Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
From quiescent to activated...
Principles of molecular MRI of...
Molecular MRI of endothelial...
Molecular MRI of endothelial...
Molecular MRI of endothelial...
Molecular MRI of endothelial...
Remaining challenges in clinical...
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(5):1195-1212. doi:10.7150/thno.22662 This issue Cite
Review
Molecular Magnetic Resonance Imaging of Endothelial Activation in the Central Nervous System
Maxime Gauberti1,2 ![]() , Antoine P. Fournier1, Fabian Docagne1, Denis Vivien1,3, Sara Martinez de Lizarrondo1
, Antoine P. Fournier1, Fabian Docagne1, Denis Vivien1,3, Sara Martinez de Lizarrondo1 ![]()
1. Normandie Univ, UNICAEN, INSERM, INSERM UMR-S U1237, Physiopathology and Imaging for Neurological Disorders (PhIND), Cyceron, 14000 Caen, France.
2. CHU Caen, Department of diagnostic imaging and interventional radiology, CHU de Caen Côte de Nacre, Caen, France.
3. CHU Caen, Clinical Research Department, CHU de Caen Côte de Nacre, Caen, France.
Received 2017-9-3; Accepted 2018-1-12; Published 2018-2-2
Abstract

Endothelial cells of the central nervous system over-express surface proteins during neurological disorders, either as a cause, or a consequence, of the disease. Since the cerebral vasculature is easily accessible by large contrast-carrying particles, it constitutes a target of choice for molecular magnetic resonance imaging (MRI). In this review, we highlight the most recent advances in molecular MRI of brain endothelial activation and focus on the development of micro-sized particles of iron oxide (MPIO) targeting adhesion molecules including intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), P-Selectin and E-Selectin. We also discuss the perspectives and challenges for the clinical application of this technology in neurovascular disorders (ischemic stroke, intracranial hemorrhage, subarachnoid hemorrhage, diabetes mellitus), neuroinflammatory disorders (multiple sclerosis, brain infectious diseases, sepsis), neurodegenerative disorders (Alzheimer's disease, vascular dementia, aging) and brain cancers (primitive neoplasms, metastasis).
Keywords: leucocytes, lymphocytes, neuroinflammation, ultra-small particles of iron oxide (USPIO), antibodies
Introduction
Inflammation is a hall-mark of most neurological disorders [1]. Following injury to the central nervous system (CNS), both innate and adaptive immune systems are at play to limit the extent of diseases and mediate repair and regeneration [2]. Yet, abnormal activation of the immune system in the brain can lead to autoimmune disorders [3] and worsen brain damages during injuries of the CNS [4, 5]. Because of its ubiquitous involvement, neuroinflammation has been one of the most targeted pathophysiological mechanisms with the aim to prevent and treat neurological disorders [6]. The ability to detect, quantify and monitor the inflammatory response of the CNS could have major implications for diagnosis and prediction of the therapeutic response [7]. Indeed, the recent application of molecular magnetic resonance imaging (MRI) to study endothelial activation appears as one of the most promising axis of research.
To detect neuroinflammation, different diagnostic modalities are available. First, plasma biomarkers can be used. Unfortunately, no reliable and specific plasma biomarker of neuroinflammation has been identified to date. This could be explained by the presence of the blood-brain barrier (BBB), which prevents the transport of biomarkers from the brain to the blood [8]. Moreover, inflammation is a natural response to injury with common pathways in different organs. Therefore, most potential biomarkers for neuroinflammation are also present in cases of peripheral inflammation, making their presence in the blood non-specific for neurological disorders[9]. An alternative is the use of cerebrospinal fluid (CSF) biomarkers of neuroinflammation, which have been used with some success [10]. For instance, C-X-C motif chemokine 13 (CXCL13) [11], chemokine (C-C motif) ligand 2 (CCL2 also known as monocyte chemoattractant protein-1, MCP-1) [12], chitinase-3-like protein 1 (CHI3L1) [13] and glial fibrillary acidic protein (GFAP) have all been associated with neuroinflammation [10]. However, repeated CSF sampling is inconvenient for patients, CSF biomarkers cannot discriminate the localization of the inflammatory process, and the alteration of the parenchymal CSF circulation may be a confounding factor by preventing the transport of biomarkers from the brain to the CSF [14].
To overcome the limitations of plasma and CSF biomarkers, molecular imaging of neuroinflammation has been developed [15]. The common principle of all molecular imaging methods is to detect in situ and by means of an imaging modality a protein that is overexpressed during neuroinflammation. To achieve this goal, i) a relevant target has to be identified, ii) a contrast agent able to reach and bind to this target with a high affinity has to be designed and iii) an imaging method able to detect this contrast agent has to be developed. To date, only nuclear medicine technology, using single photon emission computed tomography (SPECT) or positron emission tomography (PET) provides this complete framework and is currently performed in humans in a number of clinical conditions [16]. In particular, using microglial translocator protein (TSPO) ligands and PET, highly sensitive imaging of neuroinflammation can be performed, as demonstrated for instance in stroke [17], Alzheimer's disease [18] and multiple sclerosis [19]. Despite its acknowledged clinical value, these latter methods of molecular imaging are impeded by low spatial resolution, large acquisition duration, low availability, high cost and exposure to ionizing radiation. In contrast, molecular MRI is mostly devoid of these drawbacks. However, it has remained limited to proof-of-concept studies in experimental models without establishment of a given clear path towards clinical translation due to its low sensitivity and specificity [20]. Recently, the development of micro-sized particles of iron oxide (MPIO) as contrast agents has allowed a dramatic improvement of molecular MRI sensitivity and specificity and these particles may have the capacity for being a game changer for clinical translation [7, 21].
Development of molecular MRI to study neuroinflammation was previously restrained by a fundamental dilemma since the imaging targets were mostly intraparenchymal: the contrast agent should be small enough to cross the BBB but should also carry sufficiently high amounts of contrast material to induce detectable changes in MRI contrast [7]. Thus, most studies reported the use of nano-sized targeted contrast agents that are able to accumulate in the brain parenchyma in inflammatory conditions. As such, myeloperoxidase imaging using Bis-5HT-DTPA [22] or targeted ultra-small particles of iron oxide (USPIO, from 10 nm to 50 nm) with various ligands on their surface that are able to bind to inflammation specific proteins have been used [23-25]. However, a significant part of the contrast agent accumulation in the inflammatory brain parenchyma was due to a leaky BBB and increased extracellular volume in inflammatory conditions, a phenomenon known as the “enhanced permeability and retention” (EPR) effect [26]. Consequently, the additional accumulation due to the specific binding of the contrast agents to its target accounts only for a small portion of the MRI contrast changes [7]. In addition, the sensitivity of these methods is intrinsically low due to the small amount of contrast material carried by small contrast agents. Furthermore, a long delay between injection and imaging has to be observed to allow the contrast agents to accumulate in sufficiently high enough concentrations to be detected, which may restrict widespread clinical use, particularly in the acute setting. For all these reasons, although this field of research is still very active, it is now assumed that without dramatic improvement in contrast agent design or MRI detection methods, these types of small contrast agents remains of limited interest for molecular MRI of neuroinflammation in a clinically relevant context.
In 2007, the group of Robin P. Choudhury shifted the paradigm of molecular MRI of neuroinflammation [21]. Instead of trying to image intraparenchymal proteins, they chose to target vascular cell adhesion molecule-1 (VCAM-1), an adhesion molecule that is expressed on the luminal side of endothelial cells in inflammatory conditions. The target being endovascular, there was no longer a limit on the size of the contrast agent that could be used. Therefore, they used larger, micro-sized contrast agents (MPIO) harboring monoclonal antibodies directed against VCAM-1 on their surface. Using this contrast agent and standard MRI acquisition methods, they were able to unmask acute brain inflammation in a murine experimental model. This was, to our knowledge, the first demonstration of the feasibility of sensitive and specific molecular imaging of brain inflammation using MRI. Since then, their findings have been reproduced in several studies, the methodology further improved and the feasibility of unmasking CNS inflammation by targeting MPIOs to specific proteins expressed by the activated cerebrovasculature has been demonstrated in numerous neurological disorders [7, 27-30].
In the following sections, we will first present the basic principles of molecular MRI of endothelial activation and its specific challenges for brain imaging. Then, we will review the most striking advances made in the last 10 years and the remaining interrogations in molecular MRI of endothelial activation in neurovascular, neuroinflammatory, neurodegenerative and neuro-oncological disorders. Lastly, we will discuss the challenges that must be overcome before the first human studies may be envisaged.
From quiescent to activated endothelial cells in the central nervous system
Considered as an immunologically privileged organ, the CNS requires surveillance by blood-derived cells to detect and fight potential infections [31]. Central to the neuroinflammatory process is therefore the ability of circulating leukocytes to reach the brain parenchyma (Figure 1). Leukocyte diapedesis is triggered by a cascade of pro-inflammatory events: subsequent to CNS damage, resident brain cells (mainly astrocytes and microglia) release inflammatory cytokines and chemokines including tumor necrosis factor and interleukin 1-β, which activate the endothelium and lead to the luminal expression of selectins and adhesion molecules. This activated endothelium supports the adhesion of circulating leukocytes that are attracted by the gradient of chemokines originating from the CNS. Lastly, leukocytes egress from the blood to the brain to participate in the neuroinflammatory reaction. The roles of cerebral endothelial cells during the CNS inflammatory response, the related signaling pathways, and the crosstalk between cerebral endothelial cells and other resident CNS cells have been recently reviewed elsewhere [32]. Here, we will briefly describe the main mechanisms involved in endothelial activation and highlight the key proteins involved in this process that constitute potential imaging targets.
Schematic representation of the multistep process of leukocyte diapedesis. In inflammatory conditions, leukocytes first interact with the selectins expressed by the CNS endothelium, allowing their rolling to the vessel wall. Rolling leukocytes sense chemokines that induce their activation, thereby triggering the activation of their integrins and subsequent interactions with adhesion molecules (VCAM-1 and ICAM-1). Lastly, the leukocytes transmigrate across the endothelial layer, typically by a paracellular route, by a PECAM-1 dependent mechanism. LFA-1: lymphocyte function-associated antigen-1; VLA-4: very late antigen-4; PSGL-1: P-selectin glycoprotein ligand-1; VCAM-1: vascular cell adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1; PECAM-1: platelet endothelial cell adhesion molecule-1.
