (1:1000, Abcam, UK) and Cytochrome C (1:1000, Cell Signaling Technology, US), rabbit polyclonal antibodies for CD63 (1:200, Santa Cruz, US), CD9 (1:1000, Abcam, UK) or goat polyclonal antibody for Alix (1:200, Santa Cruz, US) overnight at 4°C. GAPDH (1:1000, Cell Signaling Technology, US) was used as a loading control. The bound primary antibody was detected by horseradish peroxidase-conjugated goat anti-mouse IgG (Sigma, US), goat anti-rabbit IgG (Sigma, US) or rabbit anti-goat IgG (Sigma, US), respectively. The ECL western blotting analysis system (Millipore, Billerica, MA) was applied.
Immunocytochemistry and confocal microscopy
H2K and dysferlin-deficient H2K myoblasts were seeded in 6-well plates and differentiated into myotubes as described above. Cells were washed with cold PBS twice and fixed with 4% PFA for 30 min at room temperature (RT) and subjected to permeabilization with 0.1% Triton X-100 at RT for 30 min. Subsequently, cells were blocked with 5% BSA for 1 h at RT and incubated with primary antibodies, including rabbit monoclonal antibody to dysferlin (1:50, Abcam, UK) or mouse monoclonal antibody to caveolin-3 (1:100, Santa Cruz, US), overnight at 4°C. Goat anti-rabbit Alexa Fluor 594 and goat anti-mouse Alexa Fluor 488 secondary antibodies (Invitrogen, US) were used with a dilution of 1:200 in PBS for 1 h at RT and nuclei were counterstained with DAPI (Invitrogen, US). Images were obtained using a confocal fluorescence microscope (Olympus FV1000, Olympus, Japan).
Membrane repair assay
The membrane repair assay was performed as previously reported [25, 27]. Briefly, 100 μg exosomes derived from H2K myotubes were incubated with H2K or dysferlin-deficient H2K myotubes for 48 h. Myotubes were washed twice with HBSS solution (140mM NaCl, 5mM KCl, 2mM CaCl2, 1mM MgCl2·6H2O, 10mM HEPES and 10mM glucose, pH7.4) prior to membrane wounding, and then incubated with HBSS solution containing membrane dye at 37°C for 5 min. The laser wounding experiment was performed in HBSS solution supplemented with the FM4-64 dye (2.5 μM, Invitrogen, US). A two-photon confocal laser-scanning microscope (Olympus, Japan) was used for laser injury and a 2.0μm2 area overlapping the plasma membrane was irradiated at 25% of the maximum output power of the laser operating at 850nm for 200ms. X-Y images were captured with a 63x water immersion objective at 10s intervals for 300s. The mean fluorescence intensity of FM4-64 at the site of damage was measured with the Olympus imaging software. The fluorescence intensity was normalized to the pre-wound intensity and the normalized response (ΔF/F0) was plotted as a function of time. For each treatment, at least five different myotubes per well (triplicates for each experiment, the experiment was repeated 3 times) were used for the membrane repair assay.
Scrape wounding and surface exposure of LAMP1 luminal epitopes
Dysferlin-deficient H2K myotubes were incubated with exosomes derived from H2K myotubes (100 μg) in the differentiation medium for 48 h, followed by washing with PBS twice. Cells were injured by dragging a scalpel blade four times across the surface of the dish in the presence of 1mg/mL Texas Red dextran (Invitrogen, US) in HBSS solution containing 2mM CaCl2. Subsequently, dishes were transferred to 37°C and incubated for 5 min to allow membrane repair. Cells were washed with cold PBS twice and fixed for 30 min at RT with 4% PFA and blocked with 5% BSA for 1 h at RT. Then cells were incubated with primary rat monoclonal anti-LAMP1 antibody (1:100, Santa Cruz, US) overnight at 4°C,followed by labelling with secondary antibody goat anti-mouse Alexa Fluor 488 for 1 h at RT, and then nuclei were counterstained with DAPI (Invitrogen, US). Images were obtained on an inverted fluorescence microscope (Olympus IX71, Olympus, Japan).
Imaging Ca2+ influx into cells after laser injury
H2K and dysferlin-deficient H2K myotubes were incubated with 100 μg exosomes derived from H2K myotubes for 48 h. Myotubes were washed twice with HBSS solution (140mM NaCl, 5mM KCl, 2mM CaCl2, 1mM MgCl2·6H2O, 10mM HEPES and 10mM glucose, pH7.4) and incubated with HBSS containing Fluo-3 AM (5 μM, Beyotime, China) for 30 min at 37°C. Subsequently, the dye was removed and cells were washed twice with HBSS and incubated with HBSS for another 15 min at 37°C to ensure that the Fluo-3 AM was completely transformed into Fluo-3. Cells were then laser injured in HBSS as described above. X-Y images were captured with a 63x water immersion objective at 4s intervals for 250s. The mean fluorescence intensity of Fluo-3 was measured with the Olympus imaging software. The kinetics of Ca2+ influx was normalized to the pre-wound intensity and the normalized response (ΔF/F) was plotted as a function of time. For each treatment, the detection was performed on at least six different myotubes per well (triplicates per treatment, the experiment was repeated 3 times).
Myotube counts
H2K and dysferlin-deficient H2K myoblasts were cultured in differentiation medium in the presence of 50 μg exosomes derived from H2K myotubes for 48 h. Images were obtained on an inverted fluorescence microscope (Olympus FV1000, Olympus, Japan). The number of myotubes was examined using an inverted fluorescence microscope and quantified by counting myotubes in five fields for each treatment in a double-blinded manner (triplicates for each treatment and the experiment was repeated for 3 times). Multinucleated and tube-like myotubes were counted.
