Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(1):199-211. doi:10.7150/thno.21425 This issue Cite
Research Paper
Histidine-rich Modification of a Scorpion-derived Peptide Improves Bioavailability and Inhibitory Activity against HSV-1
Zhengyang Zeng1,2*, Runhong Zhang1*, Wei Hong1*, Yuting Cheng1, Huijuan Wang1, Yange Lang1, Zhenglin Ji1, Yingliang Wu1, Wenxin Li1, Youli Xie3 ![]() , Zhijian Cao1
, Zhijian Cao1 ![]()
1. State Key Laboratory of Virology, College of Life Sciences, Wuhan University, Wuhan 430072, PR China;
2. Center for Synthetic Biology Engineering Research, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, PR China;
3. Department of General Surgery, Zhongnan Hospital of Wuhan University, Wuhan, Hubei 430071, PR China.
* These authors contributed equally to this work.
Received 2017-6-12; Accepted 2017-10-4; Published 2018-1-1
Citation:
Zeng Z, Zhang R, Hong W, Cheng Y, Wang H, Lang Y, Ji Z, Wu Y, Li W, Xie Y, Cao Z. Histidine-rich Modification of a Scorpion-derived Peptide Improves Bioavailability and Inhibitory Activity against HSV-1. Theranostics 2018; 8(1):199-211. doi:10.7150/thno.21425. https://www.thno.org/v08p0199.htm
Other stylesAbstract

Rationale: HSV is one of the most widespread human viral pathogens. HSV-1 infects a large portion of the human population and causes severe diseases. The current clinical treatment for HSV-1 is based on nucleoside analogues, the use of which is limited due to drug resistance, side effects and poor bioavailability. AMPs have been identified as potential antiviral agents that may overcome these limitations. Therefore, we screened anti-HSV-1 peptides from a scorpion-derived AMP library and engineered one candidate into a histidine-rich peptide with significantly improved antiviral activity and development potential.
Methods: A venomous gland cDNA library was constructed from the scorpion Euscorpiops validus in the Yunnan Province of China. Six putative AMPs were characterized from this cDNA library, and the synthesized peptides were screened via plaque-forming assays to determine their virucidal potential. Time of addition experiments according to the infection progress of HSV-1 were used to identify the modes of action for peptides of interest. The histidine-rich modification was designed based on structural analysis of peptides by a helical wheel model and CD spectroscopy. Peptide cellular uptake and distribution were measured by flow cytometry and confocal microscopy, respectively.
Results: The peptide Eval418 was found to have high clearance activity in an HSV-1 plaque reduction assay. Eval418 exhibited dose-dependent and time-dependent inactivation of HSV-1 and dose-dependent inhibition of HSV-1 attachment to host cells. However, Eval418 scarcely suppressed an established HSV-1 infection due to poor cellular uptake. We further designed and modified Eval418 into four histidine-rich derivative peptides with enhanced antiviral activities and lower cytotoxicities. All of the derivative peptides suppressed established HSV-1 infections. One of these peptides, Eval418-FH5, not only had strong viral inactivation activity and enhanced attachment inhibitory activity but also had high inhibitory activity against intracellular HSV-1, which was consistent with its improved intracellular uptake and distribution as confirmed by confocal microscopy and flow cytometry.
Conclusion: We successfully identified an anti-HSV-1 peptide, Eval418, from a scorpion venom peptide library and designed a histidine-rich Eval418 derivative with significantly improved potential for further development as an anti-HSV-1 drug. This successful modification can provide a design strategy to improve the bioavailability, cellular distribution and antiviral activity of peptide agents.
Keywords: Scorpion venom peptides, Anti-HSV-1 peptides, Histidine-rich modification, Cellular uptake, Bioavailability.
Introduction
Herpes simplex virus type I (HSV-1) is one of the most common human pathogens, infecting 70-90% of the human population [1]. Humans are the only natural hosts of HSV, and HSV infection is almost exclusively limited to epithelial cells and peripheral nervous system (PNS) neurons [2]. HSV can cause diseases ranging from mild conditions to lethal infections, including mucocutaneous lesions in labialis and skin, herpes keratitis and herpes encephalitis [3]. Most of the licensed treatments are based on nucleoside analogues such as acyclovir, valacyclovir and famciclovir. However, current treatments are unable to prevent establishment of latency and reactivation of latent virus [4]. Due to the drug resistance, side effects, and poor bioavailability associated with current treatments and the lack of an effective HSV-1 vaccine, the discovery and development of novel anti-HSV-1 agents are still urgent.