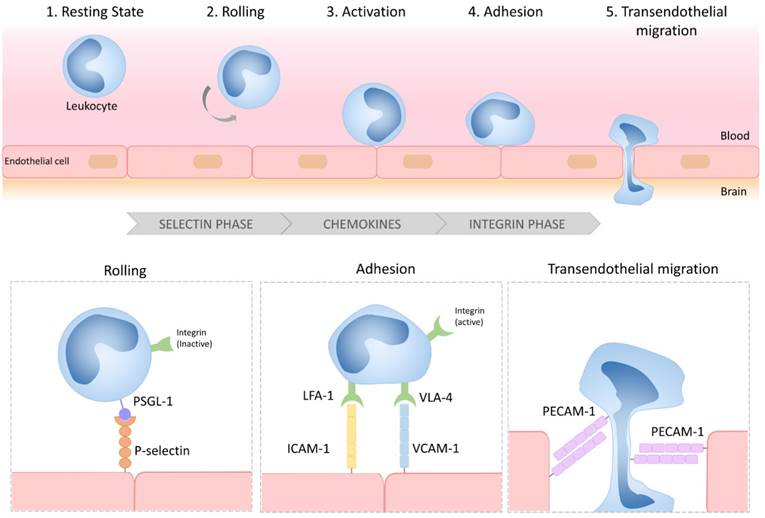
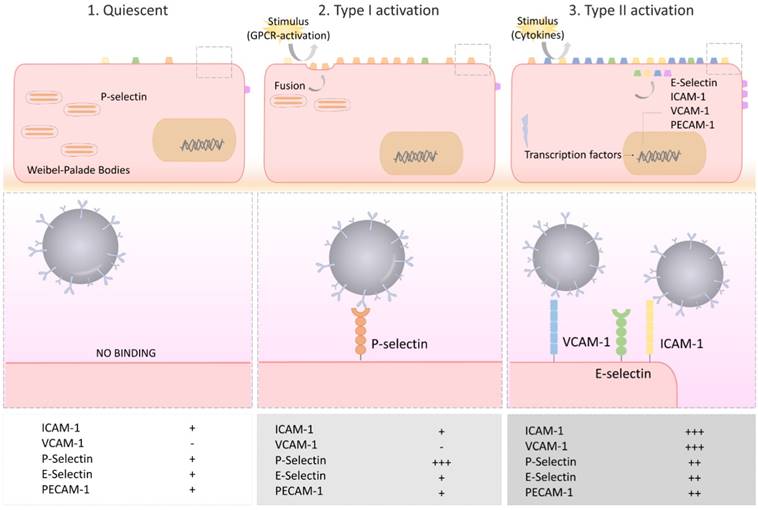
Under physiological conditions, the vascular endothelium of the CNS is in a quiescent state characterized by a limited interaction with circulating leukocytes. Accordingly, adhesion molecules such as intercellular adhesion molecule I (ICAM-1), E-selectin and P-selectin are expressed in only trace amounts at the extracellular surface of the endothelial microvasculature (Figure 2). Upon injury, the endothelial cells shift from a quiescent to an activated phenotype, involving increased vascular permeability, switch to a pro-thrombotic state, production of pro-inflammatory cytokines and exposure of adhesion molecules. According to current knowledge, endothelial activation can be divided into type I and type II[33]. Type I is characterized by an increase in cytoplasmic Ca2+ concentration typically driven by G-protein coupled receptor activation. This transient elevation in Ca2+ triggers the exocytosis of Weibel-Palade bodies (small, preformed intracytoplasmic vesicles containing essentially two proteins, von-Willebrand Factor and P-selectin) and induces the addressing of P-Selectin to the endothelial surface, which contributes to leukocyte binding on the vessel wall. Interestingly, the CNS vasculature presents a specific regulation of P-selectin expression, that was proposed to be independent on Weibel-Palade bodies [34], thus suggesting that the role of P-selectin is different in the cerebrovasculature and in other vascular beds [35]. Type II activation of endothelial cells is a more sustained inflammatory response induced by pro-inflammatory cytokines. It is characterized by an increased transcription and protein synthesis of cytokines, chemokines and adhesion molecules. In particular, type II activated endothelial cells present high expression levels of VCAM-1, ICAM-1 and E-selectin. Once engaged, leukocyte transmigration requires platelet endothelial cell adhesion molecule 1 (PECAM-1), which is expressed in endothelial cell intercellular junctions [32].
Schematic representation of the selectin and adhesion molecule expression profiles of quiescent, type I activated and type II activated endothelial cells. Type I activated endothelial cells present high surface levels of P-selectin. Type II activated endothelial cells present high surface levels of VCAM-1, ICAM-1 and E-selectin. Each of these four proteins are suitable targets for molecular MRI of endothelial activation. VCAM-1: vascular cell adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1; PECAM-1: platelet endothelial cell adhesion molecule-1.
P- and E-selectin are mainly involved in the initial recruitment of leukocytes (rolling) whereas ICAM-1 and VCAM-1 are mainly involved in firm leukocyte adhesion. The main ligands of both P- and E-selectins are P-selectin glycoprotein ligand-1 (PSGL-1) and sialylated carbohydrates, which are present on the surface of neutrophils, monocytes, platelets, eosinophils and some lymphocyte subtypes. Ligand selectivity and vascular bed expression profiles of ICAM-1 and VCAM-1 explain their differential roles [36, 37]: the main ligands of ICAM-1 are macrophage adhesion ligand-1 (Mac-1) and leukocyte associated antigen-1 (LFA-1), whereas the main ligand of VCAM-1 is very late antigen-4 (VLA-4). According to the expression profile of VLA-4, VCAM-1 is mainly involved in the diapedesis of lymphocytes, monocytes and eosinophils. Importantly, LFA-1 and VLA-4 are integrins that require a change in their conformation to acquire high affinity for ICAM-1 and VCAM-1, a process triggered by an increase in cytosolic Ca2+. As a result, only activated leukocytes can bind ICAM-1 and VCAM-1. PECAM-1 has both homophilic (binding with itself) and heterophilic ligands [38]. Except for PECAM-1 (which is not reachable by large contrast carrying particles [39]), all these proteins, which are differentially expressed by activated and quiescent endothelial cells, are potential targets for molecular MRI of endothelial activation.
Principles of molecular MRI of endothelial activation using targeted MPIOs
As a common design with other targeted contrast agents, targeted MPIOs are made of two parts. The first, the contrastophore, is the part responsible for the changes in the MRI contrast. The second, the pharmacophore, is the part responsible for the binding of the contrastophore to its target. To date, most MPIO studies have used monoclonal antibodies as pharmacophores and iron oxide particles with diameters of 1 to 4 µm as contrastophores [21, 30, 40, 41]. In particular, commercial myOne tosyl-activated MPIOs (from Invitrogen) are among the most popular contrastophores. Using these particles with monoclonal antibodies, the conjugation chemistry requires only one step and leads to the formation of amide bonds between MPIOs and antibodies (the reaction occurs mainly between the tosyl group of the MPIOs with the lysine of the antibodies). Step-by-step protocols to produce targeted MPIOs have been reported elsewhere [42, 43]. On a single 1 µm MPIO, one can expect the binding of ~109 antibodies [21]. The general structure of MPIOs and the previously reported contrastophores and pharmacophores are shown on Figure 3.
Schematic representation of the structure of a typical targeted microsized particle of iron oxide (MPIO) for molecular MRI. Targeted MPIOs are made of two parts: the first, the contrastophore, is the part responsible for the changes in the MRI contrast. The second, the pharmacophore, is the part responsible for the binding of the contrastophore to its target. In this figure are listed the most common contrastophores and pharmacophores. In the lower part of the figure is a simplified view of the biological function of the targets of the most popular pharmacophores for molecular MRI of endothelial activation.
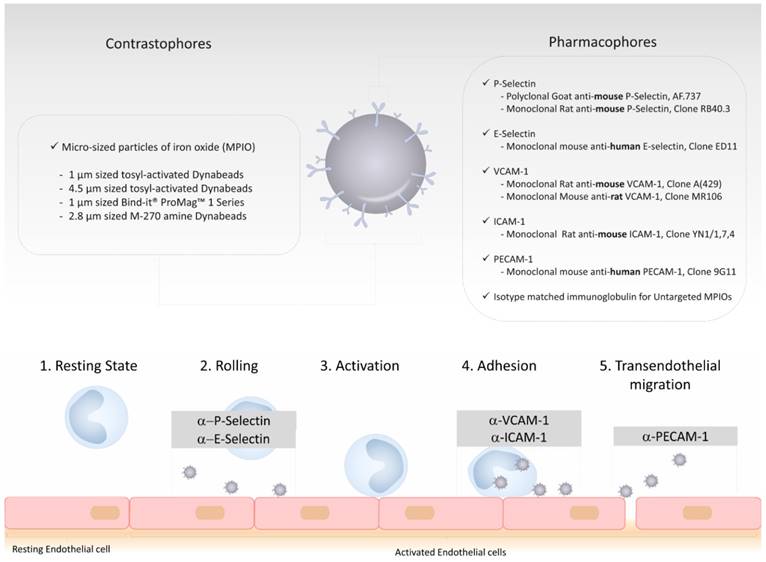
The prototypic injection-ready targeted MPIOs are therefore made of microsized spherical particles containing iron oxide that are coated with a large number of monoclonal antibodies on their surface. Compared to other contrast agents, the pharmacokinetic properties of MPIOs make them especially suitable for molecular imaging. First, because of their size, their distribution volume is small and restricted to the intravascular compartment. Even when the BBB is open, MPIOs are restricted to the vascular compartment and cannot passively extravasate into the brain parenchyma, since the size of the pores between endothelial cells do not exceed 50 nm (i.e., at least 20 times less than the size of MPIOs) [44]. This explains the very high specificity of MPIOs, as evidenced by the lack of untargeted MPIO-induced signal change in the experimental models reported so far when control IgG are used as pharmacophores [28, 43, 45]. This is a key feature of MPIOs compared to the more commonly used USPIO (diameter ranging from 10 to 50 nm), that are susceptible to passive extravasation and therefore non-specific accumulation. The absence of significant off-target binding in the CNS also suggests that MPIOs are especially well suited for quantitative imaging of endothelial activation. Second, MPIOs have a very short plasma half-life (estimated to be <30 seconds) [43]. Thus, no free MPIO can be detected in the circulation using T2*-weighted MRI as soon as 60 seconds after injection. This is explained by the rapid uptake of circulating, unbound MPIOs by the reticulo-endothelial system in the liver and the spleen [43]. Moreover, in studies reporting the use of VCAM-1 targeted MPIOs, binding of the MPIOs on activated brain endothelial cells occurred mostly during the first vascular pass and only 50% of them are unbound at between 20 minutes and 2 hours [43]. In other words, whereas the plasma half-life of VCAM-1 targeted MPIOs is estimated to be a few seconds, their “binding half-life” on activated vessels is close to 100 minutes. Therefore, difference in MRI signal between activated vessels containing VCAM-1 targeted MPIOs bound to endothelial cells and quiescent vessels is maximal in the first minutes following MPIOs injection and remains high enough for MRI during at least one hour, with a progressive decline in sensitivity. Similar results were obtained using P-selectin targeted MPIOs [27].
Another key feature of MPIOs is their ability to induce strong changes in MRI contrast. MPIOs have potent superparamagnetic properties and high positive magnetic susceptibility. Basically, MPIOs are able to strengthen the magnetic field in their close vicinity [46]. Therefore, the water protons that are close to the MPIOs experience a higher magnetic field than the water protons that are far from the MPIOs. Since the Larmor precession frequency is directly proportional to the magnetic field, this implies that the precessions of the magnetic moments of the water protons close to the MPIOs are faster than the others. Provided that the MRI signal is maximal when all the magnetic moments are in the same direction (“in phase”), the small differences in precession speed due to the presence of MPIOs induces progressive misalignment of the magnetic moments (“dephasing”) and subsequent signal drop. This explains the appearance of MPIO on T2*-weighted (or susceptibility-weighted) imaging as small spheres of signal void[46]. Interestingly, the dephasing of the protons extends to a distance of ~50 times the size of the particles (~50 µm for a single 1 µm MPIO), leading to large contrast changes extending over 50 µm. Consequently, a single isolated MPIO can be detected using high-resolution MRI (as previously reported using a 70 µm isotropic resolution at 7T), resulting in a very high MPIO detection sensitivity [29, 46]. Notably, the magnetic susceptibility effects of MPIOs are expected to be even more prominent using very high field MRI (>7T), which could be exploited to further increase the sensitivity of molecular MRI.
