Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(4):972-989. doi:10.7150/thno.22328 This issue Cite
Research Paper
The Homeotic Protein SIX3 Suppresses Carcinogenesis and Metastasis through Recruiting the LSD1/NuRD(MTA3) Complex
Yu Zheng1,2, Yi Zeng1, Rongfang Qiu1, Ruiqiong Liu1, Wei Huang1, Yongqiang Hou1, Shuang Wang1, Shuai Leng1, Dandan Feng1, Yang Yang1 ![]() , Yan Wang1
, Yan Wang1 ![]()
1. 2011 Collaborative Innovation Center of Tianjin for Medical Epigenetics, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease (Ministry of Education), Department of Biochemistry and Molecular Biology, Tianjin Medical University, Tianjin 300070, China;
2. Department of Biotherapy, Tianjin Medical University Cancer Institute and Hospital; National Clinical Research Center for Cancer; Key Laboratory of Cancer Prevention and Therapy; Tianjin's Clinical Research Center for Cancer; Key Laboratory of Cancer Immunology and Biotherapy, Tianjin 300060, China.
Received 2017-8-11; Accepted 2017-10-17; Published 2018-1-1
Citation:
Zheng Y, Zeng Y, Qiu R, Liu R, Huang W, Hou Y, Wang S, Leng S, Feng D, Yang Y, Wang Y. The Homeotic Protein SIX3 Suppresses Carcinogenesis and Metastasis through Recruiting the LSD1/NuRD(MTA3) Complex. Theranostics 2018; 8(4):972-989. doi:10.7150/thno.22328. https://www.thno.org/v08p0972.htm
Other stylesAbstract

The homeodomain transcription factor SIX3 was recently reported to be a negative regulator of the Wnt pathway and has an emerging role in cancer. However, how SIX3 contributes to tumorigenesis and metastasis is poorly understood.
Methods: We employed affinity purification and mass spectrometry (MS) to identify the proteins physically associated with SIX3. Genome-wide analysis of the SIX3/LSD1/NuRD(MTA3) complex using a chromatin immunoprecipitation-on-chip approach identified a cohort of target genes including WNT1 and FOXC2, which are critically involved in cell proliferation and epithelial-to-mesenchymal transition. Also, we used flow cytometry, growth curve analysis, EdU incorporation assay, colony formation assays, trans-well invasion assays, immunohistochemical staining and in vivo bioluminescence assay to investigate the function of SIX3 in tumorigenesis.
Results: We demonstrate that the SIX3/LSD1/NuRD(MTA3) complex inhibits carcinogenesis in breast cancer cells and suppresses metastasis in breast cancer. SIX3 expression is downregulated in various human cancers and high SIX3 is correlated with improved prognosis.
Conclusion: Our study revealed an important mechanistic link between the loss of function of SIX3 and tumor progression, identified a molecular basis for the opposing actions of MTA1 and MTA3, and may provide new potential prognostic indicators and targets for cancer therapy.
Keywords: epithelial-to-mesenchymal transition, LSD1, MTA3, SIX3, tumorigenesis.
Introduction
Sine oculis homeobox (SIX) proteins are transcription factors characterized by harboring two evolutionarily conserved domains, the homeobox nucleic acid recognition domain (homeodomain) and the SIX domain. DNA binding occurs through the homeodomain [1], while the SIX domain mostly accounts for protein-protein interactions [2]. SIX homeobox 1 (SIX1), SIX2, SIX4, and SIX5 share a remarkable similarity to a common binding sequence (TCAGGTTC) in mammals [2]; however, the site recognized by SIX3 contains the traditional ATTA homeobox binding sequence [3], which indicates SIX3 has a different spectrum of target genes than those of the other members of the family. The role of SIX3 has long been discussed in vertebrate development with a particular focus on neuronal development. During brain development, Six3 participates in telencephalon development by cooperating with Hedgehog signaling to regulate Foxg1a and repress the Wnt/catenin pathway [4]. Six3 also directly activates Sonic hedgehog expression, and its mutation results in holoprosencephaly, the most common forebrain malformation [5]. In addition, under regulation of Sox2, Six3 may have a role in forebrain patterning [6]. Outside of brain development, Six3 also functions with other transcription factors in eye formation [7]. It is of interest that transcriptional repression of SIX3 by MTA1 regulates rhodopsin during eye formation [8]. In mammary glands, SIX3 is also repressed by MTA1, which results in stimulation of Wnt1 [9]. Six family members have a distinctive role in the tumorigenesis; the most studied member, SIX1, functions as an oncogene in cancers of the breast, liver, and many other organs, whereas SIX3 is less experimentally investigated. Recent publications have indicated, however, that SIX3 might be a tumor suppressor or oncogene depending on the cell context. A comprehensive meta-analysis of SIX family members in breast cancer supported that SIX3 was a protective factor for overall survival (OS) and relapse-free survival (RFS) in basal-like breast cancer patients [10]. However, an oncogenic effect of SIX3 was indicated in lung cancer [11]. Although the role of SIX3 in tumorigenesis is unclear, hypermethylated SIX3 has been identified in several types of cancer [12-14]. How the loss of expression/loss-of-function of SIX3 contributes to carcinogenesis and metastasis remains poorly understood.
The nucleosome remodeling and deacetylation (NuRD) complex, which has chromatin-remodeling and histone deacetylase activity [15], is involved in various cellular functions, such as carcinogenesis, genomic stabilization, and senility [16]. Of the components of the NuRD complex, the metastasis-associated (MTA) family of proteins may be the most delicate functional modulator of the complex, as each member of the family can assemble a functionally independent complex [17-19]. Although all three members of this family, MTA1, MTA2, and MTA3, have been implicated in cancer progression and metastasis [17, 18, 20-22], MTA1 and MTA3 exhibit nearly opposite patterns of expression in breast cancer. MTA3 inhibits the epithelial-to-mesenchymal transition (EMT) and breast cancer metastasis [23], and its expression is progressively lost during breast cancer progression, whereas MTA1 promotes breast tumor progression and its expression progressively increases during the process [24]. Furthermore, MTA3 expression is dependent on estrogen action and MTA3 is preferentially expressed in estrogen receptor α (ERα)-positive cells [25], whereas MTA1 directly interacts with ERα and represses its transactivation activity in a histone deacetylase (HDAC)-sensitive manner [26]. In addition, MTA3 transcriptionally represses a series of EMT-promoting genes such as Snail, ZEB2, and N-cadherin [23, 27], thereby regulating an invasive pathway in breast cancer. However, the molecular mechanism that underlies the opposing effects of MTA1 and MTA3 in breast cancer development is still not understood.
Lysine-specific demethylase 1 (LSD1) is a well-characterized histone-modifying enzyme that demethylates di-methylated and mono-methylated histone H3 lysine 4 (H3K4) through a flavin adenine dinucleotide (FAD)-dependent oxidative reaction [28, 29]. LSD1 affects the growth and differentiation of human and mouse embryonic stem (ES) cells, and its deletion in mice leads to embryonic lethality [30, 31]. LSD1 has an important role in mediating the expression of genes involved in cancer as well as non-cancer diseases such as viral infections and neurodegenerative disorders [32]. LSD1 has been identified in a number of corepressor complexes including CoREST [33, 34], CtBP [35], and the NuRD complex [36]. Transcriptional target analysis revealed that the LSD1/NuRD(MTA3) complex regulates several cellular signaling pathways including the TGFβ signaling pathway, which is critically involved in cell proliferation, survival, and EMT. However, the exact transcriptional factors that recruit the LSD1/NuRD(MTA3) complex is unknown.
In this study, we investigated how SIX3 contributes to the maintenance of the epithelial state and how it inhibits carcinogenesis and metastasis through epigenetic programming. We found that SIX3 represses a cohort of genes including WNT1 and FOXC2 that are critically involved in cell proliferation and invasion through specific recruitment of the LSD1/NuRD(MTA3) complex via its direct interaction with LSD1 and MTA3. We revealed that SIX3 inhibits cell proliferation, EMT, and metastasis in vitro and in vivo. SIX3 is markedly downregulated in human carcinomas, and a higher expression level of SIX3 is associated with better prognosis. Our data identified that SIX3 is a transcriptional factor that selectively recruits the LSD1/NuRD(MTA3) complex for inhibition of tumorigenesis and metastasis, supporting the pursuit of SIX3 as a novel target for cancer therapy.
Materials and Methods
Antibodies and Reagents
The antibodies used were: anti-SIX3, anti-H3K4me1, anti-H3K4me2, anti-JAG1, anti-GLI1, anti-ZEB2, anti-ANGPTL4 (Abcam, Hong Kong, China), anti-NCOA3 (BD Biosciences, USA), anti-MTA3, anti-MBD2/3, anti-H3pan-ac (Millipore, Billerica, MD, USA), anti-LSD1, anti-HDAC1, anti-HDAC2, anti-RbAp46/48 (Sigma-Aldrich, St Louis, MO, USA), anti-MTA1, anti-MTA2, and anti-WNT1 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Human recombinant Wnt1 was purchased from Sigma-Aldrich. Protein A/G Sepharose CL-4B beads were from Amersham Biosciences (Indianapolis, IN, USA) and shRNAs were from GenePharma Co., Ltd (Shanghai, China).
Cell Culture and Transfection
HEK293T, HeLa, MCF-7, and MDA-MB-231 cell lines were purchased from ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Invitrogen. MA, USA). T47D, ZR-75-1, and U2OS cell lines were from the Chinese Academy of Medical Sciences. All media were used with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 mg/mL streptomycin (Gibco, BRL, Gaithersburg, MD, USA). Cells were cultured in a humidified incubator equilibrated with 5% CO2 at 37 °C. Transfections were performed using Lipofectamine 2000 or Lipofectamine RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Each experiment was performed in triplicate and repeated at least three times. For RNAi experiments, at least four independent shRNA sequences were tested for each gene and the one with the best efficiency was used.
Immunopurification and Mass Spectrometry
Lysates from HeLa cells expressing FLAG-SIX3 were applied to an equilibrated FLAG column. The column was then washed, followed by elution with FLAG peptides (Sigma-Aldrich). Fractions of the bed volume were collected, resolved on SDS-PAGE, and silver-stained. Gel bands then underwent LC-MS/MS sequencing and analysis.