Generation of human dysferlin-deficient myoblasts
Immortalized normal human myoblasts (kindly provided by Professor Jennifer Morgan, University College London, Institute of Child Health, London, UK) were transfected with CRISPR/Cas9-expressing lentivirus (Lentiviral CRISPR Toolbox, Zhanglab, US), followed by 2 µg/mL blasticidin selection for 1week. Cas9-expressing myoblasts were transfected with sgRNA-expressing lentivirus for 12 h, followed by 0.5 µg/mL puromycin selection for 2 weeks. sgRNAs targeting human dysferlin gene exons 22 and 55 were designed with the CRISPR/Cas9 Target Online Predictor (crispr.cos.uni-heidelberg.de, University of Heidelberg, Germany). The human dysferlin gene exon 22 and exon 55 target sgRNA sequences were 5'-GAAGGCGCAGTGCTCCACGG-3' and 5'-CTGGTGAAGCCCTTCAGCTG-3', respectively. These sgRNAs were cloned into a LentiGuide-puro vector at BsmBI site as previously reported [28].
Statistical analysis
All data are reported as mean values±SEM. Statistical differences between treatment and control groups were evaluated by SigmaStat (Systat Software, London, UK). Both parametric and non-parametric analyses were applied, in which the Mann-Whitney rank sum test (Mann-Whitney U-test) was used for samples with a non-normal distribution, whereas a two-tailed t-test was performed for samples with a normal distribution, respectively.
Results
Dysferlin is abundant in myotube-derived exosomes
To explore if exosomes carry dysferlin, we examined the presence of dysferlin in exosomes derived from different murine muscle cell lines and a non-muscle cell line 3T3 was used as a negative control. As exosomes from the immortalized muscle cell line H2K showed the highest levels of dysferlin compared to other muscle cells (Fig. S1A), H2K cells were used hereafter. Higher levels of dysferlin were detected in exosomes derived from differentiated myotubes (EXOdysf) than from undifferentiated myoblasts (EXOmyob) (Fig. 1A), synchronous with cellular expression (Fig. S1B), though the yield of EXOdysf (20 µg) was lower than EXOmyob (60 µg of protein per 3x106 cells in 48 h). EXOdysf showed a typical sauce-cup shape under transmission electron microscopy (TEM) (Fig. 1B), with a peak size of 114.3 nm as shown by nanoparticle tracking analysis (NTA) (Fig. 1C). Interestingly, levels of dysferlin protein in exosomes derived from myotubes at different time-points after induction of differentiation varied, with day 6 myotubes showing the highest expression (Fig. S1C); therefore, day 6 myotubes were used hereafter.
Figure 1
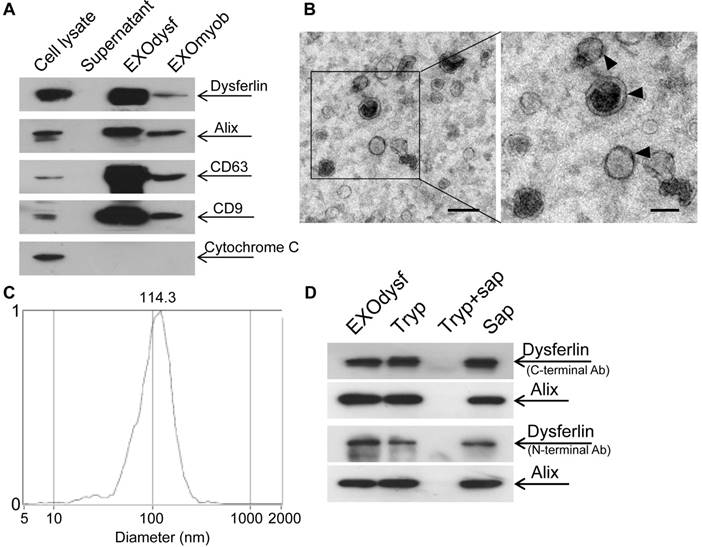
Characterization of exosomes derived from murine myotubes (EXOdysf). (A) Western blot to examine the level of dysferlin expression in EXOdysf. 20 μgor 50 μg total protein from cell lysates or exosomes was loaded and Cytochrome C was used as an organelle marker. (B) Representative transmission electron microscopy (TEM) image of EXOdysf (scale bar = 200 nm (left) or 100 nm (right)).Arrowheads point to the typical sauce-cup shape of exosomes. (C) Analysis of the size distribution of EXOdysf with a nanoparticle tracking analysis (NTA) system. (D) Western blot to evaluate the presence and localization of dysferlin protein in EXOdysf. 25 μg total protein from exosomes derived from myotubes was loaded and Alix was used as an exosomal marker. Tryp, sap, or tryp+sap refer to trypsin-, saponin- or both trypsin- and saponin-treated EXOdysf.
To investigate whether dysferlin is located inside or loosely associated on the surface of EXOdysf, we treated EXOdysf with 0.25% trypsin. Protein loosely attached to the surface of EXOdysf would be degraded. A clear dysferlin band appeared in the samples treated with trypsin when probed with antibodies targeting either the N-terminus (cytoplasmic region) or C-terminus (intracellular domain proximal to the transmembrane domain) of dysferlin protein (Fig. 1D), indicating that dysferlin is not nonspecifically attached to exosomes. In contrast, the dysferlin band diminished when EXOdysf were treated with 0.25% trypsin and 0.1% saponin, a membrane-permeabilizing detergent [29](Fig. 1D), suggesting that dysferlin is located inside the lumen of EXOdysf.
EXOdysf anchors dysferlin on the membrane of DYSF-/- myotubes
To evaluate whether EXOdysf can be taken up by and transfer dysferlin to dysferlin-deficient muscle cells (DYSF-/-), we incubated PKH67-labelled EXOdysf (5 µg) with DYSF-/- myotubes and monitored fluorescence at different time-points after unincorporated EXOdysf were removed. Fluorescence was readily detectable at 4 h and peaked at 24 h after incubation (Fig. S2A). Examination on the localization of PKH67-labelled EXOdysf in DYSF-/- myotubes 24 h after incubation indicated that the majority of EXOdysf are internalized into the cytoplasm with some present on the membrane (Fig. S2B), suggesting that EXOdysf can be efficiently absorbed into DYSF-/- myotubes. Consistently, dysferlin protein was detected in DYSF-/- myotubes after the addition of EXOdysf (Fig. 2A). Immunostaining revealed that some restored dysferlin proteins were membrane-bound and partly co-localized with Caveolin-3, a membrane protein associated with dysferlin during the formation of caveolae and repair vesicles [30] (Fig. 2B), demonstrating that EXOdysf can transfer and anchor dysferlin on DYSF-/- myotube membranes.
