Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):650-662. doi:10.7150/thno.21963 This issue Cite
Research Paper
CDK16 Phosphorylates and Degrades p53 to Promote Radioresistance and Predicts Prognosis in Lung Cancer
Jie Xie1*, Yan Li1*, Ke Jiang2*, Kaishun Hu3, Sheng Zhang1, Xiaorong Dong1, Xiaofang Dai1, Li Liu1, Tao Zhang1, Kunyu Yang1, Kai Huang4, Junjie Chen5, Shaojun Shi6, Yu Zhang6, Gang Wu1 ![]() , Shuangbing Xu1
, Shuangbing Xu1 ![]()
1. Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China;
2. Department of Thoracic Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China;
3. Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, China;
4. Clinic Center of Human Gene Research, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China;
5. Department of Experimental Radiation Oncology, The University of Texas M.D. Anderson Cancer Center, Houston, Texas 77030, USA;
6. Department of Pharmacy, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China.
* These authors contributed equally to this work
Received 2017-7-17; Accepted 2017-10-31; Published 2018-1-1
Citation:
Xie J, Li Y, Jiang K, Hu K, Zhang S, Dong X, Dai X, Liu L, Zhang T, Yang K, Huang K, Chen J, Shi S, Zhang Y, Wu G, Xu S. CDK16 Phosphorylates and Degrades p53 to Promote Radioresistance and Predicts Prognosis in Lung Cancer. Theranostics 2018; 8(3):650-662. doi:10.7150/thno.21963. https://www.thno.org/v08p0650.htm
Other stylesAbstract

Rationale: Radioresistance is considered the main cause of local relapse in lung cancer. However, the molecular mechanisms of radioresistance remain poorly understood. This study investigates the role of CDK16 in radioresistance of human lung cancer cells.
Methods: The expression levels of CDK16 were determined by immunohistochemistry in lung cancer tissues and adjacent normal lung tissues. Immunoprecipitation assay and GST pulldown were utilized to detect the protein-protein interaction. The phosphorylation of p53 was evaluated by in vitro kinase assay. Poly-ubiquitination of p53 was examined by in vivo ubiquitination assay. Cell growth and apoptosis, ROS levels and DNA damage response were measured for functional analyses.
Results: We showed that CDK16 is frequently overexpressed in lung cancer cells and tissues, and high levels of CDK16 are correlated with lymph node stage and poor prognosis in lung cancer patients. Furthermore, we provided evidence that CDK16 binds to and phosphorylates p53 at Ser315 site to inhibit transcriptional activity of p53. Moreover, we uncovered that this phosphorylation modification accelerates p53 degradation via the ubiquitin/proteasome pathway. Importantly, we demonstrated that CDK16 promotes radioresistance by suppressing apoptosis and ROS production as well as inhibiting DNA damage response in lung cancer cells in a p53-dependent manner.
Conclusion: Our findings suggest that CDK16 negatively modulates p53 signaling pathway to promote radioresistance, and therefore represents a promising therapeutic target for lung cancer radiotherapy.
Keywords: CDK16, p53, radioresistance, phosphorylation, lung cancer.
Introduction
Lung cancer continues to be the deadliest human malignancy worldwide with 5-year overall survival rate of less than 15% (1, 2). About 85% of lung cancer cases are classified as non-small cell lung cancer (NSCLC), of which adenocarcinoma accounts for 50% of histologic subtypes of NSCLC (3). Radiotherapy has been considered a promising and major local treatment strategy for lung cancer. However, local relapse occurs frequently due to radioresistance for most patients. Therefore, it is critical to elucidate the underlying mechanisms and overcome radioresistance in lung cancer.
p53, the most extensively studied tumor suppressor gene, is mutated or functionally inactive in approximately 50% of human cancers (4-6). p53 is labile and is usually ubiquitinated and targeted for degradation by MDM2 E3 ubiquitin ligase under normal physiological conditions (7, 8). Following various stress signals, such as DNA damage, hypoxia, and oncogene activation, p53 is stabilized, activated, and translocated into the nucleus, where it induces the transactivation of many downstream target genes involved in cell-cycle arrest, apoptosis, DNA repair, and senescence. Accumulating evidence suggest that p53 plays essential roles in a wide spectrum of biological processes, including apoptosis, autophagy, metabolism, immune response, DNA damage repair, and tumorigenesis (9-11). Additionally, p53 also functions as a key player that determines radiosensitivity by modulating cell cycle redistribution and DNA damage (12-14). Therefore, p53 is regarded as a potential target for radiation therapy.
CDK16, also termed PCTAIRE1 or PCTK1, is a newly identified member of the cyclin-dependent kinases family (CDK) (15). CDK16 is activated by binding to cyclin Y (CCNY) or its homologue cyclin Y-like 1 (CCNYL1) proteins (16-19). CDK16 has been found to be involved in vesicle trafficking (20, 21), neurite outgrowth (22), spermatogenesis (16), glucose transportation (23, 24), skeletal myogenesis (25), and spindle orientation (26). In addition, recent studies demonstrated indispensable roles of CDK16 in tumorigenesis. CDK16 silencing has been reported to suppress cell growth and proliferation in multiple human cancers, such as prostate cancer, breast cancer, and colorectal cancer (27-29). These studies suggest that CDK16 may act as an oncoprotein in several types of cancers. However, an in-depth exploration of its role in lung cancer has not yet been performed.
In this study, we found that CDK16 is overexpressed in lung cancer and predicts poor prognosis in patients. Furthermore, we showed that CDK16 binds to and phosphorylates p53 at Ser315 site to trigger p53 degradation via the ubiquitin/proteasome pathway. Moreover, we demonstrated that CDK16 depletion enhances radiosensitivity in a p53-dependent manner in lung cancer cells, suggesting that CDK16 is an attractive target for cancer radiotherapy.
Materials and Methods
Cell culture and transfection
Cells (BEAS, A549, H292, H1299, H358, H1975, HCC827) were obtained from ATCC and cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin and streptomycin in 5% CO2 at 37 °C. HEK293T, U2OS and MCF-7 cells were purchased from ATCC and cultured in DMEM supplemented with 10% FBS and 100 U/mL penicillin and streptomycin in 5% CO2 at 37 °C. Plasmid transfections were performed using Lipofectamine 2000 (Invitrogen).
Constructs
CDK16 plasmid was purchased from DF/HCC DNA Resource Core. The construct was subcloned into pDONR201 entry vector and transferred to destination vectors with the indicated SFB or Myc tag using Gateway Technology (Invitrogen). Plasmids encoding p53 and HA-ubiquitin have been described previously (30). Mutations were introduced using the Quik-Change Site-Directed Mutagenesis Kit (Stratagene) and verified by DNA sequencing.
Antibodies and reagents
Anti-human CDK16 antibodies were purchased from Sigma and Proteintech. Anti-p53, anti-Myc, anti-GAPDH, anti-CDK1, anti-CDK2, anti-CyclinB1, anti-CyclinD1, and anti-MDM2 antibodies were obtained from Santa Cruz Biotechnology. Anti-human p27 was from Abgent, anti-Flag antibody was from Sigma, and anti-human p53-S315 P, anti-p53-S33P, anti-p53-S46P phospho-specific antibodies and anti-HA antibody were from Cell Signaling Technology. MG132 and Cycloheximide were purchased from Calbiochem and Sigma respectively.