Fast Protein Liquid Chromatography
HeLa nuclear extracts were obtained and dialyzed against buffer D containing 20 mM HEPES (pH 8.0), 10% glycerol, 0.1 mM EDTA, and 300 mM NaCl (Applygen Technologies, Beijing, China). ~6 mg of nuclear protein was concentrated to a final volume of 0.5 mL in a Millipore Ultrafree centrifugal filter apparatus (10 kDa nominal molecular mass limit), and then applied to an 850 × 20 mm Superose 6 size exclusion column (Amersham Biosciences) that was equilibrated with buffer D containing 1 mM dithiothreitol and calibrated with protein standards (blue dextran, 2000 kDa; thyroglobulin, 669 kDa; ferritin, 440 kDa; catalase, 232 kDa; bovine serum albumin, 67 kDa; and RNase A, 13.7 kDa) (Amersham Biosciences, Little Chalfont, UK). Nuclear protein was eluted using a flow rate of 0.5 mL/min, and fractions were collected.
Glutathione S-transferase (GST) Pull-Down Experiments
GST fusion constructs were expressed in BL21 Escherichia coli cells, and crude bacterial lysates were prepared by sonication in cold PBS in the presence of a protease inhibitor mixture. The in vitro transcription and translation experiments were performed with rabbit reticulocyte lysate (Promega, Madison, WI, USA). In GST pull-down assays, ~10 μg of the appropriate GST fusion proteins was mixed with 5-8 μL of the in vitro transcribed/translated products and incubated in binding buffer (0.8% BSA in PBS with the protease inhibitor mixture). The binding reaction was then added to 30 μL of glutathione-Sepharose beads and mixed at 4 °C for 2 h. The beads were washed five times with binding buffer, resuspended in 30 μL of 2 × SDS-PAGE loading buffer, and resolved on 12% gels. Protein levels were detected with specific antibodies by western blot.
Immunoprecipitation
Cellular extracts were harvested and incubated with the appropriate primary antibody or normal mouse/rabbit immunoglobin G (IgG) at 4 °C overnight. Samples were mixed with protein A/G Sepharose CL-4B beads for 2 h at 4 °C, and following a wash, the beads underwent SDS-PAGE, followed by immunoblotting with a secondary antibody. Immunodetection was performed using enhanced chemiluminescence with an ECL System (Amersham Biosciences) according to the manufacturer's instructions.
ChIP and Re-ChIP
ChIP and re-ChIP were performed in MCF-7 cells as described previously.[36, 37] Briefly, cells were cross-linked with 1% formaldehyde, sonicated, pre-cleared, and incubated with 5-10 μg of the appropriate antibody, followed by addition of protein A/G Sepharose CL-4B beads. The beads were then washed in buffers with high and low salt concentrations, and DNA was eluted for PCR or qChIP assay. For re-ChIP, the beads were eluted with 20 mM dithiothreitol at 37 °C for 30 min, and the eluates were diluted 30-fold for further incubation with the appropriate secondary antibody and beads. The primers used are listed in Supplementary Table S3.
RT-PCR and qPCR
Total cellular RNA was extracted with Trizol under the manufacturer's instructions (Invitrogen). Potential DNA contamination was mitigated using RNase-free DNase treatment (Promega). cDNA was prepared with MMLV reverse transcriptase (Promega). Relative quantitation was performed utilizing the ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA) through the measurement of real-time SYBR green fluorescence, and the results were obtained by means of the comparative Ct method (2-ΔΔCt) using GAPDH as an internal control. This experiment was performed in triplicate. The primers used are listed in Supplementary Table S4.
Lentiviral Production and Infection
Recombinant lentiviruses expressing shSIX3, shLSD1, shMTA3, and shWNT1 were constructed by Shanghai GenePharma (Shanghai, China). Concentrated viruses were used to infect 5 × 105 cells in a 60 mm dish with 8 μg/mL polybrene. Infected cells underwent sorting for target expression. shRNA sequences are listed in Supplementary Table S5.
Cell Starvation and Flow Cytometry
MCF-7 or MDA-MB-231 cells stably expressing lentivirus-delivered shSIX3, shSCR, FLAG-SIX3, or FLAG-Vector were synchronized in G0 phase by serum deprivation for 24 h. The SIX3-overexpressing group and -downregulated group were harvested and fixed with 70% ethanol 12 h after addition of medium containing 10% FBS. The RNase A-treated and propidium iodide (Sigma-Aldrich) stained single cell suspension was analyzed using a flow cytometer equipped with CellQuest software (Becton Dickinson, Franklin Lakes, NJ, USA). The experiment was repeated three times.
EdU Incorporation Assay
MCF-7 or MDA-MB-231 cells stably expressing lentivirus-delivered shSIX3, shSCR, FLAG-SIX3, or FLAG-Vector were seeded into 6-well dishes at a density of 1 × 105 cells/ml and allowed to adhere overnight. Next, the cells were cultured with 5-ethynyl-2′-deoxyuridine (EdU) for 2 h before detection. The proliferation rate of the cells was then evaluated using a Cell-Light EdU Cell Proliferation Detection kit (RiboBio, Guangzhou, China) following the manufacturer's instructions.
Bioluminescence Assay
MDA-MB-231 cells that had been transfected to stably express firefly luciferase (Xenogen Corporation, CA, USA) were infected with lentiviruses carrying either the empty vector or the SIX3 expression construct. These cells were inoculated into the left abdominal mammary fat pad (3-4 × 106 cells) of 6-week-old female nude mice. For bioluminescence imaging, mice were abdominally injected with 200 mg/g of D-luciferin in PBS. Fifteen minutes after injection, mice were anesthetized and bioluminescence was imaged with an IVIS charge-coupled device camera (Xenogen Corporation). Bioluminescence images were obtained with a 15 cm field of view, a binning (resolution) factor of 8, 1/f stop, open filter, and an imaging time of 30 s to 2 min. Bioluminescence determined from the relative optical intensity was defined manually. Photon flux was normalized to background, which was defined from the relative optical intensity of a mouse not given an injection of luciferin. The volume of tumors was measured using a Vernier caliper and calculated according to the formula: 1/6 × length × width2. Animal handling and procedures were approved by the Tianjin Medical University Institutional Animal Care.
Quantitation of Cell Migration Based on Open-Area Analysis
The migration assay was performed using the Oris cell migration kit (Platypus Technologies, Fitchburg, WI, USA). MCF-7 or MDA-MB-231 cells (100 μL; 3 × 104 cells/well) were plated on Oris Cell Migration Assay 96-well plates coated with Collagen I. Each well contained a silicone stopper that prevented cell attachment in the center region of the well. Harmony software was used to calculate the open areas of MCF-7 and MDA-MB-231 cells pre- and post-migration. The values shown for the open areas are inversely proportional to the amount of cell migration within the Oris detection zone. Cells were seeded on Oris assay plates and allowed to adhere for 6 h, and then half of the stoppers were removed. Following a 48 h migration period, the remainder of the stoppers were removed to provide pre-migration controls. Cell migration data were acquired through a high content screening approach in the Operetta high content screening system (Perkin Elmer). The open area within the pre-migration control wells for both cell lines corresponds to a consistently sized detection zone with a diameter of 2 mm. Data shown are the mean per group (n = 6) with standard deviation (SD).
Cell Invasion Assay
Transwell chamber filters (Becton Dickinson) were coated with Matrigel. After infection with lentivirus, MDA-MB-231 cells were suspended in serum-free L-15 media and then 2 × 104 cells were seeded into the upper chamber in a volume of 500 μl. The chamber was cultured in a well containing 500 μL of L-15 media with 10% FBS at 37 °C for 18 h. Cells on the upper side of the membrane were removed by cotton swabs and those on the other side were stained and counted. Four high-powered fields were counted for each membrane.
Tissue Specimen and Immunochemistry
Samples were frozen in liquid nitrogen immediately after surgical removal and maintained at -80 °C until analyzed. Samples were fixed in 4% paraformaldehyde (Sigma-Aldrich) at 4 °C overnight, embedded in paraffin, sectioned at 8 μm onto Superfrost-Plus Slides, which were then processed per standard protocols using 3,3'-diaminobenzidine (DAB) staining and monitored microscopically. All human tissue was collected using protocols approved by the Ethics Committee of Tianjin Medical University, and informed consent was obtained from all patients.
Bisulphite Genomic Sequencing
The methylation status of the SIX3 promoter in 10 paired tumor tissues was assessed by bisulfite genomic sequencing. Genomic DNA was extracted using the QIAamp DNA kit (Qiagen, Hilden, Germany) followed by bisulfite conversion using the EpiTect Bisulfite kit (Qiagen) according to the manufacturer's protocol. The amplified product was subcloned into the pGEM-T Easy vector by TA cloning (Promega) and sequenced via automated sequencing. Primers for SIX3 amplification (+305 to +479) were forward (F) 5'-CTCTATTCCTCCCACTTCTTGTT-3' and reverse (R) 5'-AGCTGGAACATGGACAACTCTTC-3'.
Results
SIX3 is Physically Associated with the LSD1/NuRD(MTA3) Complex
To better understand the mechanistic role of SIX3, we employed affinity purification and mass spectrometry (MS) to identify the proteins physically associated with SIX3. We found that FLAG-tagged SIX3 (FLAG-SIX3) was stably expressed in human breast adenocarcinoma MCF-7 cells, and MS analysis indicated that SIX3 co-purified with Mi-2, LSD1/KDM1A, TLE1, TLE2, TLE3, MTA3, HDAC1, HDAC2, RbAp46, RbAp48, MBD3, and MBD2. Of these proteins, the association of SIX3 with members of the Transducin-like enhancer (TLE) family was previously reported [38], which validates our purification strategy. Interestingly, SIX3 selectively co-purified with all the members of the LSD1/NuRD(MTA3) complex (Figure 1A). The presence of LSD1/NuRD(MTA3) complex subunits in the SIX3 interactome was confirmed by western blot analysis of the column-bound proteins with antibodies against the corresponding proteins (Figure 1B). Detailed results of the MS analysis are provided in Supplementary Table S1.
To verify the presence of a SIX3/LSD1/NuRD(MTA3) complex in vivo, MCF-7 nuclear proteins were fractionated using fast protein liquid chromatography (FPLC), and we found that the native SIX3 from MCF-7 cell nuclear extracts was eluted with an apparent molecular mass much greater than that of the monomeric protein. SIX3 immunoreactivity was detected with a relative symmetric peak centered between ~667 and ~2000 kDa (Figure 1C). Significantly, the elution pattern of SIX3 overlapped with that of LSD1/NuRD(MTA3) complex proteins including LSD1, MTA3, HDAC1, and RbAp46/48 (Figure 1C). These observations support the existence of a SIX3/LSD1/NuRD(MTA3) complex in vivo. Moreover, examination of the expression profiles of endogenous LSD1, MTA3, and SIX3 in eight human cancer cell lines showed that, although ubiquitously expressed, LSD1/MTA3/SIX3 protein levels were higher in the non-invasive ERα-positive cell lines T47D, ZR-75-1, and MCF-7, but lower in the invasive triple-negative (ER/PR/HER2 negative) breast cancer cell lines MDA-MB-231 and MDA-MB-453 (Figure 1D).