In recent years, antimicrobial peptides (AMPs) have been suggested as a further promising strategy to develop new anti-HSV agents. A wide variety of organisms such as insects, mammals, amphibians, fish and some plants secrete AMPs as important components of their innate immune systems. These peptides can exhibit broad inhibitory activities against fungi, bacteria, parasites and viruses [5]. Many AMPs have been reported to have inhibitory activities against HSV infection, such as melittin [6, 7], cecropins [8], magainin [9] and LL-37 [10].
Scorpions are ancient terrestrial creatures that have lived on earth for more than four hundred million years. For the sake of predation and survival, they have evolved unique venom systems containing neurotoxins, antimicrobial peptides, enzymes and other components. Natural AMPs from scorpion venoms and their derivative peptides have exhibited attractive antiviral activities, and some of these peptides have been studied in previous reports. The recombinant peptide scorpine (RScp) has been reported to inhibit Dengue-2 replication in C6/36 cells [11]. A natural amphipathic α-helical peptide Hp1090 can directly interact with the HCV viral envelope and decrease viral infectivity [12]. The scorpion venom-derived synthetic peptide mucroporin-M1 exhibited virucidal activities against measles, SARS-CoV and influenza H5N1 viruses via a direct interaction with virus envelope [13]. Mucroporin-M1 also inhibited HBV replication in vitro and in vivo by activating the mitogen-activated protein kinase (MAPK) pathway and then reducing the expression of hepatocyte nuclear factor 4α (HNF4α) [14]. Kn2-7, a scorpion venom peptide derivative, can inhibit HIV-1 by direct interactions with viral particles [15]. Histidine-rich mutants of another scorpion-derived peptide, Ctry2459, showed significantly enhanced bioavailability and anti-HCV activity [16]. Two scorpion venom peptides, Hp1036 and Hp1239, inhibited HSV-1[17]. Recently, a scorpion defensin, BmKDfsin4, was reported to inhibit HBV replication in vitro [18]. Thus, the scorpion-derived AMP library is a rich source of new antiviral agents that are still awaiting further utilization.
In this study, we screened scorpion venom peptides and identified Eval418, which inhibited the initiation of HSV-1 infection. Eval418 disrupted the initial steps of infection but hardly suppressed an established HSV-1 infection, which was speculated to be a result of its poor cellular uptake. Since introducing histidine residues can improve peptide helicity and amphiphilicity, we attempted to modify Eval418 into four histidine-rich derivative peptides to improve its cellular uptake, intracellular distribution and subsequently, its antiviral activity. The helicities and amphiphilicities of the peptides were characterized by circular dichroism (CD). Anti-HSV-1 experiments showed that the modified peptides exhibited greater inhibition of HSV-1 than did the parent peptide. Especially, the modified peptide Eval418-FH5 exerted the lowest cytotoxicity and highest anti-HSV-1 activity. The results of confocal microscopy and flow cytometry analyses also revealed that Eval418-FH5 had enhanced cellular uptake and a dispersed cellular distribution.
Materials and Methods
cDNA library screening
E. validus scorpions were collected in the Yunnan Province of China. As previously described, the glands were collected 2 days after electrical extraction of the venom [19-21]. Trizol Reagent (Invitrogen) was used to prepare total RNA. Poly(A)-mRNA was purified using a Poly-A Tract mRNA Isolation System (Promega). The cDNA library was constructed according to the specifications of the Superscript Plasmid System cDNA Library Construction Kit (Gibco/BRL). The cDNA was then cloned into pSPORT1 plasmids and transformed into E. coli DH5α cells (China Center for Type Culture Collection, CCTCC). Randomly chosen cDNA clones were sequenced to obtain a reliable representation of the venom gland peptide library.
Peptide synthesis
The peptides Eval36 (GFLGNLWEGIKTAL), Eval151 (QDYNHDRDIVPPR), Eval162 (IAKTALKVLPQL), Eval418 (LWGEIWNTVKGLI), Eval655 (IWGALLSGVADLL) and Eval967 (FAFLAAIPSILSAL) were identified from the E. validus venom gland cDNA library and chemically synthesized. The peptides synthesized in this study were prepared by ChinaPeptides Company, a leading supplier of synthetic peptides in China. Briefly, the peptides were synthesized using solid-phase synthesis and C-terminal amidation in a standardized process. Fmoc-rink resin was used as the synthesis resin by Fmoc strategy. Piperidine was used for deprotecting, and HOBt/HBTU was used for coupling. The finished peptides were cleaved from the resin using trifluoroacetic acid (TFA), precipitated with ether and subjected to purification by reverse-phase HPLC on a C-18 hydrophobic resin (Elite-HPLC) in 0.1% TFA using an acetonitrile gradient. The purity of the final material was verified by reverse-phase HPLC, and the mass of the peptide was determined by mass spectrometry (Voyager-DESTR; Applied Biosystems). All peptides had purities of 95% or greater.
