Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion and Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(1):223-236. doi:10.7150/thno.21691 This issue Cite
Research Paper
An Albumin-binding Polypeptide Both Targets Cytotoxic T Lymphocyte Vaccines to Lymph Nodes and Boosts Vaccine Presentation by Dendritic Cells
Peng Wang1, Peng Zhao1, Shuyun Dong1, Tiefeng Xu2, Xiao He3, Mingnan Chen1 ![]()
1. Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, UT 84112, USA;
2. The First Affiliated Hospital and The Oncological Institute, Hainan Medical University, Haikou City, Hainan Province 570102, China;
3. Department of Pathology, University of Utah, Salt Lake City, UT 84112, USA.
Received 2017-6-29; Accepted 2017-9-23; Published 2018-1-1
Abstract

Rationale: Albumin-binding carriers have been shown to target cytotoxic T lymphocyte (CTL) vaccines to lymph nodes (LNs) and improve the efficacy of the vaccines. However, it was not clear whether the improved efficacy is solely due to the LN targeting, which prompted this study.
Methods: First, we generated a fusion protein consisting of an albumin-binding domain (ABD) and an immune-tolerant elastin-like polypeptide (iTEP). Then, we examined the binding between this fusion protein, termed ABD-iTEP, and mouse serum albumin (MSA). Next, we evaluated the accumulation of ABD-iTEP in LNs and dendritic cells (DCs) in the LNs. We also analyzed antigen presentation and in vitro T cell activation of vaccines that were delivered by ABD-iTEP and investigated possible underlying mechanisms of the presentation and activation results. Last, we measured CTL responses induced by ABD-iTEP-delivered vaccines in vivo.
Results: ABD-iTEP bound with MSA strongly with an affinity of 1.41 nM. This albumin-binding carrier, ABD-iTEP, accumulated in LNs 3-fold more than iTEP, a control carrier that did not bind with albumin. ABD-iTEP also resulted in 4-fold more accumulation in DCs in the LNs than iTEP. Most importantly, ABD-iTEP drastically enhanced the antigen presentation of its vaccine payloads and the T cell activation induced by its payloads. The enhancement was dependent on the formation of the complex between MSA and ABD-iTEP. Meanwhile, the MSA/ABD-iTEP complex was found to have increased stability in acidic subcellular compartments and increased cytosolic accumulation in DCs, which might explain the enhanced vaccine presentation resulting from the complex. Finally, when ABD-iTEP was used to deliver CTL vaccines derived from both self- and non-self-antigens, it boosted the vaccine-induced responses by 2-fold in either case.
Conclusion: ABD-iTEP not only targets vaccines to LNs but also promotes the presentation of the vaccines by DCs. Albumin-binding carriers have more than one mechanism to boost the efficacy of CTL vaccines.
Keywords: Albumin-binding domain, Cytotoxic T lymphocyte response, Lymph node targeting, Antigen presentation, Cytosolic accumulation.
Introduction
Vaccines that elicit cytotoxic T lymphocyte (CTL) responses are being developed as prophylactic and therapeutic modules to combat intracellular pathogens and cancers [1, 2]. One promising strategy to enhance the effectiveness of CTL vaccines is to target them to lymph nodes (LNs) [3-5] because LNs are abundant with dendritic cells (DCs) and DCs are required to present these vaccines to CD8+ T lymphocytes and hence to initiate CTL responses [6-8]. Further, in regard to cancer vaccines, there is an extra benefit to deliver the vaccines to LNs and enhance CTL responses there because tumors often metastasize through LNs [9]. Boosting CTL responses in LNs may be an effective approach to halt metastasis [4, 5, 10].
Among different strategies to target LNs [11-14], one takes advantage of the intrinsically high LN accumulation of serum albumin [13]. This strategy is appealing because the high LN accumulation of albumin is transferable to those molecules that bind with albumin [14]. To date, albumin-binding molecules have been used for LN targeting of imaging and therapeutic agents [15-18]. In addition, an albumin-binding lipid was used to target vaccines to LNs and enhanced CTL responses [19].
While it is clear that albumin-binding carriers are able to boost CTL responses induced by their delivered vaccines and that the boost can be attributed to the increased vaccine accumulation in LNs resulting from the carriers, it is actually not clear at all whether the boost is solely contributed by LN targeting. Whether the complex between albumin-binding carriers and albumin contribute to CTL responses through mechanisms other than LN-targeting was never reported. The speculation of such mechanisms was inspired by two discoveries: first, IgG enhances the DC-mediated presentation of the antigens that are delivered by IgG [20]; second, the enhancement is mediated by a neonatal Fc receptor (FcRn) that IgG binds with [20-22]. These two discoveries are related to albumin-binding carriers because albumin, like IgG, is a ligand of the FcRn [23]. Thus, it would be interesting to investigate whether albumin-binding carriers boost their vaccine payloads through promoting the presentation of vaccines.
To investigate interplays between albumin or albumin-binding carriers with the immune system, we designed a single polypeptide-based vaccine carrier, termed ABD-iTEP, that consists of an albumin-binding domain (ABD) and an immune-tolerant elastin-like polypeptide (iTEP). ABD is a 46-residue protein domain derived from protein G [24]. ABD has very high binding affinities with albumin of humans, monkeys, and mice and has proven biocompatibility [25-27]. iTEPs are peptide polymers with proven immuno-compatibility and reversible phase-transition property [28]. We used iTEP in this carrier for the following four reasons: (1) We used iTEP to link ABD and CTL vaccines together but with a distance so that ABD and the vaccines will be delivered to the same cell yet the vaccines will not interfere with the functions of ABD. (2) ABD, iTEP and CTL vaccines are all proteins and peptides in nature so we can generate a recombinant fusion protein consisting of all three of them just like other iTEP fusion proteins we have generated before [29], which offers us a simple method to produce highly reproducible vaccine carriers for this study. (3) Fusion proteins that contain iTEP can be purified by cycling the salt-induced, reversible phase transition [28]. Such easy purification method would facilitate the production of the carrier for this study. (4) The particular iTEP used in this study is hydrophilic and able to solubilize hydrophobic vaccines, thus avoiding the solubility problem that was associated with the previous albumin-binding vaccine carrier [19].
In this study, we found that ABD-iTEP had a high affinity with mouse serum albumin (MSA). Through the binding with MSA, ABD-iTEP showed an improved LN accumulation and DC accumulation as compared to iTEP. In addition, the association between ABD-iTEP and MSA promoted the antigen presentation of the vaccine delivered by ABD-iTEP. Meanwhile, the complex between MSA and ABD-iTEP had an increased cytosolic accumulation and an increased stability in acid intracellular compartments, which may facilitate antigen presentation. And finally, ABD-iTEP was able to enhance CTL responses of both non-self- and self-antigens.
Materials and Methods
Animals and cell lines
Female C57BL/6 mice, 4-8 weeks of age, were bred and treated in accordance with an approved protocol by the University of Utah Institutional Animal Care and Use Committee (IACUC). DC2.4 cell line (H-2Kb) was kindly provided by Dr. Kenneth Rock (University of Massachusetts). DC2.4 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1% nonessential amino acids, 10 mM HEPES, 50 µM 2-mercaptoethanol, 100 U/mL penicillin and 100 µg/mL streptomycin (Life Technologies). The B3Z T cell hybridoma specific for H-2Kb/SIINFEKL complex was kindly provided by Dr. Nilabh Shastri (University of California, Berkeley). B3Z cells were cultured with RPMI 1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 50 µM 2-mercaptoethanol, 100 U/mL penicillin and 100 µg/mL streptomycin. All cells were cultured in 37 °C with 5% CO2.
Preparation of recombinant polypeptides
Genes encoding iTEP, ABD, pOVA, and pTRP2 were synthesized (Eurofins Genomics) and inserted into the modified pET25b (+) vector using a previously described method [28]. The inserted genes were verified by DNA sequencing (Genewiz). The expression and purification of those recombinant polypeptides were conducted as previously reported [30].
