Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(2):369-383. doi:10.7150/thno.21397 This issue Cite
Research Paper
Dual Targeting of Acute Leukemia and Supporting Niche by CXCR4-Directed Theranostics
Stefan Habringer1,2#, Constantin Lapa3#, Peter Herhaus1, Margret Schottelius4, Rouzanna Istvanffy1, Katja Steiger5, Julia Slotta-Huspenina5, Andreas Schirbel3, Heribert Hänscheid3, Stefan Kircher7, Andreas K. Buck3, Katharina Götze1,2, Binje Vick2,6, Irmela Jeremias2,6, Markus Schwaiger2,8, Christian Peschel1,2, Robert Oostendorp1, Hans-Jürgen Wester4, Götz-Ulrich Grigoleit9*, Ulrich Keller1,2* ![]()
1. Internal Medicine III, Hematology and Medical Oncology, Technische Universität München, Munich, Germany;
2. German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ), Heidelberg, Germany;
3. Department of Nuclear Medicine, University Hospital Würzburg, Würzburg, Germany;
4. Institute of Pharmaceutical Radiochemistry, Technische Universität München, Garching, Germany;
5. Institute of Pathology, Technische Universität München, Munich, Germany;
6. Research Unit Gene Vectors, Helmholtz Center Munich, Germany;
7. Institute for Pathology, University of Würzburg, Würzburg, Germany;
8. Department of Nuclear Medicine, Technische Universität München, Munich, Germany;
9. Department of Internal Medicine II, Hematology and Medical Oncology, University Hospital Würzburg, Würzburg, Germany.
# These authors contributed equally.
* These authors contributed equally.
Received 2017-6-9; Accepted 2017-10-12; Published 2018-1-1
Citation:
Habringer S, Lapa C, Herhaus P, Schottelius M, Istvanffy R, Steiger K, Slotta-Huspenina J, Schirbel A, Hänscheid H, Kircher S, Buck AK, Götze K, Vick B, Jeremias I, Schwaiger M, Peschel C, Oostendorp R, Wester HJ, Grigoleit GU, Keller U. Dual Targeting of Acute Leukemia and Supporting Niche by CXCR4-Directed Theranostics. Theranostics 2018; 8(2):369-383. doi:10.7150/thno.21397. https://www.thno.org/v08p0369.htm
Other stylesAbstract

C-X-C chemokine receptor 4 (CXCR4) is a transmembrane receptor with pivotal roles in cell homing and hematopoiesis. CXCR4 is also involved in survival, proliferation and dissemination of cancer, including acute lymphoblastic and myeloid leukemia (ALL, AML). Relapsed/refractory ALL and AML are frequently resistant to conventional therapy and novel highly active strategies are urgently needed to overcome resistance.
Methods: We used patient-derived (PDX) and cell line-based xenograft mouse models of ALL and AML to evaluate the efficacy and toxicity of a CXCR4-targeted endoradiotherapy (ERT) theranostic approach.
Results: The positron emission tomography (PET) tracer 68Ga-Pentixafor enabled visualization of CXCR4 positive leukemic burden. In xenografts, CXCR4-directed ERT with 177Lu-Pentixather distributed to leukemia harboring organs and resulted in efficient reduction of leukemia. Despite a substantial in vivo cross-fire effect to the leukemia microenvironment, mesenchymal stem cells (MSCs) subjected to ERT were viable and capable of supporting the growth and differentiation of non-targeted normal hematopoietic cells ex vivo. Finally, three patients with refractory AML after first allogeneic hematopoietic stem cell transplantation (alloSCT) underwent CXCR4-directed ERT resulting in leukemia clearance, second alloSCT, and successful hematopoietic engraftment.
Conclusion: Targeting CXCR4 with ERT is feasible and provides a highly efficient means to reduce refractory acute leukemia for subsequent cellular therapies. Prospective clinical trials testing the incorporation of CXCR4 targeting into conditioning regimens for alloSCT are highly warranted.
Keywords: acute leukemia, microenvironment, C-X-C chemokine receptor 4, in vivo molecular imaging, theranostics
Introduction
C-X-C-motif chemokine receptor 4 (CXCR4) is a G-protein coupled transmembrane receptor that regulates a wide spectrum of physiologic processes in fetal organ development, hematopoiesis, and immune system function. Knock-out studies in mice have demonstrated that absence of CXCR4 or CXCL12 - its only known chemokine ligand - is embryonically lethal [1, 2]. Binding of CXCL12 to CXCR4 initiates G-protein-dependent and -independent signaling events, including MAPK, AKT and ERK pathway activation, and Ca2+ release from the endoplasmic reticulum, which ultimately coordinates chemotaxis, homing, proliferation and cell survival [3]. CXCR4 is broadly expressed in the hematopoietic system; especially hematopoietic stem and progenitor cells (HSPCs) need CXCR4 for correct localization and retention in the bone marrow (BM) microenvironment. Therefore, the CXCR4/CXCL12 axis is indispensable for homeostasis of the HSPC pool in the BM [4]. CXCR4 is also commonly expressed or overexpressed in cancer cells, regulating proliferation, neo-angiogenesis, resistance to chemotherapy and metastasis to organs with high amounts of secreted CXCL12 [5, 6]. It has been shown for several cancer types, including acute myeloid leukemia (AML) that CXCR4 expression is associated with adverse prognosis [7]. Therefore, targeting CXCR4 with small molecule inhibitors or antibodies is being investigated in several clinical trials in various cancer types [8]. This concept is particularly promising in hematological malignancies, and preclinical studies have shown that CXCR4 inhibition can both kill cancer cells directly or dislocate them from their protective microenvironment, making them more susceptible to conventional chemotherapy in combined approaches [9-11].
In AML, malignant cells arising from immature myeloid progenitors or stem cells increasingly occupy the BM space as the disease progresses, leading to rapidly fatal complications without treatment. Even in patients receiving adequate intensive therapy, most commonly a combination of cytarabine and anthracyclines, overall survival rates 3 years after therapy range from 12-66% in younger and 3-33% in older adults, depending on prognostic factors [12]. Primary refractory disease and relapse after having achieved a complete remission are the biggest challenges in treating AML. Allogeneic stem cell transplantation (alloSCT) is considered the only curative option for these patients, and highly active conditioning regimens are needed to overcome resistance and reduce leukemic burden before transplantation. Due to the lack of a standard salvage induction regimen, choice of the preferred therapeutic strategy remains an individualized decision, often varying among different centers [13].
Acute lymphoblastic leukemia (ALL) is the most common cancer in childhood, with cure rates over 95% in low risk early B-cell lineage ALL patients [14]. However, ALL in adults is more difficult to treat and outcomes are worse than in pediatric patients [15]. With the bispecific (CD19, CD3) antibody blinatumomab and chimeric antigen receptor (CAR) T-cells emerging as effective treatments in relapsed B-ALL [16], comparably effective novel treatment strategies in T-ALL have been lacking so far [17, 18].
In both AML and ALL, the BM microenvironment is believed to play an essential role in protecting leukemic cells from chemotherapy or targeted therapies. This protective activity is believed to be a major determining factor in the survival of malignant cells and relapsing disease [11, 19, 20]. The niche is a major source of CXCL12 and this chemokine has been shown to induce stem cell quiescence, which contributes to resistance of leukemic stem cells to chemotherapy [21].
We have previously shown that CXCR4-directed PET imaging with the novel, human-specific CXCR4-binding peptide tracer 68Ga-Pentixafor enables the visualization of CXCR4-expressing cells in AML and multiple myeloma patients [22, 23]. A modified version of Pentixafor, named Pentixather, allows labeling with β-emitting radionuclides (177Lutetium, 177Lu; 90Yttrium, 90Y) routinely used in clinical practice for cancer radiotherapy. This facilitates the possibility of a theranostic approach by combining CXCR4-directed imaging to select patients for CXCR4-directed endoradiotherapy (ERT) with Pentixather. This strategy would also allow targeting of the malignant cell-supportive BM microenvironment supporting malignant cells by cross-fire irradiation, which is particularly relevant in AML and ALL [11, 19, 20].
Here, we further develop this concept and apply a theranostic approach using ERT with Pentixather in preclinical models of T-ALL and AML, and ultimately in patients with relapsed AML after first alloSCT, who did not respond to conventional therapies. We investigate efficacy and toxicity, especially to the BM microenvironment, of this novel approach to treating leukemia, which provides crucial information for future prospective clinical studies.