RNAi treatment
The following target siRNA sequences were as follows: SiCDK16#1, CCACUGAGGACAUCAACAA and SiCDK16#2, GGAGAUCAGACUGGAACAU, which was previously described (27). Sip53: AAGACTCCAGTGGTAATCTAC, was described previously (31). SiRNA transfection (50 nM siRNA) was performed using Lipofectamine RNAiMAX reagent (Invitrogen). 48 h post-transfection, cells were harvested and analyzed.
CRISPR/Cas9 gene-editing technology to generate CDK16 knockout cells
This assay was performed as described previously (32). In Brief, lenti-CRISPR sgRNAs targeting CDK16 were co-transfected into HEK293T cells with the packaging plasmids pSPAX2 and pMD2G. After 48 h, supernatants were collected and used to infect A549 cells in the presence of 10 μg/mL Polybrene (Sigma). Stable CDK16 knockout A549 cells were selected in the addition of 2 μg/mL puromycin. The sgRNA sequences were as follows: CDK16-sgRNA#1: 5'-GGAGATTCAGCTACAAAAGG-3'; CDK16-sgRNA#2: 5'-ATAGGCCTGGATGAGAGTGG-3'.
Western blotting and immunoprecipitation
Cells were lysed in NETN buffer (20 mM Tris-HCl [pH 8.0], 1 mM EDTA, 100 mM NaCl, and 0.5 % Nonidet P-40), and the clarified lysates were resolved by SDS-PAGE and transferred to PVDF membranes for Western blotting using ECL detection reagents. For immunoprecipitation, the supernatants were first incubated with S-protein agarose (Novagen) overnight at 4 °C, and the precipitates were washed five times with NETN buffer. To detect endogenous interaction, the clarified supernatants were first incubated with anti-p53 antibody and then protein A/G-agaroses overnight. After being washed five times with NETN buffer, the samples were collected and analyzed by Western blotting.
GST pull down assay
GST-vector or GST-p53 fusion proteins purified from bacteria were immobilized on GST beads (GE Healthcare) and incubated with lysates prepared from HEK293T cells transiently transfected with SFB-tagged CDK16 for 2 h at 4 °C. The samples were washed five times and then analyzed by Western blotting.
Cytoplasmic and nuclear protein fractionation
A549 cells were transfected with scramble or CDK16 siRNAs. After 48 h, the cells were collected. Cytoplasmic and nuclear fractions were separated using NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Thermo Fisher Scientific) according to the manufacturer's procedures.
In vivo ubiquitination assay
A549 cells were transfected with indicated siRNAs or constructs. MG132 (10 μM) was added 4 h before harvesting. Cells were then lysed with NETN buffer. The supernatants were incubated with S-protein agarose and then analyzed by Western blotting.
In vitro kinase assay
HA-p53 and HA-p53-S315A were synthesized in vitro using the High-yield protein expression system (Promega). SFB-CDK16 was obtained by immunoprecipitation using anti-Flag agarose from lysates of cells overexpressing SFB-CDK16. The samples were then incubated with HA-p53-WT or HA-p53-S315A in 50 μL of kinase buffer (Cell Signaling Technology) containing 10 μM ATP for 30 min at 30 °C. The samples were subsequently analyzed by Western blotting.
Real-time quantitative PCR
This assay was performed as described previously (30). Briefly, total RNA from A549 cells was isolated using Trizol reagent (Invitrogen), and reverse transcription assay was carried out using the ReverTra Ace qPCR RT Kit (Toyobo, Japan). The relative mRNA levels of each gene were measured as two power values of ΔCt (Ct of GAPDH minus the Ct of the target genes). Primer sequences are listed in Table S1.
Cell clonogenic survival assays
A549 cells transfected with indicated siRNAs were seeded into 6-well plates and irradiated with doses as indicated. Cells were then cultured for 14 days. After fixation and staining, the colonies containing more than 50 cells were calculated.
Cell cycle analysis
A549 cells were transfected with scrambled or CDK16 siRNAs. 48 h later, cells were first fixed with 70% ethanol and then treated with propidium iodide (PI) and RNase A for 30 min. The samples were analyzed by flow cytometry.
Apoptosis assay
A549 cells were transfected with scrambled or CDK16 siRNAs. 48 h post-transfection, Cells were harvested by trypsin without EDTA, washed with PBS, and then stained with annexin V-EGFP and propidium iodide. The samples were analyzed by flow cytometry.
ROS measurement
A549 cells transfected with indicated siRNAs were harvested and washed three times with PBS buffer. Cells were then incubated with fresh phenol red-free DMEM containing 10% FBS and 25 mM DCF (2',7'-dichlorofluorescein diacetate) (Sigma) for 20 min and were collected. ROS levels were measured by flow cytometry.
Immunofluorescence staining
A549 Cells transfected with indicated siRNAs were cultured on coverslips and then exposed to 2 Gy IR. Cells were then fixed with 4% paraformaldehyde for 15 min at various time points after radiation (0 min, 30 min, 4 h and 24 h) and permeabilized in 0.2% Triton X-100 for 5 min. After blocking with 5% bovine serum albumin, the samples were incubated with γ-H2AX antibody overnight and secondary antibody for 1h. Cells were counterstained with DAPI for 10 min and visualized by fluorescence microscopy.
Lung Cancer Tissue Microarray and Immunohistochemistry Staining
A lung cancer tissue microarray was purchased from Shanghai Outdo Biotech (Shanghai, China), which contained 90 carcinoma tissues and paired para-carcinoma tissues. All patients had been pathologically diagnosed with adenocarcinoma after operation. The immunohistochemistry (IHC) analysis was performed as described previously (33, 34). Briefly, the tissue sections were blocked with goat serum and then incubated with anti-CDK16 antibody (Sigma) overnight at 4 ºC. The sections were stained with 3, 3-diaminobenzidine and counterstained with hematoxylin after being incubated with secondary antibody. PBS was used as a negative control. Both staining intensity and positive percentage were used to examine the expression of CDK16 in lung cancer tissue: the IHC staining score (values 0-12) was calculated by multiplying the scores for intensity of positive staining (negative = 0, weak = 1, moderate = 2, or strong = 3) and the percentage of positive-stained cells (0-25% = 1, 26-50% = 2, 51-75% = 3, >75% = 4). IHC scores ≥ 8 defined high expression, while scores < 8 defined low expression.
Data analysis
In our study, three or more independent experiments were carried out for each assay. Results are presented as the mean ± SD. An unpaired Student's t-test was used for statistical comparison in each group. χ2 test was used to determine the correlation between CDK16 expression and clinicopathologic variables. The Kaplan-Meier method was used to evaluate the overall survival. P value <0.05 was considered to be statistically significant.