Removal of endotoxin
The endotoxin present in the recombinant polypeptides was removed by the reported method [31]. The amount of residual endotoxin in each sample was determined by Limulus Amebocyte Lysate (LAL) single-test vials (Charles River Laboratories). All polypeptides used for in vitro and in vivo immune assays had an endotoxin level < 0.25 EU/mg.
Native polyacrylamide gel electrophoresis (PAGE)
20 µM MSA (Sigma-Aldrich) was incubated with ABD-iTEP of different concentrations including 10, 20, 40 and 80 µM overnight at 4 °C. 5 µL of each sample was mixed with 5 µL sample buffer and loaded into each well of the gel. The electrophoresis was run at 150 V for 2 h. The gel was then stained with Coomassie Brilliant Blue R-250 dye. The image of the gel was taken using the FluorChem FC2 imaging system (Alpha Innotech).
Size exclusion chromatography (SEC)
SEC was conducted on an Agilent 1260 infinity liquid chromatography system (Agilent Technologies) equipped with an Agilent ZORBAX GF-450 size exclusion column (diameter: 9.4 mm, length: 250 mm, particle diameter: 6.0 µm). 60 µM ABD-iTEP was incubated with 30 µM MSA overnight at 4 °C. 100 µL sample was loaded for analysis. Samples were eluted with PBS at a flow rate of 1 mL/min. Spectra were monitored by absorption at 280 nm.
Surface plasmon resonance (SPR) assay
The SPR measurement of binding affinities were measured by a MASS-1 SPR instrument (Sierra Sensors). Following a standard amine-coupling procedure, MSA was immobilized on a SPR affinity sensor (High Capacity Amine, Sierra Sensors). iTEP, ABD-iTEP, iTEP-pOVA, and ABD-iTEP-pOVA were diluted in running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% Tween-20 and 0.2 mg/mL ovalbumin) to 10 nM. The samples were injected at a constant flow rate of 25 µL/min for 3 min, followed by injection of running buffer for 30 min. After each injection cycle, the surface was regenerated with two injections of 25 µL of 10 mM HCl. After subtracting reference surface and buffer injection, the data were fitted to the one-to-one Langmuir binding model with mass transport limitations to calculate the association rate constant (ka), the dissociation rate constant (kd), and the overall affinity (KD), using the MASS-1 analysis software (Sierra Sensors).
LN accumulation, biodistribution and pharmacokinetic study
ABD-iTEP and iTEP were labelled with Alexa Fluor 488 sulfodichlorophenol ester (Life Technologies) following the manufacturer's protocol. Mice were subcutaneously injected with 1.5 nmol of ABD-iTEP or iTEP at each side of their tail bases. Draining LNs (inguinal and axillary LNs), kidney, liver, lung, spleen and blood of the mice were collected at 6, 12, 24 and 48 h after injection. The blood was left at room temperature for 30 min before centrifuging to collect the serum. The organs were then homogenized in cold PBS and sonicated using a model 500 sonic dismembrator (Fisher Scientific). Samples were centrifuged, and the supernatant was collected to measure the fluorescence intensity (excitation 494 nm, emission 518 nm) using an Infinite M1000 pro microplate reader (Tecan). The quantity of iTEP or ABD-iTEP in the tissues was determined by referencing the standard curves of fluorescence intensity of ABD-iTEP and iTEP. One compartment model for extravascular administration was used to calculate the pharmacokinetic metrics of ABD-iTEP and iTEP.
In vivo dendritic cell accumulation assay
C57BL/6 mice were subcutaneously injected with 1.5 nmol of Alexa Fluor 488 labelled iTEP or ABD-iTEP at each side of the base of tail. Inguinal and axillary LNs were isolated 6 h after injection. LNs were pooled and mechanically homogenized to prepare a single-cell suspension by passing through a 40 µm nylon mesh. Cells were then washed and stained with PE anti-mouse CD11c (Clone: N418, Biolegend) and 4′,6-diamidino-2-phenylindole (DAPI). Flow cytometric analysis was conducted using a BD Canto (BD Biosciences). The results were analyzed by FlowJo software.
In vitro dendritic cell uptake assay
This assay was conducted using a previous method with some modifications [32]. DC2.4 cells were cultured with medium containing 5 µM fluorescein-labelled polypeptides for 2 h. The medium was then removed and cells were washed. Trypan blue was added to quench extracellular fluorescence and cells were washed another two times. 0.2% Triton X-100 was added to lyse the cells and release the endocytosed polypeptides. The amount of endocytosed polypeptide was detected by measuring the fluorescence intensity.
Antigen presentation assay
DC2.4 cells were cultured in medium containing vaccines at a concentration of 5 µM for 16 h. The cells were then collected and stained with PE anti-mouse H-2Kb/SIINFEKL antibody (Clone: 25-D1.16, Biolegend) and DAPI. SIINFEKL (pOVA) presented on the cellular surface was quantified by flow cytometric analysis. The results are presented as median fluorescence intensity (MFI) relative to the MFI of untreated DC2.4 cells.
B3Z cell activation assay
B3Z cell is a CD8+ T-cell hybridoma [33]. Upon recognition of H-2Kb/SIINFEKL complex, B3Z cells will be activated to produce β-galactosidase, which can hydrolyze the substrate into red products. The level of activation of the CD8+ T cells will be reflected by the color of the solution. To do this assay, DC2.4 cells were cultured with medium containing vaccines at a concentration of 5 µM for 16 h. The cells were subsequently co-cultured with B3Z cells for 24 h. Then the cells were lysed for 4 h at 37 °C with lysis buffer (PBS with 100 mM 2-mercaptoethanol, 9 mM MgCl2, 0.2% Triton X-100 and 0.15 mM chlorophenol red-β-D-galactopyranoside). The reaction was stopped by adding the stop buffer (1 M sodium carbonate). CD8+ T cell activation were quantified by measuring the absorbance at 570 nm with 635 nm as a reference wavelength. The B3Z cell activation is shown as the normalized optical density (OD) relative to the control group.
Preparation of MSA/ABD-iTEP-pOVA complex
MSA was incubated with ABD-iTEP-pOVA overnight at 4 °C. Then MSA/ABD-iTEP-pOVA was purified by ÄKTA fast protein liquid chromatography system (GE Healthcare) equipped with a HiPrep Sephacryl S-200 HR column (diameter: 26 mm, length: 600 mm, particle diameter: 50 µm, GE Healthcare). Samples were eluted with PBS at a flow rate of 1 mL/min. The fractions containing the MSA/ABD-iTEP-pOVA complex were combined and concentrated by Vivaspin spin columns (Molecular mass cut-off: 10,000 kDa, GE Healthcare).
Subcellular accumulation study
DC2.4 cells were cultured with medium containing fluorescein-labelled ABD-iTEP-pOVA or the MSA/ABD-iTEP-pOVA complex at a concentration of 5 µM for 2 h. The cells were then washed and traced for various time as indicated in the figure. At the end of each traced time, cells were collected for subcellular fractionation by the method described previously [34] to obtain the cytosolic and vesicular fractions. The amount of ABD-iTEP-pOVA and MSA/ABD-iTEP-pOVA in these subcellular compartments were quantified by measuring the fluorescence intensity of the fractions.
Fluorescence microscopy
Poly-lysine-treated coverslips were added into wells of a 24-well plate. DC2.4 cells were incubated with medium containing either the fluorescein-labelled ABD-iTEP-pOVA or the MSA/ABD-iTEP-pOVA complex at a concentration of 15 µM for 2 h. The cells were then washed and chased for 2 h. After that, the cells were stained with LysoTracker Red DND-99 (Life Technologies). Next, the cells were fixed with 4% paraformaldehyde and stained with DAPI. The coverslips were mounted with Fluoromount-G (SouthernBiotech). Fluorescence images of the DCs were captured under Nikon Eclipse Ti microscope with 40× magnification at room temperature and analyzed by Coloc 2 plugin in Fiji software. The proportion of samples that were co-localized with vesicles including lysosomes and endosomes was determined using Manders' overlap coefficient [35].