Figure 2
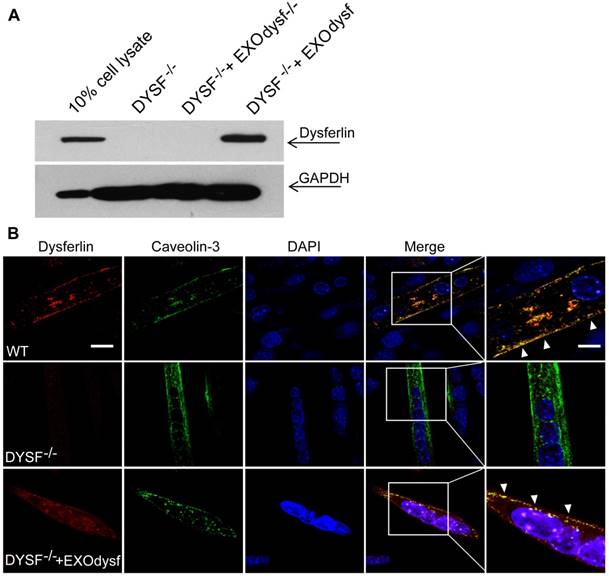
Examination of the restoration and localization of dysferlin protein in EXOdysf-treated murine dysferlin-deficient (DYSF-/-) myotubes. The restoration of dysferlin protein was examined 48 h after the addition of 100 μg/mL EXOdysf in DYSF-/- myotubes. (A) Western blot to measure the level of dysferlin protein in EXOdysf-treated DYSF-/- myotubes. GAPDH was used as a loading control and 20 μgor 200 μg total protein from cell lysates or exosomes was loaded, respectively. (B) Confocal fluorescence microscopy images to reflect the localization of restored dysferlin protein in EXOdysf-treated DYSF-/- myotubes. Nuclei were counterstained with DAPI (blue) (scale bar = 30μm; for boxed areas, scale bar= 15 μm). Arrowheads refer to the co-localization of caveolin-3 (red) and exosome (green).
Figure 3
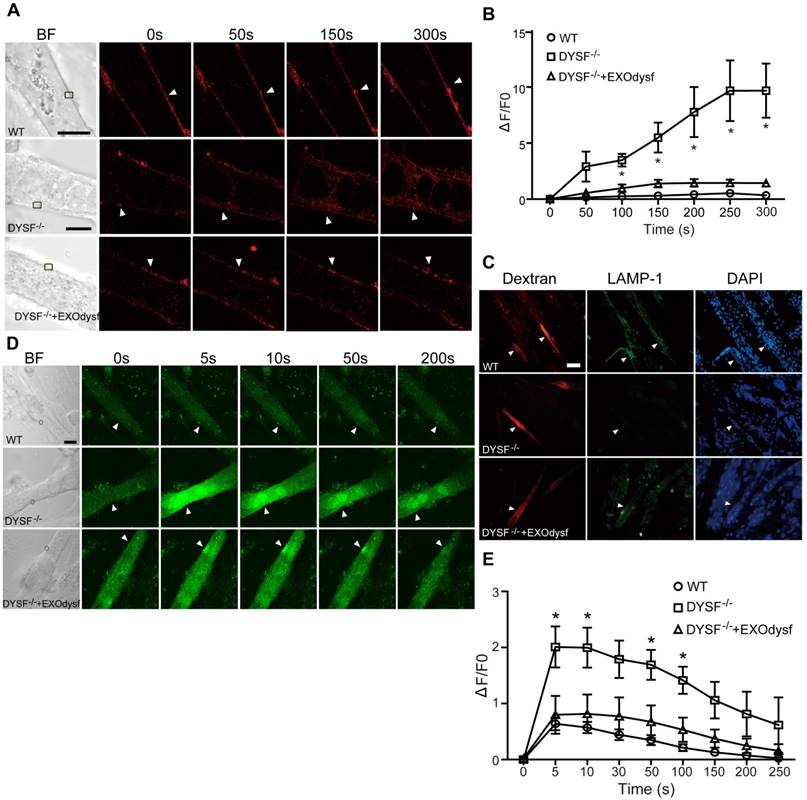
Functional restoration of EXOdysf-treated murine DYSF-/- myotubes. (A) Two-photon confocal microscopy images showing the membrane repair capacity of EXOdysf in DYSF-/- myotubes. Arrowheads point to the laser injury sites. The FM4-64 dye (red) was used to show the integrity of the membrane (scale bar = 10 μm). WT: wild-type; BF: bright field. (B) Quantitative analysis of fluorescence intensity at different time-points in WT (circles), DYSF-/- (squares) or EXOdysf-treated DYSF-/- (triangles) myotubes. Significant reductions in fluorescence intensity were achieved in EXOdysf-treated DYSF-/- myotubes compared to untreated myotubes (n=15, two-tailed test, *P<0.05). (C) Immunostaining of dysferlin and lysosome-associated membrane protein-1 (LAMP-1) in EXOdysf-treated DYSF-/- myotubes. Nuclei were counterstained with DAPI (blue). Arrowheads point to the scrape wounding sites (scale bar =100 μm). (D) Two-photon confocal microscopy images showing the Ca2+ influx in EXOdysf-treated DYSF-/- myotubes. Arrowheads point to the laser injury sites (scale bar = 10 μm). (E)Quantitative analysis of fluorescence intensity at different time-points in WT (circles), DYSF-/- (squares) or EXOdysf-treated DYSF-/- (triangles) myotubes. Significant reductions in fluorescence intensity were achieved in EXOdysf-treated DYSF-/- myotubes compared to untreated myotubes at earlier time-points (n=15, two-tailed test, *P<0.05).