To further validate the hypothesis that SIX3 interacts with LSD1/NuRD(MTA3), we performed coimmunoprecipitation experiments using cell lysates from HeLa, MCF-7, and MDA-MB-231 and found that, as predicted, although it binds to all three members of the MTA family, SIX3 prefers to interact with the LSD1/NuRD(MTA3) complex (Figure 1E and 1F).
SIX3 Directly Interacts with LSD1 and MTA3
To further support the physical association between SIX3 and the LSD1/NuRD(MTA3) complex and gain insights into the molecular details involved in the interaction of these proteins, GST pull-down experiments were performed. Incubation of GST-fused SIX3 with in vitro transcribed/translated individual components of the LSD1/NuRD complex revealed that SIX3 interacts directly with LSD1 and MTA3, but not with the other tested components of the NuRD complex (Figure 2A). Reciprocal GST pull-down experiments with GST-fused components of the LSD1/NuRD complex and in vitro transcribed/translated SIX3 yielded similar results (Figure 2B). In addition, GST pull-down assays with GST-fused N-terminal fragments harboring the SIX domain (1-181 aa, SIX3-N), the homeodomain fragments (182-264 aa, SIX3-M), or the C-terminal fragments (265-332 aa, SIX3-C) of SIX3 and in vitro transcribed/translated LSD1 or MTA3 indicated that the N-terminal region of SIX3 interacts with MTA3 and that the homeodomain of SIX3 interacts with LSD1 (Figure 2C). Similarly, GST pull-down assays with GST-fused LSD1 N-terminal fragments [1-419 aa, LSD1-N, which contains the SWIRM (Swi3-Rsc8-Moira) domain and the catalytic FAD-N region], Tower domain fragments (420-520 aa, LSD1-Tower), or C-terminal fragments (521-852 aa, LSD1-C, which contains the catalytic FAD-C region) and in vitro transcribed/translated SIX3 or MTA3 demonstrated that the Tower domain of LSD1 interacts with MTAs and SIX3 (Figure 2D). Moreover, GST pull-down assays with GST-fused N-terminal fragments (1-150 aa, MTA3-N), middle region (151-300 aa, MTA3-M), or C-terminal fragments (301-537 aa, MTA3-C) of MTA3 and in vitro transcribed/translated SIX3 or LSD1 indicate that the BAH-ELM2 (Bromo Adjacent Homology & Egl-27 and MTA1 homology 2) domains of MTA3 interact with SIX3 and that the SANT (Swi3-Ada2-N-CoR-TFIIIB) domain of MTA3 interacts with LSD1 (Figure 2E). Taken together, these results not only provided further support of the specific interaction among SIX3, LSD1, and the NuRD(MTA3) complex, but also delineated the molecular details involved in the formation of the SIX3/LSD1/NuRD(MTA3) complex, as schematically shown in Figure 2F.
Genome-Wide Transcriptional Targets for SIX3/LSD1/NuRD(MTA3) Complex
To further investigate the functional connection between SIX3 and the LSD1/NuRD(MTA3) complex and to explore the biological significance of this connection, we next analyzed genome-wide transcriptional targets of the SIX3/LSD1/NuRD(MTA3) complex using a chromatin immunoprecipitation-on-chip (ChIP-on-chip) approach, the detailed results of which are summarized in Supplementary Table S2. Using MCF-7 cells with antibodies against SIX3, LSD1, or MTA3, we found that 890 unique promoters were targeted by SIX3, which were then classified into various cellular signaling pathways using KEGG pathway analysis (http://www.kegg.jp/kegg/pathway.html) (Figure 3A). These signaling pathways included PI3K-Akt, Wnt, MAPK, and JAK-STAT, as well as pathways in cancer that are critically involved in cell growth, survival, migration, and invasion. The data from SIX3 antibodies were then analyzed with the data from LSD1 (1430 genes) and MTA3 antibodies (1323 genes) to identify common targets or co-targets (Figure 3B). We found 80 unique promoters that were co-targeted by SIX3/LSD1/MTA3. Quantitative ChIP (qChIP) analysis using specific antibodies against SIX3, LSD1, and MTA3 on 15 genes (ANGPTL4, CTCF, FOXC2, GLI1, LAMB1, MTF1, NCOA3, WNT1, WNT3, WNT5A, JAG1, PAX4, RBP1, WDR74, and ZEB2), which represent each of the classified pathways, was performed in MCF-7 cells. The results showed a strong correlation of enrichment among the three proteins, which validates the ChIP-on-chip results (Figure 3C). The qChIP results were validated by conventional DNA electrophoresis of seven representative targets, i.e., ANGPTL4, FOXC2, GLI1, NCOA3, WNT1, JAG1, and ZEB2 (Figure 3D), which have been implicated in triggering or promoting EMT. Furthermore, we examined the responses of these representative genes to SIX3, LSD1, or MTA3 depletion. MCF-7 cells were transfected with lentivirally expressed shRNAs specifically targeting SIX3, LSD1, or MTA3. SIX3, LSD1, or MTA3 depletion led to increased ANGPTL4, FOXC2, GLI1, NCOA3, WNT1, JAG1, and ZEB2 expression at both the transcriptional and protein levels (Figure 3E). Taken together, these results not only support the idea that SIX3 and the LSD1/NuRD(MTA3) complex are functionally linked but also confirm that the SIX3/LSD1/NuRD(MTA3) complex is essential in maintaining epithelial traits.
Regulation of WNT1 and FOXC2 by the SIX3/LSD1/NuRD(MTA3) Complex
Among the common target genes identified, WNT1 and FOXC2 are well-established proto-oncogenes and key EMT regulators [39, 40]. Therefore, we further investigated the transcriptional regulation of WNT1 and FOXC2 by SIX3. As expected, SIX3 knockdown by both #1 and #2 specific lentivirus mediated shRNAs in HeLa, MCF-7, and MDA-MB-231 cells led to increased WNT1 and FOXC2 at both the transcriptional and protein levels (Figure 4A, left panel), while overexpression of SIX3 in these cells resulted in a reduction of WNT1 and FOXC2 expression and protein levels (Figure 4A, right panel). Since the shSIX3#1 is more effective, we chose it for the following experiments.
Next, we investigated the regulation of WNT1 and FOXC2 by the SIX3/LSD1/NuRD(MTA3) complex. Using ChIP assays in MCF-7 cells, we found that SIX3, LSD1, and the NuRD(MTA3) complex co-occupied the promoters of WNT1 and FOXC2 (Figure 4B, upper panels). To further test our proposition that SIX3 recruits the LSD1/NuRD(MTA3) complex to WNT1 and FOXC2 promoters, sequential ChIP or ChIP/re-ChIP experiments were performed. The results showed that in precipitates, the WNT1 and FOXC2 promoters that were immunoprecipitated with antibodies against SIX3 could be re-immunoprecipitated with antibodies against LSD1, MTA3, or HDAC2 (Figure 4B, lower panels). Similar results were obtained when the initial ChIP was performed with antibodies against LSD1, MTA3, or HDAC2. These results support the argument that SIX3 and the LSD1/NuRD(MTA3) complex occupy the WNT1 and FOXC2 promoters as one functionally collaborating protein complex.
Figure 1
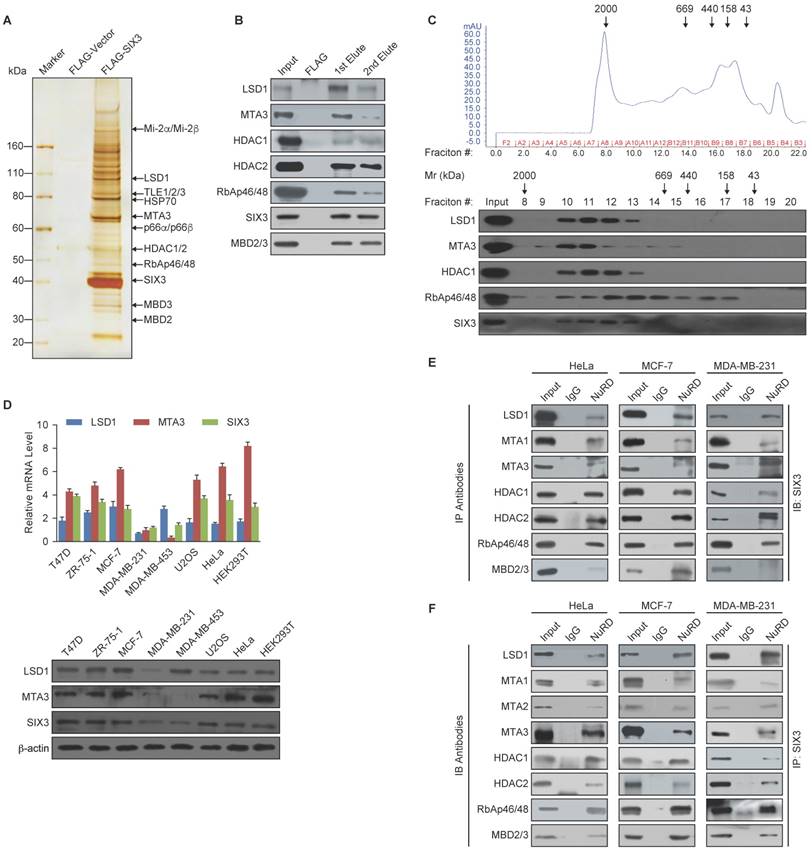
SIX3 Interacts with the LSD1/NuRD(MTA3) Complex (A) Immunoaffinity purification of SIX3-containing protein complex. Cellular extracts from MCF-7 cells stably expressing FLAG vector or FLAG-SIX3 were immunopurified with anti-FLAG affinity columns and eluted with FLAG peptide. These eluates were resolved by SDS-PAGE and silver-stained. The protein bands were retrieved and subjected to MS. (B) Western blot analysis of the identified proteins in the purified fractions, using antibodies against the identified proteins. (C) Co-fractionation of SIX3 and the LSD1/NuRD(MTA3) complex by FPLC. Nuclear extracts of MCF-7 cells underwent fractionation on Superose 6 size exclusion columns. The fractions were subjected to western blot analysis. The elution positions of calibration proteins with known molecular masses (kDa) are indicated and an equal volume from each fraction was analyzed. (D) qRT-PCR (left panel) and western blot (right panel) analysis of endogenous expression of LSD1, MTA3, and SIX3 in a series of cancer cell lines as indicated. The mRNA levels were normalized to those of GADPH (left panel) and β-actin served as a loading control for the western blot (right panel). Error bars represent mean ± SD of three independent experiments. (E) Association of SIX3 with the LSD1/NuRD(MTA3) complex including LSD1, MTA1, MTA2, MTA3, HDAC1, HDAC2, RbAp46/48, and MBD2/3 in HeLa, MCF-7, and MDA-MB-231 cells. Whole cell lysates were immunoprecipitated (IP) with antibodies against the subunits of the LSD1/NuRD(MTA3) complex indicated and immunocomplexes were immunoblotted (IB) by an antibody against SIX3 as indicated. (F) Reciprocal co-immunoprecipitated (IP) assays were performed using a SIX3 antibody and the immunocomplexes were immunoblotted (IB) by antibodies against the subunits of the LSD1/NuRD(MTA3) complex indicated in HeLa, MCF-7, and MDA-MB-231 cells.