Cell culture and virus
African green monkey kidney cells (Vero) were cultured at 37 ℃ and 5% CO2 in minimum essential medium (MEM) (Invitrogen, Foster, CA, USA) containing 10% (vol/vol) fetal bovine serum (FBS) (Gibco, Foster, CA, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma, St. Louis, MO, USA). Cells infected with HSV-1 were cultured in MEM with 2% FBS in the same environment as described above. High-titer stocks of HSV-1 (F strain) were prepared as follows. The original viral stocks were diluted in MEM and inoculated into 80%-90% confluent Vero cell cultures at a multiplicity of infection (MOI) of 0.1 in a T25 flask (Corning, New York, USA). When the cytopathic effect (CPE) reached 90%-100%, the infected monolayers were harvested and subjected to three freeze-thaw cycles using a dry ice-ethanol bath and then centrifuged at 1000 × g for 5 min. The supernatant was collected after centrifugation. After titration, the virus aliquots were stored at -80 ℃.
MTT assay
Vero cells were planted in 96-well plates (104 cells per well) and cultured at 37 ℃ for 24 h. The cell culture medium was then replaced with fresh medium containing candidate peptides in a series of concentrations, and the plates were incubated at 37 ℃ for another 48 h. Afterwards, 20 μl of MTT solution (5 mg/mL in phosphate buffered saline, PBS) was added to each well, and the plates were incubated at 37 ℃ for 4 h. The medium was removed, and 100 μl of dimethyl sulfoxide (DMSO) was added to each well, and the plates were shaken gently for 10 min at room temperature to completely dissolve the crystal purple formazan. The absorbance was then measured at 570 nm using a microplate reader (BioTek, Winooski, VT, USA).
Hemolysis assay
Human erythrocytes were obtained from fresh human blood by centrifugation for 10 min at 1000 × g and washed three times with HEPES buffer. The erythrocytes were then resuspended in normal saline and seeded in 96-well plates (107 cells per well). The peptides were added in a series of concentrations, 0.1% triton-X100 was used as a positive control and normal saline was used as a negative control. After the plates were incubated at 37 ℃ for 1 h, the supernatants were collected by centrifugation at 1000 × g for 10 min. The absorbance of hemoglobin at a wavelength of 570 nm was measured using a microplate reader (BioTek, Winooski, VT, USA). The hemolysis assay was approved by the Medical Ethical Committee at Wuhan University.
Virus titration and extracellular antiviral plaque assay
Six-well plates containing 80-90% confluent Vero cell monolayers were infected with 10-fold diluted series of HSV-1 aliquots. After incubation at 37 ℃ for 1 h, the inocula were removed. The cells were washed three times with PBS and replenished with a maintenance cover layer (MEM with 2% FBS and 0.75% carboxy-methylcellulose). After an incubation period of 72 h, the cells were stained with 1% crystal violet containing 10% methanol, and the plaque numbers were visualized. The viral titer in plaque-forming units (PFU) was then calculated according to the plaque number and dilution ratio. The details of the antiviral assays are described in the section below.
Antiviral assays
Vero cell monolayers plated in six-well plates were infected with HSV-1 aliquots to yield approximately 60 plaques per well. The peptides were dissolved in pure water over a series of concentrations. The scorpion-derived antiviral peptides were screened at 10 μg/mL. To measure the antiviral activity of Eval418, the peptide was added at a series of concentrations after a 2-fold dilution (10, 5, 2.5, 1.25 and 0.625 µg/mL). The peptides derived from Eval418 were added at a series of concentrations after a 2-fold dilution (5, 2.5, 1.25, 0.625 and 0.3125 µg/mL) to measure their antiviral activities. In all time of addition experiments as well as confocal microscopic spotting and flow cytometry analyses, the peptides were used at a concentration of 10 µg/mL. Isometric sterile water was used as a negative control in all experiments. Antiviral assays were performed by five different ways of treatments.