Molecular MRI of endothelial activation in neurovascular disorders
Inflammation is considered to be one of the major pathogenic processes occurring in the acute and subacute phases of neurovascular disorders [47, 48]. Here we will discuss the application of molecular MRI of endothelial activation in ischemic stroke, intraparenchymal hemorrhage and subarachnoid hemorrhage, the three most frequent neurovascular disorders worldwide [49]. In each of these disorders, monitoring the temporo-spatial regulation of endothelial activation in the affected brain parenchyma could have significant clinical applications, especially by identifying the patients who could potentially benefit from immunomodulatory treatments [50, 51].
Ischemic stroke is usually caused by the occlusion of a cerebral artery by a blood clot, leading to a reduction of the cerebral blood flow and subsequent brain injury [52-54]. The affected brain tissue can be divided in three regions: the ischemic core, where the brain is definitively infarcted; the “ischemic penumbra”, a region at risk that will be ultimately recruited by the ischemic core if no reperfusion occurs; a region of benign oligemia, which presents a reduction in blood flow that is not sufficient to result in brain infarction [55]. In the absence of rapid reperfusion (either spontaneous, or after treatment by intravenous thrombolysis or mechanical thrombectomy), the ischemic core will grow and recruit the ischemic penumbra [56]. This schematic representation of the suffering brain parenchyma has been the basis for the development of reperfusion strategies during the last 20 years, with significant clinical successes [57].
The pathophysiological mechanisms linking arterial occlusion to brain cell death are classically described as being excitotoxicity [58], occurring in the first minutes and due to excessive activation of glutamatergic neuronal receptors, followed by subsequent events that participate to the demise of brain tissue such as peri-infarct depolarization, apoptosis and inflammation [59, 60]. Notably, suppression of inflammation significantly reduces the final ischemic lesion size in several experimental models of ischemic stroke [48, 61]. Although the underlying mechanisms are still not fully understood, inflammation after brain ischemia is characterized by the rapid infiltration to the brain parenchyma of peripheral leucocytes, probably triggered by the release of damage associated molecular patterns by suffering cells [62]. The endothelial cells of the BBB are key players in this pathogenic mechanism by mediating the diapedesis of leucocytes from the blood to the brain, through the expression of adhesion molecules [63-65]. Molecular MRI of endothelial activation in this context brings unique information regarding the spatial and temporal regulation of leucocyte infiltration in the brain, in a noninvasive manner [7]. This is especially relevant for translational research since adhesion molecules and leucocytes are the targets of current investigation for stroke treatment (such as natalizumab or fingolimod) [66-69].
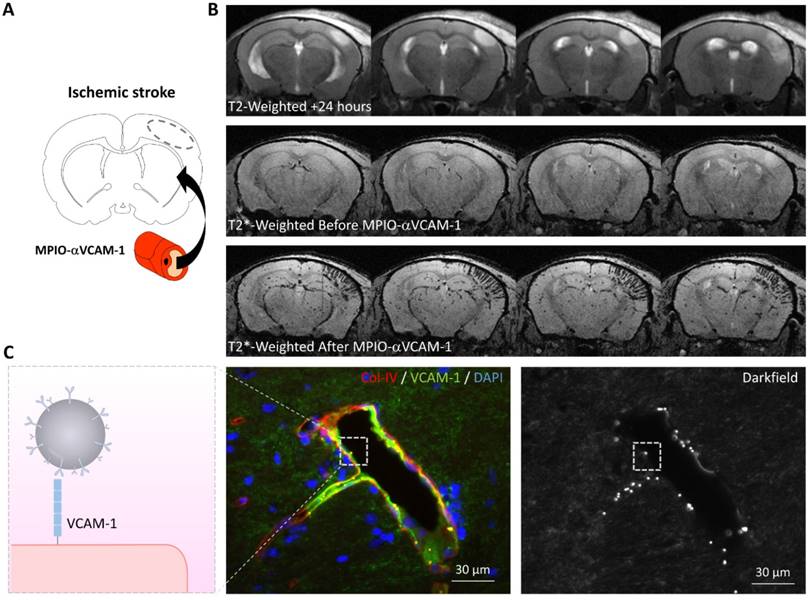
For instance, molecular MRI of VCAM-1 has provided evidence for the existence of an “inflammatory penumbra” in ischemic stroke (Figure 4) [28, 70]. It is classically considered that a few hours after ischemic onset, the ischemic core has recruited the whole ischemic penumbra and the potential beneficial effect of reperfusion is lost. However, longitudinal evaluation of ischemic lesion development in preclinical and clinical studies has revealed that the ischemic lesion continues to grow during the first 5 days after the onset of ischemia [69, 71, 72]. Current evidence suggests that this lesion growth is mediated by inflammatory mechanisms and especially infiltration by T-cells [73]. Immunohistological studies in mice revealed that T-cells were predominantly located in the peri-infarct zone, where viable cells (including neurons) were still present [28]. Therefore, these data suggested that in the peri-infarct area, endothelial cells are activated and mediate the diapedesis of T-cells, which ultimately trigger a delayed and inflammation-related expansion of the initial lesion [70]. This has been observed using molecular MRI of VCAM-1 in experimental stroke models: 24 hours after ischemic onset, MPIO-αVCAM-1 enhanced MRI revealed that there is a strong overexpression of VCAM-1 in the peri-infarct area [28]. These regions that overexpress VCAM-1 are progressively recruited by the ischemic core in the following days. Interestingly, anti-inflammatory treatments prevent this secondary lesion growth. Thus, using molecular MRI of VCAM-1, it is possible to unmask an inflammatory area at risk, surrounding the initial ischemic lesion and secondarily infiltrated by lymphocytes, that is ultimately recruited by the ischemic core. By analogy with the ischemic penumbra, the brain volume at risk of secondary infarction is called the “inflammatory penumbra”, which is defined by the mismatch between the VCAM-1 overexpressing region revealed by molecular MRI and the ischemic core revealed by diffusion-weighted imaging [28, 70]. Notably, the size of this “inflammatory penumbra” varies between experimental models [28] and probably between stroke patients. Such an observation would further support the use of molecular imaging of endothelial activation to select individuals with stroke that would benefit the most from anti-inflammatory treatment.
Molecular MRI of VCAM-1 in the subacute phase of ischemic stroke. (A) Schematic representation of the experiment. MPIO-αVCAM-1 were injected 24 hours after stroke onset (induced by electrocoagulation of the middle cerebral artery). (B) In vivo MRI before and after MPIO-αVCAM-1 injection showing cortical signal voids extending outside the ischemic lesion (as revealed by a hypersignal on T2-weighted imaging), revealing the inflammatory penumbra, which is defined by the mismatch between the VCAM-1 overexpressing region and the ischemic lesion. (C) Schematic representation (left) and immunohistological images of the activated brain vessels in the inflammatory penumbra with numerous bound MPIO-αVCAM-1 visible as white dots on the darkfield image. Further details on this experimental data are available in the paper by Gauberti et al. [28].
Whereas VCAM-1 is mainly implicated in the diapedesis of monocytes and lymphocytes, other adhesion molecules mediate the diapedesis of neutrophils, which are among the first cells to adhere to the cerebrovasculature after stroke. Deddens and co-workers performed molecular MRI of ICAM-1 in the subacute phase of stroke [40]. Using MPIO-αICAM-1 they were able to reveal the overexpression of ICAM-1 by activated endothelial cells at 24 hours after ischemic onset in a model of transient mechanical occlusion of the middle cerebral artery. They recently expanded their study by performing longitudinal molecular MRI of ICAM-1 at 1, 2, 3, 7 and 21 days post stroke [74]. They found that MPIO-αICAM-1 binding was detectable at day 1, 2 and 3 (being maximal at day 2), where ICAM-1 expression extended in the peri-infarct area, in line with the “inflammatory penumbra” concept. Interestingly, at later time points, whereas MRI failed to detect the MPIO, additional immunohistological analyses revealed that a few MPIOs were present in the ischemic tissue and were colocalized with leucocytes. This secondary finding supports previous studies reporting the expression of ICAM-1 by some leucocyte subtypes [74], suggesting that ICAM-1 targeted MPIOs could be used to track leucocytes in vivo.
Molecular imaging of endothelial activation may also provide clues about the etiology of ischemic stroke. Thrombus migration from large artery atherosclerosis is among the most common causes of acute ischemic stroke [75]. Usually, it is secondary to the rupture of an atherosclerotic plaque in the carotid artery, followed by thrombus formation on the exposed subendothelium and subsequent thrombus embolization in an intracranial artery. At the onset of stroke there is often no sign of these events on conventional imaging [76]. Therefore, determination of stroke etiology relies primarily on the severity of the arterial stenosis and elimination of other potential causes, including cardioembolism. Of note, determining the etiology of a stroke is mandatory to introduce the correct thromboprophylactic treatment (i.e., antiplatelet versus anticoagulant) [77]. In this context, the ability to detect the atherosclerotic plaque responsible for thrombus formation could be of significant clinical value. Interestingly, it has been previously demonstrated that unstable and ruptured atherosclerotic plaques present high levels of endothelial activation markers. By means of P-selectin and VCAM-1 targeted MPIOs enhanced MRI, McAteer et al. demonstrated that it is possible to detect the activated endothelium inside Atherosclerotic plaques [41]. They used atherosclerosis-prone apolipoprotein E-deficient mice and demonstrated that, after intravenous injection the targeted MPIOs rapidly accumulate in the aortic root, where the atherosclerotic lesions are the most prominent. In an acute stroke setting, it is thus tempting to speculate that molecular MRI of endothelial activation could be used to find the culprit lesion and therefore to determine stroke etiology with a greater specificity. This is also supported by a recent study showing that dual-targeted MPIO-enhanced MRI could characterize plaque vulnerability and inflammation at the molecular level, by targeting P-selectin and VCAM-1 [78].