EXOdysf restores cellular function of DYSF-/- myotubes
As dysferlin is integral in the membrane repair process in muscle cells [31], we explored if membrane resealing function can be restored by EXOdysf. We introduced FM4-64, a membrane-impermeable dye, into DYSF-/- myotubes following laser injury and monitored FM4-64 uptake at different time-points. Bright fluorescence was observed at 50s, peaking at 250s after injury in untreated DYSF-/- myotubes (Fig. 3A). Strikingly, faint signals, comparable to wild-type (WT) myotubes, were detected in DYSF-/- myotubes treated with EXOdysf (Fig. 3A). Quantification of the cellular uptake of FM4-64 indicated a significant decrease in fluorescence intensity in EXOdysf-treated DYSF-/- myotubes compared to untreated DYSF-/- myotubes (Fig. 3B), suggesting that this effect is potentially due to the restored dysferlin. Lysosomal exocytosis is associated with repair of injured cell membrane; however, docking and calcium-triggered fusion of lysosomes are impaired in DYSF-/- myotubes [32]. We investigated if intracellular lysosome-associated membrane protein-1 (LAMP-1), a lysosomal marker protein, localized to the surface of injured myotubes after repair. As expected, LAMP-1 was detectable on the surface of EXOdysf-treated DYSF-/- and WT myotubes, but not on untreated DYSF-/- myotubes 300s after scrape wounding (Fig. 3C), suggesting that dysferlin-harboring cytoplasmic vesicles aggregate to fuse with the injured membrane.
Also, dysferlin is composed of multiple Ca2+-sensitive C2 domains that are implicated in regulation of vesicle fusion, trafficking and membrane repair [33]. In the absence of dysferlin, mechanical stress or membrane injury will result in massive influx of Ca2+ and disruption of Ca2+ homeostasis [34]. To investigate whether EXOdysf can regulate the homeostasis of Ca2+ flux in DYSF-/- myotubes after laser-induced membrane injury, we monitored the level of Ca2+ change in the cytosol with Fluo-3AM, a fluorescent Ca2+ probe [35]. A sharp rise in intracellular Ca2+ (i.e., a “Ca2+ spike”) was observed in DYSF-/- myotubes 5s after injury, whereas a much milder increase in the fluorescence intensity was detected in EXOdysf-treated DYSF-/- myotubes (Fig. 3D, E). The fluorescence intensity declined gradually and reached pre-injury values for EXOdysf-treated DYSF-/- myotubes at 50s after injury, but not for untreated DYSF-/- myotubes (Fig. 3D, E), suggesting that EXOdysf enables the regulation of Ca2+ influx in DYSF-/- myotubes.
Serum exosomes restore human DYSF-/- myotube membrane repair capacity
To investigate whether dysferlin-expressing exosomes can restore dysferlin in dysferlinopathic patients, we generated human DYSF-/- myoblasts harboring different mutations in the dysferlin gene exons 22 and 55 by introducing frame-disrupting insertions or deletions with clustered, regularly interspaced short palindromic repeats (CRISPR)/Cas9 system. Analysis of genomic sequence and dysferlin protein expression confirmed that the dysferlin gene was efficiently disrupted (Fig. S3A, B), with no morphological alterations found compared to normal human myoblasts (data not shown). As dysferlin expression had been reported in peripheral blood monocytes [36, 37], we examined if the level of dysferlin expression in normal human serum exosomes may be therapeutically useful. Strikingly, dysferlin protein was also abundant in exosomes from normal human serum (EXOser) (Fig. 4A), with a typical sauce-cup shape under TEM (Fig. S3C) and a peak size of 123.4 nm in diameter (Fig. S3D). Dysferlin protein was readily detectable in human DYSF-/- myotubes 48 h after the introduction of EXOser (Fig. 4B), demonstrating that EXOser can efficiently transfer dysferlin to human DYSF-/- cells. To examine the functionality of EXOser, we added FM4-64 dyes into human DYSF EXO55-/- and EXO22-/- myotubes in the presence of EXOser following laser injury. As expected, fluorescence signals appeared around the injury sites at 50s after injury in untreated human DYSF EXO55-/- and EXO22-/- myotubes and peaked at 250s (Fig. 4C). However, much less fluorescence was found in EXOser-treated human DYSF EXO55-/- and EXO22-/- myotubes subjected to laser injury (Fig. 4C). Quantitative analysis revealed significant decreases in cellular uptake of FM4-64 dyes in EXOser-treated DYSF EXO55-/- and EXO22-/- myotubes compared to untreated myotubes at different time-points (Fig. 4D), indicating that EXOser confers the ability of membrane repair to human DYSF-/- myotubes. These findings support the conclusion that the compromised membrane repair capacity of human DYSF-/-myotubes can be restored.
Serum and urine exosomes present diagnostic value for dysferlinopathic patients
Dysferlin expression in blood monocytes was reported to correlate with dysferlin levels in skeletal muscle both in healthy controls and dysferlinopathic patients [38]. To assess if dysferlinopathic patients can be differentiated from healthy controls based on serum exosome dysferlin, we collected serum from dysferlinopathic patients and examined levels of dysferlin protein in patient serum exosomes. Strikingly, a clear dysferlin protein band appeared in exosomes from normal human serum, but no signal was detected in exosomes from dysferlinopathic patients (Fig. 5A). To validate this approach, we collected serum from another 19 dysferlinopathic patients with confirmed diagnosis by gene sequencing (Fig. 5B) and /or immunohistochemistry (Table 1). The absence of dysferlin protein in exosomes derived from 19 dysferlinopathic patients using Western blot (Fig.5C) confirmed that serum exosomes can be employed to differentiate dysferlinopathic patients from healthy controls. As dysferlin protein was found in human urine exosomes previously [39], we harvested exosomes from human urine, which showed a typical saucer-cup shape (Fig. S4A) and a peak size of 94.3 nm in diameter (Fig. S4B), and examined the expression of dysferlin. Concordantly, dysferlin was also detected in urine exosomes derived from normal controls but not from dysferlinopathic patients (Fig. 5D, E). Since dysferlin protein analysis is essential to confirm specific protein deficits and diagnosis, the data provide evidence that urine exosomes can be employed as a non-invasive diagnostic tool for dysferlinopathic patients.