Viral inactivation assay
HSV-1 was incubated with Eval418 and derivative peptides directly for 1 h at 37 ℃. The Vero cells were infected by these mixtures for 1 h at 37 ℃ and then rinsed and replenished with a cover layer that did not contain the peptide.
Cell protection assay
Vero cells were treated with Eval418 and derivative peptides for 1 h at 37 ℃ and then rinsed prior to HSV-1 infection.
Viral attachment inhibition assay
Vero cells were pre-incubated at 4 ℃ for 0.5 h. Peptides were added simultaneously with HSV-1, and the cells were incubated for 1 h at 4 ℃. The cells were then rinsed with PBS three times and replenished with a cover layer.
Viral entry inhibition assay
Vero cells were pre-incubated at 4 ℃ for 0.5 h. HSV-1 was added to the cell culture medium and incubated for 1 h at 4 ℃. The cells were rinsed with PBS three times and cultured with fresh medium containing peptides for 1 h at 37 ℃. The cells were then rinsed with PBS three times and replenished with a cover layer.
Post-entry inhibition assay (therapeutic effect on infection)
Vero cells were infected with HSV-1 for 1 h at 37 ℃. The cells were then rinsed, and the culture medium was replaced with a cover layer containing Eval418.
For all treatments, after infection at 37 ℃ for 1 h, the Vero cells were then rinsed with PBS three times and replenished with a cover layer. The inhibitory effects were determined by plaque reduction assay (PRA) after an additional 72 h.
Circular dichroism analysis
The secondary structures of Eval418 and its derivative peptides were measured by circular dichroism (CD) spectroscopy. The measurements were performed over a UV range of 250-190 nm at 25 ℃ in water or 50% TFE using a Jasco-810 spectropolarimeter at a concentration of 0.1 mg/mL. For each peptide, spectra were collected from three separate recordings and averaged after subtracting the blank spectrum of pure water.
Suppression of HSV-1 replication
Intracellular anti-HSV-1 activities of the peptides were evaluated by assessing infectivity and the DNA content of the intracellular progeny virus. After Vero cells were infected with HSV-1 at an MOI of 0.1 for 1 h at 37 ℃, the cells were washed and replenished with MEM containing the corresponding peptides. The cells and culture supernatants were harvested 24 h after infection. Intracellular viruses were collected using the freeze-thaw method. Extracellular and intracellular HSV-1 infectivity was assessed by plaque-forming assays.
DNA content was quantified by real-time PCR after total DNA extraction using the MiniBEST Viral RNA/DNA Extraction Kit (Takara, Minamikusatsu Station, JPN) with elution in 50 µL distilled water. Real-time PCR was performed using a SYBR Green PCR assay kit and an ABI7500 system. The forward primer sequence was 5'- CATCACCGACCCGGAGAGGGAC-3', and the reverse primer sequence was 5'- GGGCCAGGCGCTTGTTGGTGTA-3'. PCR products were subjected to a melting curve analysis to confirm amplification specificity.
Confocal microscopy
Vero cells were incubated with N-terminus FITC-labeled peptides at 10 μg/mL at 37 ℃ for 24 h. Afterwards, the cells were washed with PBS, fixed with 4% paraformaldehyde and rinsed twice with PBS. Cell nuclei were stained with DAPI (diluted 1: 500 in PBS) for 5 min, and the cells were rinsed six times with PBS. Peptide cellular localization was analyzed by confocal microscopy.
Flow cytometry measurement
Vero cells were treated with 10 µg/mL N-terminus FITC-labeled peptides at 37 ℃ for 24 h. The cells were then harvested by trypsin and washed six times with PBS. The average FITC intensity of the cells was measured by flow cytometry.
Statistical analysis
Data are expressed as the mean ± standard deviation from at least three separate experiments.
Results
Screening of anti-HSV-1 peptides from the venom of the scorpion E. validus
Six putative peptides from the venomous cDNA library of the scorpion E. validus were identified as candidate antimicrobial peptides by bioinformatics analysis of their sequences as well as evaluation of their amphipathy and positively charged α-helix structures, which are the classical structure features of antimicrobial peptides. Precursor structure analysis of the six scorpion-derived peptides is shown in Figure 1. The details of open-reading frames and amino acid sequences are shown. The candidate peptides were screened for viral inactivation against HSV-1 by plaque reduction assay (PRA) in Vero cells.