Results
CDK16 is frequently overexpressed and predicts poor prognosis in lung cancer
To investigate the expression of CDK16 in lung cancer, we first examined the protein level of CDK16 in different lung cancer cell lines. As shown in Figure 1A, CDK16 was significantly upregulated in most lung cancer cell lines compared to normal human lung epithelial cells BEAS-2B. Next, we performed immunohistochemical staining for CDK16 in a human lung cancer tissue microarray, which contained 90 carcinoma tissues and paired adjacent lung tissues. As shown in Figure 1B and C, CDK16 was mainly located in the cytoplasm and overexpressed in lung cancer compared to adjacent lung tissues. The positive rate of CDK16 staining in lung cancers (71.7%) was significantly higher than that in adjacent lung tissues (0%) (P < 0.01). Furthermore, we found that high CDK16 expression significantly correlated with lymph node stage in clinicopathologic characteristics (P < 0.05) (Table S2). The Kaplan-Meier analysis demonstrated that the patients with high CDK16 expression had a significantly shorter overall survival than those with low CDK16 expression (Figure 1D). Taken together, these data indicate that CDK16 overexpression may contribute to tumorigenesis and elevated levels of CDK16 predict adverse prognosis in lung cancer patients.
Figure 1
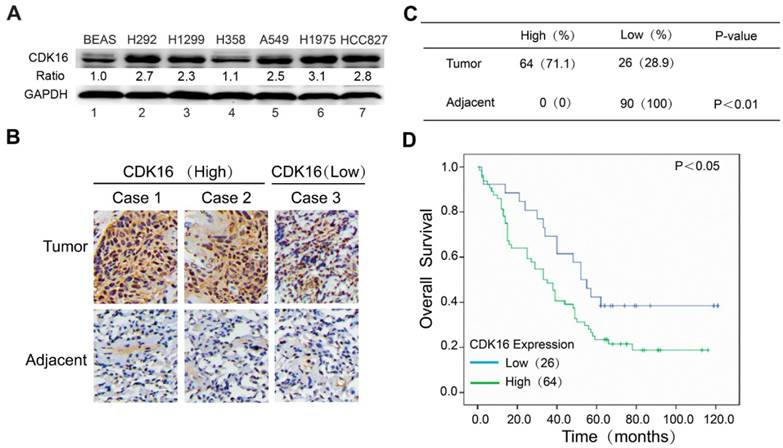
CDK16 is frequently overexpressed in lung cancer and elevated level of CDK16 predicts poor prognosis. A. Western blotting analysis of CDK16 expression in six human lung cancer cell lines and normal lung epithelial BEAS cells. The ratio shows relative CDK16 protein expression normalized for GAPDH (BEAS, set at 1). B. Representative immunohistochemical staining images of CDK16 expression (high or low) in tissue microarrays constructed from lung cancers and paired adjacent lung tissues. Scale bar, 100 μm. C. A summary of immunohistochemical staining results of CDK16 expression in tissue microarrays. D. Kaplan-Meier analysis of overall survival of lung cancer patients stratified by CDK16 expression.
CDK16 negatively regulates p53 stability in multiple p53 wild-type cell lines
Our clinical data suggested CDK16 upregulation may promote tumorigenesis in lung cancer. To further explore the underlying mechanisms, we examined a few tumour suppressive and oncogenic proteins that are known to regulate cell growth and cell cycle progression in CDK16-depleted A549 lung cancer cells. Consistent with a previous study (27), we observed increased level of p27 (Figure 2A), which has been considered a CDK16 downstream effector. Interestingly, p53 level was also substantially augmented when CDK16 was depleted (Figure 2A). Moreover, restoring siRNA-resistant CDK16 expression remarkably reduced p53 protein level in CDK16-depleted cells (Figure S1). Additionally, we employed a CRISPR/Cas9-based gene editing strategy to deplete CDK16 and found that loss of CDK16 dramatically resulted in the accumulation of endogenous p53 in A549 cells (Figure 2B). These findings strongly support the notion that the stability of p53 is regulated by CDK16 in lung cancer cells. Furthermore, we showed that ectopically expressed CDK16 led to decreased level of p53 (Figure 2C), further supporting that p53 abundance is negatively controlled by CDK16. More importantly, this phenomenon was also observed in several other cancer cell lines expressing wild-type p53 (Figure 2D), implying that p53 regulation by CDK16 may be a common feature. Notably, loss of CDK16 did not affect the mRNA level of p53 (Figure 2E), suggesting that CDK16 may control p53 protein level. Furthermore, we found that p53 protein level was not altered by CDK16 depletion in cells harboring p53 mutations (Figure 2F). Together, these results suggest that CDK16 negatively regulates p53 stability at the post-translational level.
CDK16 directly interacts with p53 in vivo and in vitro
To elucidate how the protein kinase CDK16 controls p53 stability, we first examined whether CDK16 would interact with p53 in cells. As expected, the exogenously expressed CDK16 co-immunoprecipitated with p53 and vice versa (Figure 3A). To further confirm this interaction, we performed an endogenous co-immunoprecipitation experiment and detected a complex containing CDK16 and p53 (Figure 3B). Importantly, we found that recombinant GST-tag-expressed p53 immunoprecipitated with ectopically expressed CDK16 (Figure 3C), indicating CDK16 directly associates with p53 in vitro. Next, we generated a series of p53 deletion mutations (Figure 3D) and showed that the core DNA binding domain of p53 (residues 100-300) was capable of binding to CDK16 (Figure 3E). Notably, the mobility of HA-p53 (100-393) fragment (294aa) was much faster than HA-p53 (1-300) fragment (300aa). We speculated that HA-p53 (100-393) fragment presents a lower frequency of proline since the N-terminus of p53 contains a proline-rich region (residues 61-94). In fact, this mobility difference was reported in our previous work (30). Together, these data strongly suggest that CDK16 directly binds to p53 in vivo and in vitro.
CDK16 phosphorylates p53 at Ser315 site and controls its transcriptional activity
Our data above suggest that CDK16 interacts with p53 and modulates its stability. Given that CDK16 is a protein kinase, we asked whether p53 is a substrate of CDK16. It is well established that most CDK substrates contain S/T-P-X-K/R motif (35). We first examined three known p53 phosphorylation sites that contain the S/T-P motif. As shown in Figure 4A, phospho-p53-S315 level was reduced in CDK16 knockdown cells, while phospho-p53-S33 and phospho-p53-S46 levels remained constant, suggesting that Ser315 site may be a candidate phosphorylation site by CDK16. Notably, the Ser315 site is evolutionally conserved (Figure 4B). To further investigate whether CDK16 directly phosphorylates p53, we performed an in vitro kinase assay and showed that CDK16 could phosphorylate wild-type p53 at Ser315 site (Figure 4C). Anti-phospho-p53-S315 antibody could not detect p53 when the Ser315 site was mutated to alanine (S315A) (Figure 4C), indicating that this antibody is highly specific for the S315 phosphorylated form of p53. Collectively, these findings suggest that CDK16 directly phosphorylates p53 at Ser315 site in vivo and in vitro.