Animal vaccination and enzyme-linked immunospot (ELISPOT) assay
C57BL/6 mice were subcutaneously injected with 1.5 nmol vaccines at each side of the tail base on day 0 and day 7. The mice were sacrificed on day 17 and their splenocytes and LN cells were harvested to perform the IFN-γ ELISPOT assay following a previous method [28]. Generally, splenocytes were stimulated with medium containing CTL epitopes, SIINFEKL (pOVA) or SVYDFFVWL (pTRP2) for 48 h. Cells were then transferred to a filter plate (Millipore) coated with anti-mouse IFN-γ antibody (Clone: R4-6A2, Biolegend) and cultured for 24 h. The secreted IFN-γ was detected by orderly adding biotinylated anti-mouse IFN-γ antibody (Clone: XMG1.2, Biolegend), horseradish peroxidase avidin (Avidin-HRP), and 3-amino-9-ethylcarbazole (AEC). The spots on the membranes were scanned and counted by Fiji software [35]. According to the assay design, only CD8+ T cells with T cell receptors that match the used epitopes are stimulated by the epitopes and release IFN-γ.
Statistical analysis
A two-tailed, unpaired Student's t test and one-way analysis of variance (ANOVA) with the Bonferroni post-test were used to analyze the data. P < 0.05 was considered a significant difference.
Results
ABD-iTEP binds MSA with nanomolar affinity
ABD-iTEP and its control iTEP were designed and produced as recombinant proteins using our previously developed method [28, 29]. The sequences of ABD-iTEP and iTEP are shown in Table 1. ABD-iTEP and iTEP have theoretical molecular mass of 29.7 kDa and 24.6 kDa, respectively. After we generated ABD-iTEP, we first examined the binding between ABD-iTEP and MSA through native polyacrylamide gel electrophoresis (PAGE). There was a clear association between the two proteins, evidenced as a new band of the MSA/ABD-iTEP complex on the native gel (Fig. 1A). We observed that the intensity of the new band increased along with an increase in the ratio of ABD-iTEP to MSA. Meanwhile, the intensity of the MSA band decreased with an increase in the ratio. The MSA band disappeared completely when the ratio reached 4:1, suggesting that MSA was consumed by complexing with ABD-iTEP. We noticed that ABD-iTEP couldn't be stained by Coomassie Brilliant Blue R-250 in the native gel (Fig. 1A-lane 1). This is likely due to the scarcity of positively charged and hydrophobic residues in ABD-iTEP (11 among all 268 residues, Table 1). Coomassie dye molecules bind to proteins through positively charged and hydrophobic residues in the proteins [36]. However, the MSA/ABD-iTEP complex can be stained because of MSA. Together, these results suggested that ABD-iTEP bound MSA. This conclusion was affirmed by the result of the size exclusion chromatography (SEC) analysis of the mixture of ABD-iTEP and MSA. On a chromatogram recorded at 280 nm absorbance (Fig. 1B), MSA emerged as a peak at 10.04 min; ABD-iTEP did not have a peak at the injected amount (6.0 nmol) because of its low extinction coefficient (2980 M-1 cm-1) at 280 nm; the mixture of ABD-iTEP and MSA resulted in a new peak at 9.16 min, presumably the MSA/ABD-iTEP complex.
Names and sequences of polypeptides used in current study
| Polypeptides | Sequences (from N- to C-terminus) | Molecular mass (kDa) |
|---|---|---|
| iTEP a | (GVGVPG)35-(GVLPGVG)16 | 24.6 |
| ABD-iTEP b | LAEAKVLANRELDKYGVSDFYKRLINKAKTVEGVEALKLHILAALP-(GVGVPG)35-(GVLPGVG)16 | 29.7 |
| iTEP-pOVA c | (GVGVPG)35-(GVLPGVG)16-GSIINFEKL | 25.6 |
| ABD-iTEP-pOVA | LAEAKVLANRELDKYGVSDFYKRLINKAKTVEGVEALKLHILAALP-(GVGVPG)35-(GVLPGVG)16-GSIINFEKL | 30.7 |
| iTEP-pTRP2 c | (GVGVPG)35-(GVLPGVG)16-GSVYDFFVWL | 25.9 |
| ABD-iTEP-pTRP2 | LAEAKVLANRELDKYGVSDFYKRLINKAKTVEGVEALKLHILAALP-(GVGVPG)35-(GVLPGVG)16-GSVYDFFVWL | 30.9 |
a The number in the subscript represents the repeating number of the sequences in parentheses.
b ABD was fused at the N-terminus of iTEP. The letters in bold represent positively charged or hydrophobic residues.
c pOVA and pTRP2 were fused at the C-terminus of iTEP. A glycine (G) was inserted between the iTEP and the epitopes to facilitate cleavage of the epitopes from iTEP.
Association between ABD-iTEP and MSA. (A) The image of a native PAGE gel showed the binding between ABD-iTEP and MSA. ABD-iTEP was incubated with MSA at different ratios as listed in the lanes 2-6. Coomassie Brilliant Blue R-250 can stain the MSA/ABD-iTEP complex in the lanes 3-6 but not ABD-iTEP alone in lane 1. Other lightly stained-bands represent impurities in MSA samples. (B) An overlay of SEC chromatographs of ABD-iTEP, MSA, and the mixture of ABD-iTEP and MSA. The mixture was incubated overnight before analysis. ABD-iTEP had no apparent absorbance at 280 nm. (C) Representative SPR sensorgrams of ABD-iTEP and iTEP after they were flowed separately over a chip surface immobilized with MSA.
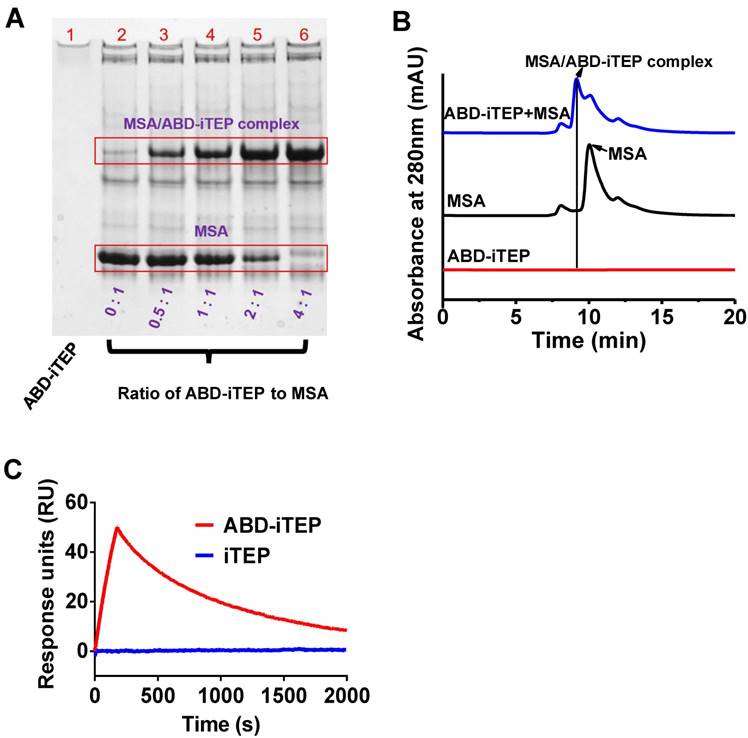
We quantified the binding affinity between ABD-iTEP and MSA by using the surface plasmon resonance (SPR) method (Fig. 1C). According to binding kinetic data, the association rate constant (ka) between the two proteins was calculated as 2.17±0.07 ×106 M-1s-1; the dissociation rate constant (kd) was 3.05±0.01 ×10-3 s-1. The dissociation constant (KD) between ABD-iTEP and MSA was calculated as 1.41±0.04 nM, which is similar to the reported KD value between ABD and MSA (1.24±0.01 nM) [25]. The similar KD values suggested that fusing iTEP to ABD did not interfere with the interaction between ABD and MSA. As a control, iTEP did not bind with MSA (Fig. 1C).