Figure 2
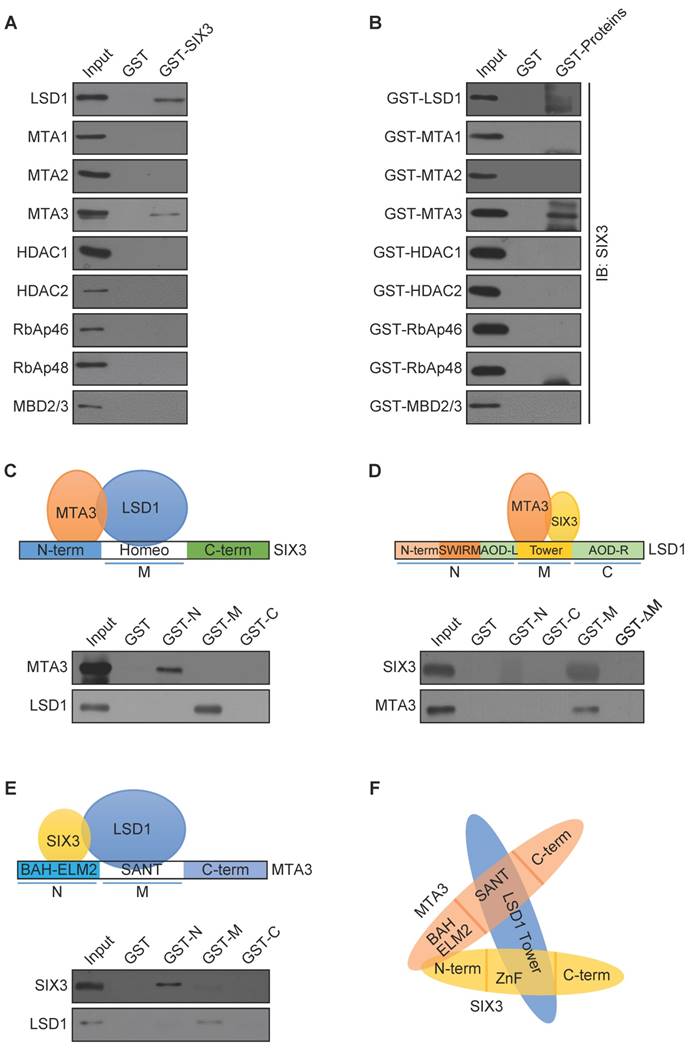
Molecular Interaction Between SIX3 and the LSD1/NuRD(MTA3) Complex (A) GST pull-down assays with GST-fused SIX3 and in vitro transcribed/translated components of the LSD1/NuRD complex as indicated. (B) GST pull-down assays with the indicated GST-fused proteins and in vitro transcribed/translated SIX3. (C) GST pull-down experiments with GST-fused SIX3 deletion constructs and in vitro transcribed/translated LSD1 and MTA3. (D) GST pull-down experiments with GST-fused LSD1 deletion constructs and in vitro transcribed/translated SIX3 and MTA3. (E) GST pull-down experiments with GST-fused MTA3 deletion constructs and in vitro transcribed/translated SIX3 and LSD1. (F) Schematic diagram depicting the molecular interaction between SIX3, LSD1, and MTA3.
Figure 3
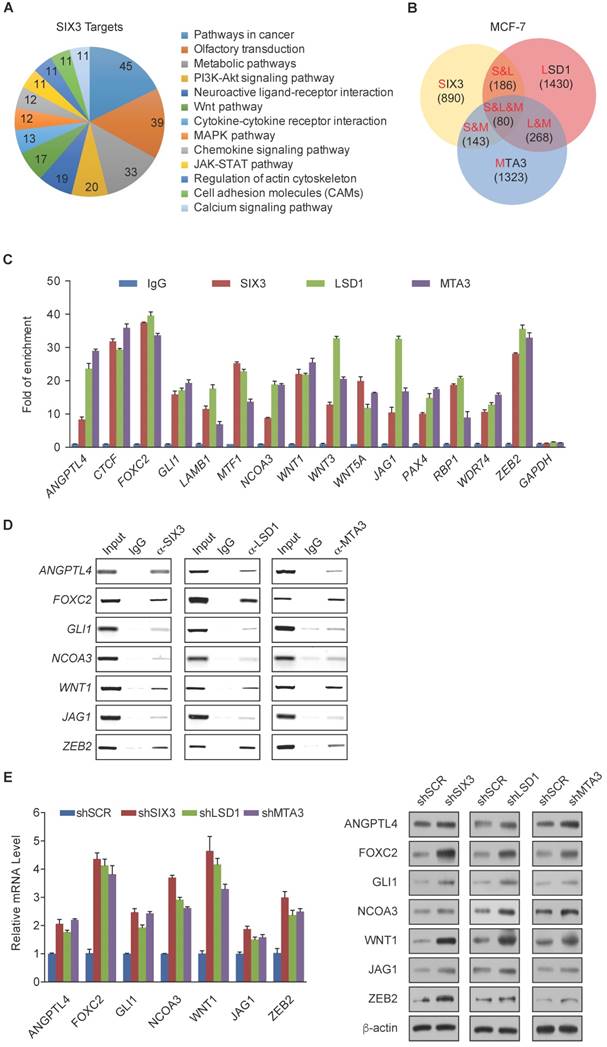
Genome-Wide Transcription Target Analysis of the SIX3/LSD1/NuRD(MTA3) Complex (A) Clustering of the 890 overlapping target genes of SIX3 into functional groups. (B) Venn diagram of overlapping promoters bound by SIX3, LSD1, and MTA3 in MCF-7 cells. The numbers represent the number of promoters that were targeted by the indicated proteins. The detailed results of the ChIP-on-chip experiments are summarized in Supplementary Table S2. (C) Verification of the ChIP-on-chip results via qChIP analysis of the indicated genes in MCF-7 cells. Results are represented as fold change over control with GAPDH as a negative control. Error bars represent mean ± SD for three independent experiments. (D) Verification of the ChIP-on-chip results by conventional DNA electrophoresis. IgG served as a negative control. (E) MCF-7 cells were treated with specific lentivirally expressed shRNAs, and the levels of mRNA (upper panel) and protein (lower panel) of ANGPTL4, FOXC2, GLI1, NCOA3, WNT1, JAG1, and ZEB2 were measured. The mRNA levels were normalized to those of GADPH (upper panel), and β-actin served as a loading control for the western blot (lower panel). Error bars represent mean ± SD of three independent experiments.
Figure 4
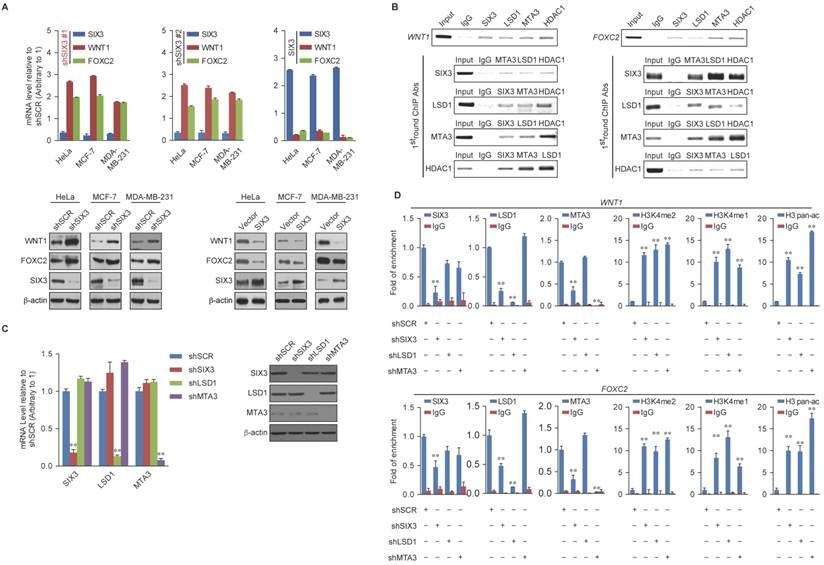
WNT1 and FOXC2 are Co-Targeted by the SIX3/LSD1/NuRD(MTA3) Complex (A) Clones in which SIX3 was stably knocked down using two lentivirally expressed anti-SIX3 shRNAs (left panel) or overexpressed (right panel) using a SIX3 expression construct. The knockdown and overexpressing cells were compared with the parental cell line with respect to the levels of mRNA (upper panel) and protein (lower panel) of WNT1 and FOXC2 in HeLa, MCF-7, and MDA-MB-231 cells. The mRNA levels were normalized to those of GADPH (left panel), and β-actin served as a loading control for the western blot (right panel). Error bars represent mean ± SD of three independent experiments. (B) SIX3 and the LSD1/NuRD(MTA3) complex exist in the same protein complex on WNT1 and FOXC2 promoters. ChIP and re-ChIP experiments were performed in MCF-7 cells with the indicated antibodies. (C) The lentivirus-mediated stable knockdown efficiencies of SIX3, LSD1, and MTA3 were confirmed by qPCR (left panel) or western blot (right panel). mRNA levels were normalized to those of GADPH (left panel), and β-actin served as a loading control for western blot (right panel). Error bars represent mean ± SD of three independent experiments. *p < 0.05; **p < 0.01 (two-tailed t test). (D) qChIP analysis of the recruitment of the indicated proteins on WNT1 and FOXC2 promoters in MCF-7 cells after infection with control lentivirus-mediated shRNA, or shRNAs targeting SIX3, LSD1, or MTA3. Purified rabbit IgG was used as a negative control. Error bars represent mean ± SD of three independent experiments. * p < 0.05 and **p < 0.01 (two-tailed t test).