As shown in Figure 2a, compared with the other five candidate peptides, Eval418 exerted a significant inhibitory activity against plaque formation and cleared almost 100% of plaques. As the control group was set as 100%, the average amounts of plaques in each group are shown as percentages in Figure 2b. Eval418 exerted a 97.47% plaque clearance at a concentration of 10 μg/mL. Sequence alignments of the 6 scorpion peptides were performed using Genedoc software (Figure 2c).
Eval418 cytotoxicity and hemolysis
To assess whether Eval418 is suitable for further development as a candidate anti-HSV-1 agent, its cytotoxicity to Vero cells was determined by MTT assay (Figure 3a). Vero cells were treated by Eval418 at a series of concentrations ranging from 0 to 200 μg/mL. After incubation for 48 h, the 50% cytotoxicity concentration (CC50) of Eval418 to Vero cells was 68.50 μg/mL. After treatment with Eval418 at 10 μg/mL, the cell viability of the peptide-treated Vero cells was greater than 95%. Hemolysis assay showed that the cell viability of erythrocytes was also more than 95% at concentrations of Eval418 of 10 μg/mL or less (Figure 3b). These data indicated that 10 μg/mL or less, Eval418 was minimally cytotoxic to Vero cells and suitable for further anti-HSV-1 experiments.
Figure 1

Precursor structure analysis of six scorpion-derived peptides. Six scorpion peptides from the venomous cDNA library of the scorpion E. validus were characterized as candidate antimicrobial agents, and their precursor structures were analyzed. The amino acid sequences from open-reading frames are shown. The signal peptide and pro-peptide regions are underlined and highlighted with a gray background, respectively. The cutting signals are shown in frames, and the mature peptide regions are bolded.
Figure 2
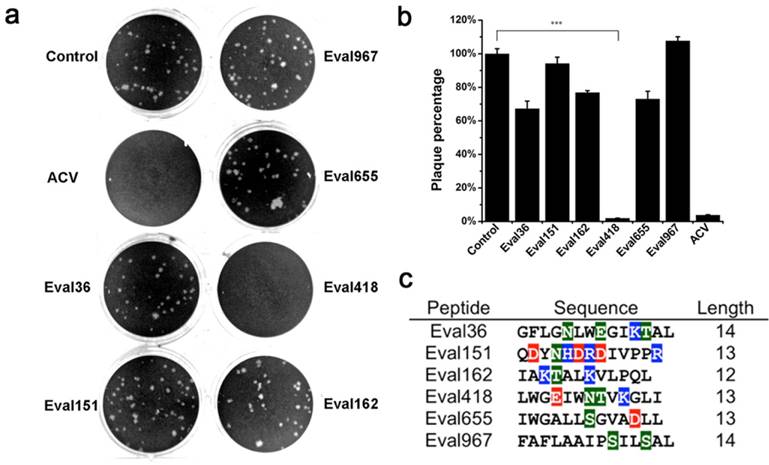
Screening of anti-HSV-1 peptides from the venomous cDNA library of the scorpion E. validus. The six scorpion peptides were prepared by chemical synthesis and screened for their abilities to inactivate HSV-1. (a) The inhibitory activities of the six candidate peptides at 10 μg/mL against HSV-1 proliferation were measured by plaque reduction assay in Vero cells. (b) Comparison of inhibitory rates of the six scorpion peptides against HSV-1 proliferation. The plaques in each well were counted, and the inhibitory rates were calculated. The values represent the mean ± SEM of five independent samples. The drug ACV (10 μg/mL) was used as a positive control. ***P< 0.001. (c) Sequence alignments of the six scorpion peptides. The sequence alignments of the candidate antimicrobial peptides were performed using Genedoc software. Hydrophilic and hydrophobic residues are marked by colorful and colorless backgrounds, respectively.
Figure 3
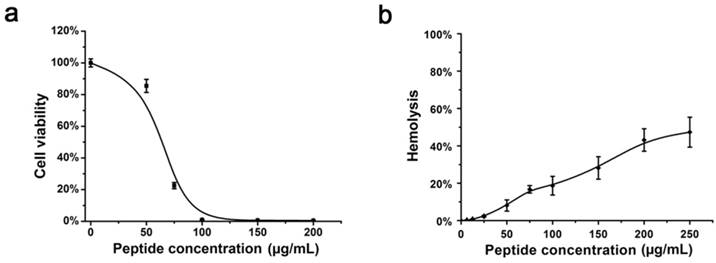
Cytotoxicity and hemolytic activity of the Eval418 peptide. (a) Cytotoxicity of Eval418 peptide was measured on Vero cells by MTT assay. The concentrations ranged from 0 to 200 μg/mL. The 50% cytotoxicity concentration (CC50) of Eval418 to Vero cells was 68.50 μg/mL. (b) The hemolytic activity of Eval418 was evaluated in human erythrocytes by hemolytic assay. The concentrations also ranged from 0 to 200 μg/mL. The hemolysis rate of human erythrocytes was less than 50% when the concentration of Eval418 was as high as 200 μg/mL.