ABD increases the LN accumulation of ABD-iTEP
We compared the accumulation of ABD-iTEP and iTEP in draining LNs (inguinal and axillary LNs) at 6, 12, 24, and 48 h post injection of the samples at the tail base of mice. Both ABD-iTEP and iTEP reached their peak accumulation in LN at 6 h after injection, 1.31 ± 0.11% and 0.36 ± 0.02% (P < 0.01) of injected doses (ID), respectively (Fig. 2A). At all of the time points, the LN accumulation of ABD-iTEP was at least 3.5-fold higher than that of iTEP. More strikingly, ABD-iTEP accumulated more in LNs than in those organs where macromolecules typically accumulate: liver, kidney, spleen and lung [37, 38]. The LN accumulation of ABD-iTEP per tissue mass was 4- to 10-fold higher than that of kidneys across all observed time points (Fig. 2B). The superior LN accumulation of ABD-iTEP was even more prominent when the accumulation was compared to the accumulation in liver, spleen, and lungs (Fig. 2B). In sharp contrast, iTEP had the highest accumulation in kidneys (Fig. 2C). The higher renal accumulation of iTEP (42.20 %ID/g at 12 h post injection) than ABD-iTEP (22.99 %ID/g at 12 h post injection) may be attributed to the factor of molecular weights (MW) of polypeptides and the renal filtration threshold. The renal filtration threshold for proteins is ~70 kDa: those proteins with MW >70 kDa have significantly decreased clearance from blood to the glomerular capsule of the kidney [39]. iTEP has a MW of 24.6 kDa, whereas ABD-iTEP, when complexed with MSA, has a MW of 98.45 kDa. Thus, it is easier for iTEP than the MSA/ABD-iTEP complex to accumulate in the glomerular capsule of the kidney, which explains why iTEP apparently has a higher renal accumulation than ABD-iTEP.
The biodistribution and pharmacokinetics of ABD-iTEP. (A) LN accumulation of ABD-iTEP and iTEP. The data are expressed as percentage of injected dose, %ID, (N=3). Biodistribution of (B) ABD-iTEP and (C) iTEP in various organs. The data were expressed as percentage of injected dose per gram of tissue mass, %ID/g, (N=3). (D) The percentage of DCs in LNs that internalized iTEP or ABD-iTEP. DCs that internalized iTEP or ABD-iTEP (Alexa Fluor 488 positive) were quantified by flow cytometry, (N=3). (E) In vitro uptake of iTEP, ABD-iTEP and MSA/ABD-iTEP by DCs. DC2.4 cells were incubated with iTEP, ABD-iTEP or MSA/ABD-iTEP for 2 h before the analysis. The experiment was performed in triplicate. (F) The serum concentration of ABD-iTEP and iTEP, (N=3). All data in this figure are shown as mean ± standard error of the mean (SEM). Data in (D) was analyzed by Student's t test, data in (E) was analyzed by one-way ANOVA with Bonferroni post-test, **P < 0.01, NS = not significant.
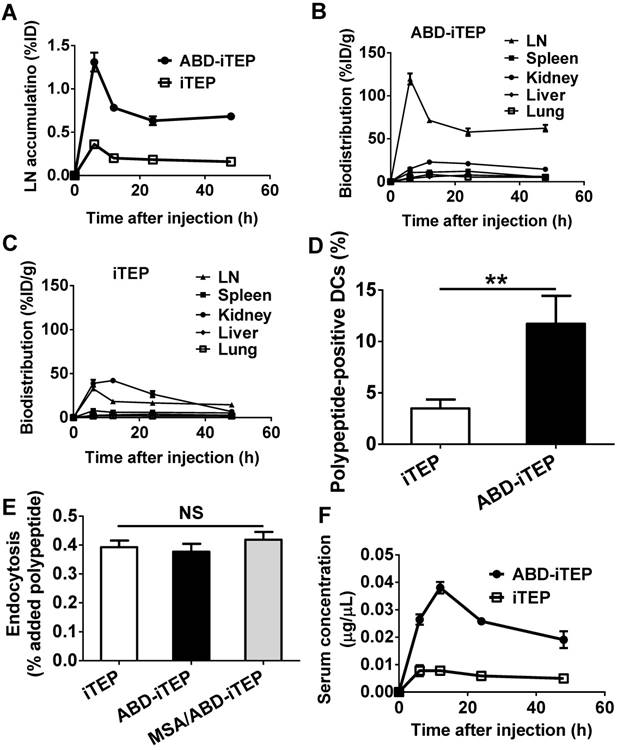
We next investigated whether the increased LN accumulation of ABD-iTEP led to an increased accumulation of ABD-iTEP by DCs in LNs. The accumulation of ABD-iTEP in the DCs is critical for the vaccine delivery function of ABD-iTEP. We collected and analyzed DCs from draining LNs of the ABD-iTEP- and iTEP-treated mice and found that the ABD-iTEP-positive fraction among all collected DCs from the ABD-iTEP-treated mice was 13.1 ± 1.4%, which was significantly greater than the iTEP-positive fraction from the iTEP-treated mice, 3.1 ± 0.5% (P < 0.05, Fig. 2D). Therefore, DCs in draining LNs accumulate more ABD-iTEP than iTEP. The greater accumulation of ABD-iTEP was not likely due to DCs preferentially internalizing ABD-iTEP or its complex with MSA. Indeed, DCs internalized ABD-iTEP, iTEP, and MSA/ABD-iTEP equally in vitro (Fig. 2E). Thus, the greater accumulation of ABD-iTEP by DCs in LNs is likely due to the increased accumulation of ABD-iTEP in LNs and the consequently increased exposure of ABD-iTEP to the DCs.
We also analyzed pharmacokinetics (PK) of ABD-iTEP and iTEP based on serum concentration changes of the two samples after administration (Fig. 2F). We compared the PK by five metrics, AUC (area under the curve), Cmax (the peak serum concentration after administration), Tmax (the time to reach Cmax), t1/2(abs) (absorption half-life), and t1/2(elim) (elimination half-life) (Table S1). The two polypeptides are different in AUC, Cmax, and t1/2(elim): the AUC of ABD-iTEP is ~4-fold higher than that of iTEP (1.20 µg·h/µL versus 0.29 µg·h/µL); the Cmax of ABD-iTEP is also ~4-fold higher than that of iTEP (0.0328 µg/µL versus 0.0083 µg/µL); ABD-iTEP has a longer t1/2(elim) than iTEP (51.6 h versus 24.1 h). In contrast, ABD-iTEP and iTEP have similar Tmax (12.4 h versus 12.8 h) and t1/2(abs) (2.8 h versus 4.2 h). ABD-iTEP is expected to have longer t1/2(elim) than iTEP because albumin-binding molecules have long t1/2(elim) [25, 27, 40]. Consistently, the long half-life of ABD-iTEP leads to the greater AUC and Cmax of ABD-iTEP. The similar Tmax and t1/2(abs) of ABD-iTEP and iTEP suggest that ABD doesn't affect the absorption of ABD-iTEP. The greater AUC of ABD-iTEP is advantageous for its vaccine delivery function as this allows ABD-iTEP to deliver more vaccine payloads to spleen and other lymphatic organs [41]. Putting together these results, we concluded that ABD increases the LN accumulation and plasma half-life of ABD-iTEP and boosts the accumulation of ABD-iTEP in DCs in LNs.
ABD-iTEP promotes the antigen presentation of its vaccine payload through complexing with MSA
To examine the effectiveness of ABD-iTEP as a vaccine carrier, we generated a fusion polypeptide consisting of ABD-iTEP and pOVA (sequence: SIINFEKL), a CTL epitope derived from chicken ovalbumin (Table 1). pOVA was fused to the C-terminus of ABD-iTEP. The fusion was termed ABD-iTEP-pOVA. ABD-iTEP-pOVA had a similar binding affinity with MSA as ABD-iTEP (KD =1.4±0.1 nM, Fig. 3A). We also generated a control fusion, iTEP-pOVA.