Because p53 phosphorylation has been reported to associate with its nuclear localization (36), we therefore speculated that CDK16 might also affect this process. Indeed, CDK16 knockdown led to p53 accumulation in the nucleus (Figure 4D). Nuclear p53 functions as a transcriptional factor and induces the expression of p53 downstream genes. As shown in Figure 4E, CDK16 knockdown clearly increased the expression levels of MDM2 and PUMA by over 2-fold, while those of NOXA and P21 were not remarkably altered. Notably, CDK16 depletion did not affect the protein level of MDM2, which functions as ubiquitin ligase to target p53 for degradation (Figure S2), suggesting that MDM2 may not feedback to degrade p53 under this condition. Taken together, our results indicate that the phosphorylation of p53 by CDK16 inhibits its nuclear accumulation and activation of its transcriptional activity.
Figure 2
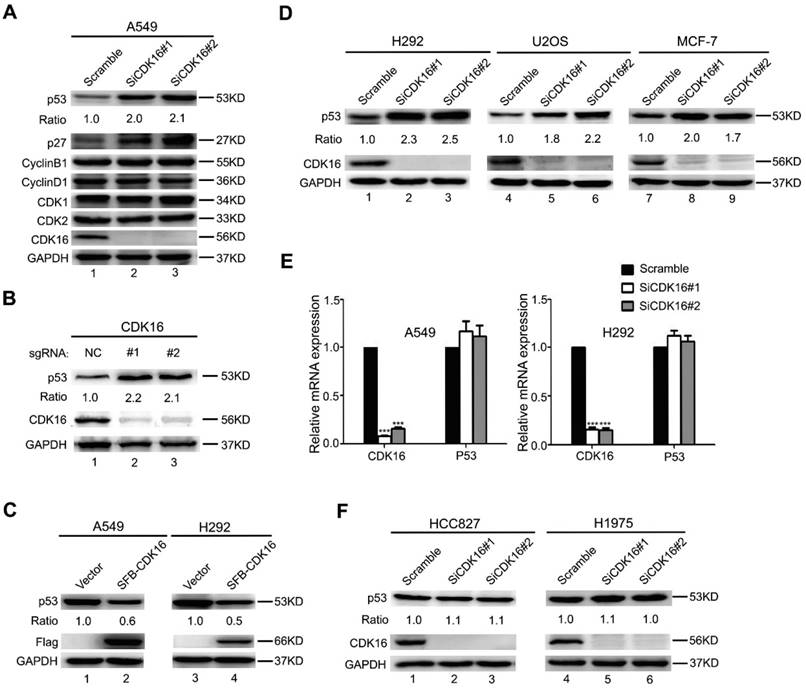
CDK16 knockdown leads to p53 accumulation. A. A549 cells were transfected with indicated siRNAs for 48 h, and samples were collected and analyzed by Western blotting with the indicated antibodies. The ratio shows relative p53 protein expression normalized for GAPDH (scramble, set at 1). B. A549 cells infected with lenti-CRISPR sgRNAs targeting CDK16 or control were incubated with puromycin for one week, and endogenous CDK16 and p53 were analyzed by western blotting. C. A549 and H292 cells were transfected with constructs encoding SFB-CDK16. Cells were collected 24 h later and cell lysates were analyzed by Western blotting using indicated antibodies. D. H292, U2OS, and MCF-7 cells were transfected with indicated siRNAs for 48 h, and analyzed as described in (A). E. A549 and H292 cells were transfected with scrambled or CDK16 siRNAs for 48 h. The mRNA levels for the indicated genes were determined by Real-time quantitative PCR (n=3). F. HCC827 and H1975 cells were transfected with scrambled or CDK16 siRNAs as indicated for 48 h and analyzed as described in (A).
CDK16 regulates p53 stability via the ubiquitin/proteasome pathway
Considering that CDK16 negatively regulates p53 accumulation and p53 is a labile protein degraded by the proteasome, we speculated that CDK16 may affect p53 stability via the ubiquitin/proteasome pathway. Indeed, there was no further increase of p53 protein level when proteasome inhibitor MG132 was added in CDK16-depleted cells (Figure 5A), suggesting that CDK16 knockdown inhibits p53 degradation by the ubiquitin/proteasome pathway. Moreover, we showed that the half-life of endogenous p53 protein was longer in CDK16-knockdown cells when compared to that in control cells (Figure 5B). We also performed in vivo ubiquitination assays and showed that p53 poly-ubiquitination was decreased when CDK16 was inhibited (Figure 5C), indicating that p53 degradation promoted by CDK16 is ubiquitination-dependent.
Since CDK16 phosphorylates p53 and then leads to its degradation, we speculated that the CDK16-dependent degradation of p53 also requires its ability to phosphorylate p53 at Ser315 site. As shown in Figure 5D, the S315A mutant displayed significantly reduced p53 ubiquitination when compared to wild-type p53 in cells with ectopic expression of CDK16. Moreover, wild-type CDK16, but not its kinase dead mutant (K194M) (26, 27), promoted poly-ubiquitination of p53 (Figure 5E). Taken together, our results suggest that CDK16 negatively controls the stability of p53 by promoting its polyubiquitination and degradation in a phosphorylation-dependent manner.
Knockdown of CDK16 enhanced radiosensitivity of lung cancer cells
To explore the biological significance of CDK16 in lung cancer cells, we examined cell proliferation and apoptosis since the p53 target genes MDM2 and PUMA were upregulated when CDK16 was inhibited. As shown in Figure 6A, cell colony formation was substantially decreased in CDK16-depleted cells, suggesting that CDK16 contributes to cell proliferation in lung cancer. Moreover, apoptosis was significantly increased after CDK16 depletion (Figure 6B), which was further confirmed by elevated sub-G1 population (Figure 6C), indicating that knockdown of CDK16 promotes apoptosis.
p53 has been shown to modulate reactive oxygen species (ROS) generation (37); therefore, we measured ROS levels in CDK16 knockdown lung cancer cells. As shown in Figure 6D, CDK16 silencing led to increased production of ROS. p53 also participates in DNA damage response (38, 39). Therefore, we carried out a γH2AX foci assay and showed that IR-induced γ-H2AX foci were significantly increased 4 h and 24 h after irradiation in CDK16-depleted lung cancer cells (Figure 6E). Additionally, loss of CDK16 significantly enhanced cellular sensitivity to radiation (Figure 6F). Together, these findings indicate that CDK16 promotes radioresistance by suppressing apoptosis and ROS production as well as inhibiting DNA damage response in lung cancer cells.
p53 mediates the biological effects of CDK16 in lung cancer cells
To confirm the functional roles of p53 in radioresistance caused by CDK16 knockdown, A549 cells were transfected with siRNAs targeting CDK16, p53 or both (Figure 7A). As shown in Figure 7B, p53 knockdown partially rescued the defects in cell colony formation in CDK16-depleted cells. Furthermore, we found that apoptosis caused by CDK16 silencing was inhibited by p53 depletion (Figure 7C). Moreover, the increased ROS levels and γ-H2AX foci induced by CDK16 depletion were partially reversed when p53 was co-depleted (Figure 7D and E). We also showed that p53 knockdown partially restored cell survival after irradiation in CDK16-depleted cells (Figure 7F). Together, these data suggest that CDK16 exerts its functions mainly via its ability to inhibit p53.