Results
CXCL12/CXCR4 signaling is crucial for ALL and AML establishment
The CXCR4/CXCL12 axis is known to be involved in disease initiation, migration and treatment resistance in murine AML and ALL models, and in patients [20, 24]. We generated T-ALL (ALL0 and ALL230) and AML (AML356 and AML346) PDX mice (Fig. S1, S2 and Table S1) and used an orthotopic cell line xenograft model of human AML with moderate or enforced CXCR4 expression (OCI-AML3-eGFP and OCI-AML3-CXCR4, Fig. S5a, b). To evaluate if the CXCR4/CXCL12 axis is functionally relevant in ALL and AML xenografts, we performed transwell migration assays with AML and ALL cells. CXCL12-induced migration and inhibition of CXCR4 with the clinically approved CXCR4 inhibitor AMD3100 (Plerixafor) resulted in significant reduction of the migrated cell fraction in OCI-AML3 cells, and in ALL0 and ALL230 PDX leukemia (Fig. 1a). Furthermore, immunoblot analysis confirmed that phosphorylation of AKT, a known downstream target and surrogate marker for CXCR4 activation, was induced (Fig. S3). To test if in vivo homing of human leukemic blasts to the BM and spleen of NSG mice is CXCR4-dependent, we pre-incubated T-ALL PDX (ALL230 and ALL0) and OCI-AML3 with the established CXCR4-blocking antibody 12G5 or an appropriate isotype control antibody, and injected these cells into NSG mice (Fig. 1b). After 48 h, BM and splenic infiltration by human leukemic cells was significantly lower when CXCR4 was blocked with 12G5, indicating that CXCR4-mediated homing is necessary for disease initiation in these acute leukemia models (Fig. 1c).
Thus, these leukemia models are suitable for testing CXCR4-directed theranostics in vivo with regard to efficacy and microenvironment effects.
Figure 1
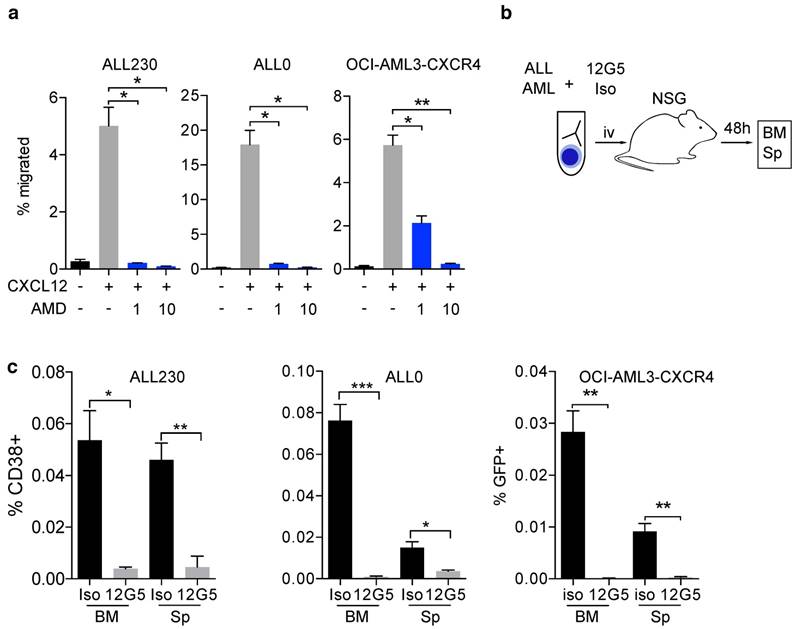
CXCR4/CXCL12 signaling is crucial for establishing acute leukemia PDX. a) Migration of OCI-AML3-CXCR4 and ALL PDX (ALL230 and ALL0) towards 100ng/μl CXCL12 alone or combined with 1μM and 10μM AMD3100 (n=3 replicates). b) Schematic of experimental setup for experiments shown in c). c) OCI-AML3-CXCR4 and ALL PDX homing in vivo after pre-incubation with CXCR4 antibody clone 12G5 or isotype control. Cells were injected into NSG mice and infiltration of organs was assessed after 48h (n=3 mice per group). Statistical significance was determined by two-sided t-tests, *p<0.05, **p<0.01, ***p<0.001, AMD: AMD3100, BM: bone marrow, Sp: spleen, NSG: NOD-SCID-gamma.
In vivo molecular CXCR4 imaging reflects surface expression of CXCR4 and represents the first step in CXCR4 theranostics
We next sought to determine if leukemic PDX cells expressing CXCR4 could be imaged in vivo with the human-specific CXCR4-binding peptide PET tracer 68Ga-Pentixafor as an initial component of CXCR4 theranostics. The grafts of the employed PDX models of T-ALL and AML clearly displayed different levels of CXCR4 surface expression (Fig. 2a). Upon NSG recipient injection with PDX, peripheral blood (PB) blast counts were monitored with flow cytometry. 68Ga-Pentixafor PET imaging was performed when human blasts could be detected in PB or when mice displayed symptoms of leukemia (weight loss, behavioral abnormalities). 68Ga-Pentixafor enabled approximate visualization of leukemic burden of T-ALL (ALL230, ALL0) and AML PDX (ALL356) in spleens and bones of NSG mice (Fig. 2b) and correlated with CXCR4 surface expression of PDX (Fig. 2c). Histology and immunohistochemistry of imaged mice confirmed CXCR4 expression on human infiltrating blasts (Fig. 2d). 69Ga-Pentixafor PET images of control NSG mice without leukemic burden are shown in Fig. S4 in different intensities.
To determine if Pentixather, the structurally modified therapeutic counterpart of Pentixafor, binds to human leukemia cells in vivo, we injected AML356 PDX recipients with 125Iodine-Pentixather three, four and five weeks after injection of PDX cells. Binding of Pentixather to splenic AML blasts was detected by ex vivo autoradiographic imaging of spleens. Progressive splenic infiltration could be visualized by autoradiography, indicating that 125I-Pentixather binds to AML356 PDX in vivo (Fig. 2e).
Thus, the CXCR4-directed therapeutic peptide Pentixather represents a pre-clinically applicable means to target human CXCR4+ PDX in vivo, and may serve as a surrogate imaging diagnostic for assessing human leukemia infiltration in BM and other organs.
Figure 2
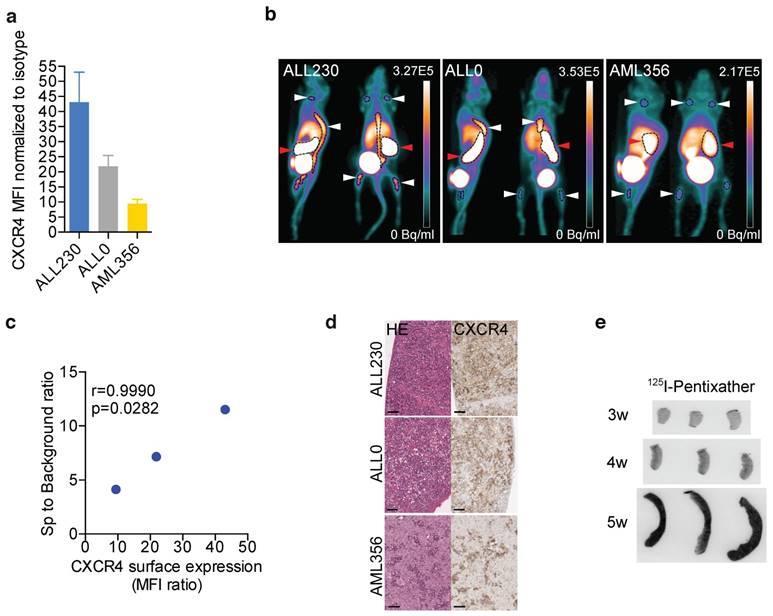
CXCR4 diagnostics in preclinical models of acute leukemia. a) CXCR4 surface expression in ALL and AML PDX (n=3 replicates). b) Representative PET images of 68Ga-Pentixafor scans in mice bearing ALL and AML PDX. Red arrows: spleen, white arrows: bone marrow (n=6 ALL230, n=6 ALL0, n=5 AML356). c) Correlation between Ga-Pentixafor PET uptake and mean CXCR4 surface expression. d) HE staining and CXCR4 immunohistochemistry of spleens of ALL230, ALL0 and AML356 mice. Scale bars: 50 μm. e) Representative images of 125I-Pentixather autoradiography of AML356 spleens. MFI: mean fluorescence intensity, HE: Hematoxylin and eosin, r: Pearson correlation coefficient.
Therapeutic CXCR4-targeted peptide effectively reduces leukemia in PDX mice
To test the efficacy of CXCR4 targeting in vivo we chose peptide labeling with the well-established therapeutic beta-emitter 177Lu [25, 26]. Two CXCR4high T-ALL PDX models and two cell line models of AML (OCI-AML-eGFP and OCI-AML-CXCR4) with different infiltration characteristics of BM, spleen and blood (Fig. S1) were subjected to 177Lu-Pentixather (Lu-P) treatment (d0) or unlabeled Pentixather as control. Mice were sacrificed 3 and 7 days after injection of Lu-P (Fig. 3a). To determine the distribution of Lu-P in treated mice, we measured radioactivity in PB, BM and spleen of treated mice and found that Lu-P distributed to and strongly accumulated in leukemia-harboring organs (Fig. 3b).