Discussion
In this study, we demonstrated that exosomes derived from myotubes or human serum carry dysferlin and can efficiently transport functional dysferlin to murine and human dysferlin-deficient muscle cells, restoring their membrane repair capacities in response to injury. Importantly, the lack of dysferlin protein in exosomes from dysferlinopathic patients' serum and urine enables the identification of dysferlinopathic patients non-invasively, which is particularly attractive compared to the current standard - muscle biopsy. This is the first proof-of-concept study showing the capability of exosomes as carriers for endogenous dysferlin protein not only for treatment but also as a non-invasive tool for diagnosis of dysferlinopathy.
Figure 4
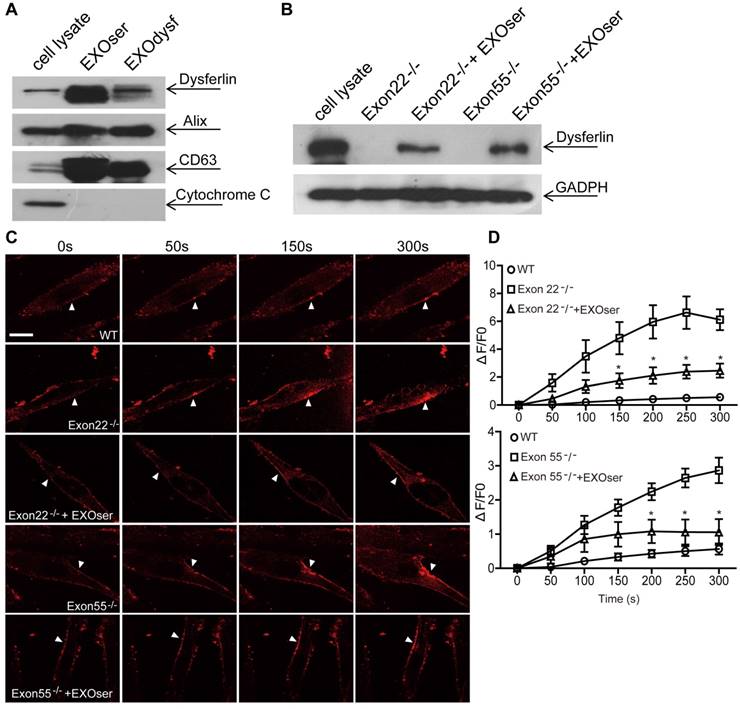
Restoration of dysferlin protein and membrane repair capacities in human DYSF-/- myotubes treated with human serum exosomes (EXOser). (A) Western blot to examine the level of dysferlin protein in human serum exosomes. 20 μgor 50 μg total protein from cell lysates or exosomes was loaded and Cytochrome C was used as an organelle marker. EXOdysf refers to exosomes derived from normal human myotubes. (B) Western blot to examine the level of dysferlin restoration in human DYSF-/- myotubes at 48 h after the addition of 100 μg/mL human EXOser. GAPDH was used as a loading control and 100 μg or 200 μg total protein from cell lysates or exosomes was loaded, respectively. (C) Two-photon confocal microscopy image showing the membrane repair capacity of EXOser-treated DYSF-/- myotubes. Arrowheads point to the laser injury sites (scale bar =20 μm). (D) Quantitative analysis of fluorescence intensity at different time-points in WT (circles), DYSF Exon22-/- or Exon55-/- (squares) or EXOser-treated DYSF-/- (triangles) myotubes. Significant reductions in fluorescence intensity were achieved in EXOser-treated DYSF Exon22-/- or Exon55-/- myotubes compared to untreated myotubes (n=15, two-tailed test, *P<0.05).
Figure 5
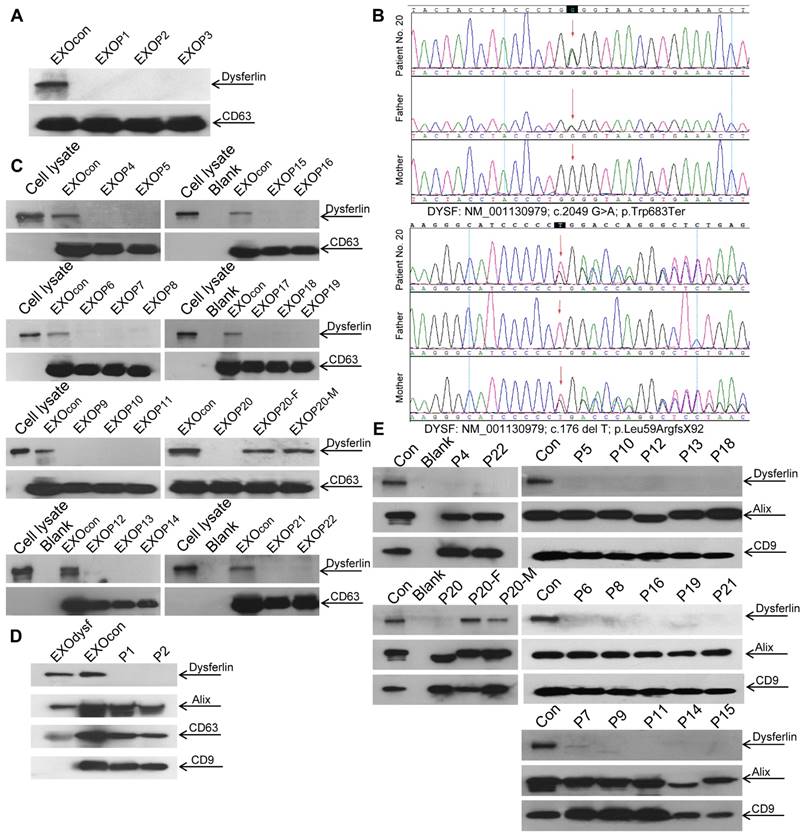
Examination of dysferlin proteinin exosomes from normal controls' and dysferlinopathic patients' serum and urine. (A) Western blot to examine levels of dysferlin protein in exosomes from normal controls' and dysferlinopathic patients' serum. 30 μg total protein from exosomes was loaded. Pn stands for the patient No. EXOcon refers to exosomes from normal controls' serum. (B) Representative gene sequencing results for dysferlinopathic patients (patient no. 20 and her parents). (C) Western blot to analyze the expression of exosomes derived from a spectrum of dysferlinopathic patients' serum. P20-F and P20-M mean patient no.20's father and mother. (D) Western blot to examine levels of dysferlin protein in exosomes from normal controls' and dysferlinopathic patients' urine. 50 μg total protein from exosomes was loaded. EXOcon refers to exosomes derived from normal human urine. (E) Western blot to analyze the expression of dysferlin in urine exosomes derived from a spectrum of dysferlinopathic patients.