The MSA/ABD-iTEP-pOVA complex enhanced T cell activation and antigen presentation. (A) Representative SPR sensorgram showed the binding between ABD-iTEP-pOVA and MSA. (B) ABD-iTEP-pOVA and iTEP-pOVA induced comparable T cell activation in vitro as determined by a B3Z cell activation assay, while the carrier only, ABD-iTEP, didn't induce any T cell activation. The optical density (OD) at 570 nm was used as an indicator of activation levels of B3Z cells. (C) The MSA/ABD-iTEP-pOVA complex induced greater T cell activation than free ABD-iTEP-pOVA according to the B3Z activation assay. (D) The MSA/ABD-iTEP-pOVA complex led to greater pOVA presentation by DCs than free ABD-iTEP-pOVA. An antibody that recognized pOVA/MHC class I complex on the DC surface was used to determine the presentation of pOVA on the DC surface. The results are shown as median fluorescence intensity (MFI) relative to the MFI of untreated DC2.4 cells. Experiments in this figure were repeated in triplicate. Data are shown as mean ± SEM and analyzed by one-way ANOVA with Bonferroni post-test, ****P < 0.0001. NS = not significant.
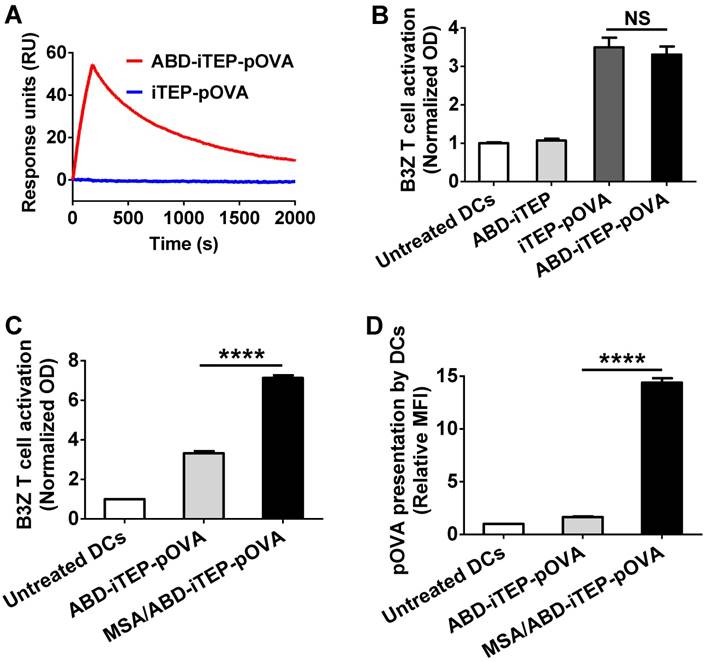
Using ABD-iTEP-pOVA and iTEP-pOVA, we first examined whether the carrier ABD-iTEP, without forming a complex with MSA, enhanced immune responses toward its vaccine payload, pOVA. We used the B3Z cell-based in vitro T cell activation assay to measure the immune responses because pOVA, when presented by DCs, activates B3Z cells. B3Z is a CD8+ T cell clone that is restricted to pOVA. We found that ABD-iTEP-pOVA and iTEP-pOVA, after being incubated and presented by DCs, were equally effective in activating B3Z cells (Fig. 3B). The data suggested that ABD-iTEP, alone, was not different to iTEP in enhancing immune responses toward its vaccine payload. The empty carrier, ABD-iTEP, didn't activate any B3Z cells as compared to the untreated B3Z cells (Fig. 3B).
We next investigated whether the carrier ABD-iTEP would enhance pOVA-induced immune responses differently when it was and was not associated with MSA. This is an unanswered yet important question for albumin-binding carriers because the majority of administered carriers likely complex with MSA before they interact with DCs. To answer this question, we compared the purified MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA using the aforementioned B3Z cell activation assay. MSA/ABD-iTEP-pOVA was found to be 2-fold more potent than free ABD-iTEP-pOVA in activating B3Z cells (Fig. 3C). The superior activation could have two possible causes: the presence of the MSA/ABD-iTEP-pOVA complex and the mere presence of MSA in the assay. To investigate which cause is true, we compared iTEP-pOVA with the mixture of MSA and iTEP-pOVA using the B3Z activation assay. We found that the two samples have the same capacity to activate B3Z cells (Fig. S1). Thus, the mere presence of MSA does not improve B3Z activation results of the MSA/ABD-iTEP-pOVA complex. Thus, the superior activation of the complex is due to interactions between MSA and ABD-iTEP-pOVA.
We further explored how the MSA/ABD-iTEP-pOVA complex was able to lead to stronger B3Z activation than free ABD-iTEP-pOVA. We suspected that the MSA/ABD-iTEP-pOVA complex led to greater pOVA presentation by DCs than ABD-iTEP-pOVA because greater epitope presentation leads to stronger B3Z cell activation. Thus, we investigated pOVA presentation by DCs after we incubated the DCs with the MSA/ABD-iTEP-pOVA complex or free ABD-iTEP-pOVA. We examined the presentation by using an antibody that specifically binds with pOVA presented with MHC class I molecules on the DC surface and a flow cytometry assay. Indeed, we found that the MSA/ABD-iTEP-pOVA complex led to a 5-fold greater pOVA presentation than free ABD-iTEP-pOVA, as indicated by the 5-fold higher relative median fluorescence intensity (MFI) from the DCs incubated with the MSA/ABD-iTEP-pOVA complex than the DCs incubated with free ABD-iTEP-pOVA (Fig. 3D).
In summary, the MSA/ABD-iTEP-pOVA complex is advantageous not only for LN targeting but also for CD8+ T cell activation. Further, the superior T cell activation resulting from the complex can be attributed to the increased presentation of vaccines that are delivered by the complex.
The MSA/ABD-iTEP-pOVA complex has increased cytosolic accumulation and endosomal/lysosomal stability
Inspired by the fact that the MSA/ABD-iTEP-pOVA complex enhanced antigen presentation by DCs, we expanded our exploration on interactions between the complex and DCs with a focus on those interactions that may affect the antigen presentation.
We first compared the endocytosis of the MSA/ABD-iTEP-pOVA complex and ABD-iTEP-pOVA by DCs because greater endocytosis should facilitate vaccine accumulations in DCs and hence increase antigen presentation. We found that DCs internalized a similar amount of the MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA (Fig. 4A). This result agrees with the data showing that DCs have the same uptake of the empty carrier, ABD-iTEP, whether the carrier is associated with MSA or not (Fig. 2E). Therefore, endocytosis is not a reason for the greater antigen presentation resulting from the MSA/ABD-iTEP-pOVA complex (Fig. 3D).
The MSA/ABD-iTEP-pOVA complex has increased cytosolic accumulation and greater endosomal/lysosomal stability than free ABD-iTEP-pOVA. (A) DCs internalized comparable amounts of the MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA. (B) Cytosolic accumulation of the MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA in DCs during 4 h of sample tracing. The primary quantity data of the two samples in the cytosol is shown in Fig. S2A. This figure shows the quantity data after normalization against the cytosolic quantity of the samples at the beginning of the tracing. (C) The accumulation of the MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA in vesicles (endosomes and lysosomes) of DCs during 4 h of sample tracing. The primary quantity data is shown in Fig. S2B. This figure shows the quantity data after normalization against the vesicular quantity of the samples at the beginning of the tracing. (D) The proportion of the MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA that were overlapped with vesicles (endosomes and lysosomes). Colocalization analysis was performed using all cells in Fig S3A and S3B. The proportion was calculated based on Manders' overlap coefficient. The data are shown as mean ± SEM, and analyzed by Student's t test, ***P < 0.001.