To determine a functional connection between SIX3 and the LSD1/NuRD(MTA3) complex on WNT1 and FOXC2 promoters, MCF-7 cells were transfected with lentivirus-mediated shRNAs specifically against SIX3, LSD1, or MTA3. Each of these shRNAs led to a significant reduction in the expression of its target gene without causing detectable changes in non-target genes (Figure 4C). qChIP analyses showed that SIX3 knockdown led to a significant reduction in the binding of LSD1 and MTA3 to the promoters of WNT1 and FOXC2, whereas depletion of LSD1 and MTA3 expression resulted in only a marginal decrease in the association of SIX3 with the promoters of WNT1 and FOXC2 (Figure 4D). Notably, knockdown of either SIX3 or MTA3 led to a severe increase in H3K4me1/2 at the promoters of WNT1 and FOXC2, whereas knockdown of either SIX3 or LSD1 resulted in a significant elevation in H3 pan-acetylation (H3pan-ac) at the promoters of WNT1 and FOXC2 (Figure 4D). These findings suggest that the LSD1/NuRD(MTA3) complex epigenetically represses WNT1 and FOXC2 upon recruitment of SIX3.
SIX3 Suppresses Breast Carcinogenesis and Invasion
Based on the known role of the LSD1/NuRD(MTA3) complex in cancer development and progression and our observations that SIX3 and the LSD1/NuRD(MTA3) complex are physically and functionally associated, we next investigated what role, if any, SIX3 has in tumorigenesis. For this purpose, we first analyzed the effect of gain of function and loss of function of SIX3 on the cell cycle profile of MCF-7 (higher SIX3 expression) or MDA-MB-231 cells (lower SIX3 expression). We found that compared to the control, SIX3 depletion was associated with a decreased cell population in G1 and an increased cell population in the S-phase, whereas MDA-MB-231 cells exposed to SIX3 overexpression exhibited an increase in the proportion of cells in G1 and a decrease in the proportion of cells in the S-phase (Figure 5A). We found from growth curve assays that the S-phase accumulation by SIX3 depletion in MCF-7 was not due to S-phase arrest. Specially, the proliferation-promoting effect of SIX3 depletion was blocked by the combination of SIX3 and WNT1 knockdown (Figure 5B). Furthermore, using an enhanced BrdU (EdU) incorporation assay, we found that SIX3 depletion was associated with an increased mitotic rate compared to the control, while MDA-MB-231 cells exposed to SIX3 overexpression exhibited a decreased mitotic rate (Figure 5C). In addition, using colony formation assays, we found that in MCF-7 cells, SIX3 knockdown was associated with a marked increase in colony number (Figure 5D, left panels), whereas SIX3 overexpression in MDA-MB-231 cells was associated with a significant decrease in colony number (Figure 5D, right panels). Furthermore, in agreement with the functional link between SIX3 and WNT1 described previously, the increase in colony formation resulting from SIX3 knockdown was partially reversed by co-knockdown of WNT1 (Figure 5D). We next investigated the role of SIX3 in tumor progression in vivo. For this purpose, we performed a bioluminescence assay to measure tumor growth in situ in 6-week-old female nude athymic BALB/c mice. We found that tumor growth was greatly suppressed by SIX3 overexpression, which indicates that SIX3 inhibits tumor growth (Figure 5E). These findings support the notion that SIX3 suppresses breast cancer cell proliferation and it does so, at least in part, through cooperation with LSD1/NuRD(MTA3) and via repression of WNT1.
Next, we investigated whether SIX3 has a role in EMT and tumor metastasis. For this purpose, SIX3 was depleted in MCF-7 cells or overexpressed in MDA-MB-231 cells, and the impact of the loss of function or gain of function of SIX3 on the migration potential and invasive potential of these cells was assessed. Using a cell migration assay, we found that the amount of open area remaining after 48 h of migration differed; SIX3 knockdown in MCF-7 cells was associated with an increased migration rate, whereas SIX3 overexpression in MDA-MB-231 cells was associated with a decreased migration rate (Figure 5F). Furthermore, in the trans-well invasion assay, we found that a significant increase in invasiveness was associated with SIX3 depletion, which could be restored by combining with WNT1 depletion (Figure 5G, left panel). Moreover, overexpression of SIX3 resulted in a decrease of ~3-fold in invading cells, and this decrease was reversed by addition of human recombinant WNT1. In addition, exogenous addition of WNT1 completely masked the metastatic inhibition effect of SIX3 overexpression (Figure 5G, right panel). We confirmed the efficiency of lentivirus-mediated shRNAs targeting SIX3 or SIX3 overexpression using western blot analysis (Figure 5H). Collectively, these results indicate that SIX3 inhibits the proliferation and invasive potential of breast cancer cells possibly by acting in conjunction with LSD1/NuRD(MTA3) and by repressing WNT1 expression.
SIX3 is Downregulated in Breast Cancer through Promoter Aberrant Hypermethylation
To further observe the role of SIX3 in tumorigenesis, we collected 109 breast cancer samples from patients, 33 of which were paired with adjacent normal tissues, and performed tissue arrays by immunohistochemical staining. SIX3 was significantly downregulated in tumors, and its expression was negatively correlated with histological grade (Figure 6A and 6B). In addition, SIX3 expression was significantly downregulated in tumors compared to adjacent normal breast tissue (Figure 6C). In 14 of 15 paired samples of each cancer grade, the level of SIX3 mRNA was lower in tumor tissue than in adjacent tissue (Figure 6D), as measured by qPCR. In addition, statistical analysis revealed a significant negative correlation when the relative expression levels of WNT1 and FOXC2 were plotted against those of SIX3 in the 30 samples of grade II (Figure 6E), which indicates a significant negative correlation between the expression of SIX3 and WNT1/FOXC2 in these samples. Furthermore, the negative correlation between WNT1 expression and SIX3 expression was verified in three published datasets, GSE7882, GSE19615 and GSE70905, which supports our findings that WNT1 is transcriptionally regulated by SIX3 (Figure 6F). Moreover, clinical data from both our group (Figures 6G, left panel) and GSE1456 (Figures 6G, right panel) revealed that SIX3 expression also gradually decreases in breast cancer subtypes including luminal A, luminal B, HER2-enriched and basal-like. Importantly, CpGs residing within SIX3 were strongly hypermethylated in tumor tissues compared to adjacent normal tissues from 10 paired grade II or III samples (Figure 6H). Collectively, our findings are consistent with a role of SIX3 in promoting carcinogenesis and support the observation that WNT1/FOXC2 is a downstream target of SIX3. In addition, aberrant promoter hypermethylation that leads to a gradual loss of SIX3 with breast cancer malignancy may be the main reason for the epithelial-to mesenchymal-transition and the initial cause of breast carcinogenesis.
Figure 5
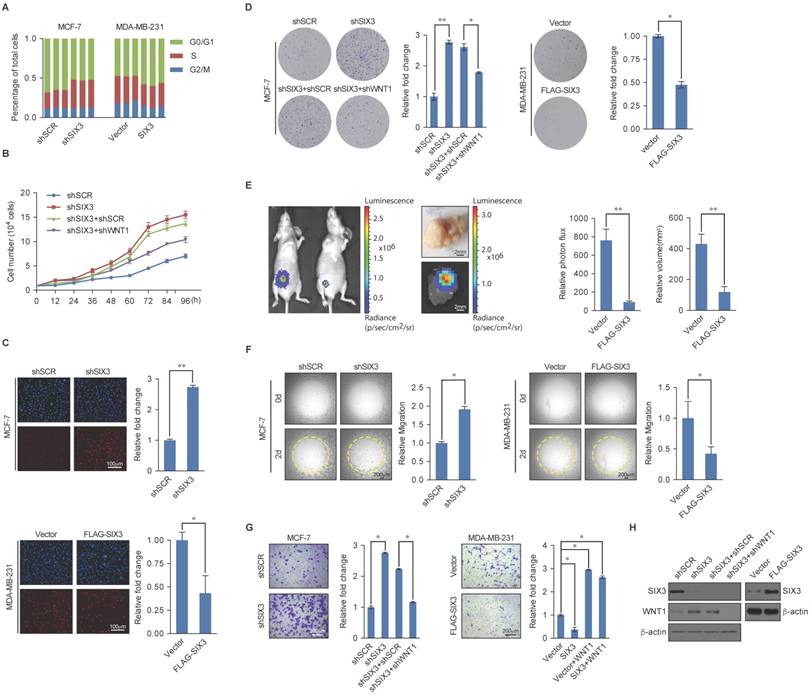
SIX3 Inhibits Breast Carcinogenesis and Metastasis (A) SIX3 suppresses cellular proliferation. MCF-7 cells stably expressing SIX3 or stably transfected with SIX3 shRNA were subjected to cell cycle analysis using flow cytometry. (B) SIX3 inhibits cellular proliferation. Growth curve analysis of non-invasive SIX3 high-expressing MCF-7 cells infected with lentivirus mediated shSCR, shSIX3, and/or shWNT1. (C) Non-invasive SIX3 high-expressing MCF-7 cells were infected with lentivirus mediated shSCR, shSIX3, and/or shWNT1 (left panels), and invasive SIX3 low-expressing MDA-MB-231 cells were infected with lentivirus mediated SIX3 and/or human recombinant WNT1 (right panels). After 48 h of plasmid transfection, the EdU incorporation assay were performed using a fluorescence method. For each group, six different fields were randomly chosen and counted under fluorescence microscopy with 10-fold magnification. Representative photos are shown on the left and statistically analyzed on the right. *p < 0.05 and **p < 0.01 (two-tailed t test). (D) MCF-7 cells or MDA-MB-231 cells infected with the indicated lentivirus were maintained in culture media for 14 days prior to being stained with crystal violet. Representative photos are shown on the left and statistically analyzed on the right. (E) MDA-MB-231-Luc-D3H2LN cells infected with lentiviruses expressing vector or SIX3 were inoculated orthotopically into the abdominal mammary fat pad of 6-week-old female nude mice (n = 8). Eight tumors were quantified using bioluminescence imaging 4 weeks after the initial implantation. Tumors were measured using Vernier calipers, and the volume was calculated according to the formula: 1/6 × length × width2. Error bars indicate mean ± SD. *p < 0.05; **p < 0.01 (two-tailed t test). Representative in vivo and in vitro bioluminescent images are shown. (F) SIX3 represses the migration rate of breast cancer cells. MCF-7 cells (100 μL, 3 × 104 cells/well) with shSCR/shSIX3 or MDA-MB-231 cells with vector/SIX3 overexpression (3 × 104 cells/well) were seeded on Oris assay plates and allowed to adhere for 6 h. At this point, half of the stoppers were removed. Following a 48 h migration period, the remainder of the stoppers were removed to provide pre-migration controls. The open area within the pre-migration control wells for both cell lines corresponds to a consistent-sized detection zone with a diameter of 2 mm. Data shown are the means per group (n = 6) ± SD. *p < 0.05 and **p < 0.01 (two-tailed t test). (G) SIX3 represses the invasiveness of breast cancer cells. MCF-7 cells or MDA-MB-231 cells infected with the indicated lentivirus were starved for 18 h before cell invasion assays were performed using Matrigel transwell filters. The invaded cells were stained and counted. The images represent one field under microscopy (10× magnification). (H) The overexpression and knockdown efficiencies of SIX3 and WNT1 were confirmed by qPCR. (A-B, D-E, G) Each bar represents the mean ± SD for triplicate measurements. *p < 0.05 and **p < 0.01 (two-tailed t test).