Eval418 blocks the initial steps of HSV-1 infection
To determine the exact steps in the life cycle of HSV-1 that were inhibited by Eval418, a time of addition experiment for Eval418 against HSV-1 was performed. HSV-1 or Vero cells were incubated with Eval418 at 10 μg/mL for different periods of time, and the inhibitory effects were measured by PRA (Figure 4a). After pre-incubation with HSV-1 or following addition to the cells during the attachment step of HSV-1, Eval418 exhibited potent inhibition of plaque formation, suggesting an initial inhibitory activity against HSV-1 infection. On the contrary, Eval418 scarcely reduced plaque quantity when pre-incubated with Vero cells or added to cells after infection and showed a weak inhibition against viral entry. These results suggested that Eval418 only exerted a viral inactivation effect and prevented initiation of HSV-1 infection but did not affect intracellular anti-HSV-1 activity.
Figure 4
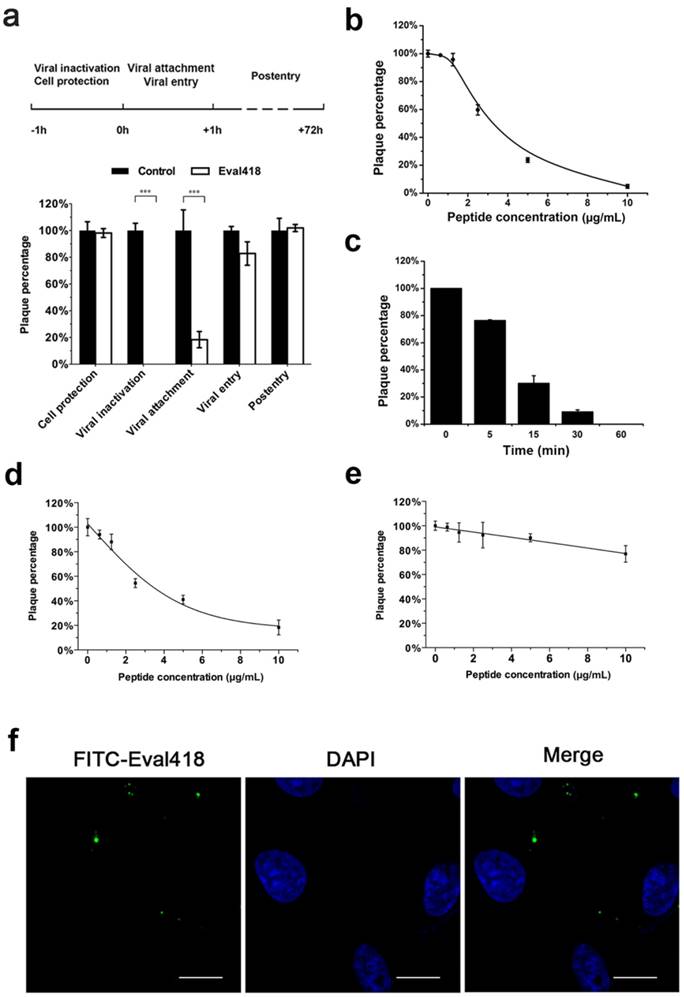
Comprehensive anti-HSV-1 activities and cellular localization of the Eval418 peptide. (a) Time of addition experiments were conducted to determine the anti-HSV-1 activity of the Eval418 peptide. The upper panel is a schematic overview of the operational approach for the time of addition experiments. The cells or viruses were treated with Eval418 peptide at a final concentration of 10 μg/mL under five diverse treatment modes. The lower panel shows the anti-HSV-1 activity assays of Eval418 in the time of addition experiments. The inhibitory effects of Eval418 in each treatment mode were determined by plaque reduction assay. ***P< 0.001. (b) Concentration-dependent inactivation activity of the Eval418 peptide. HSV-1 (60 PFU/well) was directly incubated with the Eval418 peptide. The virus-peptide mixture was then diluted to the indicated concentrations and applied to plaque reduction assay on Vero cells. (c) Time-dependent inactivation activity of the Eval418 peptide. HSV-1 (60 PFU/well) was directly incubated with the Eval418 peptide over serial durations, and the mixture was applied to a plaque reduction assay on Vero cells. (d) Inhibitory activity of the Eval418 peptide against HSV-1 viral attachment. Eval418 was added to the cell culture medium at a series of concentrations simultaneously with HSV-1 at the viral attachment step and rinsed out before viral entry. Then, the inhibitory effect was measured by plaque reduction assay. (e) The inhibitory activity of the Eval418 peptide against HSV-1 at the viral entry step. After viral attachment, Eval418 was added and co-incubated with cells during the viral entry step. The inhibitory effect was then measured by plaque reduction assay. (f) Cellular localization of the Eval418 peptide in Vero cells. Eval418 was labeled by FITC, and the cellular localization of Eval418 was determined by confocal microscopy after incubation for 24 h with Vero cells. Scale bars: 10 µM.