Another recent study demonstrated that molecular MRI of endothelial activation could be used to diagnose a transient ischemic attack (TIA), a focal neurological symptom of ischemic origin resolving spontaneously without any abnormality on MRI [76], a virtue that could lead to better stroke prevention [27]. Indeed, it is currently admitted that about 20% of patients with ischemic stroke have had a preceding TIA. In a TIA, the duration of brain ischemia is too short to induce substantial cell death and thus to induce changes on unenhanced MRI. The diagnosis is often difficult because the ischemic nature of the event cannot be ascertained and numerous alternative diagnosis can be misleading [79]. Consequently, diagnosis of TIA may be done by excess (exposing patients to thrombo-prophylactic drugs with potentially serious side effects) or by default (exposing patients to the risk of a recurrent and disabling stroke) [80]. Using molecular imaging of P-selectin, experimental evidence suggests that it is possible to detect the endothelial activation triggered by the TIA and therefore to distinguish TIA from stroke mimics, such as epilepsy or migraine [27]. In a mouse model of TIA induced by transient mechanical compression of the middle cerebral artery, Quenault et al. have shown that molecular MRI performed 24 hours after the TIA and enhanced by MPIOs-αP-selectin revealed the affected vascular territory that presents an overexpression and increased surface exposure of P-selectin [27]. In a clinical setting, this could provide a large diagnostic window to make a positive diagnosis of TIA and therefore, to select the patients who will benefit the most from thrombo-prophylactic treatment.
As reported for ischemic stroke, several studies demonstrated a deleterious role for inflammation after hemorrhagic stroke [81, 82]. In the first days following intraparenchymal hemorrhage, there is activation of the cerebrovasculature and subsequent infiltration of leucocytes, which have a role in the removal of the blood degradation products. Interestingly, patients that have high levels of VCAM-1 in their cerebrospinal fluid have a poor stroke outcome [83]. Moreover, recent small clinical trials reported a beneficial effect of immunomodulatory treatment, aimed at lowering infiltration of lymphocytes in the brain parenchyma on hemorrhagic stroke outcome [84]. Altogether, these results suggest that exacerbated cerebrovascular activation after hemorrhagic stroke could have a deleterious role. In a mouse model of intraparenchymal hemorrhage induced by in situ injection of collagenase, molecular MRI of VCAM-1 allows noninvasive and longitudinal monitoring of cerebrovascular inflammation [28, 85]. This method allows one to observe that in hemorrhagic stroke, endothelial activation occurs in the whole brain, i.e., in areas far from the primary insult, although the activation appears most intense in the close periphery of the hematoma. Noteworthy, in the context of hemorrhage, previous exposure of the animals to a short normobaric hyperoxia reduces the susceptibility effect of the hematoma (by converting deoxyhemoglobin into oxyhemoglobin), therefore allowing one to differentiate MPIOs induced signal voids from the susceptibility effects of the hematoma [85].
In subarachnoid hemorrhage, a subtype of hemorrhagic stroke usually caused by the rupture of an intracranial aneurysm in the subarachnoid space, endothelial activation is suspected to play a key role during delayed cerebral ischemia, the second phase of neurological injuries [86]. After rupture of an intracranial aneurysm, blood flows into the subarachnoid space at high pressure, leading to the first phase of brain injury related to the transient global ischemia caused by the rise in intracranial pressure and the direct toxicity of the blood [87]. Once the cause of the subarachnoid hemorrhage has been identified and treated (for instance by coil placement in a ruptured saccular aneurysm), patients remain at risk of delayed cerebral ischemia, which is thought to be caused by an interaction of delayed onset of angiographic vasospasm, impaired cerebral autoregulation, capillary and arteriolar microthrombosis, capillary transit time heterogeneity and cortical spreading ischemia [88, 89]. Notably, endothelial activation is central to all these alleged and probably co-operating pathophysiological mechanisms. Since we cannot predict the occurrence of delayed cerebral ischemia using currently available diagnostic tools, selection of at-risk patients for prophylactic treatment remains challenging. In this regard, the use of molecular MRI of endothelial activation may help to identify these patients. In line with this hypothesis, patients presenting delayed cerebral ischemia have higher CSF level of endothelial activation biomarkers, including VCAM-1 and ICAM-1 [86]. Interestingly, Siler et al. demonstrated that 24 hours after subarachnoid hemorrhage in a mouse model, MPIO-αVCAM-1 enhanced MRI reveals widespread overexpression of VCAM-1 [90]. Notably, soluble epoxide hydrolase knockout mice (which had less severe neurological deficits following subarachnoid hemorrhage), present less endothelial activation as assessed by molecular MRI. Altogether, these data suggest that molecular MRI of endothelial activation could be used to identify patients who present severe forms of subarachnoid hemorrhage and to discharge the other patients.
Molecular MRI of endothelial activation in primary neuroinflammatory disorders
Primary neuroinflammatory disorders are the consequence of autoimmune or infectious processes. As the most common autoimmune CNS disorders, multiple sclerosis is a chronic inflammatory disease of the central nervous system that leads to demyelination in the brain and spinal cord [91]. The most frequent form of multiple sclerosis is the relapsing-remitting subtype, characterized by unpredictable relapses occurring between periods of clinically-silent disease activity [92]. Immunomodulatory and immunosuppressive drugs are the bases of patient management, reducing the rate of relapses and appearance of new lesions as assessed by brain imaging [93]. Multiple sclerosis CNS lesions are currently identified using T2-weighted (for vasogenic edema and gliosis) and gadolinium-enhanced MRI (for BBB breakdown) [94]. Of note, these MRI protocols allow one to quantify lesion load but the results obtained cannot predict whether, nor when, a relapse will occur. Moreover, in some patients, even during clinically silent periods, the underlying activity of the disease can remain substantial and may lead to progressive increase in the CNS lesion load [95]. In this context, the ability to monitor disease activity during clinically silent periods and to predict when a relapse may occur could dramatically impact the management of multiple sclerosis patients by allowing physicians to start new treatments or intensify disease modifying therapies before CNS damage. On the other hand, patients who present with a less-active form of the disease could benefit from lowering treatment intensity to avoid the side effects associated with immunomodulation. Recent studies in experimental models of multiple sclerosis demonstrated that molecular MRI of endothelial activation could be used to further that aim [29, 30, 45, 96].
Although the etiology remains unknown, leukocyte recruitment to CNS tissues plays a major role in multiple sclerosis, with endothelial activation being a critical mediator of this process [97]. Therefore, endothelial activation can be used as a surrogate biomarker for multiple sclerosis activity. In line with this hypothesis, expression of adhesion molecules expression is upregulated in the CNS of multiple sclerosis patients[98] and their plasma levels correlate with the activity of the disease [99], although, at the individual level, these biomarkers are of insufficient clinical utility. Furthermore, one of the most effective treatments for multiple sclerosis is a monoclonal antibody directed against VLA-4 (natalizumab) that prevents its interaction with VCAM-1, thereby blocking lymphocyte diapedesis and significantly reducing disease activity [67]. Since these data pointed toward a central role for VCAM-1 in multiple sclerosis activity, the first molecular MRI studies of endothelial activation using MPIOs in experimental models were performed using VCAM-1 targeted MPIOs [29, 30, 96]. These studies demonstrated that MPIO-αVCAM-1 enhanced MRI detects widespread endothelial activation in the brains of mice with experimental autoimmune encephalomyelitis, the most widely used model for multiple sclerosis. Moreover, there is a correlation between disease activity (as assessed by a clinical score) and the intensity of MPIO-αVCAM-1 binding, supporting the use of molecular MRI of endothelial activation as a marker for multiple sclerosis activity. Interestingly, even at the pre-symptomatic stage, MPIO-αVCAM-1 detected endothelial activation, suggesting that it enables detection of currently invisible disease activity. Importantly, these results have been independently replicated using different MPIO-αVCAM-1 formulations [29, 30]. Mardiquian et al. also demonstrated that MPIO-αVCAM-1 enhanced MRI could be used to monitor the efficacy of immunomodulatory treatment, as evidenced in mice receiving an anti-IL-17A treatment that presented lower levels of both neurological deficit and endothelial activation compared to control mice [96].
Subsequently, other adhesion molecules have been studied. Like VCAM-1, ICAM-1 is overexpressed by the CNS endothelium during early lesion formation in experimental autoimmune encephalomyelitis and in patients with multiple sclerosis. However, ICAM-1 presents specific features, such as its ability to drive the crawling of T-cells against the direction of blood flow to sites of diapedesis, supporting its relevance as an imaging target in neuroinflammatory disorders [37]. Using MPIOs targeted against ICAM-1, Blezer et al. demonstrated that ICAM-1 was particularly overexpressed in the subarachnoid space [100]. In contrast to what was observed using MPIO-αVCAM-1, the volume of MPIO-αICAM-1 induced signal voids did not differ between the phases of experimental autoimmune encephalomyelitis with similar findings reported in the acute and chronic phases.
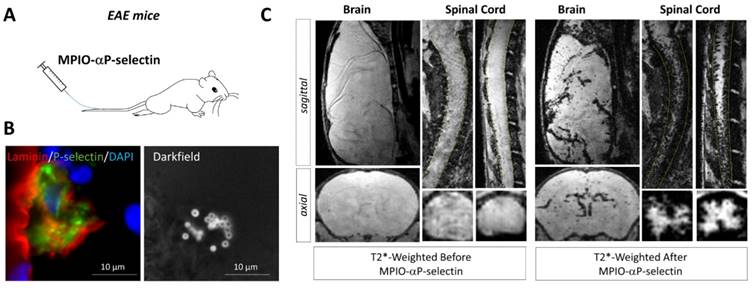
More recently, P-selectin has been studied as an imaging target in experimental models of multiple sclerosis [45]. P-selectin is rapidly expressed at the surface of activated endothelial cells where it mediates crawling of leukocytes [35]. Fournier et al. recently demonstrated that molecular MRI using MPIO-αP-selectin unmasks the pathological events that take place in the spinal cord and brain of mice subjected to chronic or relapsing experimental autoimmune encephalomyelitis (Figure 5) [45]. Overall, they accumulated evidence to suggest that the expression of P-Selectin on the endothelial surface of the CNS is strongly correlated to disease activity. First, they have shown that MPIO-αP-selectin binding in the spinal cord increases with the clinical score in two experimental models characterized by either chronic or relapsing-remitting disease courses. Moreover, MPIO-αP-selectin enhanced molecular MRI was able to predict relapse in initially asymptomatic animals in the relapsing-remitting subtype of experimental autoimmune encephalomyelitis (as well as remission in symptomatic animals). Indeed, the presence of MPIOs-αP-selectin-induced signal void in asymptomatic animals predicted the appearance of relapse. Conversely, the disappearance of MPIOs-αP-selectin-induced signal void in symptomatic animals predicted their remission [45]. If successfully translated to the clinic, such methodology would enable one to modulate the intensity of the treatment according to the activity of the disease. To our knowledge, this latter study was also the first to demonstrate the feasibility of in vivo molecular MRI in the mouse spinal cord.
Molecular MRI of P-selectin in the brain and spinal cord of experimental autoimmune encephalomyelitis (EAE) mice. (A) Schematic representation of the experiment. MPIO-αP-Selectin were injected several days after EAE induction and MRI of the brain and the spinal cord were acquired. (B) Immunohistological images revealing numerous MPIO-αP-Selectin on an activated endothelium in the brain of an EAE mouse. (C) In vivo MRI of the brain and spinal cord of an EAE mouse before and after MPIO-αP-Selectin injection. Molecular MRI reveals strong endothelial activation in the whole CNS, predominating in the spinal cord. Further details on this experiment are available in the paper by Fournier et al. [45].