Figure 6
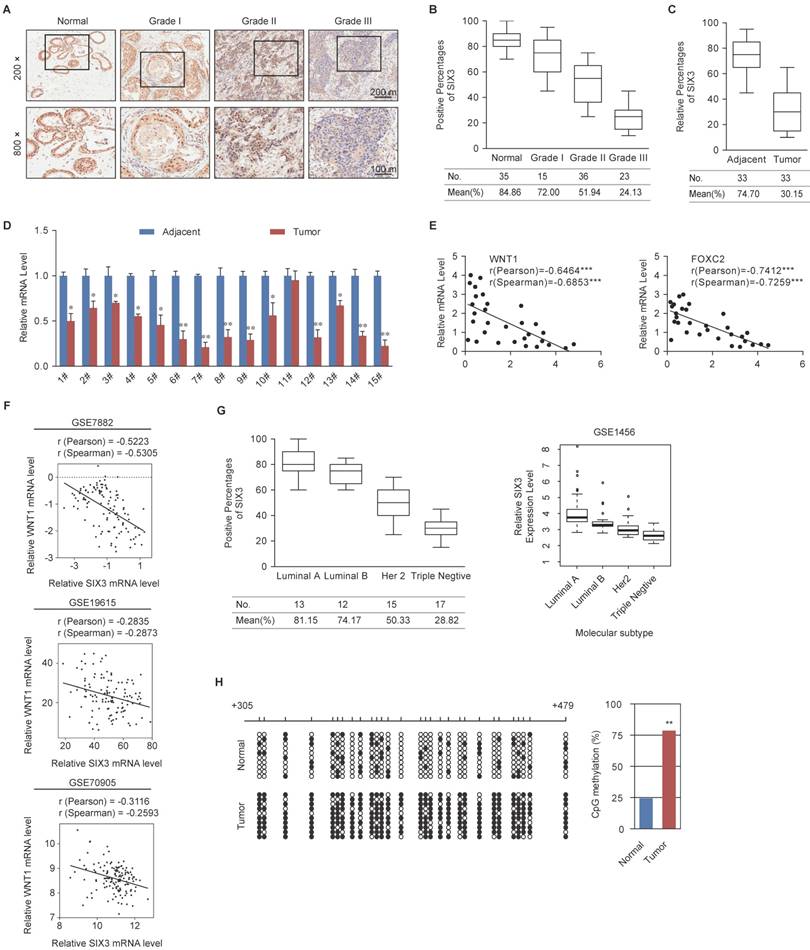
Expression of SIX3 is Negatively Correlated with Breast Cancer Malignancy (A) Immunohistochemical staining of SIX3 in normal breast tissue and breast carcinomas (histological grades I, II, and III). For each grade, representative photos of two specimens are shown. (B, C) The positively stained nuclei (%) in grouped samples (B) or 30 paired samples (C) were analyzed by two-tailed t test. (D) SIX3 mRNA is downregulated in breast cancer. Total RNA in paired samples of breast cancer versus adjacent normal breast tissue were extracted and the expression of each gene was measured by qPCR. mRNA levels were normalized to those of GADPH. Each bar represents the mean ± SD for triplicate experiments (*p < 0.05; **p < 0.01; ***p < 0.001, two-tailed t test). (E) SIX3 mRNA level is negatively correlated with the level of WNT1 or FOXC2 mRNA. The relative level of SIX3 expression was plotted against the relative level of WNT1 or FOXC2 expression (***p < 0.001, two-tailed t test). (F) Analysis of public data sets (GSE7882, GSE19615, and GSE70905) for expression of SIX3 and WNT1 in breast cancer. The relative expression level of WNT1 was plotted against that of SIX3. (G) Analysis of clinical data collected for this study (left panel) or public dataset GSE1456 (right panel) for the expression of SIX3 by molecular subtype. (H) Bisulfite sequencing shows that SIX3 promoter methylation is increased at a number of CpG sites in tumor tissue compared to adjacent normal tissue. Each row represents the analysis of ten individual paired samples, and each square is a single CpG site. White and black squares represent unmethylated and methylated CpGs, respectively. *p < 0.05 and **p < 0.01 (two-tailed t test).
SIX3 is Downregulated in Multiple Carcinomas and Associated with Better Overall Survival
To investigate whether the effect of SIX3 could be extended to a broader range of cancers, we collected a series of carcinoma samples from patients with esophageal, stomach, colon, lung, and prostate cancer. At least 15 samples of each type of carcinoma were collected and paired with adjacent normal tissue samples. Tissue microarray analysis by immunohistochemical staining showed statistically significant downregulation of SIX3 in carcinomas from multiple tissues compared to that in the adjacent normal tissues (Figures 7A and 7B). Representative results from the Oncomine database (https://www.oncomine.com/) also confirmed that SIX3 expression was decreased in breast carcinoma, cervical cancer, tongue carcinoma, gastric cancer, pancreatic carcinoma, liver cancer, myeloma, lymphoma, and renal carcinoma (Figure 7C). Furthermore, analysis of nine published clinical datasets revealed that the expression of WNT1 was statistically significantly and negatively correlated with SIX3 expression in multiple carcinomas (Figure 7D). In addition, we found using Kaplan-Meier survival analysis that higher expression of SIX3 was associated with improved overall survival in patients with breast cancer and gastric cancer (Figure 7E). Taken together, our findings support a role for SIX3 in repressing tumorigenesis and suggest that SIX3 could serve as a novel biomarker for cancer diagnosis and as a potential target for cancer therapy.
Discussion
Our findings from this study demonstrate that SIX3 acts as a transcriptional repressor that recruits the chromatin remodeling LSD1/NuRD(MTA3) complex to inhibit the expression of a set of genes including WNT1, FOXC2, ANGPTL4, GLI1, NCOA3, JAG1, and ZEB2, which are known to be critically involved in EMT, a hallmark of cancer metastasis [41]. Through a physical interaction, the transcriptional regulators SIX3, LSD1, and NuRD(MTA3) cooperate to operate a transcriptional repression pathway that maintains mammary epithelial hemostasis by controlling the hierarchical molecular network of EMT.
Among the signaling pathways in which SIX3 is involved, previous studies have found that the Wnt pathway is particularly important [42, 43], a conclusion that is supported by our observation that SIX3 binds to the WNT1 promoter in breast cancer cells and represses WNT1 expression. Notably, we also demonstrated that the SIX3/LSD1/NuRD(MTA3) complex targets a group of Wnt ligands including WNT1, WNT3, and WNT5A, which suggests that SIX3 is a novel massive negative regulator of the Wnt pathway. In addition, the target genes include the Hedgehog pathway (GLI1), Notch pathway (JAG1), EMT transcription factors (FOXC2, ZEB2), and ANGPTL4, which affects breast cancer lung seeding by disrupting endothelial cell junctions [44]. Our finding that SIX3 and LSD1/NuRD(MTA3) function as an integral co-repressive complex marks the first time that SIX3 has been identified as a component of a protein complex exerting control over epigenetic transcriptional activity. Based on this study, many more potential regulatory functions may be added to the SIX3 transcription network to show its true complexity. Through direct interactions between SIX3, LSD1, and MTA3, the SIX3/LSD1/NuRD(MTA3) complex is formed, which can mediate coordinative demethylation of H3K4me1/2 and histone acetylation on target promoters for epigenetic repression of gene activity (Figure 7F upper panel). We showed that the abundance of the epigenetic modifications H3K4me1, H3K4me2, and H3 pan-ac on the promoters of target genes was greatly elevated upon deletion of SIX3, LSD1, or MTA3, and these findings support the hypothesis that SIX3, LSD1, and MTA3/NuRD act as an integrated complex. Previously, our group found that LSD1 is an integral component of the NuRD complex, and that LSD1/NuRD(MTA3) can transcriptionally repress a series of EMT-promoting genes, such as TGFβ1, to inhibit breast cancer metastasis [36]. The finding that SIX3 selectively recruits the LSD1/NuRD(MTA3) complex for transactional repression and inhibition of breast carcinogenesis effectively supplements our previous findings. In addition, our results also proved that aberrant promoter hypermethylation that leads to a gradual loss of SIX3 with breast cancer malignancy may be the main reason for the epithelial-to-mesenchymal transition and the initial cause of breast carcinogenesis (Figure 7F, lower panel). Because SIX3 was found to be downregulated and reported to be involved in promoter hypermethylation in several types of solid tumors [13, 14, 45, 46], we expect that this mechanism is suitable not only for understanding breast tumorigenesis, but also for evaluation of more types of SIX3 governing epithelium carcinogenesis.