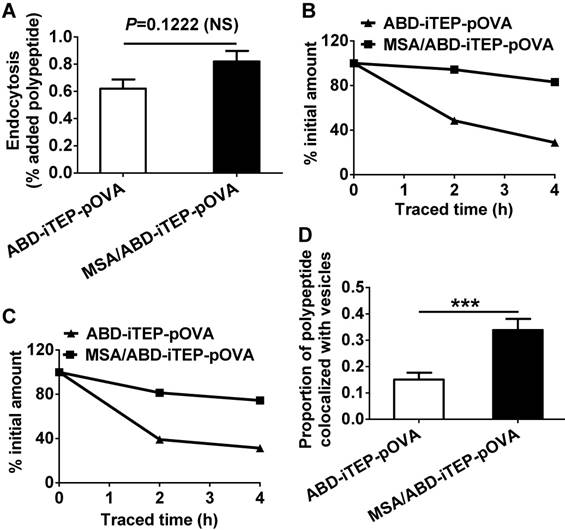
We then compared cytosolic accumulation of the MSA/ABD-iTEP-pOVA complex and ABD-iTEP-pOVA in DCs. It is in the cytosol of DCs where CTL vaccines are processed and CTL epitopes are released from the vaccines before the epitopes are presented by DCs on the cell surface [42]. Thus, more CTL vaccines reaching the DC cytosol after internalization means that more vaccines are available for cytosolic processing and subsequent epitope presentation. For comparison, we traced the amount of the complex and free ABD-iTEP-pOVA in DC cytosol after incubated them with DCs. At the beginning of the tracing, the complex and free ABD-iTEP-pOVA had comparable cytosolic accumulation (Fig. S2A). However, while the cytosolic ABD-iTEP-pOVA decreased rapidly within 4 h of tracing, the amount of the complex remained almost unchanged (Fig. S2A). After we normalized the quantity of cytosolic complex and free ABD-iTEP-pOVA against their quantity at the beginning of the tracing, we found that cytosolic ABD-iTEP-pOVA decreased by ~60% and ~70% during the first 2 h and 4 h of the tracing, while the complex only decreased by ~20% and ~25% during the same period (Fig. 4B). Interestingly, by using the same tracing approach, we also found that the MSA/ABD-iTEP-pOVA complex was more stable than free ABD-iTEP-pOVA in endosomes and lysosomes (Fig. S2B). During the 4 h tracing, the quantity of free ABD-iTEP-pOVA decreased by 70%, while the quantity of the complex in endosomes and lysosomes decreased by 25% (Fig. 4C). In addition, the major decrease of free ABD-iTEP-pOVA happened in the first 2 h of tracing. These tracing results are consistent with the fluorescence microscopy data we obtained at 2 h after the tracing started (Fig. S3). Analysis of these fluorescence imaging data (Fig. S3) indicated that the proportion of the MSA/ABD-iTEP-pOVA complex that was overlapped with endosomes and lysosomes was significantly higher than the proportion of free ABD-iTEP-pOVA that was overlapped with these vesicles (Fig. 4D). Putting together all the interactions between the complex and DCs that we have discovered so far, it is likely that the greater cytosolic accumulation of the MSA/ABD-iTEP-pOVA complex over free ABD-iTEP-pOVA may contribute to the greater pOVA presentation caused by the complex. The high cytosolic accumulation of the MSA/ABD-iTEP-pOVA complex may be related to the increased stability of the complex in endosomes and lysosomes, although a clear elucidation of the relationship between the stability of the complex and the cytosolic accumulation of the complex requires further investigation.
ABD-iTEP enhances the CTL responses of both non-self- and self-antigens in vivo
Since ABD-iTEP had increased LN accumulation and boosted the DC presentation of vaccines it delivered, we next examined whether ABD-iTEP enhanced in vivo CTL responses toward vaccines it delivered. To this end, we compared ABD-iTEP-pOVA, iTEP-pOVA, and ABD-iTEP for their ability to induce pOVA-specific CTL responses using the IFN-γ based ELISPOT assay [43, 44]. We found that ABD-iTEP-pOVA induced 2-fold stronger CTL responses than iTEP-pOVA: splenocytes from the ABD-iTEP-pOVA-immunized mice generated 78.4±12.7 spots per 2×105 cells while splenocytes from the iTEP-pOVA-immunized mice only generated 39.2±6.5 spots per 2×105 cells (P < 0.01, Fig. 5A). The splenocytes from the ABD-iTEP-immunized mice only generated several spots per 2×105 cells (Fig. 5A), suggesting the carrier itself did not induce any CTL response. Taken together, these results point to the conclusion that ABD-iTEP enhanced CTL responses as a vaccine carrier. Next, we expanded the investigation beyond pOVA by using ABD-iTEP to deliver self-antigens. Self-antigens are generally poorly immunogenic because of self-tolerance. However, these antigens are important in immunotherapies for cancer [2, 45], thus it is worthwhile to examine whether ABD-iTEP also enhance CTL responses against these antigens. To this end, we fused an epitope, pTRP2 (sequence: SVYDFFVWL) to ABD-iTEP and iTEP. pTRP2 is derived from a melanoma-associated self-antigen, tyrosinase-related protein 2 (TRP2) [46]. The fusions were termed ABD-iTEP-pTRP2 and iTEP-pTRP2, respectively (Table 1). According to CTL vaccination results, we found that ABD-iTEP-pTRP2 induced 2-fold stronger CTL response than iTEP-pTRP2 based on the IFN-γ ELISPOT result (57.25±11.16 spots per 2×105 cells versus 27.00±2.04 spots per 2×105 cells, P < 0.05, Fig. 5B). In addition to the systemic CTL responses using splenocytes, we also checked the local CTL responses toward the antigen by analyzing lymphocytes from draining LNs. The results showed that ABD-iTEP-pTRP2 also induced ~2-fold stronger CTL responses than iTEP-pTRP2 in the LNs (52.33±3.48 spots per 2×105 cells versus 24.33±2.67 spots per 2×105 cells, P < 0.01, Fig. 5C). An enhancement of CTL responses in draining LNs is particularly important to tumor immunotherapy. Because tumors utilize the draining LNs to metastasize [9], and thus, a strong anti-tumor CTL response in the LNs could help to halt metastasis [4, 5, 10].
ABD-iTEP enhanced the CTL responses of its vaccine payloads in vivo. (A) ABD-iTEP increased the CTL responses against the non-self-antigen pOVA. C57BL/6 mice (N=5) were subcutaneously immunized with ABD-iTEP, iTEP-pOVA or ABD-iTEP-pOVA. Splenocytes were harvested to determine antigen-specific CD8+ T cell responses by IFN-γ ELISPOT assay. ABD-iTEP increased the systemic (B) and local (C) CTL responses against the self-antigen pTRP2. C57BL/6 mice (N=4) were subcutaneously immunized with iTEP-TRP2 or ABD-iTEP-TRP2. Splenocytes (B) and LN cells (C) were collected to determine antigen-specific CD8+ T cell responses by IFN-γ ELISPOT assay. Data are shown as mean ± SEM. Data in (A) were analyzed by one-way ANOVA with Bonferroni post-test. Data in (B) and (C) were analyzed by Student's t test. *P < 0.05, **P < 0.01.
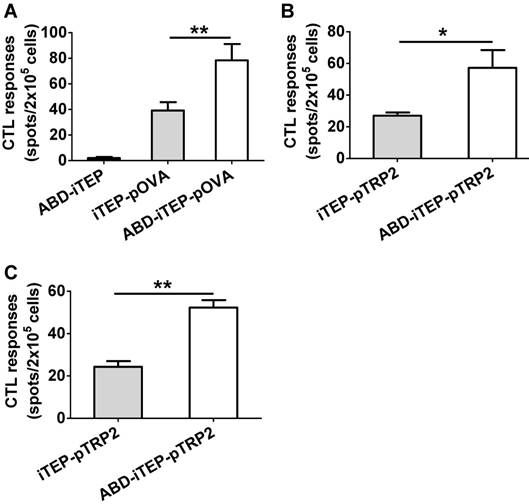
Taking all in vivo CTL vaccination results together, we found that ABD-iTEP boosted CTL responses of both non-self-antigens and self-antigens it delivered. Such boosts can be detected at both the systemic and the regional LN levels.