Figure 3
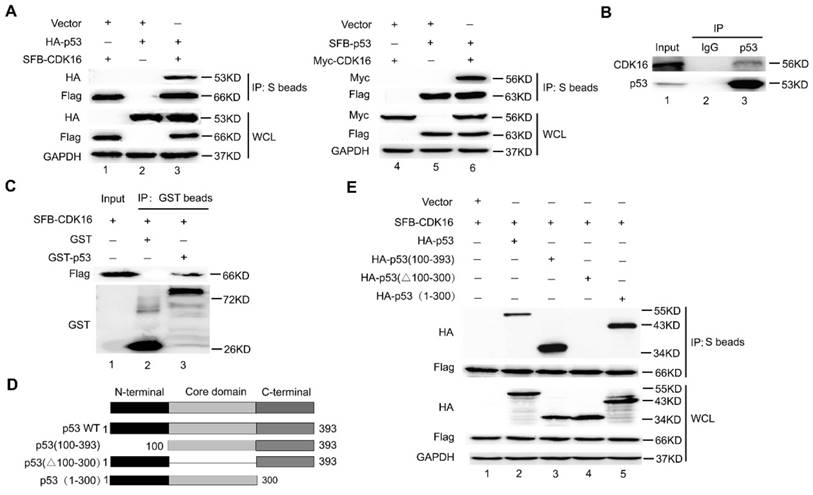
CDK16 directly interacts with p53 in vivo and in vitro. A. HEK293T cells transfected with the indicated constructs were collected 24 h later. Cells were lysed with NETN buffer. Immunoprecipitation (IP) using S-protein agarose were performed and Western blotted with the indicated antibodies. WCL: whole cell lysate. B. Endogenous CDK16 associated with p53 in A549 cells. A549 cells were lysed and subjected to immunoprecipitation using anti-IgG or anti-p53 as indicated, and were analyzed by Western blotting using indicated antibodies. C. Beads coated with GST or GST-p53 fusion proteins were incubated with SFB-CDK16 protein overnight. GST pulldown was immunoblotted with indicated antibodies. D. Schematic description of p53 domains and deletion mutants used in this study. E. HEK293T cells co-transfected with constructs encoding SFB-CDK16 and indicated p53 mutants were collected 24 h later. Cells were lysed, the supernatants were incubated with S-protein agarose beads, and then analyzed by Western blotting using antibodies as indicated.
Figure 4
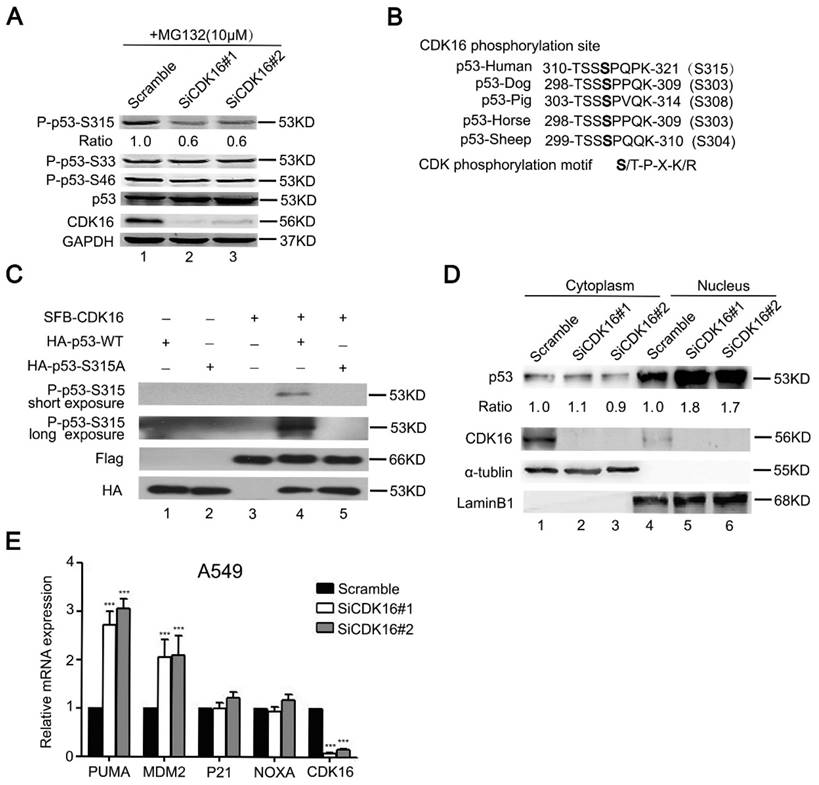
CDK16 phosphorylates p53 at Ser315 site and inhibits p53 transcriptional activity. A. A549 cells were transfected with the indicated siRNA for 48 h, then incubated with 10 μM MG132 for 4 h prior to collection. Cell lysates were analyzed by Western blotting using indicated antibodies. The ratio shows relative phosphorylated p53 protein expression normalized for GAPDH (scramble, set at 1). B. Alignment of CDK16 candidate phosphorylation site in p53 from different species. C. CDK16 phosphorylates p53 at Ser315 site in vitro. HA-p53-WT or S315A proteins were incubated in vitro with immunoprecipitates isolated from HEK293T cells transfected with constructs encoding SFB-CDK16 and then analyzed by Western blotting using indicated antibodies. D. CDK16 depletion increases p53 abundance in the nucleus. A549 cells were fractionated into cytoplasmic and nuclear fractions, and the indicated proteins in each compartment were analyzed by Western blotting. α-tubulin and LaminB1 were used respectively as cytoplasmic and nuclear loading controls. E. The mRNA levels of the indicated genes were analyzed by Real time quantitative PCR in A549 cells transfected with scramble or CDK16 siRNAs. *** P < 0.001 (n=3).
Discussion
In this study, we demonstrated that CDK16 phosphorylates p53 at Ser315 and promotes ubiquitination and subsequent degradation of p53. We also showed that CDK16 promotes radioresistance by suppressing apoptosis and ROS production as well as inhibiting DNA damage response in a p53-dependent manner. Importantly, CDK16 is overexpressed in lung cancer and predicts unfavorable prognosis, implying that CDK16 may be a promising therapeutic target for lung cancer radiotherapy.
As a key tumor suppressor protein, p53 and its associated activities are tightly controlled by its interactions with other proteins, its subcellular localization and its post-translational modifications (34, 40, 41). For example, our previous study has shown that hSSB1 interacts with p53 and regulates the transcriptional activity and stability of p53 (30). Moreover, phosphorylation of p53 at Ser15 site by ATM kinase leads to p53 stabilization by blocking p53 ubiquitination in response to DNA damage (41). In this study, we identified CDK16 as a key upstream kinase responsible for p53 phosphorylation at Ser315 site. We further demonstrated that p53 phosphorylation by CDK16 leads to destabilization of p53 and inhibits p53 transcriptional activity. In addition, MDM2 ubiquitin ligase has been shown to be mainly responsible for p53 degradation (41). Thus, we speculate that MDM2 E3 ligase may also be involved in the ubiquitination and degradation of p53 promoted by CDK16. Collectively, our data suggest that CDK16 is a key regulator of p53 and promotes the degradation of p53 in a phosphorylation-dependent manner.