The retinal determination gene network (RDGN, Dach-Eya-Six network), initially discovered in the process of Drosophila eye specification, is attracting increasing attention for its role in tumorigenesis [47]. In RDGN, the antagonist effect of SIX on DACH1 is well established [47], and the DACH/EYA/SIX network is aberrantly expressed in adult cancers in a seemingly coordinated fashion, in which the Eya and Six genes are up-regulated and the Dach genes are down-regulated [48]. DACH1 has been confirmed to inhibit FOXC2, ZEB1, SNAIL [49, 50], and has been shown to be recruited onto chromatin DNA along with other transcriptional factors, such as Six1, Jun, and Smad4 [51-53]. SIX1 functioned as an oncogene in cancer, but we found that the function of SIX3 was different from that of SIX1, as SIX3 could recruit the LSD1/NuRD(MTA3) complex to inhibit FOXC2, WNT1, and ZEB2 for inhibition of tumorigenesis and metastasis. It is interesting that both DACH1 and SIX3 can inhibit FOXC2; therefore, one possibility is that DACH1 and SIX3 may be functionally correlated. To our surprise, our results showed that SIX3 can interact with all three members of the MTA family while only directly binding to MTA3, thus constituting a facultative component of the NuRD complex. It is believed that the NuRD complex contains several subunits whose pattern of expression is heterogeneous in various cell and tissue types [23], and it has been proposed that subunit heterogeneity confers these complexes with additional regulatory capacity and with unique functional properties. For example, NuRD(MTA1) promotes carcinogenesis and metastasis, whereas NuRD(MTA3) inhibits EMT and tumor metastasis [24, 54]. The specific binding of SIX3 and MTA3 is similar to our previous finding that GATA3 selectively binds to NuRD/(MTA3) for transcriptional repression [27]. We propose that the “mesenchymal promoters” such as WNT1, FOXC2, ZEB2, SNAIL, and TWIST preferentially interact with NuRD(MTA1) to promote the EMT and metastasis, whereas the “epithelium maintainers” such as SIX3 and GATA3 maintain epithelial traits. Interestingly, WNT1, FOXC2, and ZEB2 are all SIX3 transcriptional targets. Based on the complexity of the EMT regulatory network, we expect that SIX3-regulated targets would elicit negative feedback to repress SIX3. Our current study not only provides a molecular basis for the opposing action of MTA3 and MTA1 in breast cancer progression, but also adds to the understanding of the molecular interplay involved in the sophisticated regulatory network of EMT. The exact molecular mechanism underlying the recruitment specificity for the NuRD complex by the transcription factors that control EMT processes remains to be delineated. It is possible that SIX3 interacts with MTA1 and MTA2 indirectly for a specific purpose such as regulatory feedback. For example, it has been previously reported that SIX3 physically interacts with MTA1 to transcriptionally repress itself [9].
We also showed that the SIX3/LSD1/NuRD(MTA3) complex is a potent suppressor of breast cancer metastasis through targeting of the promoters of an array of genes, which comprise several important cellular signaling pathways that regulate cell migration and invasion. Several of these genes, including WNT1, FOXC2, ANGPTL4, GLI1, and ZEB2, have been implicated in the development and progression of a variety of human malignancies [40, 44, 55-57]. WNT1 and FOXC2 are well-known as EMT promoters through their influence on cellular signaling pathways including Wnt and TGFβ to promote carcinogenesis and metastasis [58, 59]. Our finding that the SIX3/LSD1/NuRD(MTA3) complex transcriptionally represses these genes positions the SIX3/LSD1/NuRD(MTA3) complex upstream of these genes and places the complex at the node of the regulatory network of EMT. This realization provides important mechanistic insights into the functional similarity and interplay of the above-described genes in the development and progression of breast cancer. With the aid of LSD1/NuRD(MTA3), SIX3 inhibits the expression of mesenchymal-promoting genes and maintains luminal cell fate in the mammary epithelium. Aberrant promoter hypermethylation and gene silencing of SIX3 are observed in many cancers, such as glioma, lung adenocarcinoma, gastric cancer, and breast cancer, and are associated with poor clinical outcomes [13, 14, 45, 46]. Upon loss of function of SIX3 resulting from aberrant promoter hypermethylation, the expression of WNT1, FOXC2, and other mesenchymal promoters are depressed, whereas elevated mesenchymal promoters would promote the EMT and tumor metastasis. As the complexity of the function of SIX3 emerged, we linked the transcriptional activity of SIX3 with its pathological impact on not only breast cancer but also other tumor types, which suggests SIX3 could be a potentially valuable biomarker for cancer diagnosis and prognosis.
In summary, our results demonstrate that the SIX3/LSD1/NuRD(MTA3) complex maintains mammary epithelial hemostasis, dictates physical epithelial cell fate, and governs the dynamics of epithelial cell plasticity in the mammary gland, the dysfunction of which affects the fate of mammary epithelial cells and contributes to the carcinogenesis and metastasis of breast cancer. Our data indicate a mechanistic link between the loss of function of SIX3 and cancer progression, and a molecular mechanism underlying the opposing action of MTA1 and MTA3 in the development and progression of breast cancer. Furthermore, our findings significantly add to the understanding of the complex hierarchical regulatory network of the EMT and support the pursuit of SIX3 and MTA1/MTA3 as potential prognostic indicators and/or targets for cancer therapy.
Figure 7
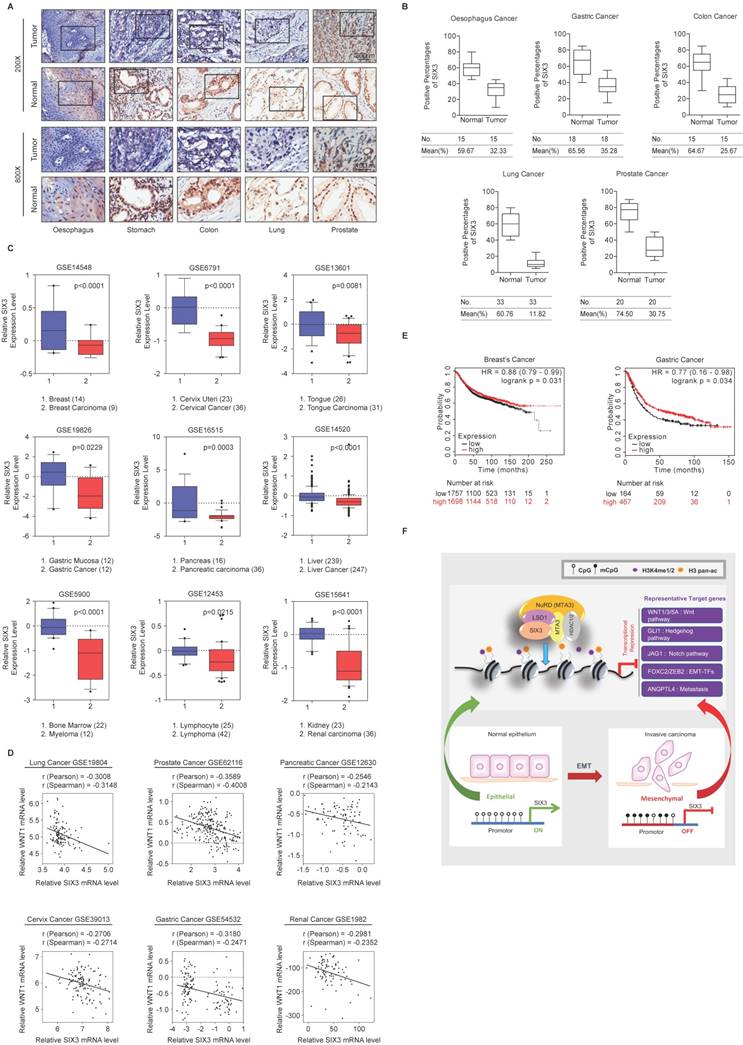
SIX3 is Downregulated in Multiple Carcinomas and Positively Correlated with Better Prognosis (A, B) SIX3 is downregulated in multiple carcinomas. Immunohistochemical staining of SIX3 in paired samples of esophageal, stomach, colon, lung, and prostate carcinoma versus adjacent normal tissue samples. Representative images of 200-fold magnifications of each type of paired tumor section are presented. Each bar represents the mean ± SD for triplicate experiments (*p < 0.05 and ** p < 0.01). (C) Analysis of public datasets for the expression of SIX3 by two-tailed t test (*p < 0.05, **p < 0.01, ***p < 0.001). (D) Analysis of public data sets for the expression of SIX3 and WNT1 in multiple carcinomas. The relative level of WNT1 is plotted against that of SIX3. (E) Kaplan-Meier survival analysis of the relationship between survival time and SIX3 signature in breast cancer and gastric cancer using KM plotter (http://kmplot.com/analysis/). (F) Graphic model as discussed in the text. DNA (black line); nucleosomes with single N terminus of H3 (blue ball); unmethylated CpG sites (hollow circle); methylated CpG sites (solid circle); mono- and di-methylated H3K4 (purple circle); pan-acetylated H3 (orange circle).
Abbreviations
SIX: Sine oculis homeobox; NuRD: The nucleosome remodeling and deacetylation; EMT: Epithelial-to-mesenchymal transition; LSD1: Lysine-specific demethylase 1; TLE: Transducin-like enhancer; FPLC: Fast protein liquid chromatography; RDGN: Retinal Determination Gene Network (Dach-Eya-Six network).
Supplementary Material
Supplementary Table S1: Mass spectrometry analysis of SIX3-containing protein complex. Supplementary Table S3: The primers used in ChIP and qChIP assays. Supplementary Table S4: The primers used in quantitative real-time PCR (qPCR). Supplementary Table S5: The shRNA sequences.
Supplementary Table S2: ChIP-on-chip results of SIX3, LSD1 and MTA3.
Acknowledgements
This work was supported by grant (2016YFA0102400 to Y. W.) from the Major State Basic Research Development Program of China, grants (81472733 and 81322032 to Y. W., 81402334 to Y. Y. and 81502446 to R. Q.) from the National Natural Science Foundation of China, grants (NCET-12-1067, FANEDD-201231, and RFDP-20131202110012 to Y. W.) from the Ministry of Education of China, and grant from the Excellent Talent Project of Tianjin Medical University to Y. W.