In the ALL230 cohort, mice in the control group had large spleens (254.7 mg and 551.7 mg in the d3 and d7 groups, respectively). Lu-P therapy significantly reduced spleen size and weight in treated animals. In mice receiving the less aggressive ALL0 PDX, spleens were less enlarged ~4 weeks after PDX injection. Again, Lu-P therapy resulted in significantly reduced spleen weight as compared to control mice (Fig. 3c, d). In order to investigate ALL involvement of PB as well as spleen and BM, we performed flow cytometry. ALL230 displayed pronounced reduction of blast populations in spleen, BM and PB (Fig. 3e, f). Histology and immunohistochemistry of spleen and BM of control mice revealed an extensive infiltration with CXCR4+ neoplastic cells. In contrast, in all organs of Lu-P-treated mice, cellularity was significantly reduced and leukemic cells were effectively targeted by ERT (Fig. 3g). In BM and spleen of heavily infiltrated mice, although single neoplastic blasts were still detectable, hemorrhage and necrotic tissue damage were apparent, indicating effective targeting of the infiltrative tumor cell population as a result of therapy.
In summary, these experiments show that CXCR4-directed ERT effectively targeted CXCR4+ tumor cells and reduced leukemic burden in T-ALL PDX recipient mice. The data also indicate that damage occurred to the remaining functional BM, most likely as a result of cross fire effects by Lu-P.
To answer the question whether the intensity of CXCR4 surface expression affected treatment efficacy, we used OCI-AML3-CXCR4, with a ~2.5-fold overexpression of surface CXCR4 (Fig. S5a) as compared to empty vector control-eGFP transduced cells. These cells were injected intravenously into NSG mice to establish xenografts, followed by treatment with Lu-P or unlabeled Pentixather. Three days after treatment we observed a significant reduction of spleen weight irrespective of the level of CXCR4 expression (moderate vs. enforced) (Fig. S5c). Importantly, treatment of AML346, the PDX line with the lowest CXCR4 expression, did not result in reduction of leukemic burden (Fig. S6.). These experiments with AML models with low (AML346), moderate (OCI-AML3-eGFP) and elevated (OCI-AML3-CXCR4-eGFP) CXCR4 surface expression suggested that a certain degree of target expression, i.e., CXCR4 surface expression, is necessary for treatment efficacy. This finding should be relevant with regard to clinical benefit for AML patients with various extent of CXCR4 expression.
Cross-fire originating from Lu-P targeting impairs normal HSPCs and the BM niche
We hypothesized that, owing to the β-emitting properties of 177Lu, radionuclide targeting of CXCR4 expressing leukemic cells would result in damage to the surrounding tissue, including murine HSPCs and other cellular components of the BM niche. The PDX models in combination with a targeting peptide specific for human CXCR4 [27, 28] thus provided an ideal model to address cross-fire effects on murine recipient HSPCs and the host cellular microenvironment.
To directly assess damage inflicted to murine HSPCs, we performed colony forming unit (CFU) assays with murine growth factors on BM harvests of ERT-treated and control ALL230 and ALL0 PDX mice (Fig. 4a). In both PDX models, the proliferative potential of murine BM HPCs was significantly reduced with treatment. In ALL230 mice, which had subtotal infiltration of the BM by human ALL (Fig. 3g), Lu-P treatment even resulted in complete ablation of CFU growth (Fig. 4b). Using flow cytometry, we observed a significantly reduced fraction of lineage negative (Lin-), Sca1 positive, cKit positive stem cells (LSKs), and BM myeloid progenitors (MPs) (Fig. 4c, gating strategy in Fig. S7). In Lu-P treated mice, LSKs and MPs were almost completely absent in flow cytometry, further emphasizing the toxicity of radioactive targeting for the neighboring hematopoietic population.
To test if the BM niche, especially the MSC population crucial for reconstitution of hematopoiesis, is affected by ERT, we isolated and analyzed endosteal niche cells from collagenase treated bones of Lu-P treated and untreated mice and performed flow cytometry analyses as described earlier [29, 30]. MSC were still present in both ERT-treated and control mice after isolation of endosteal niche cells (Fig. 4d). To further assess if the BM MSC population was still viable after Lu-P treatment, we performed CFU-F assays to isolate MSCs from bones of treated and control mice (representative images in Fig. S8). MSCs are defined as adherent cells emerging from bones, which form colonies and proliferate in vitro [31]. Colony numbers were not significantly different in Lu-P vs. control with a trend towards reduction in the treatment group (Fig. 4e). Further growth of the isolated MSCs was significantly reduced in the Lu-P group, indicating targeting of the BM niche by the cross-fire effect (Fig. 4f).
Figure 3
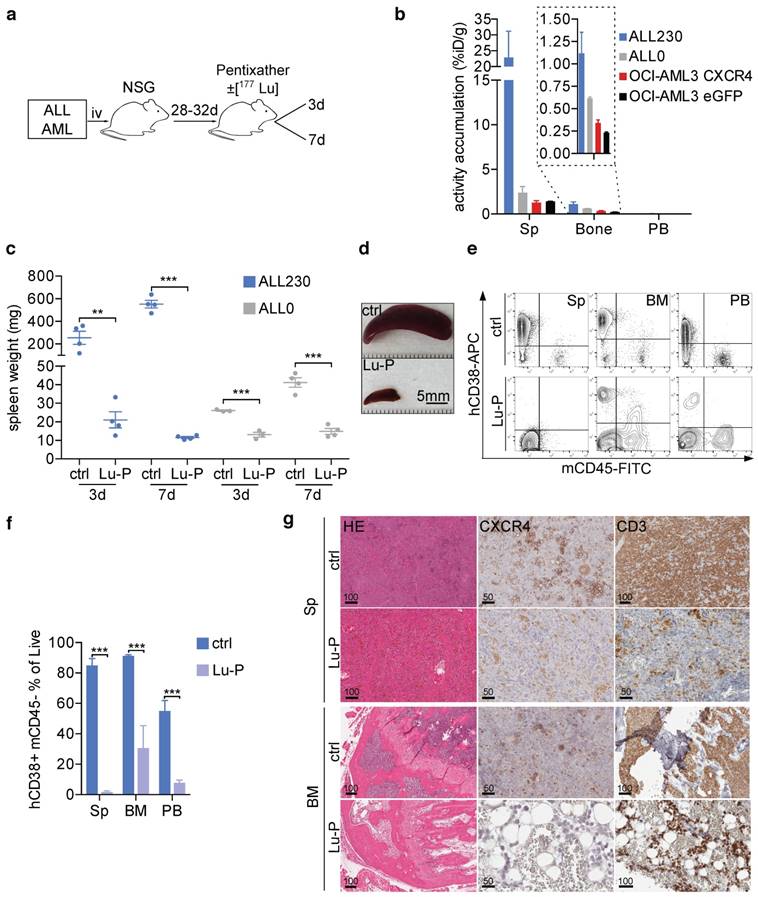
CXCR4-directed theranostics effectively reduce leukemia burden. a) Schematic of experimental setup. b) Activity accumulation in percent of injected dose per gram (%iD/g) of Sp, bone and PB 3d post injection of Lu-P in mice bearing ALL PDX or OCI-AML3 cells overexpressing CXCR4 or eGFP empty vector (n=4 for ALL230, OCI-AML3 CXCR4, n=3 for ALL0, OCI-AML3 eGFP). c) Spleen weight of ctrl and Lu-P treated ALL0 and ALL230 mice after 3 and 7d (n=3 for ALL0 3d, n=4 for all other groups). d) Representative images of ALL230 spleens treated for 3d with ctrl or Lu-P. e) Representative FACS plots of Sp, BM and PB of ALL230 mice treated for 7d with ctrl or Lu-P, gated on propidium-iodide negative, live cells. Human ALL cells are human CD38 positive and murine CD45 negative. f) Quantification of e), percentage of human blasts is shown. g) Sp and BM histology of ALL230 mice treated with ctrl or Lu-P with HE stain, CXCR4 and CD3 immunohistochemistry, scale bar length in μm as indicated. Statistical significance was determined by two-sided t-tests, *p<0.05, **p<0.01, ***p<0.001, ctrl: control, Lu-P: 177Lu-Pentixather, PB: peripheral blood.