As shown in Figure 4b, HSV-1 was incubated with Eval418 over a series of concentrations, and the mixture was then used to infect Vero cells. The data from PRA experiments showed that plaque formation was inhibited by Eval418 in a dose-dependent manner and that the 50% inhibition concentration (IC50) was 2.48 μg/mL. The viral inactivation effect of Eval418 was also time-dependent at 10 μg/mL (Figure 4c). When treated with Eval418 for 5 min, the PFU of HSV-1 decreased by 20%, suggesting a rapid-onset inactivation. After treatment with Eval418 for 15 min, the average reduction increased to 67.87%. The reduction was more than 90% after 30 min and almost 100% following Eval418 treatment for 1 h.
To further investigate the inhibitory activity of Eval418 against HSV-1, Vero cells were treated with Eval418 over a series of concentrations during the viral attachment or entry steps. Similar to viral inactivation, the inhibitory activity against viral attachment was also dose-dependent, and the IC50 was 3.70 μg/mL (Figure 4d). Eval418 also showed weak inhibition of viral entry. The inhibition rate was only 23.08% at 10 µg/mL (Figure 4e). Therefore, Eval418 showed a dose-dependent and time-dependent inhibition against HSV-1 plaque formation during the viral inactivation step and a dose-dependent inhibition of viral attachment. In addition, compared with its CC50 to Vero cells, the selection index (SI) of Eval418 was 27.62 during the viral inactivation step and 18.51 during the viral attachment step (Table 1).
Considering the significant difference between the extracellular and intracellular anti-HSV-1 activities of Eval418, we speculated that Eval418 may possess low cellular uptake in Vero cells and/or unfavorable intracellular localization. As shown in Figure 4f, Vero cells were incubated with FITC-labeled Eval418 at 10 μg/mL for 24 h at 37 ℃ and then examined by confocal microscopy. The result showed that FITC-labeled Eval418 hardly entered Vero cells. The poor cellular uptake of Eval418 possibly explained its low intracellular anti-HSV-1 activity.
Design of histidine-rich peptides based on the molecular template of Eval418
The structure model using Swiss PDB Viewer software showed that Eval418 adopts an α-helix structure, which was then verified by circular dichroism (CD) spectroscopy (Figure 5a). The results of CD spectroscopy revealed that Eval418 adopts an α-helix structure in 50% TFE and a random coil structure in water. Eval418 exhibits an amphipathic α-helix in the helical wheel, which can be divided into a hydrophobic face and a hydrophilic face (Figure 5a). These data indicated that Eval418 is an amphipathic peptide molecule with an α-helix structure.
It has been reported that an artificial modified histidine-rich amphipathic peptide LAH4 exhibits efficient DNA delivery into mammalian cells [22]. The presence of histidine residues and a well-defined hydrophobic face have been identified as determinants of peptide delivery efficiency. The modification of a scorpion-derived anti-HCV peptide Ctry2459 into histidine-rich derivative peptides also significantly enhanced bioavailability and anti-HCV activity [16]. Referring to the design strategy of LAH4 and Ctry2459, several histidine residues were added in the derivative peptides, which were named Eval418-FH2 (Figure 5b), Eval418-FH3 (Figure 5c), Eval418-FH4 (Figure 5d) and Eval418-FH5 (Figure 5e). The secondary structures of the derivative peptides were predicted and determined using Swiss PDB Viewer software and circular dichroism (CD) spectroscopy (Figure 5b-e), respectively. The addition of histidine residues did not disrupt the pattern and stabilization of secondary structures, and the amphiphilicity was enhanced. The sequence alignments of Eval418 and its derivative peptides are shown in Figure 5f.