Dysferlin-expressing exosomes can restore the expression of dysferlin and cellular function of human dysferlin-deficient muscle cells bearing different mutations in the dysferlin gene, indicating the clinical relevance and general applicability of this approach. Further functional evaluation and toxicological studies on dysferlin-expressing exosomes in dysferlin-null mouse models are warranted to develop this therapeutic modality further. Dysferlin was particularly abundant in human serum exosomes with a concentration of 200 µg exosomes per mL of blood. This supports the notion that serum exosomes can be used as a treatment option for dysferlinopathic patients considering that a large volume of serum (at least 400 mL) can be drawn from a healthy donor or close relatives to lower the chances of potential immunogenicity and thus provide a timely intervention for dysferlinopathic patients.
Dysferlinopathy is a rare disease with an estimated prevalence of 1 in 15,000 individuals (data from Jain foundation), varying geographically [40]. In this study, we were able to recruit 22 dysferlinopathic patients with confirmed diagnosis characterized by typical clinical manifestations, defined dysferlin gene mutations, and muscle biopsies in one single medical center, and this is a relatively large cohort of patients. The dysferlin gene mutation types identified here are similar to dysferlinopathic cases deposited in the database such as UMD-DYSF mutations and Leiden Muscular Dystrophy databases, further confirming that there is no racial difference for dysferlinopathy. Consistent with the clinical data, dysferlin is absent in serum or urine exosomes from dysferlinopathic patients but present in exosomes from their parents (Patient No. 20), supporting the conclusion that exosomes from serum or urine can be used as a reliable non-invasive diagnostic tool, particularly for urine exosomes.
We demonstrated that dysferlin protein is specifically sorted into exosomes and can be efficiently transferred to and localized correctly on the membrane of dysferlin-deficient muscle cells and confer the membrane repair capability of dysferlin-deficient myotubes in response to injury. It was reported previously that the absence of dysferlin delays the differentiation of human dysferlin-deficient myoblasts in vitro [41]; the presence of more myotubes when dysferlin-expressing exosomes were added in the dysferlin-deficient myoblasts supports the notion that dysferlin contribute to the differentiation of myoblasts (Fig. S5), though other secondary effects cannot be excluded. Also, our results imply that transplantation of dysferlin-positive myotubes may be able to “treat” surrounding muscle cells via exosomes, making cell therapy more plausible.
In conclusion, we demonstrate that dysferlin-expressing exosomes can be employed as carriers for endogenous dysferlin protein and can restore cellular functions of dysferlin-deficient muscle cells irrespective of mutations. Lack of dysferlin in urine exosomes from dysferlinopathic patients provides evidence for the use of urine exosomes as a non-invasive diagnostic tool for dysferlinopathy.