Figure 4
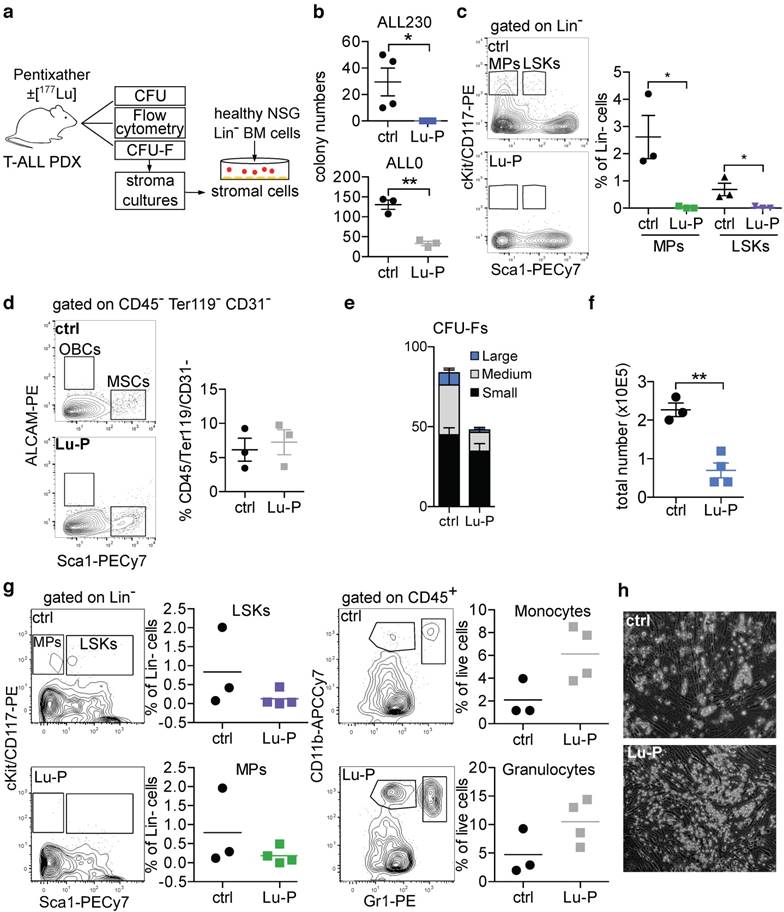
Cross-fire effect on non-malignant hematopoiesis and BM microenvironment. a) Schematic of experimental setup. b) Methylcellulose CFU assay for murine progenitors of BM from ALL230 and ALL0 mice 3d after treatment with ctrl or Lu-P (n=4 mice for ALL230, n=3 mice for ALL0). c) Flow cytometry of BM isolated from ctrl and Lu-P treated ALL0 mice. LSKs and MPs were analyzed (n=3 mice per group). d) Comparison of MSCs (Sca1+, ALCAM- fraction of CD45-, Ter119-, CD3-, live cells) of treated and control ALL0 mice (n=3 mice per group). e) Quantification of CFU-F colony numbers (small: <20 cells, medium: 20-200 cells, large >200 cells) of ctrl and Lu-P treated ALL230 mice (n=4 mice per group). f) Total number of stromal cells from d) after 18d of culture. g) Coculture of MSCs from Lu-P and ctrl ALL20 mice (3d) and lineage- BM cells from WT NSG mice. LSKs, MPs, Gr1+CD11b+ granulocytes and Gr1medCD11b+ monocytes were analyzed after 7d of coculture (n=3 for ctrl, n=4 for Lu-P). h) Representative microscopic images of coculture experiment as described in g). Statistical significance was determined by two-sided t-tests, *p<0.05, **p<0.01, ***p<0.001, colony forming unit (CFU), LSKs: Lineage-, Sca1+, cKit+ cells, MPs: myeloid progenitors, MSCs: mesenchymal stem cells, CFU-F: colony forming unit-fibroblasts.
Support by the BM niche is essential for engraftment of HSPCs after myeloablative conditioning. We therefore investigated whether the isolated MSCs were still supportive of healthy HSPCs by co-culturing MSCs with MACS-purified immature (Lin-) murine HSPCs and analyzed MP and LSK number and frequency, and differentiation into mature myeloid cells. In both Lu-P treated and control stroma co-cultures, MPs and LSKs were supported and differentiation into granulocytes and monocytes was induced (Fig. 4g). Furthermore, HSPCs in co-culture formed cobblestone-like areas on both Lu-P-treated and control stroma (Fig. 4h). To model the effect of Lu-P on human BM MSCs, a small subset of which are known to express CXCR4 [32], we isolated MSCs from BM samples of healthy individuals and treated MSCs with Lu-P or unlabeled Pentixather. After treatment, we co-cultured purified healthy human CD34+ cells with the MSCs and assessed viability and potential to form colonies in methylcellulose (Fig. S9). There was no significant impairment of Lu-P pre-treated MSCs' ability for subsequent support of normal CD34+ cells.
In summary, the cross-fire effect caused by β-emitter ERT results in significant damage to HSPCs and substantially targets proliferative activity of BM niche cells, while the functional capacity of stromal cells to support the growth and differentiation of in vitro co-cultured healthy immature hematopoietic cells was not abrogated.
Pentixather treatment in very advanced human acute leukemia: ERT followed by second alloSCT
Finally, the concept of targeting CXCR4 in acute leukemia by ERT was translated into a clinical setting. Three patients with relapsed AML were referred for assessment of further treatment option, e.g., Pentixather treatment. All patients were heavily pretreated, including first alloSCT, but had experienced early relapse (for patient characteristics and previous treatments see Table S2). Given the lack of alternative treatment options in this advanced disease stage, experimental CXCR4-directed treatment was offered on a compassionate use basis (German Drug Act, §13,2b) in compliance with §37 of the Declaration of Helsinki. All subjects gave written informed consent prior to therapy.
Patient #1 (46-year-old male) presented with AML without maturation (AML M1). After standard induction chemotherapy and salvage therapy due to primary refractory disease first alloSCT was performed. Three months after SCT, relapse was diagnosed and the patient was referred. Given the early relapse, investigation of CXCR4 expression as a putative therapy target was considered. 68Ga-Pentixafor-PET demonstrated receptor expression (Fig. S10a), which qualified for CXCR4-targeted ERT. Pre-therapeutic dosimetry resulted in tolerable activities of 5 GBq (with the kidneys being the dose-limiting organ) of 90Y-Pentixather (Y-P), and achievable BM doses of ~11 Gy. 90Y as radionuclide was chosen due to its higher energy and longer range as compared to 177Lu, and because its shorter half-life allows earlier SCT. ERT with 4.72 GBq of Y-P was performed and well tolerated. In order to maximize the therapeutic effect of ERT, we decided to increase internal radiation by adding a course of 153Sm-labelled ethylene diamine tetramethylene phosphonate (EDTMP), which, in contrast to Pentixather, localizes to the surface of cortical and trabecular bone, to the conditioning regimen. Based on previous studies investigating 153Sm-EDTMP for BM ablation [33, 34], 15.9 GBq was intravenously administered five days after Y-P and resulted in additional BM irradiation as high as 8 Gy for the red marrow and 53 Gy in osteogenic cells in the cortical bones. Conditioning was completed by fludarabine (40 mg/m2; d-8 to d-5), thiotepa (5 mg/kg body weight; d-4; twice), melphalan (70 mg/m2; d-3 to d-2) and antithymocyte globulin (ATG; 10/20/30 mg/kg; d-10 to d-8). 19 days after the initial Y-P treatment, the patient received a second alloSCT (day 0). Besides prolonged epistaxis in aplasia requiring temporary intubation, he did not experience major complications. On day +11, recovery of neutrophils, on day +14, recovery of thrombocytes could be recorded (Fig. 5d). On day +30, donor chimerism in peripheral blood was 99.89%.
Patient #2 (67-year-old female) suffered from therapy-related AML (t-AML) that arose after exposure to polychemotherapy for breast cancer 18 years earlier. After induction therapy and first alloSCT (Table S2) the patient was in complete remission for 16 months until relapse. At the time of presentation, 50% AML infiltration of the BM as well as multiple extramedullary disease (EMD) lesions in the soft tissue of the pelvis and abdomen were present. 68Ga-Pentixafor PET revealed a rather modest receptor expression of the BM but intense tracer uptake in all EMD lesions (Fig. S10b). Given the lack of established treatment options, a second alloSCT after a combined conditioning approach using Y-P for both the intra- as well as especially EMD lesions (administered activity of 4.5 GBq; d-20), 188Re-labelled anti-CD66 antibodies for BM ablation (5.2 GBq; d-14) and conventional agents including rituximab (375 mg/m2; d-6), total body irradiation (TBI, 2 Gy; d-5); ATG (10 mg/kg; d-4; resulting in anaphylactic shock), and melphalan (70 mg/m2; d-3 to d-2) was chosen. Pre-therapeutic dosimetry in this patient yielded estimated BM doses of up to 17 Gy and EMD doses of 23 Gy. Achieved doses for 188Re-anti-CD66 antibodies could not be calculated due to technical problems; however, post-therapeutic whole-body scintigraphy proved high antibody retention in the bone marrow (Fig. S10b). Unfortunately, this patient died after hematological recovery from septic complications on day +17 after second SCT (Fig. 5d). Chimerism analysis had not yet been performed.
Patient #3 (39-year-old male) had been diagnosed with AML M0 nine months prior to presentation (Table S2). He had experienced leukemia relapse only five months after haploidentical first alloSCT (Fig. 5a-c) and was referred for salvage therapy. In analogy to patient #2, combined CXCR4-directed ERT (with 2.7 GBq of Y-P; d-33) and anti-CD66 therapy (with 7.7 GBq of 188Re-anti-CD66 antibodies; d-28) as part of conditioning prior to second alloSCT was performed and resulted in red BM doses of 20 Gy. During aplasia, severe mucositis and a septic episode could be successfully managed. After neutrophil reconstitution on d+20, 99.9% peripheral blood donor chimerism were recorded on d+26. The patient was discharged the day after. Repeated BM biopsy 6 months after alloSCT confirmed complete remission.
Discussion
Our data provide first evidence for efficacy of CXCR4-directed ERT with Pentixather in preclinical models of T-ALL and AML, and a limited number of patients treated within individual therapy approaches for very advanced disease.