As another etiology of neuroinflammatory processes, infectious diseases of the CNS represent a frequent pathological condition and can be difficult to diagnose using conventional imaging. To date, few molecular MRI studies have focused on infectious diseases. Von zur Muhlen et al. used MPIOs directed against activated platelets to reveal murine cerebral pathology [101]. Indeed, cerebral malaria is characterized by a sequestration of platelets in the cerebral vasculature which is thought to be caused by overexpression of selectins by activated endothelial cells. By revealing wall-adherent platelets, the authors demonstrated that the CNS endothelial activation induced by Plasmodium berghei offers an opportunity to detect cerebral malaria, which is otherwise undetectable using conventional imaging. Although still unproven, it is likely that molecular MRI of endothelial activation in the CNS could improve the sensitivity of conventional MRI for the diagnosis of meningitis, meningoencephalitis (including herpetic CNS infection), cerebral abscesses and empyema. Further investigations are needed in these pathological conditions.
Molecular MRI of endothelial activation in neurodegenerative disorders
Alzheimer's disease (AD) and vascular dementia are the two most common neurodegenerative disorders. In both conditions, a growing body of evidence supports a role for endothelial activation in disease progression [102]. Accordingly, molecular MRI could help identifying patients with more inflammatory forms of the disorders and who may benefit from targeted therapies.
AD is an age-related disorder characterized by progressive cognitive decline leading to dementia, with a suspected role for amyloid beta deposits and tau tangles resulting in neurological dysfunction and subsequent neuronal cell death. The cerebrovasculature of AD patients shows structural and functional abnormalities such as increased pinocytic vesicles, increased permeability and decreased vascular density [103]. Interestingly, imaging data in humans suggest that endothelial activation precedes neurological dysfunction in AD [104]. Accordingly, AD patients present elevated plasma levels of endothelial dysfunction markers such as E-selectin and VCAM-1 [105]. It has been postulated that, in some experimental models of AD, the vascular damage is the first event to occur and that subsequent amyloid deposition only exacerbates this vascular damage. In line with this hypothesis, numerous studies link vascular risk factors to cognitive decline and dementia in the elderly [103, 104]. If this claim is true, endothelial activation could become a key therapeutic target to prevent neuronal cell death in AD patients. Therefore, early detection of endothelial activation in the brain could be of important diagnostic value in paucisymptomatic subjects. Moreover, longitudinal monitoring of endothelial activation could ascertain the response to therapeutic intervention aimed at preventing further cerebrovascular damages. In a proof-of-concept study in a mouse model of AD, molecular MRI of VCAM-1 revealed a significantly higher level of endothelial activation in AD than in age-matched control mice, confirming the feasibility of this approach [29].
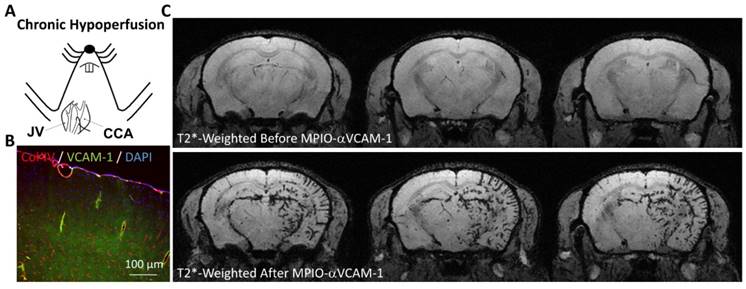
As the second cause of dementia worldwide, vascular dementia is defined as a progressive neurodegenerative disease attributable to cerebrovascular factors [106]. The common pathophysiological mechanism of all vascular dementia subtypes is cerebral hypoperfusion [107]. In a mouse model of vascular dementia induced by ligature of the right common carotid artery, Montagne et al. demonstrated that the hypoperfused cerebral vessels present a high-level of endothelial activation, as revealed by MPIO-αVCAM-1 enhanced MRI (Figure 6) [29]. Although still unexplored, these data suggest that one could use molecular imaging of endothelial activation to monitor the effectiveness of therapies aimed at preventing further cerebrovascular damages. As one of the most common subtype of vascular dementia, cerebral amyloid angiopathy is characterized by the presence of amyloid deposit in the walls of cerebral blood vessels [108]. It is associated with microvascular modifications, microinfarcts and microbleeds. Endothelial activation in cerebral amyloid angiopathy is thought to play a key role in the progression of cerebrovascular damage, but still very little is known regarding the interaction between amyloid deposition, endothelial activation and subsequent brain damage [109]. The development of non-invasive means to monitor endothelial activation brings, therefore, great promise for a further understanding of this complex condition.
Molecular MRI of VCAM-1 in a model of vascular dementia induced by chronic cerebral hypoperfusion. (A) Schematic representation of the experimental model. The right common carotid artery (CCA) was ligated (after its isolation from the jugular vein, JV), inducing chronic hypoperfusion of the right brain hemisphere. (B) Immunohistological images revealing VCAM-1 expression by the endothelium of the hypoperfused brain. (C) In vivo MRI of the brain before and after MPIO-αVCAM-1 injection 48 hours after CCA ligation revealing significant endothelial activation in the hypoperfused hemisphere. Further details on this experiment are available in Montagne et al. [29].
Among the other causes of neurodegenerative brain disorders is chronic alcohol consumption [110]. Indeed, human studies consistently found that alcoholic patients have a smaller brain volume compared to moderate drinkers [111]. In line with these clinical studies, experimental studies in rodents found a direct neurotoxic effect of alcohol, leading to neuronal loss in multiple brain regions [112]. Although a direct toxic effect of alcohol on endothelial cells is probably not at the center of the pathophysiological process [113], neuronal cell death leads to the release of pro-inflammatory molecules (especially damage associated molecular patterns) which can trigger endothelial activation. In line with this hypothesis, in a mouse model of acute ethanol intoxication, Montagne et al. demonstrated that the cerebrovasculature becomes activated as revealed by MPIO-αVCAM-1 enhanced MRI [29]. Further studies are needed to determine whether molecular MRI of endothelial activation can unmask the deleterious effects of chronic alcohol consumption on the CNS.
Inflammation associated with aging can also be detected using molecular MRI of VCAM-1 [29]. Indeed, several studies reported that apparently healthy humans present a chronic low-level of inflammation associated with aging, a phenomenon known as “inflammaging” (a contraction of inflammation and aging) [114]. Although still not fully understood, the impact of chronic inflammation on brain functions is suspected to be deleterious and to favor the progression of cognitive impairment associated with aging [115]. Interestingly, MPIO-αVCAM-1 revealed a significant activation of the cerebrovasculature in otherwise healthy, aged mice [29, 43]. Longitudinal monitoring of this chronic endothelial activation could be useful to evaluate the effectiveness of therapeutic interventions (such as anti-hypertensive drugs, statins, specific diets, meditation) to prevent age-associated cognitive impairment.
Altogether, these data demonstrate the potential interest of molecular MRI of endothelial activation in neurodegenerative disorders. Since there is renewed interest in the role of neuroinflammation in these disorders, including clinical development of an immunomodulatory treatment, one can expect the clinical need for a sensitive means of monitoring endothelial activation. It is also tempting to speculate that molecular MRI of endothelial activation could have applications in other neurodegenerative diseases such as Parkinson's disease or amyotrophic lateral sclerosis.
Molecular MRI of endothelial activation in brain cancers
Brain cancers are classically divided into primitive brain cancers and brain metastasis [116]. Interestingly, it has been demonstrated that the latter can be detected using molecular MRI of endothelial activation with a higher sensitivity than conventional imaging [117]. Since the survival of patients presenting brain metastasis is typically inferior to one year after diagnosis [118], the possibility to diagnose brain metastases at their earliest stages is mandatory to improve patient survival. Indeed, surgery and radiotherapy (especially using Gamma knife radiosurgery) can cure brain metastasis if they are sufficiently small and limited to a few lesions [119]. To date, diagnosis of brain metastasis is performed using contrast enhanced computed tomography (CT) or by the more sensitive, current gold standard, gadolinium-enhanced MRI [120]. Both methods rely on the breakdown of the BBB associated with CNS metastasis. Since BBB breakdown appears in the latest stages of metastasis progression, only large metastases can be detected, delaying the introduction of potentially lifesaving treatments [121]. In contrast, endothelial activation is an early event in the development of metastasis [122]. To evaluate whether molecular MRI of endothelial activation could reveal brain metastasis before conventional imaging, Serres et al. injected cancer cells into the circulation of mice and performed longitudinal MPIO-αVCAM-1 enhanced imaging [117]. They found that MPIO-αVCAM-1 were frequently co-localized with tumor colonies made of 103 cells, 4 orders of magnitude smaller than the metastases detected by gadolinium-enhanced MRI. In a clinical setting, this would allow detection of brain metastases at their earliest developmental stages and would thus help to start appropriate treatment early (e.g., whole-brain irradiation, Gamma knife radiosurgery, chemotherapy or minimally invasive surgery). Since brain metastasis remains a challenging clinical problem, the improvement in our ability to detect early metastatic stages using molecular MRI of endothelial activation is likely to yield substantial improvement in patient survival.
To date, there is no published experimental study on the use of molecular MRI of endothelial activation, regarding primary brain cancer. However, there is a growing body of evidence which suggests that monitoring of endothelial activation could have considerable value for diagnosis, treatment response prediction and prognosis assessment in primitive brain neoplasms [123]. Indeed, high-grade glioma (especially glioblastoma) are characterized by a marked neoangiogenesis [124]. This is thought to be triggered by the secretion of endothelial growth factor by the glioma stem cells and is one of the key criteria to differentiate indolent glioma from highly invasive glioma [125]. Interestingly, this endothelial proliferation is associated with endothelial dysfunction and overexpression of adhesion molecules, including VCAM-1 [126]. Accordingly, it is tempting to speculate that molecular imaging of endothelial activation could allow differentiating high-grade from low-grade gliomas. Moreover, recurrent and residual portions of the tumor (after initial treatment) remain difficult to detect using currently available methods [127]. In this context, molecular imaging of endothelial activation could offer a sensitive way to detect early recurrence, high-grade transformation or residual tumor-associated endothelial activation. It should however be acknowledged that radiation injuries may cause false positive findings in this setting, as demonstrated by Zhu et al. who showed that ICAM-1 targeted MPIOs bind to irradiated endothelium both in vitro and in vivo [128].
Another still unexplored research field is the ability of molecular imaging of endothelial activation to predict the response to immune checkpoint inhibitors. Indeed, the recent development of immunotherapy for cancer treatment has provided significant benefits for the patient. Immunotherapy using immune checkpoint inhibitor aims at releasing endogenous T cells from the inhibitory signals that prevent them to attack cancer cells [129]. Unfortunately, only a small proportion of cancers are responsive to these treatments. Among the factors influencing cancer response to immune checkpoint inhibitors is the ability of T cells to reach the tumor, which is highly dependent on the expression of adhesion molecules by the tumor-associated endothelial cells [130]. In this regard, the ability to monitor the expression of adhesion molecules using molecular MRI of endothelial activation could provide a means to select the patients with the highest probability to respond to immunotherapy. This is especially relevant to brain metastases, since immune checkpoint inhibitors are largely used against lung cancer and melanoma, in which brain metastases are common. Further studies are needed to unveil the complete picture of the potential application of molecular MRI of endothelial activation for brain cancers.