Elevated expression of CDK16 has been observed in some types of human cancer (27-29). We demonstrated that CDK16 is overexpressed in lung cancer and is associated with poor clinical outcomes. Our follow-up functional studies have revealed that CDK16 accelerates cell proliferation and impairs apoptosis, further supporting that CDK16 may act as an oncoprotein and overexpression of CDK16 could contribute to tumorigenesis. Furthermore, we uncovered a new role for CDK16 in the regulation of radioresistance although CDK16 may not be a DNA damage-responsive protein (Figure S3). We uncovered that CDK16 depletion enhanced radiosensitivity of lung cancer cells by promoting apoptosis and ROS production as well as inhibiting DNA damage repair. Importantly, p53 knockdown could partially rescue these phenotypes caused by CDK16 depletion, suggesting that p53 is the major downstream effector of CDK16, which functions in radioresistance in lung cancer.
In conclusion, our study identified p53 as a novel substrate of CDK16 and elucidated the mechanism by which p53 is regulated by CDK16 in lung cancer. Furthermore, we demonstrated that CDK16 promotes radioresistance in lung cancer by facilitating p53 degradation, as proposed in Figure 8. Several CDK16 inhibitors have been developed and exhibited anti-tumor activities (42, 43). Our data further support the development of CDK16 inhibitors and suggest that these inhibitors may be effective for cancer radiotherapy, especially for lung cancer displaying elevated levels of CDK16.
Figure 5
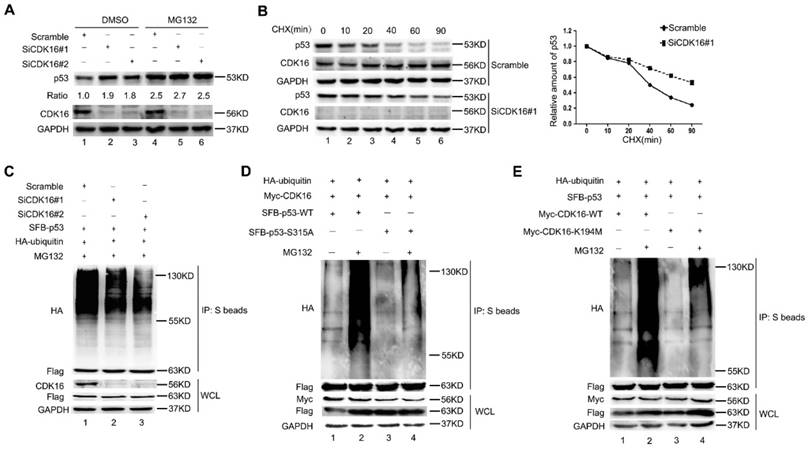
CDK16 regulates p53 stability via the ubiquitin/proteasome pathway. A. A549 cells were transfected with the indicated siRNA for 48 h, then incubated with 10 μM MG132 for 4 h prior to harvesting. Cell lysates were analyzed by Western blotting using indicated antibodies. The ratio shows relative p53 protein expression normalized for GAPDH (scramble, set at 1). B. Left panel: A549 cells transfected with indicated siRNA for 48 h were treated with CHX (20 mg/mL) for the indicated times and then analyzed by Western blotting. Right panel: quantification of p53 band intensities is shown. C. A549 cells were transfected with indicated constructs after cells were transfected with CDK16 siRNA for 24 h. Cells were harvested after treatment with MG132 (10 μM) for 4 h. The samples were subjected to immunoprecipitation using S-protein agarose beads followed by Western blotting analysis as indicated. D and E. A549 cells were transfected with indicated constructs for 24 h and collected after treatment with MG132 (10 μM) for 4 h. The samples were subjected to immunoprecipitation using S-protein agarose beads and analyzed by Western blotting using indicated antibodies.
Figure 6
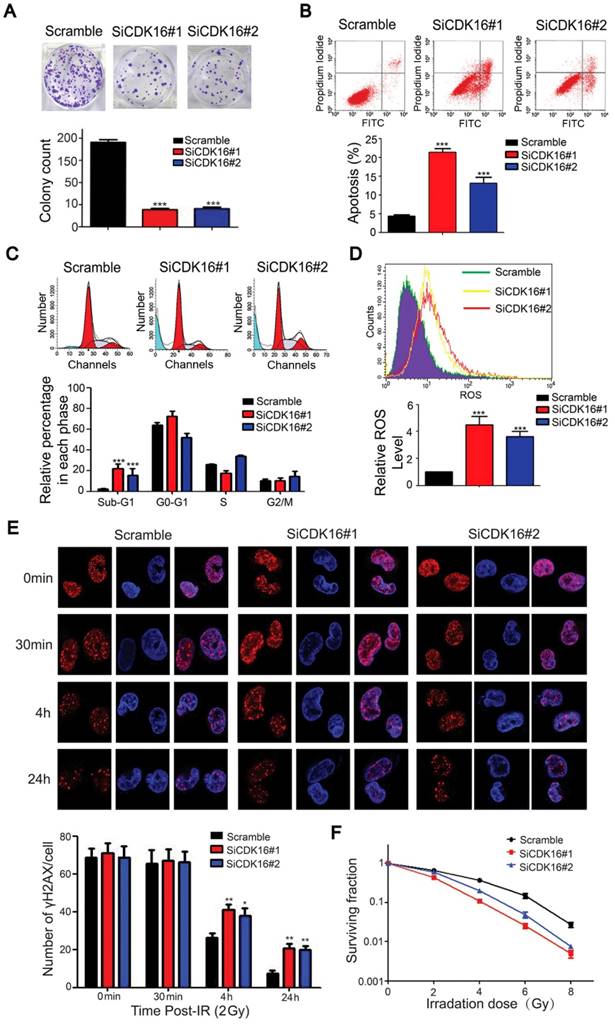
CDK16 silencing results in enhanced radiosensitivity of lung cancer cells. A. Colony formation ability was significantly reduced in CDK16-depleted A549 cells. *** P < 0.001 compared with controls cells (n=3). B. Apoptosis was remarkably increased in CDK16-depleted A549 cells. A549 cells were transfected with indicated siRNAs. 48 h later, cells were subjected to annexin V-EGFP/propidium iodide staining, and analyzed by flow cytometry (n=3). C. A549 cells were transfected with indicated siRNAs for 48 h, and cells were stained with propidium iodide (PI) and analyzed by flow cytometry (n=3). D. Knockdown of CDK16 promoted cellular ROS production. A549 cells were transfected with indicated siRNAs. 48 h later, cells were collected and ROS levels were measured by flow cytometry (n=3). E. Upper panel: A549 cells were irradiated with 2 Gy of ionizing radiation and harvested at indicated time points. Immunostaining staining was performed to determine γ-H2AX foci formation. Scale bar, 50 μm. Lower panel: quantification result of γ-H2AX foci in A549 cells is shown. *P < 0.05, **P < 0.01 compared with controls cells (n=3). F. Radiation sensitivity of A549 cells lacking CDK16. A549 cells transfected with indicated siRNAs were irradiated with indicated doses of IR. The percentages of surviving colonies were evaluated two weeks later (n=3).