Figure 5
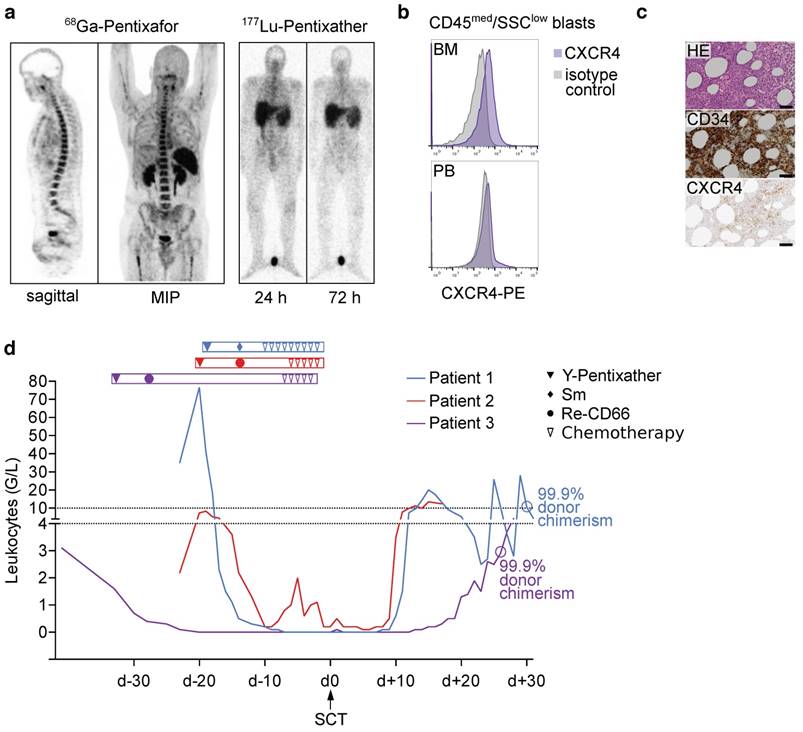
Proof-of-concept evaluation in advanced human acute leukemia: ERT followed by allogeneic stem cell transplantation. a) 68Ga-Pentixafor PET imaging and planar whole-body scintigraphic images 24h and 72h after injection of 200 MBq Lu-P in patient 3 (activity injected for pre-therapeutic dosimetry). b) Blast CXCR4 surface expression in flow cytometry in patient 3 (gated on CD45med/SSClow blasts) c) HE stain, CD34 and CXCR4 BM immunohistochemistry of patient 3, scale bars: 50 μm. d) Leukocyte counts and % donor chimerism of patients undergoing Y-Pentixather based conditioning and allogeneic stem cell transplantation. MIP: maximum intensity projection, Sm: 153Samarium-EDTMP, Re-CD66: 166Rhenium-anti-CD66 antibody.
When AML or ALL relapse or are refractory to treatment, a multitude of challenges arises. Both relapse and refractoriness are a manifestation of cells having developed resistance to therapy or re-emerging after a period of dormancy. In such a scenario, salvage therapy and pre-transplant conditioning needs to be of the highest possible efficacy with regard to eradicating leukemic cells without compromising engraftment or causing excessive or irreversible damage to non-hematopoietic organs, which would further increase treatment-related morbidity and mortality.
Despite the prominent role of cell-intrinsic mechanisms in T cell as well as myeloid HSPC transformation, ALL/AML cell growth is not fully cell autonomous. In the BM, T-ALL lymphoblasts establish stable contacts with vascular endothelial niche cells expressing CXCL12 and are dependent on cues from the microenvironment for cell proliferation and survival [20, 35, 36]. A similar role for the CXCR4/CXCL12 axis has been established for AML [11, 19, 37], indicating that CXCR4/CXCL12 activity in the BM microenvironment marks a highly beneficial local sanctuary for ALL and AML cells. Despite tremendous efforts and the fact that several drugs are available and have already entered the clinical phase of development, neither small molecule or peptide inhibitors nor CXCR4-targeted antibodies have yet shown convincing efficacy [38]. There are several reasons why such pharmacological approaches (antibodies, inhibitors) may not result in long-term benefit. Potential mechanisms of resistance include downregulation or internalization of surface CXCR4, heterogeneous expression on cancer cells resulting in incomplete targeting, and potential competition with locally increased CXCL12 concentrations [39, 40].
Our therapeutic preclinical PDX data indicate that the CXCR4 ERT concept provides substantial efficacy via the cross-fire effect, overcoming the requirement that every single cancer cell is reached und thus also providing a strong niche-targeted impact. This however comes with a major intricacy, namely targeting basically all niches where CXCR4-expressing cells, whether malignant or not, reside. Due to the limited number of patients treated with the experimental protocol presented in this work, and the fact that only AML patients received Pentixather ERT, we cannot conclude at this time whether the level of expression of CXCR4 is predictive of efficacy or toxicity. The preclinical data regarding level of CXCR4 expression (Fig. 2a) and correlation of CXCR4 expression with 68Ga-Pentixafor PET imaging however could hint towards a correlation between organ-bound dosage and local effectiveness as well as toxicity (Fig. 3, Fig. 4, Fig. S9).
Direct and indirect targeting of the interaction of leukemic cells with cellular components of the BM microenvironment represents an attractive strategy to induce the best possible remission before immunological intervention. In fact, various studies have shown that AML patients who enter conditioning and alloSCT have significantly better survival as compared to those with residual disease [41]. Severe impairment of the hematopoietic niche before alloSCT may result in severely prolonged pancytopenia or engraftment failure. Therefore, careful dosage and detailed preclinical evaluation of a potential niche-targeting agent is warranted. Our study of the BM microenvironment of PDX recipient mice, whose HSPC and MSC cannot bind Pentixather, and our experiments with human MSC showed that the number of niche cells is not affected by treatment. However, proliferation of niche cells after in vivo ERT is severely impaired. Despite this strong collateral damage observed within the BM microenvironment of PDX recipient mice, whose HSPC and MSC cannot bind Pentixather, patients receiving radionuclide-labeled Pentixather showed engraftment well within the expected range. Clearly, only a prospective clinical trial will allow determining the full extent of short and long-term effects of Pentixather treatment on the BM niche and other organs. Our results suggest that despite proliferative impairment, recipient MSC are well capable of supporting HSPC maintenance when cultured ex vivo.
Uptake of the imaging tracer Pentixafor within the liver of mice undergoing 68Ga-Pentixafor PET imaging is not reflected in patients [42]. Due to the high affinity to human compared to murine CXCR4 [28], this phenomenon is most likely due to unspecific binding or an idiosyncrasy of the murine metabolism of Pentixafor.
TBI has been applied in pre-transplant regimens as early as the 1960s, and is still used routinely in the treatment of leukemia due to its efficacy and ability to penetrate sanctuary sites [43]. Toxicity to non-hematopoietic organs is a major limitation of this technique. Approaches delivering radioactivity directly and selectively to the hematopoietic system, for instance radiolabeled antibodies against CD45 or CD66, have been used in pre-transplant conditioning [44, 45]. Targeting CXCR4 with a radioactive peptide might facilitate killing particularly therapy-resistant and otherwise difficult to treat leukemic cells in their protective niche. Especially in T-ALL, CXCR4 was recently shown to be essential for leukemia-initiating cells and maintenance of the disease in the BM [20, 24], further supporting potential benefit of CXCR4 targeting in this malignancy. We would expect that the same applies for CXCR4+ AML. We also would expect that Pentixather ERT could be provided to patients in need of non-chemotherapy salvage therapy before alloSCT who are not candidates for full-dose TBI due to age and/or comorbidities.
Due to the flexibility and various possibilities to label Pentixather, the choice of the radionuclide is not limited to 177Lu or 90Y [26]. Alpha emitters like 213Bismuth or 225Actinium have a much shorter range and deliver higher amounts of energy, potentially increasing specificity of targeting tumor cells and their immediate surroundings [46, 47]. We chose 177Lu in animal experiments and 90Y in patients due to different maximum ranges (0.8 mm and 11 mm, respectively), with the rationale to deliver appropriate doses to affected organs and spare healthy tissue. Other theranostic strategies currently being used in clinical practice include the treatment of midgut neuroendocrine tumors using 177Lu-DOTATATE, with excellent results in a phase 3 trial [48].
For a CXCR4-directed theranostic concept, screening with 68Ga-Pentixafor PET/CT or MRI will determine patients eligible for incorporation of Pentixather treatment in the conditioning regimen. A clinical phase I/II study (COLPRIT, EudraCT: 2015-001817-28) to evaluate safety of a Pentixafor/Pentixather based concept in the treatment of relapsed/refractory Non-Hodgkin lymphoma and multiple myeloma followed by autologous SCT is planned to be performed. Whether CXCR4 PET imaging is required in leukemia or can be replaced by flow cytometry of BM needs to be evaluated in the controlled prospective clinical setting. Based on the data presented here however, a clinical trial testing the incorporation of Pentixather into conditioning regimens before alloSCT is urgently warranted.