Remaining challenges in clinical molecular MRI of endothelial activation
Besides the many unexplored potential applications of molecular MRI of endothelial activation using MPIOs including imaging of traumatic brain injury, spinal cord compression, CNS infection or psychiatric disorders, the main challenges for the years to come are related to the clinical translation of this technique. Here, we will discuss the four main steps that will have to be addressed before the first clinical trials can be envisaged: selection of a suitable pharmacophore; design of a biocompatible MPIO; development of a sensitive MRI procedure to detect MPIOs and demonstration of the added diagnostic value of molecular MRI of endothelial activation over conventional imaging.
To date, most studies investigating molecular MRI of endothelial activation were performed in rodents, using monoclonal antibodies as pharmacophores [7]. Since rodent and human adhesion molecules present different epitopes, the pharmacophores should be adequately selected to allow MPIO binding in humans. This selection step is mandatory for the efficient translational development of molecular MRI of endothelial activation. Indeed, it has been demonstrated in rodent that the sensitivity of molecular MRI is heavily dependent on the targeting antibody, as evidenced for mouse VCAM-1 [29] and P-Selectin [27]. Therefore, in vitro and in vivo selection steps should be performed to optimize the choice of the antibody. So far, in vitro experiments are performed using cultured endothelial cells exposed to a pro-inflammatory cytokine to trigger endothelial activation [21]. Then, targeted MPIOs are incubated in the presence of the activated cells and the number of bound MPIOs remaining after a washing step is measured [131]. The use of human cell lines offers a practical way to screen pharmacophores and optimize their binding efficiency before undertaking clinical trials. Moreover, it allows one to test easily whether binding of the MPIOs has toxic or pro-inflammatory effects on the endothelium [131]. Thereafter, in vivo proof-of-concept experiments may require the use of non-human primates that share epitope sequences with humans. However, to date, the practicality of performing molecular MRI of endothelial activation in non-human primates has not been addressed.
Once the pharmacophore has been satisfactorily selected, the next step is to develop a biocompatible MPIO. Indeed, most experimental studies used commercial MPIOs that accumulate in the liver and the spleen [43]. Development of a large, biodegradable and biocompatible (i.e. non-toxic) MPIO sets a number of technical challenges. Recently, Perez-Balderas et al. developed a strategy based on the covalent assembly of nanoparticles as a peptidase-degradable platform for molecular MRI [132]. In this paper, the authors addressed one of the most significant issues for clinical translation of MPIOs. Basically, these MPIOs are made of multiple colloidal iron nanoparticles measuring ~65 nm that are linked together by peptides that can be degraded by cathepsins, which are lysosomal proteases. Thus, once cleared from the blood by the reticulo-endothelial system, the particles can be degraded inside macrophages and the iron recycled. The authors also demonstrated that the particles that associated with their target on activated endothelial cells (they used antibodies against VCAM-1 as pharmacophores) remained unaltered for a sufficiently long period of time to allow MRI to be performed. However, the sensitivity of these particles appears significantly lower than conventional MPIOs, making their suitability uncertain as molecular contrast agents for MRI in clinically relevant conditions. Moreover, other hurdles should be considered before translational development of any contrastophores, such as stability, toxicity and reproducibility of the chemical lots [133].
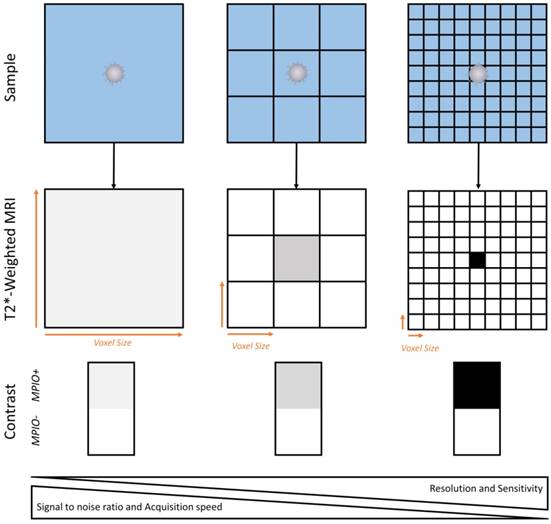
Before undertaking the first trials in humans, it is mandatory to develop new imaging sequences to detect MPIOs using magnets at clinically relevant field strengths and using standard coils. Several peculiarities of MPIOs as contrast agents are critical in this regard, especially the fact that their repartition in the imaged tissue is heterogeneous [46]. Therefore, there is a correlation between the imaging resolution and the sensitivity of the imaging procedure (Figure 7). As a general concept (when using MPIOs and T2* weighted MRI), the higher the image resolution (i.e., the smaller the voxel size), the higher the apparent concentration of MPIOs in MPIO-positive voxels and therefore, the higher the sensitivity of the imaging procedure. At very high resolution (~50 µm isotropic), this explains why single isolated MPIO can be detected using MRI [46, 134]. This comes at the cost of a lower signal-to-noise ratio (small voxels give less signal than large voxels) and an increased acquisition time. To achieve high sensitivity without impairing signal to noise ratio, it is possible to acquire high resolution images (high sensitivity) and to perform a minimum intensity projection (minIP) over several slices to increase the signal to noise ratio. This method is already performed in humans to visualize cerebral veins or microbleeds using susceptibility weighted imaging [135]. Further investigations are needed to select the best imaging strategy to detect MPIOs in a way that would be convenient and suitable for patients.
Schematic representation of the relationship between spatial resolution and MPIO detection sensitivity. Given the heterogenous repartition of targeted MPIOs in the inflamed CNS, increase in imaging resolution allows isolation of MPIO-labeled (MPIO+) from non-labeled tissue (MPIO-) and therefore increases the sensitivity of MRI. This comes at the price of a lower signal to noise ratio and a slower image acquisition.
As for therapeutic clinical trials, diagnostic trials are made of different phases which aim to collectively demonstrate the safety and superiority of the new diagnostic method over the current gold standard. As a first step, the safety of molecular MRI and its ability to detect endothelial activation in humans will first have to be demonstrated. In a small number of patients presenting a pathology known to be associated with endothelial activation, contrast enhanced MRI will have to demonstrate its ability to detect its target. This will also allow pharmacokinetic data to be obtained. If successful, the contrast agent will have to go through a second phase that aims to identify the best dose and administration regimen (e.g., the amount of MPIOs to be administered, the timing between the injection and imaging, the best suited MRI sequences) and provide the first assessment of the diagnostic performance. Then, further studies will have to be performed to evaluate the sensitivity and specificity of the molecular MRI method over a gold standard. For instance, in a population of patients presenting a clinically isolated syndrome, what is the sensitivity and specificity of molecular MRI of endothelial activation to predict which patients will ultimately develop multiple sclerosis (as ascertained by validated imaging criteria after a few months or years of evolution)? Lastly, even if the new contrast agent succeeds and shows a high sensitivity and specificity, demonstration of its clinical utility would be mandatory. Continuing with the same example in multiple sclerosis patients: will the opportunity to get a better early diagnostic of multiple sclerosis translate into a better outcome for patients? Completion of all these diagnostic trials will take years before the actual benefits of applying molecular MRI of endothelial activation can be put into practice and its potential usefulness realized for clinical diagnosis. Cooperation between chemists, radiologists, pharmaceutical companies and health authorities will be mandatory to achieve this goal.
Conclusion
The recent development of targeted MPIOs dramatically improved the sensitivity and specificity of molecular MRI of endothelial activation compared to other reported methods. The potential applications are numerous for neurovascular, neuroinflammatory, neurodegenerative and neurooncological disorders. This imaging strategy opens new avenues for the experimental study of endothelial activation in preclinical models by allowing sensitive and longitudinal detection of adhesion molecules. The suitability of MPIOs as contrast agents for molecular MRI of endothelial activation has now been confirmed by numerous laboratories throughout the world. This is a critical point to consider, given the reproducibility issues currently arising in most research fields. Now that the preclinical feasibility of molecular MRI of endothelial activation is established, the ongoing research aims at promoting the clinical translation of this promising technology. This is a new challenge for the community of molecular MRI, since despite decades of research, gadolinium chelates remain the only clinically approved contrast agents for MRI. Overall, molecular MRI of endothelial activation has the potential to drastically improve the diagnostic value of MRI in a number of CNS disorders.
Abbreviations
AD: Alzheimer's disease; BBB: blood brain barrier; CCL2: chemokine (C-C motif) ligand 2; CHI3L1: chitinase-3-like protein 1; CNS: central nervous system; CSF: cerebrospinal fluid; CXCL13: C-X-C motif chemokine 13; EPR: enhanced permeability and retention; GFAP: glial fibrillary acidic protein; ICAM-1: intercellular adhesion molecule I (ICAM-1); LFA-1: leukocyte associated antigen-1; MAC-1: macrophage adhesion ligand-1; MRI: magnetic resonance imaging; MPIO: microsized particles of iron oxide; PECAM-1: platelet endothelial cell adhesion molecule 1; PET: positron emission tomography; PSGL-1: P-selectin glycoprotein ligand-1; SPECT: single photon emission computed tomography; TIA: transient ischemic attack; TSPO: microglial translocator protein; USPIO: ultra-small particles of iron oxide; VCAM-1: vascular cell adhesion molecule-1.
Acknowledgements
We thank Dr Alan R. Young for help in editing the manuscript.
This work was supported by the RHU MARVELOUS (ANR-16-RHUS-0009) of Université de Lyon, within the program “Investissements d'Avenir” operated by the French National Research Agency (ANR).
Competing Interests
MG, APF, FD and DV are co-authors of a patent “Imaging method for predicting the onset of multiple sclerosis” (WO 2017134178 A1). SM has declared that she has no competing interest.