Figure 7
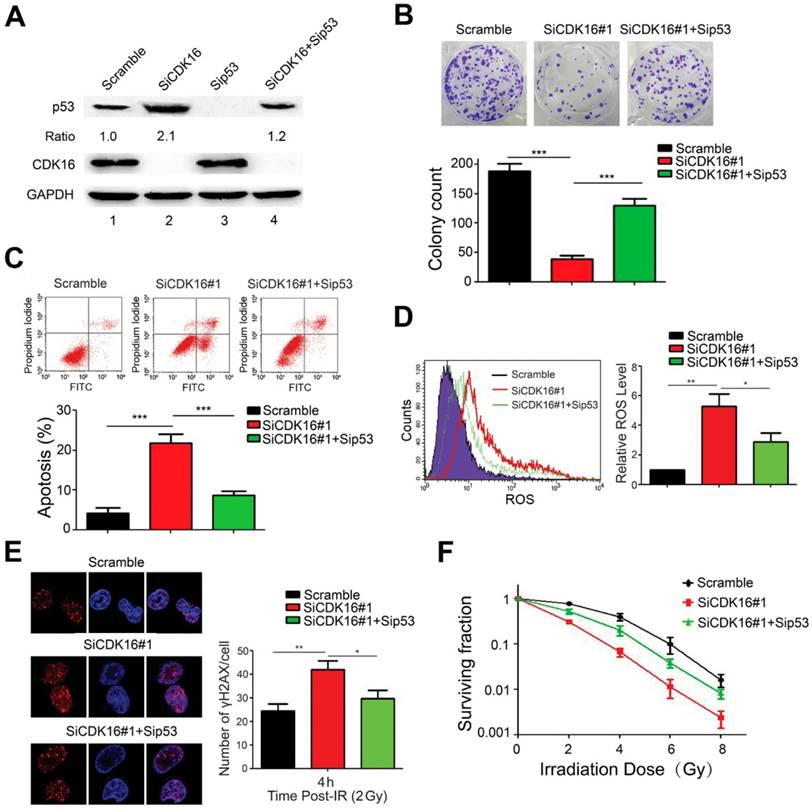
p53 mediates the biological effects of CDK16 depletion in lung cancer cells. A. A549 cells were transfected with indicated siRNAs for 48 h. Cell lysates were analyzed by immunoblotting as indicated. B. A549 cells transfected with indicated siRNAs were seeded and cultured for two weeks. The colonies were stained and then counted. Representative pictures are shown. *** P < 0.001 (n=3). C. A549 cells were transfected with indicated siRNAs. 48 h later, cells were subjected to annexin V-EGFP/propidium iodide staining and analyzed by flow cytometry. *** P < 0.001 (n=3). D. A549 cells were transfected with indicated siRNAs and ROS levels were measured by flow cytometry. *P < 0.05, **P < 0.01 (n=3). E. Left panel: A549 cells were irradiated with 2 Gy of ionizing radiation and harvested at 4 h post IR. Immunostaining was performed to determine γ-H2AX foci formation. Scale bar, 50 μm. Right panel: quantification result of γ-H2AX foci in A549 cells is shown. *P < 0.05, **P < 0.01 (n=3). F. A549 cells transfected with indicated siRNAs were irradiated with indicated doses. After two weeks, colonies containing more than 50 cells were counted. (n=3).
Figure 8
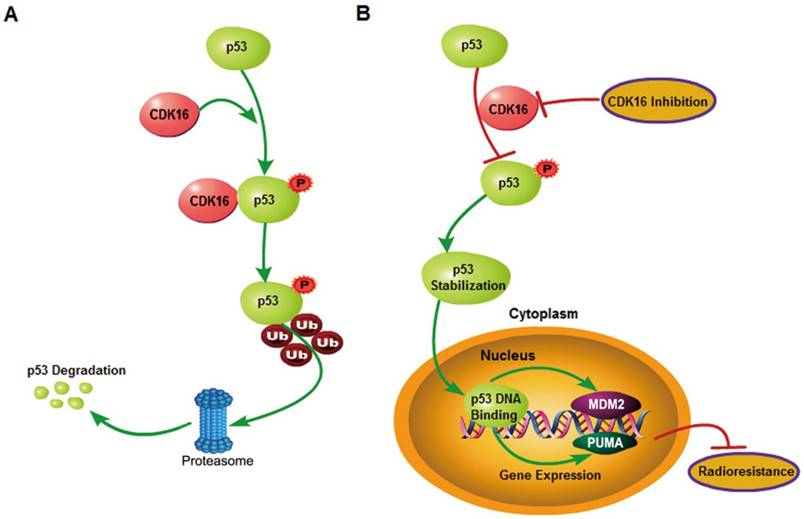
A proposed model shows mechanistically how CDK16 participates in radioresistance by phosphorylating and destabilizing p53. A. High level of CDK16 leads to its binding to p53, which subsequently phosphorylates p53 at Ser315 site and promotes p53 degradation via the ubiquitin/proteasome system. B. Loss of CDK16 or CDK16 inhibition prevents p53 phosphorylation, which leads to p53 stabilization and transcriptional activation of p53 that ultimately inhibits radioresistance in lung cancer.
Abbreviations
CDK16: cyclin-dependent kinase 16; NSCLC: non-small cell lung cancer; CCNY: cyclin Y; PI: propidium iodide; IHC: immunohistochemistry; ROS: reactive oxygen species; IP: immunoprecipitation.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81672289 to S.X), the Natural Science Foundation of Hubei Province of China (2016CFB377 to S.X), the National Natural Science Foundation of China (81672979 to G.W), National Key R&D Program of China (2016YFC0106700 to G.W and S.Z), National Key R&D Program of China (2017YFC0909904 to Y.Z, S.S and S.X), and Department of Defense Era of Hope research scholar award (W81XWH-09-1-0409 to J.C).
Author contributions
S.X conceived and designed the study, S.X and G.W supervised the study, J.X, Y.L, K.J and K.H performed experiments and analyzed the data, S.Z, X.D, X.D, L.L, T.Z, K.Y, K.H, S.S, Y.Z and J. C provided advice and technical assistance, S.X wrote the manuscript. All authors have contributed to and approved the final manuscript.