Materials and Methods
Cell culture and cell lines
The human AML cell line OCI-AML3 was cultured in high glucose (4.5 g/L) DMEM supplemented with 10% FCS, 100 U/mL penicillin and 100 µg/mL streptomycin. Cells were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Leibniz, Germany) and routinely re-authenticated. Cells were maintained at 37°C in a 5% CO2 humidified atmosphere. All media and supplements were obtained from Gibco/Life Technologies (Carlsbad, CA, USA). CXCR4 overexpression was achieved by lentiviral transduction of OCI-AML3 cells with pHIV-CXCR4-eGFP, with cDNA of human CXCR4 cloned into pHIV-eGFP (Addgene plasmid ID #21373).
Migration assay
Migration assays were performed as described previously [23]. Briefly, cells were incubated with AMD3100 (Selleckchem, Houston, TX, USA) or DMSO and placed in the top chamber of transwell plates with 5 µm pore size (Corning Inc., Corning, NY, USA) with 100 ng/mL CXCL12 in the lower chamber (R&D Systems, Minneapolis, MN, USA). Cells were then incubated at 37°C for 4 h and the total number of cells migrated to the lower chamber was measured using CountBright beads (Thermo Fisher, Waltham, MA, USA).
CFU, CFU-F and MSC coculture
For CFUs, BM of treated and untreated mice was mixed with methylcellulose with murine growth factors M3434 (Stemcell Technologies, Vancouver, Canada) and processed according to the manufacturer's protocol.
CFU-Fs (MSCs) were isolated by placing bone fragments in stromal cell culture medium and cultured on plastic surfaces coated with 0.1% gelatin as described earlier [49]. Lineage negative cells (2500 total) from BM of NSG mice were isolated by MACS (magnetic cell separation, lineage depletion kit, Miltenyi Biotec, Bergisch Gladbach, Germany) and cultured on confluent MSCs for 7 days.
Flow cytometry
Experiments were performed on a Cyan ADP (Beckman Coulter, Brea, CA, USA). For surface markers used in routine diagnostics (Fig. S2), a Cytomics FC500 flow cytometer was used (Beckman Coulter). The following antibodies were used: human CD3-FITC (clone UCHT1), human CD4-PECy5 (clone 13B8.2), human CD5-PECy7 (clone BC1a), human CD7-PECy5 (clone 8H8.1), human CD8-ECD (clone SFC/Thy2D3), human CD13-PE (clone SJ1D1), human CD14-PE (clone RM052), human CD15-FITC (clone 80H5), human CD30-PE (clone HRS-4), human CD33-PE (clone D3HL60.251), human CD34-FITC (clone 581), human CD56-PE (clone N901(NHK-1)), human CD64-FITC (clone 22), human CD117-PE (clone 104D2D1), human HLADR-ECD (clone Immu357), human MPO-FITC (clone CLB-MPO-1), human TdT-FITC (HT1,HT4,HT4,HT9) from Beckman Coulter; human CXCR4-PE/BV421 (clone 12G5), BV421-isotype, human CD38-APC (clone HB7) from BD Biosciences (Franklin Lakes, NJ, USA), human CD45-eFluor450 (clone HI30), murine CD45-FITC/APC-eFluor780 (clone 30F11), murine CD3-PECy5.5 (clone 145-2C11), murine CD4-PECy5 (clone GK1.5), murine CD8a-PECy5 (clone 53-6.7), murine CD117/cKit-PE (clone 2B8), murine Sca1-PECy7 (clone D7), mB220-PECy7 (clone RA3-6B2), mGr1-PE (clone RB6-8C5), mCD11b-APC-eFluor780 (clone M1/70), murine Ter119-eFluor450 (clone TER-119), murine CD166/ALCAM-PE (clone ALC48), murine CD31-APC (clone 390), murine Lineage (CD3, B220, Gr1, CD11b, Ter119)-biotin from eBioscience (San Diego, CA, USA). Cells were incubated with respective antibodies and buffer (phosphate buffered saline with 0.5% bovine serum albumin) in 4°C in the dark for 15 minutes. Data were analyzed using FlowJo (Ashland, OR, USA).
Immunohistochemistry
Human and mouse tissues were fixed in 10% neutral-buffered formalin solution for maximum 48 h, dehydrated under standard conditions (Leica ASP300S, Wetzlar, Germany) and embedded in paraffin. BM specimen were decalcified in Osteosoft® (Merck Millipore, Darmstadt, Germany). Serial 2 µm thin sections prepared with a rotary microtome (HM355S, ThermoFisher Scientific, Waltham, USA) were collected and subjected to histological and immunohistochemical analysis. Hematoxylin-Eosin (H.-E.) staining was performed on deparaffinized sections with Eosin and Mayer's Haemalaun according to a standard protocol. For immunohistochemistry, slides were deparaffinized in xylene and rehydrated by alcohol washes of decreasing concentration (100%, 96%, 70%). After heat-induced antigen retrieval (target retrieval solution, pH 6 (Dako, Glostrup, Denmark, S1699)), unspecific protein and peroxidase binding was blocked with 3% hydrogen peroxide and 3% normal goat serum (Abcam, Cambridge, UK, 7481). Immunohistochemistry was performed with a Dako autostainer (Dako, Glostrup, Denmark) (CD3, CXCR4) or a Ventana Benchmark XT (Roche, Basel, Switzerland) (CD34) using antibodies against CD3 (clone SP7, DCS, Hamburg, Germany, CI597C01), CD34 (human specimen, (clone QBEnd/10, CellMarque, Darmstadt, Germany) and CXCR4 (clone UMB-2, Abcam, Cambridge, UK, 124824). For antibody detection, the Dako Envision-HRP rabbit labeled polymer (Dako, Glostrup, Denmark) or the UltraView Detection Kit (Roche, Basel, Switzerland) was used. Antibody binding was visualized by diaminobenzidine (DAB) giving a brown precipitate (Medac Diagnostica, Wedel, Germany, BS04-500). Counterstaining was performed using hematoxylin and slides were dehydrated by alcohol washes of increasing concentration (70%, 96%, 100%) and xylene and coverslipped using Pertex® mounting medium (Histolab, Goeteborg, Sweden, 00801).
Mice and patient-derived xenografts
For xenograft experiments, immunocompromised NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ (NSG) mice were purchased from Charles River (Charles River Laboratories Inc., Wilmington, MA, USA) and kept in a pathogen free environment in our animal facility. All experiments were approved by the regional authorities (Regierung von Oberbayern) in compliance with the German law on the protection of animals. Patient-derived xenograft models (PDX) were generated as previously described [50, 51].
Synthesis of Pentixafor and Pentixather
Synthesis of all used radiopharmaceuticals was performed in a fully automated, GMP-compliant procedure using a GRP® module (SCINTOMICS GmbH, Fürstenfeldbruck, Germany) equipped with disposable single-use cassette kits (ABX, Radeberg, Germany), using the method [28, 52] and standardized labeling sequence previously described [53]. Prior to injection, the quality of 68Ga-Pentixafor was assessed according to the standards described in the European Pharmacopoeia for 68Ga-Edotreotide (European Pharmacopoeia; Monograph 01/2013:2482; available at www.edqm.eu).
CXCR4 PET imaging in mice
PET imaging of leukemia-bearing mice was performed as previously described [23]. Briefly, mice were anesthetized with isoflurane and 12 MBq 68Ga-Pentixafor was injected via the tail vein. After 75 min, static images were obtained for 15 min on a µPET-system (Inveon, Siemens, Erlangen, Germany).
Pentixather treatment in vitro
Whole bone marrow from healthy donors was cultured in DMEM (1 g/L Glucose) with 10% pooled human platelet lysate, Heparin (50 U/mL), 2 mM L-Glutamine, 1% Penicillin/Streptomycin to isolate plastic-adherent MSCs for in-vitro Pentixather treatment. Briefly, cells were treated for 10 min, 1 h and 6 h in 12-well plates with 1.5 MBq/mL Lu-P (comparable to 30 MBq distributed in a mouse weighing 20 g) or unlabeled Pentixather. For co-culture, CD34+ cells were isolated with CD34 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) via magnetic separation and ~1.5x104 cells were added to treated/control MSCs. After 4d of co-culture, flow cytometry and colony forming unit assays (StemMACS HSC-CFU complete with EPO, Miltenyi Biotec) were performed.
Pentixather treatment in mice
When leukemia was apparent in peripheral blood (4-5 weeks after injection), mice were subjected to treatment with Lu-P or ctrl (unlabeled Pentixather) via tail vein injection, and then transferred to a radiation-restricted area for surveillance before they were sacrificed and relevant organs were harvested for further analysis. Mean injected radioactivity in ALL230, ALL0, OCI-AML3-eGFP and OCI-AML3-CXCR4 mice was 27.5 MBq, 23.4 MBq, 26.2 MBq and 25.2 MB1, respectively. Sample sizes were as follows: n=4 treated with Lu-P for 3d, n=4 treated for 7d with respective controls for ALL230, n=3 treated for 3d, n=4 treated for 7d with respective controls for ALL0, n=4 treated and n=4 control for AML346, n=3 treated and n=3 control for OCI-AML3-pHIV, n=4 treated and n=4 control for OCI-AML3-CXCR4.