References
1. Wee Yong V. Inflammation in neurological disorders: a help or a hindrance? Neuroscientist. 2010;16:408-20
2. Kyritsis N, Kizil C, Zocher S, Kroehne V, Kaslin J, Freudenreich D. et al. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science. 2012;338:1353-6
3. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015;14:406-19
4. Becker KJ, Kalil AJ, Tanzi P, Zierath DK, Savos AV, Gee JM. et al. Autoimmune responses to the brain after stroke are associated with worse outcome. Stroke. 2011;42:2763-9
5. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777-83
6. Skaper SD. The brain as a target for inflammatory processes and neuroprotective strategies. Ann N Y Acad Sci. 2007;1122:23-34
7. Gauberti M, Montagne A, Quenault A, Vivien D. Molecular magnetic resonance imaging of brain-immune interactions. Front Cell Neurosci. 2014;8:389
8. Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol. 2013;9:201-10
9. Reichsoellner M, Raggam RB, Wagner J, Krause R, Hoenigl M. Clinical evaluation of multiple inflammation biomarkers for diagnosis and prognosis for patients with systemic inflammatory response syndrome. J Clin Microbiol. 2014;52:4063-6
10. Novakova L, Axelsson M, Khademi M, Zetterberg H, Blennow K, Malmestrom C. et al. Cerebrospinal fluid biomarkers of inflammation and degeneration as measures of fingolimod efficacy in multiple sclerosis. Mult Scler. 2017;23:62-71
11. Khademi M, Kockum I, Andersson ML, Iacobaeus E, Brundin L, Sellebjerg F. et al. Cerebrospinal fluid CXCL13 in multiple sclerosis: a suggestive prognostic marker for the disease course. Mult Scler. 2011;17:335-43
12. Conductier G, Blondeau N, Guyon A, Nahon JL, Rovere C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93-100
13. Comabella M, Fernandez M, Martin R, Rivera-Vallve S, Borras E, Chiva C. et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain. 2010;133:1082-93
14. Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N. et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci. 2015;35:518-26
15. Jacobs AH, Tavitian B. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012;32:1393-415
16. Ory D, Celen S, Verbruggen A, Bormans G. PET radioligands for in vivo visualization of neuroinflammation. Curr Pharm Des. 2014;20:5897-913
17. Gulyas B, Toth M, Schain M, Airaksinen A, Vas A, Kostulas K. et al. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: a PET study with the TSPO molecular imaging biomarker [((11))C]vinpocetine. J Neurol Sci. 2012;320:110-7
18. Varrone A, Oikonen V, Forsberg A, Joutsa J, Takano A, Solin O. et al. Positron emission tomography imaging of the 18-kDa translocator protein (TSPO) with [18F]FEMPA in Alzheimer's disease patients and control subjects. Eur J Nucl Med Mol Imaging. 2015;42:438-46
19. Herranz E, Gianni C, Louapre C, Treaba CA, Govindarajan ST, Ouellette R. et al. Neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol. 2016;80:776-90
20. Hengerer A, Grimm J. Molecular magnetic resonance imaging. Biomedical Imaging and Intervention Journal. 2006:2
21. McAteer MA, Sibson NR, von Zur Muhlen C, Schneider JE, Lowe AS, Warrick N. et al. In vivo magnetic resonance imaging of acute brain inflammation using microparticles of iron oxide. Nat Med. 2007;13:1253-8
22. Chen JW, Pham W, Weissleder R, Bogdanov A Jr. Human myeloperoxidase: a potential target for molecular MR imaging in atherosclerosis. Magn Reson Med. 2004;52:1021-8
23. Kelly KA, Allport JR, Tsourkas A, Shinde-Patil VR, Josephson L, Weissleder R. Detection of vascular adhesion molecule-1 expression using a novel multimodal nanoparticle. Circ Res. 2005;96:327-36
24. Nahrendorf M, Jaffer FA, Kelly KA, Sosnovik DE, Aikawa E, Libby P. et al. Noninvasive vascular cell adhesion molecule-1 imaging identifies inflammatory activation of cells in atherosclerosis. Circulation. 2006;114:1504-11
25. Frechou M, Beray-Berthat V, Raynaud JS, Meriaux S, Gombert F, Lancelot E. et al. Detection of vascular cell adhesion molecule-1 expression with USPIO-enhanced molecular MRI in a mouse model of cerebral ischemia. Contrast media & molecular imaging. 2013;8:157-64
26. Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189-207
27. Quenault A, Martinez de Lizarrondo S, Etard O, Gauberti M, Orset C, Haelewyn B. et al. Molecular magnetic resonance imaging discloses endothelial activation after transient ischaemic attack. Brain. 2017;140:146-57
28. Gauberti M, Montagne A, Marcos-Contreras OA, Le Behot A, Maubert E, Vivien D. Ultra-sensitive molecular MRI of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke. 2013;44:1988-96
29. Montagne A, Gauberti M, Macrez R, Jullienne A, Briens A, Raynaud JS. et al. Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. Neuroimage. 2012;63:760-70
30. Serres S, Mardiguian S, Campbell SJ, McAteer MA, Akhtar A, Krapitchev A. et al. VCAM-1-targeted magnetic resonance imaging reveals subclinical disease in a mouse model of multiple sclerosis. FASEB J. 2011;25:4415-22
31. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48-65
32. Wu F, Liu L, Zhou H. Endothelial cell activation in central nervous system inflammation. J Leukoc Biol. 2017;101:1119-32
33. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803-15
34. Barkalow FJ, Goodman MJ, Gerritsen ME, Mayadas TN. Brain endothelium lack one of two pathways of P-selectin-mediated neutrophil adhesion. Blood. 1996;88:4585-93
35. Kerfoot SM, Kubes P. Overlapping roles of P-selectin and alpha 4 integrin to recruit leukocytes to the central nervous system in experimental autoimmune encephalomyelitis. J Immunol. 2002;169:1000-6
36. Norman MU, James WG, Hickey MJ. Differential roles of ICAM-1 and VCAM-1 in leukocyte-endothelial cell interactions in skin and brain of MRL/faslpr mice. J Leukoc Biol. 2008;84:68-76
37. Steiner O, Coisne C, Cecchelli R, Boscacci R, Deutsch U, Engelhardt B. et al. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood-brain barrier endothelium. J Immunol. 2010;185:4846-55
38. Privratsky JR, Newman PJ. PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res. 2014;355:607-19
39. Deddens LH, van Tilborg GA, van der Toorn A, de Vries HE, Dijkhuizen RM. PECAM-1-targeted micron-sized particles of iron oxide as MRI contrast agent for detection of vascular remodeling after cerebral ischemia. Contrast media & molecular imaging. 2013;8:393-401
40. Deddens LH, van Tilborg GA, van der Toorn A, van der Marel K, Paulis LE, van Bloois L. et al. MRI of ICAM-1 upregulation after stroke: the importance of choosing the appropriate target-specific particulate contrast agent. Mol Imaging Biol. 2013;15:411-22
41. McAteer MA, Schneider JE, Ali ZA, Warrick N, Bursill CA, von zur Muhlen C. et al. Magnetic resonance imaging of endothelial adhesion molecules in mouse atherosclerosis using dual-targeted microparticles of iron oxide. Arterioscler Thromb Vasc Biol. 2008;28:77-83
42. McAteer MA, von Zur Muhlen C, Anthony DC, Sibson NR, Choudhury RP. Magnetic resonance imaging of brain inflammation using microparticles of iron oxide. Methods Mol Biol. 2011;680:103-15
43. Belliere J, Martinez de Lizarrondo S, Choudhury RP, Quenault A, Le Behot A, Delage C. et al. Unmasking Silent Endothelial Activation in the Cardiovascular System Using Molecular Magnetic Resonance Imaging. Theranostics. 2015;5:1187-202
44. Robinson PJ, Rapoport SI. Size selectivity of blood-brain barrier permeability at various times after osmotic opening. Am J Physiol. 1987;253:R459-66
45. Fournier AP, Quenault A, Martinez de Lizarrondo S, Gauberti M, Defer G, Vivien D. et al. Prediction of disease activity in models of multiple sclerosis by molecular magnetic resonance imaging of P-selectin. Proc Natl Acad Sci U S A. 2017;114:6116-21
46. Shapiro EM, Skrtic S, Sharer K, Hill JM, Dunbar CE, Koretsky AP. MRI detection of single particles for cellular imaging. Proc Natl Acad Sci U S A. 2004;101:10901-6
47. Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U. et al. Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol. 2011;10:471-80
48. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796-808
49. Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2:43-53
50. Veltkamp R, Gill D. Clinical Trials of Immunomodulation in Ischemic Stroke. Neurotherapeutics. 2016;13:791-800
51. del Zoppo GJ. Acute anti-inflammatory approaches to ischemic stroke. Ann N Y Acad Sci. 2010;1207:143-8
52. van der Worp HB, van Gijn J. Clinical practice. Acute ischemic stroke. N Engl J Med. 2007;357:572-9
53. Vivien D, Gauberti M, Montagne A, Defer G, Touze E. Impact of tissue plasminogen activator on the neurovascular unit: from clinical data to experimental evidence. J Cereb Blood Flow Metab. 2011;31:2119-34
54. Martinez de Lizarrondo S, Gakuba C, Herbig BA, Repesse Y, Ali C, Denis CV. et al. Potent Thrombolytic Effect of N-Acetylcysteine on Arterial Thrombi. Circulation. 2017
55. Manning NW, Campbell BC, Oxley TJ, Chapot R. Acute ischemic stroke: time, penumbra, and reperfusion. Stroke. 2014;45:640-4
56. Saver JL. Time is brain-quantified. Stroke. 2006;37:263-6
57. Goyal M, Menon BK, van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM. et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. 2016;387:1723-31
58. Gakuba C, Gauberti M, Mazighi M, Defer G, Hanouz JL, Vivien D. Preclinical evidence toward the use of ketamine for recombinant tissue-type plasminogen activator-mediated thrombolysis under anesthesia or sedation. Stroke. 2011;42:2947-9
59. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391-7
60. Marcos-Contreras OA, Martinez de Lizarrondo S, Bardou I, Orset C, Pruvost M, Anfray A. et al. Hyperfibrinolysis increases blood-brain barrier permeability by a plasmin- and bradykinin-dependent mechanism. Blood. 2016;128:2423-34
61. Gauberti M, Martinez de Lizarrondo S, Orset C, Vivien D. Lack of secondary microthrombosis after thrombin-induced stroke in mice and non-human primates. J Thromb Haemost. 2014;12:409-14
62. Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401-10
63. Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N. et al. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol. 2013;23:34-44
64. Kim HA, Whittle SC, Lee S, Chu HX, Zhang SR, Wei Z. et al. Brain immune cell composition and functional outcome after cerebral ischemia: comparison of two mouse strains. Front Cell Neurosci. 2014;8:365
65. Shichita T, Ito M, Yoshimura A. Post-ischemic inflammation regulates neural damage and protection. Front Cell Neurosci. 2014;8:319
66. Brunkhorst R, Vutukuri R, Pfeilschifter W. Fingolimod for the treatment of neurological diseases-state of play and future perspectives. Front Cell Neurosci. 2014;8:283
67. McCormack PL. Natalizumab: a review of its use in the management of relapsing-remitting multiple sclerosis. Drugs. 2013;73:1463-81
68. Hammond MD, Ambler WG, Ai Y, Sansing LH. alpha4 integrin is a regulator of leukocyte recruitment after experimental intracerebral hemorrhage. Stroke. 2014;45:2485-7
69. Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G. et al. Combination of the Immune Modulator Fingolimod With Alteplase in Acute Ischemic Stroke: A Pilot Trial. Circulation. 2015;132:1104-12