Supplementary Material
Supplementary figures (S1-S3) and tables (S1-S2).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
3. Eberhardt WE, Stuschke M. Multimodal treatment of non-small-cell lung cancer. Lancet. 2015;386:1018-20
4. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10
5. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749-58
6. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359-70
7. Lee JT, Gu W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010;17:86-92
8. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83-96
9. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413-31
10. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393-405
11. Munoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol. 2016;16:741-50
12. Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis. 2009;14:597-606
13. Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714-23
14. Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016:6
15. Malumbres M, Harlow E, Hunt T. et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol. 2009;11:1275-76
16. Mikolcevic P, Sigl R, Rauch V. et al. Cyclin-dependent kinase 16/PCTAIRE kinase 1 is activated by cyclin Y and is essential for spermatogenesis. Mol Cell Biol. 2012;32:868-79
17. Mikolcevic P, Rainer J, Geley S. Orphan kinases turn eccentric: a new class of cyclin Y-activated, membrane-targeted CDKs. Cell Cycle. 2012;11:3758-68
18. Zi Z, Zhang Z, Li Q. et al. CCNYL1, but Not CCNY, Cooperates with CDK16 to Regulate Spermatogenesis in Mouse. Plos Genet. 2015;11:e1005485
19. Shehata SN, Deak M, Morrice NA. et al. Cyclin Y phosphorylation- and 14-3-3-binding-dependent activation of PCTAIRE-1/CDK16. Biochem J. 2015;469:409-20
20. Palmer KJ, Konkel JE, Stephens DJ. PCTAIRE protein kinases interact directly with the COPII complex and modulate secretory cargo transport. J Cell Sci. 2005;118:3839-47
21. Liu Y, Cheng K, Gong K, Fu AK, Ip NY. Pctaire1 phosphorylates N-ethylmaleimide-sensitive fusion protein: implications in the regulation of its hexamerization and exocytosis. J Biol Chem. 2006;281:9852-8
22. Mokalled MH, Johnson A, Kim Y, Oh J, Olson EN. Myocardin-related transcription factors regulate the Cdk5/Pctaire1 kinase cascade to control neurite outgrowth, neuronal migration and brain development. Development. 2010;137:2365-74
23. Tang X, Guilherme A, Chakladar A. et al. An RNA interference-based screen identifies MAP4K4/NIK as a negative regulator of PPARgamma, adipogenesis, and insulin-responsive hexose transport. Proc Natl Acad Sci U S A. 2006;103:2087-92
24. Chen XY, Gu XT, Saiyin H. et al. Brain-selective kinase 2 (BRSK2) phosphorylation on PCTAIRE1 negatively regulates glucose-stimulated insulin secretion in pancreatic beta-cells. J Biol Chem. 2012;287:30368-75
25. Shimizu K, Uematsu A, Imai Y, Sawasaki T. Pctaire1/Cdk16 promotes skeletal myogenesis by inducing myoblast migration and fusion. Febs Lett. 2014;588:3030-7
26. Iwano S, Satou A, Matsumura S. et al. PCTK1 regulates integrin-dependent spindle orientation via protein kinase A regulatory subunit KAP0 and myosin X. Mol Cell Biol. 2015;35:1197-208
27. Yanagi T, Krajewska M, Matsuzawa S, Reed JC. PCTAIRE1 phosphorylates p27 and regulates mitosis in cancer cells. Cancer Res. 2014;74:5795-807
28. Yanagi T, Matsuzawa S. PCTAIRE1/PCTK1/CDK16: a new oncotarget? Cell Cycle. 2015;14:463-4
29. Yanagi T, Tachikawa K, Wilkie-Grantham R. et al. Lipid Nanoparticle-mediated siRNA Transfer Against PCTAIRE1/PCTK1/Cdk16 Inhibits In Vivo Cancer Growth. Mol Ther Nucleic Acids. 2016;5:e327
30. Xu S, Wu Y, Chen Q. et al. hSSB1 regulates both the stability and the transcriptional activity of p53. Cell Res. 2013;23:423-35
31. Ma H, Pederson T. Depletion of the nucleolar protein nucleostemin causes G1 cell cycle arrest via the p53 pathway. Mol Biol Cell. 2007;18:2630-5
32. Wu Y, Zhou L, Wang X. et al. A genome-scale CRISPR-Cas9 screening method for protein stability reveals novel regulators of Cdc25A. Cell Discov. 2016;2:16014
33. Wang Q, Ma J, Lu Y. et al. CDK20 interacts with KEAP1 to activate NRF2 and promotes radiochemoresistance in lung cancer cells. Oncogene. 2017;36:5321-30
34. Lu Y, Ma J, Li Y. et al. CDP138 silencing inhibits TGF-β/Smad signaling to impair radioresistance and metastasis via GDF15 in lung cancer. Cell Death Dis. 2017;8:e3036
35. Cho HJ, Oh YJ, Han SH. et al. Cdk1 protein-mediated phosphorylation of receptor-associated protein 80 (RAP80) serine 677 modulates DNA damage-induced G2/M checkpoint and cell survival. J Biol Chem. 2013;288:3768-76
36. Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528-36
37. Humpton TJ, Vousden KH. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb Perspect Med. 2016:6
38. Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128-36
39. Speidel D. The role of DNA damage responses in p53 biology. Arch Toxicol. 2015;89:501-17
40. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793-805
41. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609-22
42. Cwiek P, Leni Z, Salm F. et al. RNA interference screening identifies a novel role for PCTK1/CDK16 in medulloblastoma with c-Myc amplification. Oncotarget. 2015;6:116-29
43. Dixon-Clarke SE, Shehata SN, Krojer T. et al. Structure and inhibitor specificity of the PCTAIRE-family kinase CDK16. Biochem J. 2017;474:699-713
Author contact
![]() Corresponding authors: Prof. Shuangbing Xu or Prof. Gang Wu, Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China. Email: xsb723edu.cn or xhzlwgcom
Corresponding authors: Prof. Shuangbing Xu or Prof. Gang Wu, Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China. Email: xsb723edu.cn or xhzlwgcom
Citation styles
APA
Xie, J., Li, Y., Jiang, K., Hu, K., Zhang, S., Dong, X., Dai, X., Liu, L., Zhang, T., Yang, K., Huang, K., Chen, J., Shi, S., Zhang, Y., Wu, G., Xu, S. (2018). CDK16 Phosphorylates and Degrades p53 to Promote Radioresistance and Predicts Prognosis in Lung Cancer. Theranostics, 8(3), 650-662. https://doi.org/10.7150/thno.21963.
ACS
Xie, J.; Li, Y.; Jiang, K.; Hu, K.; Zhang, S.; Dong, X.; Dai, X.; Liu, L.; Zhang, T.; Yang, K.; Huang, K.; Chen, J.; Shi, S.; Zhang, Y.; Wu, G.; Xu, S. CDK16 Phosphorylates and Degrades p53 to Promote Radioresistance and Predicts Prognosis in Lung Cancer. Theranostics 2018, 8 (3), 650-662. DOI: 10.7150/thno.21963.
NLM
Xie J, Li Y, Jiang K, Hu K, Zhang S, Dong X, Dai X, Liu L, Zhang T, Yang K, Huang K, Chen J, Shi S, Zhang Y, Wu G, Xu S. CDK16 Phosphorylates and Degrades p53 to Promote Radioresistance and Predicts Prognosis in Lung Cancer. Theranostics 2018; 8(3):650-662. doi:10.7150/thno.21963. https://www.thno.org/v08p0650.htm
CSE
Xie J, Li Y, Jiang K, Hu K, Zhang S, Dong X, Dai X, Liu L, Zhang T, Yang K, Huang K, Chen J, Shi S, Zhang Y, Wu G, Xu S. 2018. CDK16 Phosphorylates and Degrades p53 to Promote Radioresistance and Predicts Prognosis in Lung Cancer. Theranostics. 8(3):650-662.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.