Author Contributions
Conceptualization, Y.Z., Y.Y., and Y.W.; Methodology, Y.Y., Yi Z., and Y.W.; Software, Formal Analysis, and Data Curation, R.L., S.W., and Y. Y.; Investigation, Y.Z., Yi Z., R. Q., R.L., H.W., Y.H., S.W., S.L., and D.F.; Writing, Y.Y. and Y.W.; Funding Acquisition, Y.Y., R.Q. and Y.W.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Scott MP, Tamkun JW, Hartzell GW 3rd. The structure and function of the homeodomain. Biochimica et biophysica acta. 1989;989:25-48
2. Kumar JP. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cellular and molecular life sciences: CMLS. 2009;66:565-83
3. Zhu CC, Dyer MA, Uchikawa M, Kondoh H, Lagutin OV, Oliver G. Six3-mediated auto repression and eye development requires its interaction with members of the Groucho-related family of co-repressors. Development. 2002;129:2835-49
4. Carlin D, Sepich D, Grover VK, Cooper MK, Solnica-Krezel L, Inbal A. Six3 cooperates with Hedgehog signaling to specify ventral telencephalon by promoting early expression of Foxg1a and repressing Wnt signaling. Development. 2012;139:2614-24
5. Geng X, Speirs C, Lagutin O, Inbal A, Liu W, Solnica-Krezel L. et al. Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Developmental cell. 2008;15:236-47
6. Beccari L, Conte I, Cisneros E, Bovolenta P. Sox2-mediated differential activation of Six3.2 contributes to forebrain patterning. Development. 2012;139:151-64
7. Masse K, Bhamra S, Eason R, Dale N, Jones EA. Purine-mediated signalling triggers eye development. Nature. 2007;449:1058-62
8. Manavathi B, Peng S, Rayala SK, Talukder AH, Wang MH, Wang RA. et al. Repression of Six3 by a corepressor regulates rhodopsin expression. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13128-33
9. Kumar R, Balasenthil S, Manavathi B, Rayala SK, Pakala SB. Metastasis-associated protein 1 and its short form variant stimulates Wnt1 transcription through promoting its derepression from Six3 corepressor. Cancer research. 2010;70:6649-58
10. Xu HX, Wu KJ, Tian YJ, Liu Q, Han N, He XL. et al. Expression profile of SIX family members correlates with clinic-pathological features and prognosis of breast cancer: A systematic review and meta-analysis. Medicine. 2016;95:e4085
11. Liu Q, Li A, Tian Y, Liu Y, Li T, Zhang C. et al. The expression profile and clinic significance of the SIX family in non-small cell lung cancer. Journal of hematology & oncology. 2016;9:119
12. Jin B, Yao B, Li JL, Fields CR, Delmas AL, Liu C. et al. DNMT1 and DNMT3B modulate distinct polycomb-mediated histone modifications in colon cancer. Cancer research. 2009;69:7412-21
13. Rauscher GH, Kresovich JK, Poulin M, Yan L, Macias V, Mahmoud AM. et al. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC cancer. 2015;15:816
14. Zhang Z, Tang H, Wang Z, Zhang B, Liu W, Lu H. et al. MiR-185 targets the DNA methyltransferases 1 and regulates global DNA methylation in human glioma. Molecular cancer. 2011;10:124
15. Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Molecular cell. 1998;2:851-61
16. Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nature reviews Cancer. 2011;11:588-96
17. Manavathi B, Kumar R. Metastasis tumor antigens, an emerging family of multifaceted master coregulators. The Journal of biological chemistry. 2007;282:1529-33
18. Singh RR, Kumar R. MTA family of transcriptional metaregulators in mammary gland morphogenesis and breast cancer. Journal of mammary gland biology and neoplasia. 2007;12:115-25
19. Manavathi B, Singh K, Kumar R. MTA family of coregulators in nuclear receptor biology and pathology. Nuclear receptor signaling. 2007;5:e010
20. Kumar R, Wang RA, Bagheri-Yarmand R. Emerging roles of MTA family members in human cancers. Semin Oncol. 2003;30:30-7
21. Kumar R. Another tie that binds the MTA family to breast cancer. Cell. 2003;113:142-3
22. Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26:5433-8
23. Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207-19
24. Zhang H, Stephens LC, Kumar R. Metastasis tumor antigen family proteins during breast cancer progression and metastasis in a reliable mouse model for human breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:1479-86
25. Fujita N, Kajita M, Taysavang P, Wade PA. Hormonal regulation of metastasis-associated protein 3 transcription in breast cancer cells. Mol Endocrinol. 2004;18:2937-49
26. Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M. et al. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nature cell biology. 2001;3:30-7
27. Si W, Huang W, Zheng Y, Yang Y, Liu X, Shan L. et al. Dysfunction of the Reciprocal Feedback Loop between GATA3- and ZEB2-Nucleated Repression Programs Contributes to Breast Cancer Metastasis. Cancer cell. 2015;27:822-36
28. Lan F, Nottke AC, Shi Y. Mechanisms involved in the regulation of histone lysine demethylases. Current opinion in cell biology. 2008;20:316-25
29. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941-53
30. Adamo A, Sese B, Boue S, Castano J, Paramonov I, Barrero MJ. et al. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nature cell biology. 2011;13:652-9
31. Foster CT, Dovey OM, Lezina L, Luo JL, Gant TW, Barlev N. et al. Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Molecular and cellular biology. 2010;30:4851-63
32. Yang Y, Liu R, Qiu R, Zheng Y, Huang W, Hu H. et al. CRL4B promotes tumorigenesis by coordinating with SUV39H1/HP1/DNMT3A in DNA methylation-based epigenetic silencing. Oncogene. 2015;34:104-18
33. Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Molecular cell. 2005;19:857-64
34. Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432-5
35. Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG. et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882-7
36. Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W. et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660-72
37. Yang Y, Liu R, Qiu R, Zheng Y, Huang W, Hu H. et al. CRL4B promotes tumorigenesis by coordinating with SUV39H1/HP1/DNMT3A in DNA methylation-based epigenetic silencing. Oncogene. 2013
38. Lopez-Rios J, Tessmar K, Loosli F, Wittbrodt J, Bovolenta P. Six3 and Six6 activity is modulated by members of the groucho family. Development. 2003;130:185-95
39. Ayyanan A, Civenni G, Ciarloni L, Morel C, Mueller N, Lefort K. et al. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3799-804
40. Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR. et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer research. 2013;73:1981-92
41. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
42. Lagutin OV, Zhu CC, Kobayashi D, Topczewski J, Shimamura K, Puelles L. et al. Six3 repression of Wnt signaling in the anterior neuroectoderm is essential for vertebrate forebrain development. Genes & development. 2003;17:368-79
43. Lavado A, Lagutin OV, Oliver G. Six3 inactivation causes progressive caudalization and aberrant patterning of the mammalian diencephalon. Development. 2008;135:441-50
44. Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR. et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66-77
45. Mo ML, Okamoto J, Chen Z, Hirata T, Mikami I, Bosco-Clement G. et al. Down-regulation of SIX3 is associated with clinical outcome in lung adenocarcinoma. PloS one. 2013;8:e71816
46. Rajkumar T, Vijayalakshmi N, Gopal G, Sabitha K, Shirley S, Raja UM. et al. Identification and validation of genes involved in gastric tumorigenesis. Cancer cell international. 2010;10:45
47. Liu Y, Han N, Zhou S, Zhou R, Yuan X, Xu H. et al. The DACH/EYA/SIX gene network and its role in tumor initiation and progression. International journal of cancer. 2016;138:1067-75
48. Miller SJ, Lan ZD, Hardiman A, Wu J, Kordich JJ, Patmore DM. et al. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene. 2010;29:368-79
49. Zhou J, Liu Y, Zhang W, Popov VM, Wang M, Pattabiraman N. et al. Transcription elongation regulator 1 is a co-integrator of the cell fate determination factor Dachshund homolog 1. The Journal of biological chemistry. 2010;285:40342-50
50. Wu K, Chen K, Wang C, Jiao X, Wang L, Zhou J. et al. Cell fate factor DACH1 represses YB-1-mediated oncogenic transcription and translation. Cancer research. 2014;74:829-39
51. Li X, Perissi V, Liu F, Rose DW, Rosenfeld MG. Tissue-specific regulation of retinal and pituitary precursor cell proliferation. Science. 2002;297:1180-3
52. Wu K, Yang Y, Wang C, Davoli MA, D'Amico M, Li A. et al. DACH1 inhibits transforming growth factor-beta signaling through binding Smad4. The Journal of biological chemistry. 2003;278:51673-84
53. Wu K, Liu M, Li A, Donninger H, Rao M, Jiao X. et al. Cell fate determination factor DACH1 inhibits c-Jun-induced contact-independent growth. Molecular biology of the cell. 2007;18:755-67
54. Kumar R, Wang RA. Structure, expression and functions of MTA genes. Gene. 2016;582:112-21
55. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G. et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature cell biology. 2008;10:593-601
56. Stemmer V, de Craene B, Berx G, Behrens J. Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene. 2008;27:5075-80
57. Sun Y, Wang Y, Fan C, Gao P, Wang X, Wei G. et al. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Molecular cancer. 2014;13:137
58. Gu B, Watanabe K, Dai X. Epithelial stem cells: an epigenetic and Wnt-centric perspective. Journal of cellular biochemistry. 2010;110:1279-87
59. Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N. et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:10069-74
Author contact
![]() Corresponding authors: Yan Wang, PhD, 2011 Collaborative Innovation Center of Tianjin for Medical Epigenetics, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease (Ministry of Education), Department of Biochemistry and Molecular Biology, Tianjin Medical University, Tianjin 300070, China, Phone and Fax: 86-22-83336946, Email: yanwangedu.cn; or Yang Yang, PhD, Department of Biochemistry and Molecular Biology, Tianjin Medical University, Tianjin 300070, China, Phone and Fax: 86-22-83336948, Email: yangyang8668edu.cn.
Corresponding authors: Yan Wang, PhD, 2011 Collaborative Innovation Center of Tianjin for Medical Epigenetics, Tianjin Key Laboratory of Cellular and Molecular Immunology, Key Laboratory of Immune Microenvironment and Disease (Ministry of Education), Department of Biochemistry and Molecular Biology, Tianjin Medical University, Tianjin 300070, China, Phone and Fax: 86-22-83336946, Email: yanwangedu.cn; or Yang Yang, PhD, Department of Biochemistry and Molecular Biology, Tianjin Medical University, Tianjin 300070, China, Phone and Fax: 86-22-83336948, Email: yangyang8668edu.cn.
Citation styles
APA
Zheng, Y., Zeng, Y., Qiu, R., Liu, R., Huang, W., Hou, Y., Wang, S., Leng, S., Feng, D., Yang, Y., Wang, Y. (2018). The Homeotic Protein SIX3 Suppresses Carcinogenesis and Metastasis through Recruiting the LSD1/NuRD(MTA3) Complex. Theranostics, 8(4), 972-989. https://doi.org/10.7150/thno.22328.
ACS
Zheng, Y.; Zeng, Y.; Qiu, R.; Liu, R.; Huang, W.; Hou, Y.; Wang, S.; Leng, S.; Feng, D.; Yang, Y.; Wang, Y. The Homeotic Protein SIX3 Suppresses Carcinogenesis and Metastasis through Recruiting the LSD1/NuRD(MTA3) Complex. Theranostics 2018, 8 (4), 972-989. DOI: 10.7150/thno.22328.
NLM
Zheng Y, Zeng Y, Qiu R, Liu R, Huang W, Hou Y, Wang S, Leng S, Feng D, Yang Y, Wang Y. The Homeotic Protein SIX3 Suppresses Carcinogenesis and Metastasis through Recruiting the LSD1/NuRD(MTA3) Complex. Theranostics 2018; 8(4):972-989. doi:10.7150/thno.22328. https://www.thno.org/v08p0972.htm
CSE
Zheng Y, Zeng Y, Qiu R, Liu R, Huang W, Hou Y, Wang S, Leng S, Feng D, Yang Y, Wang Y. 2018. The Homeotic Protein SIX3 Suppresses Carcinogenesis and Metastasis through Recruiting the LSD1/NuRD(MTA3) Complex. Theranostics. 8(4):972-989.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.