70. Gauberti M, De Lizarrondo SM, Vivien D. The “inflammatory penumbra” in ischemic stroke: From clinical data to experimental evidence. European Stroke Journal. 2016;1:20-7
71. Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S. et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134:704-20
72. Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ. et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111:18315-20
73. Gill D, Veltkamp R. Dynamics of T cell responses after stroke. Curr Opin Pharmacol. 2015;26:26-32
74. Deddens LH, van Tilborg GAF, van der Marel K, Hunt H, van der Toorn A, Viergever MA. et al. In Vivo Molecular MRI of ICAM-1 Expression on Endothelium and Leukocytes from Subacute to Chronic Stages After Experimental Stroke. Translational stroke research. 2017
75. Le Behot A, Gauberti M, Martinez De Lizarrondo S, Montagne A, Lemarchand E, Repesse Y. et al. GpIbalpha-VWF blockade restores vessel patency by dissolving platelet aggregates formed under very high shear rate in mice. Blood. 2014;123:3354-63
76. Albers GW, Caplan LR, Easton JD, Fayad PB, Mohr JP, Saver JL. et al. Transient ischemic attack-proposal for a new definition. N Engl J Med. 2002;347:1713-6
77. Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD. et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160-236
78. Chan JMS, Monaco C, Wylezinska-Arridge M, Tremoleda JL, Cole JE, Goddard M. et al. Imaging vulnerable plaques by targeting inflammation in atherosclerosis using fluorescent-labeled dual-ligand microparticles of iron oxide and magnetic resonance imaging. J Vasc Surg. 2017
79. Nadarajan V, Perry RJ, Johnson J, Werring DJ. Transient ischaemic attacks: mimics and chameleons. Pract Neurol. 2014;14:23-31
80. Chompoopong P, Rostambeigi N, Kassar D, Maud A, Qureshi IA, Cruz-Flores S. et al. Are We Overlooking Stroke Chameleons? A Retrospective Study on the Delayed Recognition of Stroke Patients. Cerebrovasc Dis. 2017;44:83-7
81. Rolland WB, Lekic T, Krafft PR, Hasegawa Y, Altay O, Hartman R. et al. Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp Neurol. 2013;241:45-55
82. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25-44
83. Kraus J, Oschmann P, Leis S, Neundorfer B, Heckmann JG. High concentrations of sVCAM-1 and sICAM-1 in the cerebrospinal fluid of patients with intracerebral haemorrhage are associated with poor outcome. J Neurol Neurosurg Psychiatry. 2002;73:346-7
84. Fu Y, Hao J, Zhang N, Ren L, Sun N, Li YJ. et al. Fingolimod for the treatment of intracerebral hemorrhage: a 2-arm proof-of-concept study. JAMA neurology. 2014;71:1092-101
85. Gaberel T, Gakuba C, Hebert M, Montagne A, Agin V, Rubio M. et al. Intracerebral hematomas disappear on T2*-weighted images during normobaric oxygen therapy. Stroke. 2013;44:3482-9
86. Frijns CJ, Fijnheer R, Algra A, van Mourik JA, van Gijn J, Rinkel GJ. Early circulating levels of endothelial cell activation markers in aneurysmal subarachnoid haemorrhage: associations with cerebral ischaemic events and outcome. J Neurol Neurosurg Psychiatry. 2006;77:77-83
87. Lawton MT, Vates GE. Subarachnoid Hemorrhage. N Engl J Med. 2017;377:257-66
88. Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet. 2017;389:655-66
89. Gaberel T, Gakuba C, Goulay R, Martinez De Lizarrondo S, Hanouz JL, Emery E. et al. Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: a new target for fibrinolysis? Stroke. 2014;45:3092-6
90. Siler DA, Berlow YA, Kukino A, Davis CM, Nelson JW, Grafe MR. et al. Soluble Epoxide Hydrolase in Hydrocephalus, Cerebral Edema, and Vascular Inflammation After Subarachnoid Hemorrhage. Stroke. 2015;46:1916-22
91. Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502-17
92. Comi G, Radaelli M, Soelberg Sorensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017;389:1347-56
93. Sormani MP, Laroni A. Approved drugs for multiple sclerosis: the challenge of choice. Lancet Neurol. 2017;16:252-3
94. Brownlee WJ, Hardy TA, Fazekas F, Miller DH. Diagnosis of multiple sclerosis: progress and challenges. Lancet. 2017;389:1336-46
95. Scalfari A, Neuhaus A, Degenhardt A, Rice GP, Muraro PA, Daumer M. et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010;133:1914-29
96. Mardiguian S, Serres S, Ladds E, Campbell SJ, Wilainam P, McFadyen C. et al. Anti-IL-17A treatment reduces clinical score and VCAM-1 expression detected by in vivo magnetic resonance imaging in chronic relapsing EAE ABH mice. Am J Pathol. 2013;182:2071-81
97. Rossi B, Angiari S, Zenaro E, Budui SL, Constantin G. Vascular inflammation in central nervous system diseases: adhesion receptors controlling leukocyte-endothelial interactions. J Leukoc Biol. 2011;89:539-56
98. Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann Neurol. 1995;37:424-35
99. Elovaara I, Ukkonen M, Leppakynnas M, Lehtimaki T, Luomala M, Peltola J. et al. Adhesion molecules in multiple sclerosis: relation to subtypes of disease and methylprednisolone therapy. Arch Neurol. 2000;57:546-51
100. Blezer EL, Deddens LH, Kooij G, Drexhage J, van der Pol SM, Reijerkerk A. et al. In vivo MR imaging of intercellular adhesion molecule-1 expression in an animal model of multiple sclerosis. Contrast media & molecular imaging. 2015;10:111-21
101. von Zur Muhlen C, Sibson NR, Peter K, Campbell SJ, Wilainam P, Grau GE. et al. A contrast agent recognizing activated platelets reveals murine cerebral malaria pathology undetectable by conventional MRI. J Clin Invest. 2008;118:1198-207
102. Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D. et al. Inflammation in neurodegenerative diseases-an update. Immunology. 2014;142:151-66
103. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL. et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388-405
104. Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. J Neuroinflammation. 2011;8:26
105. Ewers M, Mielke MM, Hampel H. Blood-based biomarkers of microvascular pathology in Alzheimer's disease. Exp Gerontol. 2010;45:75-9
106. Skrobot OA, O'Brien J, Black S, Chen C, DeCarli C, Erkinjuntti T. et al. The Vascular Impairment of Cognition Classification Consensus Study. Alzheimer's & dementia: the journal of the Alzheimer's Association. 2017;13:624-33
107. O'Brien JT, Thomas A. Vascular dementia. Lancet. 2015;386:1698-706
108. Grinberg LT, Korczyn AD, Heinsen H. Cerebral amyloid angiopathy impact on endothelium. Exp Gerontol. 2012;47:838-42
109. Ghiso J, Fossati S, Rostagno A. Amyloidosis associated with cerebral amyloid angiopathy: cell signaling pathways elicited in cerebral endothelial cells. J Alzheimers Dis. 2014;42(Suppl 3):S167-76
110. Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med. 2008;45:1542-50
111. Paul CA, Au R, Fredman L, Massaro JM, Seshadri S, Decarli C. et al. Association of alcohol consumption with brain volume in the Framingham study. Arch Neurol. 2008;65:1363-7
112. Saito M, Chakraborty G, Hui M, Masiello K, Saito M. Ethanol-Induced Neurodegeneration and Glial Activation in the Developing Brain. Brain sciences. 2016:6
113. Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78:1223-32
114. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and 'Garb-aging'. Trends Endocrinol Metab. 2017;28:199-212
115. Liang Z, Zhao Y, Ruan L, Zhu L, Jin K, Zhuge Q. et al. Impact of aging immune system on neurodegeneration and potential immunotherapies. Prog Neurobiol. 2017
116. Ricard D, Idbaih A, Ducray F, Lahutte M, Hoang-Xuan K, Delattre JY. Primary brain tumours in adults. Lancet. 2012;379:1984-96
117. Serres S, Soto MS, Hamilton A, McAteer MA, Carbonell WS, Robson MD. et al. Molecular MRI enables early and sensitive detection of brain metastases. Proc Natl Acad Sci U S A. 2012;109:6674-9
118. Nussbaum ES, Djalilian HR, Cho KH, Hall WA. Brain metastases. Histology, multiplicity, surgery, and survival. Cancer. 1996;78:1781-8
119. Bindal AK, Bindal RK, Hess KR, Shiu A, Hassenbusch SJ, Shi WM. et al. Surgery versus radiosurgery in the treatment of brain metastasis. J Neurosurg. 1996;84:748-54
120. Suzuki K, Yamamoto M, Hasegawa Y, Ando M, Shima K, Sako C. et al. Magnetic resonance imaging and computed tomography in the diagnoses of brain metastases of lung cancer. Lung Cancer. 2004;46:357-60
121. Katakami N, Inaba Y, Sugata S, Tsurusaki M, Itoh T, Machida T. et al. Magnetic resonance evaluation of brain metastases from systemic malignances with two doses of gadobutrol 1.0 m compared with gadoteridol: a multicenter, phase ii/iii study in patients with known or suspected brain metastases. Invest Radiol. 2011;46:411-8
122. Carbonell WS, Ansorge O, Sibson N, Muschel R. The vascular basement membrane as "soil" in brain metastasis. PLoS One. 2009;4:e5857
123. Charalambous C, Chen TC, Hofman FM. Characteristics of tumor-associated endothelial cells derived from glioblastoma multiforme. Neurosurg Focus. 2006;20:E22
124. Das S, Marsden PA. Angiogenesis in glioblastoma. N Engl J Med. 2013;369:1561-3
125. Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845-8
126. Zheng Y, Yang W, Aldape K, He J, Lu Z. Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (VCAM-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J Biol Chem. 2013;288:31488-95
127. Shah R, Vattoth S, Jacob R, Manzil FF, O'Malley JP, Borghei P. et al. Radiation necrosis in the brain: imaging features and differentiation from tumor recurrence. Radiographics. 2012;32:1343-59
128. Zhu Y, Ling Y, Zhong J, Liu X, Wei K, Huang S. Magnetic resonance imaging of radiation-induced brain injury using targeted microparticles of iron oxide. Acta Radiol. 2012;53:812-9
129. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
130. Tan LY, Martini C, Fridlender ZG, Bonder CS, Brown MP, Ebert LM. Control of immune cell entry through the tumour vasculature: a missing link in optimising melanoma immunotherapy? Clin Transl Immunology. 2017;6(e):134
131. Jefferson A, Ruparelia N, Choudhury RP. Exogenous microparticles of iron oxide bind to activated endothelial cells but, unlike monocytes, do not trigger an endothelial response. Theranostics. 2013;3:428-36
132. Perez-Balderas F, van Kasteren SI, Aljabali AA, Wals K, Serres S, Jefferson A. et al. Covalent assembly of nanoparticles as a peptidase-degradable platform for molecular MRI. Nature communications. 2017;8:14254
133. Mahmoudi M, Sheibani S, Milani AS, Rezaee F, Gauberti M, Dinarvand R. et al. Crucial role of the protein corona for the specific targeting of nanoparticles. Nanomedicine (London, England). 2015;10:215-26
134. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z. et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296-302
135. Duyn JH. Studying brain microstructure with magnetic susceptibility contrast at high-field. Neuroimage. 2017
Author contact
![]() Corresponding authors: Maxime Gauberti or Sara Martinez de Lizarrondo, INSERM U1237 “Physiopathology and Imaging for Neurological Disorders (PhIND)”, GIP Cyceron, Bd Henri Becquerel, BP5229, 14074 Caen, France, Phone: 0033.231.47.01.55, Fax: 33.231.47.02.22, Mail: gaubertifr or smartinezfr
Corresponding authors: Maxime Gauberti or Sara Martinez de Lizarrondo, INSERM U1237 “Physiopathology and Imaging for Neurological Disorders (PhIND)”, GIP Cyceron, Bd Henri Becquerel, BP5229, 14074 Caen, France, Phone: 0033.231.47.01.55, Fax: 33.231.47.02.22, Mail: gaubertifr or smartinezfr