Pentixather-based conditioning therapy in patients
Three patients with relapsed AML after alloSCT were referred for further therapy. Given the lack of alternative treatment options in this advanced disease stage, experimental CXCR4-directed treatment (with additional internal irradiation with 153Sm-EDTMP or 188Re-anti-CD66 antibodies for BM ablation) was offered on a compassionate use basis (German Drug Act, §13,2b) in compliance with §37 of the Declaration of Helsinki. Treatment was approved by the clinical ethics committee of our institution. All subjects gave written informed consent prior to therapy.
A pre-therapy dosimetry study using SPECT/CT and serial planar imaging was performed in patients scheduled for CXCR4-directed ERT after intravenous injection of ~200 MBq of Lu-P. This was done to i) record sites of unexpected tracer accumulation that may denote potential toxicity, ii) determine the organ radiation doses, and iii) to estimate the achievable tumor doses. The absorbed doses in tumors and organs were assessed by analyzing regions of interest in multiple planar total body images to obtain pharmacokinetic data and a single SPECT/CT scan to scale the pharmacokinetic curve.
All images were acquired using dual head gamma cameras (Siemens Symbia E for planar imaging, Siemens Symbia T2 calibrated from phantom measurements with 177Lu activity standards for SPECT/CT acquisition) equipped with medium energy collimators. Pharmacokinetic data were fitted by bi-exponential functions. SPECT/CT data were reconstructed using a 3D-OSEM (6 subsets, 6 iterations, Gauss 6mm) algorithm with corrections for scatter and attenuation to obtain absolute activity quantification in voxels sized 0.11 cm³. Estimates for the absorbed doses from treatment with Y-P were calculated from the 1 mL volumes with highest activity concentrations in dosimetry with Lu-P.
Based on their individual dosimetry, patients were treated by intravenous injection of 90Y- labeled Pentixather. ERT was performed 4 and 7 days after pre-therapy dosimetry, respectively. To prevent renal toxicity, 2 L of a solution containing arginine and lysine (25 g/L each) was co-infused in analogy to the joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy in neuroendocrine tumors [54]. Vital signs, complete blood count, and blood chemistry were documented during the infusion and within 7 days after administration.
In order to enhance treatment effects, 90Y-Pentixather therapy was followed by myeloablation by 153Sm-EDTMP (patient #1) and 188Re-anti-CD66-directed antibodies (patients #2 and #3).
Statistics
Statistics were performed with GraphPad Prism (GraphPad Software, La Jolla, CA). A two-tailed student's T-test was used to determine statistical significance (p value <0.05). Error bars represent mean ± standard error of the mean (SEM). Statistical significance is depicted as follows: * p<0.05, ** p<0.01, *** p<0.001.
Abbreviations
ALL: acute lymphoblastic leukemia; alloSCT: allogeneic hematopoietic stem cell transplantation; AML: acute myeloid leukemia; ATG: antithymocyte globulin; BFU-E: burst-forming unit, erythrocyte; BM: bone marrow; CFU: colony-forming unit; CFU-F: colony-forming unit fibroblast; CR: complete remission; ctrl: control; CXCR4: C-X-C chemokine receptor 4; EDTMP: ethylene diamine tetramethylene phosphonate; EMD: extramedullary disease; ERT: endoradiotherapy; EV: empty-vector control; iso: isotype antibody control; FLAG-IDA: fludarabine, cytarabine, G-CSF, idarubicine; FLAMSA-Bu-Cy-ATG: fludarabine, amsacrine, busulfane, cyclophosphamide, antithymocyte globulin; Flu-Bu: fludarabine, busulfane; G: granulocyte; HE: hematoxylin and eosin; GEMM: granulocyte, erythrocyte, monocyte, megakaryocyte; GM: granulocyte, monocyte; HSPC: hematopoietic stem and progenitor cells; ICE: idarubicine, cytarabine, etoposide; ID: initial diagnosis; ITD: internal tandem duplication; LSK: lineage negative, Sca1 positive, cKit positive stem cell; Lu-P: 177Lu-Pentixather; M: monocyte; MFI: mean fluorescence intensity; MIP: maximum-intensity projection; MP: myeloid progenitor cell; MRD: minimal residual disease; MSC: mesenchymal stem cell; NSG: NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ; OBC: osteoblastic cell; PI: propidium iodide; PB: peripheral blood; PDX: patient-derived xenograft; PET: positron emission tomography; PTD: partial tandem duplication; RR: relapsed/refractory; S-HAM: sequential high-dose cytarabine and mitoxantrone; Sp: spleen; TBI: total body irradiation; Y-P: 90Y-Pentixather.
Supplementary Material
Table S1: Clinical characteristics of patients who provided primary material for PDX. Table S2: Characteristics of patients treated with Pentixather. Fig. S1: PDX establishment. Fig. S2: PDX surface markers. Fig. S3: P-AKT immunoblotting. Fig. S4: NSG control 68Ga-Pentixafor imaging. Fig. S5: Lu-P in AML xenografts with enforced CXCR4 expression. Fig. S6: Lu-P in AML346. Fig. S7: Flow cytometry gating strategy. Fig. S8: Images of cultured stromal cells. Fig. S9: Lu-P treatment of primary human MSCs. Fig. S10: 68Ga-Pentixafor imaging and planar whole-body scintigraphic images in patients 1 and 2.
Acknowledgements
We thank Tanja Weißer, Jolanta Slawska and Sybille Reeder for providing expert technical assistance. We thank Johannes Notni, Alexander Wurzer and Stephanie Robu for synthesis of 68Ga-Pentixafor. We thank the members of the transplant teams at the sites in Würzburg and Munich (TUM) for dedicated clinical care and critical input during the preclinical and clinical work. This work received support from the German Cancer Consortium (DKTK) and the German Cancer Research center (DKFZ).
Author contributions
S.H., C.L., P.H., U. G and U.K. designed the study and wrote the manuscript. C.L., A.K.B., A.S., H.H., and U.G. performed patient dosimetry and ERT. S.H., P.H., M.S., R.I., R.O. performed pre-clinical experiments, S.K. and S.K. performed histology and IHC. B.V. and I.J. established and provided PDX. K.G. provided patient data and interpretation and C.P. provided critical input. H.J.W. provided Pentixather and Pentixafor. All authors interpreted data, critically reviewed and approved the final manuscript.
Funding
U.K., H.J.W., M. Schwaiger, M. Schottelius and R.A.J.O. received support from the Deutsche Forschungsgemeinschaft (DFG; SFB824, FOR2033, KE 222/7-1, OO 8/9-1). U.K. was further supported by Deutsche Krebshilfe (111305, 111944). This work received support from the German Cancer Consortium (DKTK).
Competing Interests
H.J.W. is shareholder of Scintomics (Germany). All other authors have no relevant conflicts of interest to declare.