Table 1
Clinical information for dysferlinopathic patients
| Patient No. | Sex | DOB | Clinical diagnosis | Gene mutations | IHC |
|---|---|---|---|---|---|
| 1 | M | 1984-4-25 | LGMD2B | c.1033+1G>A (Intron11, het) | N/A |
| 2 | F | 1989-10-27 | LGMD2B | c.1721T>C (p.Leu574Pro, het) c.6241C>T (p.Arg2081Cys, het) | Dysferlin(-) |
| 3 | M | 1997-2-10 | Miyoshi | c.959A>T (p.Asp320Val, het) c.1721T>C (p.Leu574Pro, het) | Dysferlin(-) |
| 4 | M | 1974-11-25 | Miyoshi | c.5905T>C(p.Trp1969Arg, het) c.5509G>A(p.Asp1837Asn, het) | Dysferlin(-) |
| 5 | M | 1979-7-30 | Miyoshi | c.4939G>T(p.Glu1647Ter, het) | Dysferlin(±) |
| 6 | M | 1983-5-19 | LGMD2B | c.3166C>T(p.Arg1056Ter, het); c.1577-2A>C(Intron17, het) | N/A |
| 7 | M | 1983-9-5 | LGMD2B | c.847A>G(p.Thr283Ala,hom) | N/A |
| 8 | F | 1993-7-4 | LGMD2B | c.1468_1469insA(p.Met490AsnfsX15,hom) | Dysferlin(-) |
| 9 | F | 1983-4-21 | LGMD2B | c.1560delT(p.Pro520fs, het); c.3093G>T(p.Trp1031Cys, het) | Dysferlin(-) |
| 10 | F | 1981-4-13 | LGMD2B | c.5657dupG(p.Trp1886fs,hom) | N/A |
| 11 | F | 1979-8-11 | Miyoshi | N/A | Dysferlin(-) |
| 12 | F | 1987-1-3 | Miyoshi | c.1523-1G>A(intron17, het) c.5525G>A(p.Gly1842Asp, het) | Dysferlin(-) |
| 13 | F | 2002-10-1 | LGMD2B | c.4532delA(p.Glu1512SerfsX13, het) c.5626G>T(p.Asp1876Tyr, het) | N/A |
| 14 | F | 1996-12-16 | LGMD2B | c.3112C>T(p.Arg1038Ter,hom) | Dysferlin(-) |
| 15 | F | 1981-11-22 | LGMD2B | c.1033+1G>A(Intron11, het) c.3575-1G>T(Intron32, het) | N/A |
| 16 | M | 1990-8-22 | Miyoshi | c.1033+1G>A (Intron11, het) c.3575-1G>T (Intron32, het) | N/A |
| 17 | M | 2001-4-23 | Miyoshi | c.5830C>T(p.Arg1944Ter, het) c.5346delG(p.Leu1782LeufsX81, het) | Dysferlin(-) |
| 18 | F | 1989-2-28 | LGMD2B | c.1461C>A(p.Cys487Ter, het) c.3033delG(p.Val1011ValfsX77, het) | N/A |
| 19 | M | 1988-12-10 | LGMD2B | c.703C>T(p.Arg235Ter,hom) | N/A |
| 20 | F | 1996-3-26 | LGMD2B | c.2049G>A(p.Trp683Ter, het) c.176delT(p.Leu59ArgfsX92, het) | N/A |
| 21 | M | 1992-2-25 | Miyoshi | c.703C>T(p.Arg235Ter,hom) | N/A |
| 22 | F | 1986-10-1 | Miyoshi | N/A | Dysferlin(-) |
*N/A: not available; DOB: date of birth; IHC: immunohistochemistry; Dysferlin (±) means very faint staining. Miyoshi: miyoshi myopathy; LGMD2B: limb-girdle muscular dystrophy type 2B; Gene mutations: transcript accession numbers NM_001130987; 001130979; 003494.
Abbreviations
DYSF-/-: dysferlin-deficient; LGMD2B: limb-girdle muscular dystrophy type 2B; MM: miyoshi myopathy; DMAT: distal myopathy with anterior tibial; NTA: nanoparticle tracking analysis; EXOdysf: exosomes derived from differentiated myotubes; EXOmyob: exosomes derived from undifferentiated myoblasts; LAMP-1: lysosome-associated membrane protein-1; EXOser: exosomes from normal human serum.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors acknowledge Dr. Xiaoming Feng and Ms. Wanzhu Yang (Tianjin Institute of Hematology, Chinese Academy of Medical Sciences, Tianjin, China) for assistance with two-photon experiments and Dr. Yiqi Seow (Biomedical Sciences Institutes, A* STAR, Singapore) for critical review of the manuscript. This study was funded by Chinese National Basic Research Program (973) (No.2017YFC1001902), National Natural Science Foundation of China (Grant No. 81672124, 81671528, 81501531 and 81361128013), Tianjin Municipal Science and Technology Key Project (Grant No. 14JCZDJC36000 and 14JCQNJC11000) and Tianjin Municipal 13th five-year plan (Tianjin Medical University Talent Project).
Author contributions
X.D. and H. Y. designed the research and analyzed the data. X. D., X. G., Y. D. and N. R. performed the experiments. H. Y. supervised the research. H. Y. wrote the manuscript with input from all authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456-71
2. Cardenas AM, Gonzalez-Jamett AM, Cea LA, Bevilacqua JA, Caviedes P. Dysferlin function in skeletal muscle: Possible pathological mechanisms and therapeutical targets in dysferlinopathies. Exp Neurol. 2016;283:246-54
3. Patel NJ, Van Dyke KW, Espinoza LR. Limb-girdle muscular dystrophy 2B and miyoshi presentations of dysferlinopathy. Am J Med Sci. 2017;353:484-91
4. Lee JJ, Yokota T. Antisense therapy in neurology. J. Pers Med. 2013;3:144-76
5. Cotta A, Carvalho E, da-Cunha-Junior AL, Paim JF, Navarro MM, Valicek J. et al. Common recessive limb girdle muscular dystrophies differential diagnosis: why and how? Arquivos de Neuro-psiquiatria. 2014;72:721-34
6. Kobayashi K, Izawa T, Kuwamura M, Yamate J. Dysferlin and animal models for dysferlinopathy. J Toxicol Pathol. 2012;25:135-47
7. Krahn M, Labelle V, Borges A, Bartoli M, Levy N. Exclusion of mutations in the dysferlin alternative exons 1 of DYSF-v1, 5a, and 40a in a cohort of 26 patients. GenetTest Mol Biomarkers. 2010;14:153-4
8. Lostal W, Bartoli M, Roudaut C, Bourg N, Krahn M, Pryadkina M. et al. Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy. PLoS One. 2012;7:e38036
9. Sondergaard PC, Griffin DA, Pozsgai ER, Johnson RW, Grose WE, Heller KN. et al. AAV.dysferlin overlap vectors restore function in dysferlinopathy animal models. Ann Clin Transl Neurol. 2015;2:256-70
10. Llanga T, Nagy N, Conatser L, Dial C, Sutton RB, Hirsch ML. Structure-based designed nano-dysferlin significantly improves dysferlinopathy in BLA/J mice. Mol Ther. 2017;25:2150-62
11. Escobar H, Schowel V, Spuler S, Marg A, Izsvak Z. Full-length dysferlin transfer by the hyperactive sleeping beauty transposase restores dysferlin-deficient muscle. Mol Ther Nucleic Acids. 2016;5:e277
12. Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol life Sci. 2017 DOI 10.1007/s00018-017-2595-9
13. Juan T, Furthauer M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Sem Cell Dev Biol. 2017 DOI: 10.1016/j.semcdb.2017.08.022
14. Lasser C. Exosomes in diagnostic and therapeutic applications: biomarker, vaccine and RNA interference delivery vehicle. Expert Opin Biol Ther. 2015;15:103-17
15. Rao Q, Zuo B, Lu Z, Gao X, You A, Wu C. et al. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology. 2016;64:456-72
16. Bastos N, Ruivo CF, da Silva S, Melo SA. Exosomes in cancer: Use them or target them?. Sem Cell Dev Biol. 2017 DOI.10.1016/j.semcdb.2017.08.009
17. Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L. et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42
18. Yuyama K, Igarashi Y. Exosomes as carriers of alzheimer's amyloid-ss. Front Neurosci. 2017;11:229
19. Forterre A, Jalabert A, Berger E, Baudet M, Chikh K, Errazuriz E. et al. Proteomic analysis of C2C12 myoblast and myotube exosome-like vesicles: a new paradigm for myoblast-myotube cross talk? PLoS One. 2014;9:e84153
20. Prada I, Meldolesi J. Binding and fusion of extracellular vesicles to the plasma membrane of their cell targets. Int J Mol Sci. 2016:17
21. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569-79
22. Barile L, Vassalli G. Exosomes: Therapy delivery tools and biomarkers of diseases. PharmacolTher. 2017;174:63-78
23. Cohen TV, Cohen JE, Partridge TA. Myogenesis in dysferlin-deficient myoblasts is inhibited by an intrinsic inflammatory response. Neuromus Disord. 2012;22:648-58
24. Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H. et al. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet. 2004;13:1999-2010
25. Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R. et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168-72
26. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ. et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161-72
27. Philippi S, Bigot A, Marg A, Mouly V, Spuler S, Zacharias U. Dysferlin-deficient immortalized human myoblasts and myotubes as a useful tool to study dysferlinopathy. PLoS Curr. 2012;4:RRN1298
28. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783-4
29. Jamur MC, Oliver C. Permeabilization of cell membranes. Methods in Mol Biol. 2010;588:63-6
30. Hernandez-Deviez DJ, Howes MT, Laval SH, Bushby K, Hancock JF, Parton RG. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J Biol Chem. 2008;283:6476-88
31. Glover L, Brown RH Jr. Dysferlin in membrane trafficking and patch repair. Traffic. 2007;8:785-94
32. Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157-69
33. Kerr JP, Ziman AP, Mueller AL, Muriel JM, Kleinhans-Welte E, Gumerson JD. et al. Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc Natl AcadSci U S A. 2013;110:20831-6
34. Kerr JP, Ward CW, Bloch RJ. Dysferlin at transverse tubules regulates Ca(2+) homeostasis in skeletal muscle. Frontiers in Physiol. 2014;5:89
35. Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem. 1989;264:8171-8
36. Gallardo E, Ankala A, Nunez-Alvarez Y, Hegde M, Diaz-Manera J, Luna ND. et al. Genetic and epigenetic determinants of low dysferlin expression in monocytes. Hum Mutat. 2014;35:990-7
37. De Luna N, Freixas A, Gallano P, Caselles L, Rojas-Garcia R, Paradas C. et al. Dysferlin expression in monocytes: a source of mRNA for mutation analysis. Neuromus Disord. 2007;17:69-76
38. Meznaric M, Gonzalez-Quereda L, Gallardo E, de Luna N, Gallano P, Fanin M. et al. Abnormal expression of dysferlin in skeletal muscle and monocytes supports primary dysferlinopathy in patients with one mutated allele. Eur JNeurol. 2011;18:1021-3
39. Gonzales PA, Pisitkun T, Hoffert JD, Tchapyjnikov D, Star RA, Kleta R. et al. Large-scale proteomics and phosphoproteomics of urinary exosomes. J Am SocNephrol. 2009;20:363-79
40. Urtizberea JA, Bassez G, Leturcq F, Nguyen K, Krahn M, Levy N. Dysferlinopathies. Neurol India. 2008;56:289-97
41. de Luna N, Gallardo E, Soriano M, Dominguez-Perles R, de la Torre C, Rojas-Garcia R. et al. Absence of dysferlin alters myogenin expression and delays human muscle differentiation "in vitro". J Biol Chem. 2006;281:17092-8
Author contact
![]() Corresponding author: HaiFang Yin, Email: haifangyinedu.cn; Tel: +86 (0)22 83336537; Fax: +86 (0)22 83336537; Address: Tianjin Medical University, Qixiangtai Road, Heping District, Tianjin, 300070, China.
Corresponding author: HaiFang Yin, Email: haifangyinedu.cn; Tel: +86 (0)22 83336537; Fax: +86 (0)22 83336537; Address: Tianjin Medical University, Qixiangtai Road, Heping District, Tianjin, 300070, China.
Citation styles
APA
Dong, X., Gao, X., Dai, Y., Ran, N., Yin, H. (2018). Serum exosomes can restore cellular function in vitro and be used for diagnosis in dysferlinopathy. Theranostics, 8(5), 1243-1255. https://doi.org/10.7150/thno.22856.
ACS
Dong, X.; Gao, X.; Dai, Y.; Ran, N.; Yin, H. Serum exosomes can restore cellular function in vitro and be used for diagnosis in dysferlinopathy. Theranostics 2018, 8 (5), 1243-1255. DOI: 10.7150/thno.22856.
NLM
Dong X, Gao X, Dai Y, Ran N, Yin H. Serum exosomes can restore cellular function in vitro and be used for diagnosis in dysferlinopathy. Theranostics 2018; 8(5):1243-1255. doi:10.7150/thno.22856. https://www.thno.org/v08p1243.htm
CSE
Dong X, Gao X, Dai Y, Ran N, Yin H. 2018. Serum exosomes can restore cellular function in vitro and be used for diagnosis in dysferlinopathy. Theranostics. 8(5):1243-1255.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.