Discussion and Conclusions
Targeting CTL vaccines to LNs is an attractive strategy to improve the efficacy of CTL vaccines. Here, we developed a polypeptide-based, albumin-binding carrier to target CTL vaccines to LNs. More importantly, we established a new mechanism by which albumin-binding vaccine carriers facilitate CTL vaccines. Through this study, we gained new insights in the three following aspects.
First, ABD-iTEP, as an albumin-binding vaccine carrier, enhanced a wide range of vaccines, whether they are from self-antigens or non-self-antigens. Equally important, we were able to unambiguously attribute the enhancement to the association between the carrier and albumin because of our experimental design: we compared vaccine-induced CTL responses between ABD-iTEP and iTEP, an otherwise very similar carrier that does not bind with albumins. Such conclusiveness sets our study apart from the previous study of albumin-binding vaccine carriers where CTL responses were compared between free peptide vaccines and peptide vaccines delivered by an albumin-binding, micellar carrier [19]. These compared vaccines are different for not only the albumin-binding factor but also the micellar carrier factor. Thus, it is difficult to attribute different CTL responses solely to the factor of albumin-binding, although this factor likely contributed to the difference. Indeed, it was reported that peptide vaccines with or without a carrier have different capacities to induce responses [28, 47, 48]. In addition to the distinct experimental design differences between our study and the previous study, it is worth mentioning four interrelated features of ABD-iTEP that make it a robust delivery platform to realize advantages of albumin-binding carriers: (1) ABD-iTEP is a recombinant protein that is easy to produce, and no conjugation is needed to generate this carrier; (2) ABD-iTEP has absolute reproducibility and homogeneity across production batches; (3) ABD-iTEP is biodegradable and biocompatible; and (4) ABD-iTEP has no solubility issues, as reported in the previous albumin-binding vaccine carrier [19].
Second, our data have shown that albumin-binding vaccine carriers not only target vaccines to LNs but also promote the antigen presentation of vaccines. Further, we elucidated, by using a series of control treatments, that the improved antigen presentation was not due to the presence of the albumin-binding domain in the carriers or albumin in the experimental system. Instead, the improvement relies on the formation of the complex between MSA and ABD-iTEP-pOVA. Thus, albumin-binding vaccine carriers, for the first time, were found to have a second mechanism to boost vaccine-induced immune responses. This mechanism very likely contributed to the previously reported enhancement of CTL responses that was caused by albumin-binding carriers [19], although this mechanism was never proposed or investigated. The discovery of this new mechanism not only supports a new utility of albumin-binding carriers to improve antigen presentation, but also inspires more efforts to explore mechanisms that these carriers possess to support their delivery function.
Last, we discovered three interesting relationships between the MSA/ABD-iTEP-pOVA complex and DCs that may or may not explain the improved antigen presentation. Discovery one, DCs internalize a similar amount of the MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA, suggesting the improved presentation was not due to an increased uptake of the complex. Discovery two, the complex has a steady presence in DC cytosol as compared to free ABD-iTEP-pOVA. Because both the MSA/ABD-iTEP-pOVA complex and free ABD-iTEP-pOVA are transferred from endosomes to the cytosol and they are both degraded inside the cytosol before epitopes from them are presented by DCs [6-8], the stable presence of the complex may be caused by one of two following possible reasons, or both: (1) the endosome-to-cytosol translocation process replenishes more complex than free ABD-iTEP-pOVA into the cytosol, and (2) the complex is more stable than free ABD-iTEP-pOVA in the cytosol. While the idea of the increased translocation agrees with the improved presentation, the idea of the increased cytosolic stability disagrees with the improved presentation because cytosolic vaccines need to be degraded before the antigen presentation of the vaccines by DCs. Discovery three, the complex is more stable than free ABD-iTEP-pOVA in acidic organelles in DCs. This result is consistent with the above proposed translocation idea because more complex in endosomes and lysosomes would allow more complex to be transported to the cytosol. The increased stability in acidic organelles has been reported for IgG-complexed vaccines and was used to explain the improved presentation of these vaccines [20-22]. The increased stability was credited to interactions between IgG and FcRn in acidic organelles such as endosomes and lysosomes. Indeed, FcRn binds with albumin and rescues it from degradation in acidic organelles [49, 50]. Interactions between albumin and FcRn was previously proposed as one explanation for the extraordinary stability of albumin in the body [27, 40]. Putting together all three discoveries, a simple and plausible mechanism to explain the improved antigen presentation is as follows: the MSA/ABD-iTEP-pOVA complex interacts with FcRn in endosomes and lysosomes and hence becomes more stable and abundant there; consequently, more complex is transferred from these acidic organelles to the cytosol; eventually, more of the complex is available for processing in the cytosol so that more epitopes are released from the complex and presented by DCs. It would be interesting to test whether this hypothetical mechanism is true or not in future studies.
In summary, we developed a novel albumin-binding carrier, ABD-iTEP, that enhanced the effectiveness of CTL vaccines through two mechanisms: (1) increasing the LN and DC accumulation of the vaccines; (2) promoting the antigen presentation of the vaccines.
Abbreviations
CTL: cytotoxic T lymphocyte; LN: lymph node; DC: dendritic cell; ABD: albumin-binding domain; iTEP: immune-tolerant elastin-like polypeptide; MSA: mouse serum albumin; IACUC: institutional animal care and use committee; FBS: fetal bovine serum; LAL: limulus amebocyte lysate; PAGE: polyacrylamide gel electrophoresis; SEC: size exclusion chromatography; SPR: surface plasmon resonance; RU: response units; DAPI: 4',6-diamidino-2-phenylindole; MFI: median fluorescence intensity; OD: optical density; ELISPOT: enzyme-linked immunospot; Avidin-HRP: horseradish peroxidase avidin; AEC: 3-amino-9-ethylcarbazole; %ID: percentage of injected dose; %ID/g: percentage of injected dose per gram of an organ; SEM: standard error of the mean; NS: not significant; ANOVA: analysis of variance; pOVA: a CTL peptide derived from chicken ovalbumin; pTRP2: a CTL peptide derived from tyrosinase-related protein 2.
Acknowledgements
We thank Kenneth Rock (University of Massachusetts) for providing the DC2.4 cell line; and Nilabh Shastri (University of California, Berkeley) for providing the B3Z cell line. We appreciate Xiaomin Wang and Hu Dai for their assistance in breeding mice and purifying protein samples. We thank Michael Kay and Debbie Eckert for their assistance in the SPR study. We also recognize the Core Facility of University of Utah for their service with flow cytometry and cell imaging. The work was partially supported by the Ministry of Human Resources and Social Security of People's Republic of China Fund [grant number 2013-277] and the National Science Foundation of People's Republic of China Fund [grant number 81160297] to T.X. The work was primarily supported by the University of Utah Start-up Fund, the Huntsman Cancer Institute Pilot Grant [grant number 170301], and the National Institutes of Health grant [grant numbers R00CA153929 and R21EB024083] to M.C.