References
1. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595-9
2. Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T. et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U S A. 1998;95:9448-53
3. Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochim Biophys Acta. 2007;1768:952-63
4. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977-88
5. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-6
6. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927-31
7. Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109:786-91
8. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG. et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219-30
9. Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C. et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19:357-66
10. Cho B-S, Zeng Z, Mu H, Wang Z, McQueen T, Protopopova M. et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood. 2015;126:222-32
11. Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK. et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206-14
12. Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP. et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012;30:4515-23
13. Tsirigotis P, Byrne M, Schmid C, Baron F, Ciceri F, Esteve J. et al. Relapse of AML after hematopoietic stem cell transplantation: methods of monitoring and preventive strategies. A review from the ALWP of the EBMT. Bone Marrow Transplant. 2016;51:1431-8
14. Schultz KR, Pullen DJ, Sather HN, Shuster JJ, Devidas M, Borowitz MJ. et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood. 2007;109:926-35
15. Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011;29:532-43
16. Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC. et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57-66
17. Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14:e205-17
18. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017
19. Parmar A, Marz S, Rushton S, Holzwarth C, Lind K, Kayser S. et al. Stromal niche cells protect early leukemic FLT3-ITD+ progenitor cells against first-generation FLT3 tyrosine kinase inhibitors. Cancer Res. 2011;71:4696-706
20. Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y. et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell. 2015;27:755-68
21. Cashman J, Clark-Lewis I, Eaves A, Eaves C. Stromal-derived factor 1 inhibits the cycling of very primitive human hematopoietic cells in vitro and in NOD/SCID mice. Blood. 2002;99:792-9
22. Philipp-Abbrederis K, Herrmann K, Knop S, Schottelius M, Eiber M, Luckerath K. et al. In vivo molecular imaging of chemokine receptor CXCR4 expression in patients with advanced multiple myeloma. EMBO Mol Med. 2015;7:477-87
23. Herhaus P, Habringer S, Philipp-Abbrederis K, Vag T, Gerngross C, Schottelius M. et al. Targeted positron emission tomography imaging of CXCR4 expression in patients with acute myeloid leukemia. Haematologica. 2016
24. Passaro D, Irigoyen M, Catherinet C, Gachet S, Da Costa De Jesus C, Lasgi C. et al. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell. 2015;27:769-79
25. Herrmann K, Schottelius M, Lapa C, Osl T, Poschenrieder A, Hanscheid H. et al. First-in-Human Experience of CXCR4-Directed Endoradiotherapy with 177Lu- and 90Y-Labeled Pentixather in Advanced-Stage Multiple Myeloma with Extensive Intra- and Extramedullary Disease. J Nucl Med. 2016;57:248-51
26. Schottelius M, Osl T, Poschenrieder A, Hoffmann F, Beykan S, Hanscheid H. et al. [177Lu]pentixather: Comprehensive Preclinical Characterization of a First CXCR4-directed Endoradiotherapeutic Agent. Theranostics. 2017;7:2350-62
27. Wester HJ, Keller U, Schottelius M, Beer A, Philipp-Abbrederis K, Hoffmann F. et al. Disclosing the CXCR4 expression in lymphoproliferative diseases by targeted molecular imaging. Theranostics. 2015;5:618-30
28. Gourni E, Demmer O, Schottelius M, D'Alessandria C, Schulz S, Dijkgraaf I. et al. PET of CXCR4 expression by a (68)Ga-labeled highly specific targeted contrast agent. J Nucl Med. 2011;52:1803-10
29. Nakamura Y, Arai F, Iwasaki H, Hosokawa K, Kobayashi I, Gomei Y. et al. Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood. 2010;116:1422-32
30. Schreck C, Istvanffy R, Ziegenhain C, Sippenauer T, Ruf F, Henkel L. et al. Niche WNT5A regulates the actin cytoskeleton during regeneration of hematopoietic stem cells. J Exp Med. 2017;214:165-81
31. Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I. et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324-36
32. Wynn RF, Hart CA, Corradi-Perini C, O'Neill L, Evans CA, Wraith JE. et al. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood. 2004;104:2643-5
33. Bartlett ML, Webb M, Durrant S, Morton AJ, Allison R, Macfarlane DJ. Dosimetry and toxicity of Quadramet for bone marrow ablation in multiple myeloma and other haematological malignancies. Eur J Nucl Med Mol Imaging. 2002;29:1470-7
34. Dispenzieri A, Wiseman GA, Lacy MQ, Litzow MR, Anderson PM, Gastineau DA. et al. A phase I study of 153Sm-EDTMP with fixed high-dose melphalan as a peripheral blood stem cell conditioning regimen in patients with multiple myeloma. Leukemia. 2005;19:118-25
35. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16:494-507
36. Passaro D, Irigoyen M, Catherinet C, Gachet S, De Jesus CD, Lasgi C. et al. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell. 2015;27:769-79
37. Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O. et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215-24
38. Cho BS, Kim HJ, Konopleva M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J Intern Med. 2017
39. Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1 alpha)-CXCR4/CXCR7 Pathway Inhibition: An Emerging Sensitizer for Anticancer Therapies? Clin Cancer Res. 2011;17:2074-80
40. Scala S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis-Untapped Potential in the Tumor Microenvironment. Clin Cancer Res. 2015;21:4278-85
41. Wattad M, Weber D, Dohner K, Krauter J, Gaidzik VI, Paschka P. et al. Impact of salvage regimens on response and overall survival in acute myeloid leukemia with induction failure. Leukemia. 2017
42. Herrmann K, Lapa C, Wester HJ, Schottelius M, Schiepers C, Eberlein U. et al. Biodistribution and radiation dosimetry for the chemokine receptor CXCR4-targeting probe 68Ga-pentixafor. J Nucl Med. 2015;56:410-6
43. Hill-Kayser CE, Plastaras JP, Tochner Z, Glatstein E. TBI during BM and SCT: review of the past, discussion of the present and consideration of future directions. Bone Marrow Transplant. 2011;46:475-84
44. Pagel JM, Appelbaum FR, Eary JF, Rajendran J, Fisher DR, Gooley T. et al. 131I-anti-CD45 antibody plus busulfan and cyclophosphamide before allogeneic hematopoietic cell transplantation for treatment of acute myeloid leukemia in first remission. Blood. 2006;107:2184-91
45. Bunjes D, Buchmann I, Duncker C, Seitz U, Kotzerke J, Wiesneth M. et al. Rhenium 188-labeled anti-CD66 (a, b, c, e) monoclonal antibody to intensify the conditioning regimen prior to stem cell transplantation for patients with high-risk acute myeloid leukemia or myelodysplastic syndrome: results of a phase I-II study. Blood. 2001;98:565-72
46. Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F. et al. 225Ac-PSMA-617 for PSMA-Targeted alpha-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J Nucl Med. 2016;57:1941-4
47. Sathekge M, Knoesen O, Meckel M, Modiselle M, Vorster M, Marx S. 213Bi-PSMA-617 targeted alpha-radionuclide therapy in metastatic castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging. 2017
48. Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B. et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med. 2017;376:125-35
49. Oostendorp RA, Harvey KN, Kusadasi N, de Bruijn MF, Saris C, Ploemacher RE. et al. Stromal cell lines from mouse aorta-gonads-mesonephros subregions are potent supporters of hematopoietic stem cell activity. Blood. 2002;99:1183-9
50. Vick B, Rothenberg M, Sandhofer N, Carlet M, Finkenzeller C, Krupka C. et al. An advanced preclinical mouse model for acute myeloid leukemia using patients' cells of various genetic subgroups and in vivo bioluminescence imaging. PLoS One. 2015;10:e0120925
51. Ebinger S, Ozdemir EZ, Ziegenhain C, Tiedt S, Castro Alves C, Grunert M. et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell. 2016;30:849-62
52. Demmer O, Gourni E, Schumacher U, Kessler H, Wester HJ. PET Imaging of CXCR4 Receptors in Cancer by a New Optimized Ligand. Chemmedchem. 2011;6:1789-91
53. Martin R, Juttler S, Muller M, Wester HJ. Cationic eluate pretreatment for automated synthesis of [(6)(8)Ga]CPCR4.2. Nuclear medicine and biology. 2014;41:84-9
54. Bodei L, Mueller-Brand J, Baum RP, Pavel ME, Horsch D, O'Dorisio MS. et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. European journal of nuclear medicine and molecular imaging. 2013;40:800-16
Author contact
![]() Corresponding author: Ulrich Keller, Email: ulrich.kellerde Internal Medicine III, Technische Universität München, Ismaningerstraße 22, Munich, Germany Phone: +49-89-4140-7435 Fax: +49-89-4140-4879
Corresponding author: Ulrich Keller, Email: ulrich.kellerde Internal Medicine III, Technische Universität München, Ismaningerstraße 22, Munich, Germany Phone: +49-89-4140-7435 Fax: +49-89-4140-4879
Citation styles
APA
Habringer, S., Lapa, C., Herhaus, P., Schottelius, M., Istvanffy, R., Steiger, K., Slotta-Huspenina, J., Schirbel, A., Hänscheid, H., Kircher, S., Buck, A.K., Götze, K., Vick, B., Jeremias, I., Schwaiger, M., Peschel, C., Oostendorp, R., Wester, H.J., Grigoleit, G.U., Keller, U. (2018). Dual Targeting of Acute Leukemia and Supporting Niche by CXCR4-Directed Theranostics. Theranostics, 8(2), 369-383. https://doi.org/10.7150/thno.21397.
ACS
Habringer, S.; Lapa, C.; Herhaus, P.; Schottelius, M.; Istvanffy, R.; Steiger, K.; Slotta-Huspenina, J.; Schirbel, A.; Hänscheid, H.; Kircher, S.; Buck, A.K.; Götze, K.; Vick, B.; Jeremias, I.; Schwaiger, M.; Peschel, C.; Oostendorp, R.; Wester, H.J.; Grigoleit, G.U.; Keller, U. Dual Targeting of Acute Leukemia and Supporting Niche by CXCR4-Directed Theranostics. Theranostics 2018, 8 (2), 369-383. DOI: 10.7150/thno.21397.
NLM
Habringer S, Lapa C, Herhaus P, Schottelius M, Istvanffy R, Steiger K, Slotta-Huspenina J, Schirbel A, Hänscheid H, Kircher S, Buck AK, Götze K, Vick B, Jeremias I, Schwaiger M, Peschel C, Oostendorp R, Wester HJ, Grigoleit GU, Keller U. Dual Targeting of Acute Leukemia and Supporting Niche by CXCR4-Directed Theranostics. Theranostics 2018; 8(2):369-383. doi:10.7150/thno.21397. https://www.thno.org/v08p0369.htm
CSE
Habringer S, Lapa C, Herhaus P, Schottelius M, Istvanffy R, Steiger K, Slotta-Huspenina J, Schirbel A, Hänscheid H, Kircher S, Buck AK, Götze K, Vick B, Jeremias I, Schwaiger M, Peschel C, Oostendorp R, Wester HJ, Grigoleit GU, Keller U. 2018. Dual Targeting of Acute Leukemia and Supporting Niche by CXCR4-Directed Theranostics. Theranostics. 8(2):369-383.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.