Supplementary Material
Supplementary figures and tables: Figure S1 shows the impact of MSA on T cell activation; Figure S2 shows accumulation dynamics of the MSA/ABD-iTEP-pOVA complex and ABD-iTEP-pOVA in cytosol and vesicles; Figure S3 includes fluorescent images of the MSA/ABD-iTEP-pOVA complex and ABD-iTEP-pOVA in cytosol and vesicles; Table S1 summarizes pharmacokinetic parameters of ABD-iTEP and iTEP.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Berzofsky JA, Ahlers JD, Belyakov IM. Strategies for designing and optimizing new generation vaccines. Nat Rev Immunol. 2001;1:209-19
2. Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015;125:3401-12
3. Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH. et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125:2532-46
4. Jeanbart L, Ballester M, de Titta A, Corthesy P, Romero P, Hubbell JA. et al. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol Res. 2014;2:436-47
5. Smith KA, Meisenburg BL, Tam VL, Pagarigan RR, Wong R, Joea DK. et al. Lymph node-targeted immunotherapy mediates potent immunity resulting in regression of isolated or metastatic human papillomavirus-transformed tumors. Clin Cancer Res. 2009;15:6167-76
6. Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev. 2005;207:145-57
7. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557-69
8. Wagner CS, Grotzke JE, Cresswell P. Intracellular events regulating cross-presentation. Front Immunol. 2012;3:138
9. Giuliano AE, Kirgan DM, Guenther JM, Morton DL. Lymphatic mapping and sentinel lymphadenectomy for breast cancer. Annals of Surgery. 1994;220:391-401
10. Bourquin C, Anz D, Zwiorek K, Lanz AL, Fuchs S, Weigel S. et al. Targeting CpG oligonucleotides to the lymph node by nanoparticles elicits efficient antitumoral immunity. J Immunol. 2008;181:2990-8
11. Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Control Release. 2006;112:26-34
12. Wu F, Bhansali SG, Law WC, Bergey EJ, Prasad PN, Morris ME. Fluorescence imaging of the lymph node uptake of proteins in mice after subcutaneous injection: molecular weight dependence. Pharm Res. 2012;29:1843-53
13. Bedrosian I, Scheff AM, Mick R, Callans LS, Bucky LP, Spitz FR. et al. 99mTc-human serum albumin: an effective radiotracer for identifying sentinel lymph nodes in melanoma. J Nucl Med. 1999;40:1143-8
14. Harrell MI, Iritani BM, Ruddell A. Lymph node mapping in the mouse. J Immunol Methods. 2008;332:170-4
15. Tsopelas C, Bevington E, Kollias J, Shibli S, Farshid G, Coventry B. et al. 99mTc-Evans blue dye for mapping contiguous lymph node sequences and discriminating the sentinel lymph node in an ovine model. Ann Surg Oncol. 2006;13:692-700
16. Wang Y, Lang L, Huang P, Wang Z, Jacobson O, Kiesewetter DO. et al. In vivo albumin labeling and lymphatic imaging. Proc Natl Acad Sci U S A. 2015;112:208-13
17. Pollaro L, Raghunathan S, Morales-Sanfrutos J, Angelini A, Kontos S, Heinis C. Bicyclic peptides conjugated to an albumin-binding tag diffuse efficiently into solid tumors. Mol Cancer Ther. 2015;14:151-61
18. Schmid B, Warnecke A, Fichtner I, Jung M, Kratz F. Development of albumin-binding camptothecin prodrugs using a Peptide positional scanning library. Bioconjug Chem. 2007;18:1786-99
19. Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519-22
20. Baker K, Qiao SW, Kuo TT, Aveson VG, Platzer B, Andersen JT. et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc Natl Acad Sci U S A. 2011;108:9927-32
21. Qiao SW, Kobayashi K, Johansen FE, Sollid LM, Andersen JT, Milford E. et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci U S A. 2008;105:9337-42
22. Baker K, Rath T, Pyzik M, Blumberg RS. The Role of FcRn in Antigen Presentation. Front Immunol. 2014;5:408
23. Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J Immunol. 2015;194:4595-603
24. Jonsson A, Dogan J, Herne N, Abrahmsen L, Nygren PA. Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng Des Sel. 2008;21:515-27
25. Levy OE, Jodka CM, Ren SS, Mamedova L, Sharma A, Samant M. et al. Novel exenatide analogs with peptidic albumin binding domains: potent anti-diabetic agents with extended duration of action. PLoS One. 2014;9:e87704
26. Orlova A, Jonsson A, Rosik D, Lundqvist H, Lindborg M, Abrahmsen L. et al. Site-specific radiometal labeling and improved biodistribution using ABY-027, a novel HER2-targeting affibody molecule-albumin-binding domain fusion protein. J Nucl Med. 2013;54:961-8
27. Andersen JT, Pehrson R, Tolmachev V, Daba MB, Abrahmsen L, Ekblad C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J Biol Chem. 2011;286:5234-41
28. Cho S, Dong S, Parent K, Chen M. Immune-tolerant elastin-like polypeptides (iTEPs) and their application as CTL vaccine carriers. Journal of drug targeting. 2016;24:328-39
29. Dong S, Xu T, Zhao P, Parent KN, Chen M. A Comparison Study of iTEP Nanoparticle-Based CTL Vaccine Carriers Revealed a Surprise Relationship between the Stability and Efficiency of the Carriers. Theranostics. 2016;6:666-78
30. MacEwan SR, Hassouneh W, Chilkoti A. Non-chromatographic Purification of Recombinant Elastin-like Polypeptides and their Fusions with Peptides and Proteins from Escherichia coli. Journal of Visualized Experiments: JoVE. 2014:51583
31. Liu S, Tobias R, McClure S, Styba G, Shi Q, Jackowski G. Removal of endotoxin from recombinant protein preparations. Clin Biochem. 1997;30:455-63
32. Wan CP, Park CS, Lau BH. A rapid and simple microfluorometric phagocytosis assay. J Immunol Methods. 1993;162:1-7
33. Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci U S A. 1992;89:6020-4
34. Baghirova S, Hughes BG, Hendzel MJ, Schulz R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX. 2015;2:440-5
35. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T. et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676-82
36. Tal M, Silberstein A, Nusser E. Why does Coomassie Brilliant Blue R interact differently with different proteins? A partial answer. Journal of Biological Chemistry. 1985;260:9976-80
37. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271-84
38. Zhao P, Xia G, Dong S, Jiang ZX, Chen M. An iTEP-salinomycin nanoparticle that specifically and effectively inhibits metastases of 4T1 orthotopic breast tumors. Biomaterials. 2016;93:1-9
39. Knauf MJ, Bell DP, Hirtzer P, Luo ZP, Young JD, Katre NV. Relationship of effective molecular size to systemic clearance in rats of recombinant interleukin-2 chemically modified with water-soluble polymers. J Biol Chem. 1988;263:15064-70
40. Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D. et al. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. Journal of Biological Chemistry. 2002;277:35035-43
41. Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013;39:806-18
42. Rock KL, Farfan-Arribas DJ, Shen LJ. Proteases in MHC Class I Presentation and Cross-Presentation. Journal of Immunology. 2010;184:9-15
43. Ghanekar SA, Nomura LE, Suni MA, Picker LJ, Maecker HT, Maino VC. Gamma Interferon Expression in CD8(+) T Cells Is a Marker for Circulating Cytotoxic T Lymphocytes That Recognize an HLA A2-Restricted Epitope of Human Cytomegalovirus Phosphoprotein pp65. Clinical and Diagnostic Laboratory Immunology. 2001;8:628-31
44. Ranieri E, Popescu I, Gigante M. CTL ELISPOT Assay. In: (ed.) Ranieri E. Cytotoxic T-Cells: Methods and Protocols. New York, NY: Springer New York. 2014 p. 75-86
45. Vigneron N. Human Tumor Antigens and Cancer Immunotherapy. Biomed Res Int. 2015;2015:948501
46. Schreurs MW, Eggert AA, de Boer AJ, Vissers JL, van Hall T, Offringa R. et al. Dendritic cells break tolerance and induce protective immunity against a melanocyte differentiation antigen in an autologous melanoma model. Cancer Res. 2000;60:6995-7001
47. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787-96
48. Ludewig B, Barchiesi F, Pericin M, Zinkernagel RM, Hengartner H, Schwendener RA. In vivo antigen loading and activation of dendritic cells via a liposomal peptide vaccine mediates protective antiviral and anti-tumour immunity. Vaccine. 2000;19:23-32
49. Nilvebrant J, Hober S. The albumin-binding domain as a scaffold for protein engineering. Comput Struct Biotechnol J. 2013;6:e201303009
50. Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC. et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197:315-22
Author contact
![]() Corresponding author: Mingnan Chen, Mailing address: 30 South 2000 East, Skaggs Pharmacy Hall, Room 301, Salt Lake City, UT 84112, USA Phone: (+1) 801-581-7616 Email: mingnan.chenedu
Corresponding author: Mingnan Chen, Mailing address: 30 South 2000 East, Skaggs Pharmacy Hall, Room 301, Salt Lake City, UT 84112, USA Phone: (+1) 801-581-7616 Email: mingnan.chenedu