Anti-HSV-1 activities and cellular uptake of Eval418 derivative peptides
The cytotoxicities of the Eval418 derivative peptides to Vero cells were evaluated by MTT assay (Figure 6a). The CC50 values for Eval418-FH2, Eval418-FH3, Eval418-FH4 and Eval418-FH5 were 27.60, 26.83, 27.58 and 106.68 μg/mL, respectively, indicating that Eval418-FH5 had a lower cytotoxicity than the wild type peptide and other derivatives.
Table 1
Pharmacological profiles of Eval418 peptide and its derivative peptides
| Peptide | IC50 (µg/mL) | CC50 (µg/mL) | SI | ||||
|---|---|---|---|---|---|---|---|
| Viral inactivation | Viral attachment | Viral Entry | Viral inactivation | Viral attachment | Viral Entry | ||
| Eval418 | 2.48 | 3.70 | 31.71 | 68.50 | 27.62 | 18.51 | 2.16 |
| Eval418-FH2 | 1.50 | 1.43 | 8.63 | 27.60 | 18.40 | 19.30 | 3.20 |
| Eval418-FH3 | 1.01 | 0.86 | 4.23 | 26.83 | 26.56 | 31.20 | 6.34 |
| Eval418-FH4 | 0.87 | 0.63 | 4.37 | 27.58 | 31.70 | 43.78 | 6.31 |
| Eval418-FH5 | 0.86 | 0.67 | 2.88 | 106.68 | 124.05 | 159.22 | 37.04 |
Figure 5
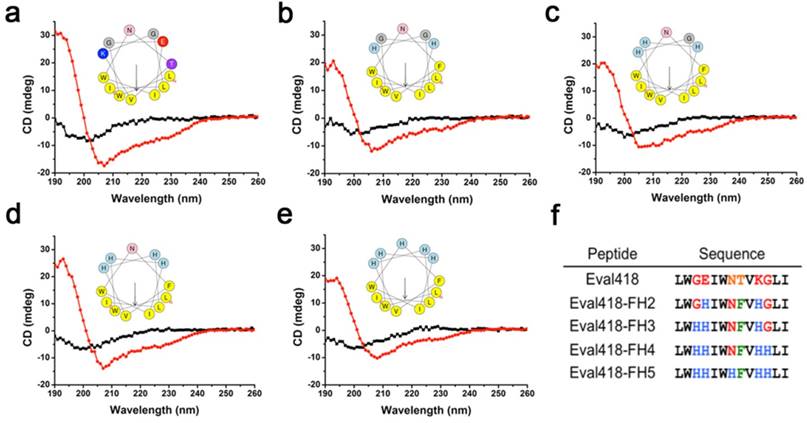
Design of Eval418 peptide derivatives. The CD spectra measurements were performed over a UV range of 250-190 nm at 25 ℃ in water on a jasco-810 spectropolarimeter (a-e). The helical wheels showed the hydrophilic and hydrophobic faces of Eval418 (a) and its derivative peptides, which were designated Eval418-FH2 (b), Eval418-FH3 (c), Eval418-FH4 (d) and Eval418-FH5 (e). (f) Sequence alignments of the Eval418 peptide and its derivative peptides.
The modes of action for these derivative peptides were identified by time of addition experiments as described previously (Figure 6b). All four derivative peptides maintained inhibitory activities against HSV-1 proliferation when pre-incubated with HSV-1 and added to cells at the viral attachment step. In addition, these derivative peptides showed greater inhibition of viral entry than did the wild type peptide Eval418. Interestingly, the derivative peptides also reduced plaque formation in PRA experiments when added to the cells at 5 μg/mL after HSV-1 infection. The inhibitory rates were 21.34%, 24.07%, 21.83% and 29.72%, respectively, whereas the wild type peptide Eval418 hardly exerted such an effect.
The viral inactivation capabilities of the derivative peptides were then further tested. Viral inactivation by these four derivative peptides was also dose-dependent, and the IC50 values of Eval418-FH2, Eval418-FH3, Eval418-FH4 and Eval418-FH5 were 1.50, 1.01, 0.87 and 0.86 μg/mL, respectively, which were significantly lower than that of the wild type peptide (Figure 6c). The SIs of the four derivative peptides were 18.40, 26.56, 31.70 and 124.05, respectively (Table 1). The derivative peptides showed a more rapid inactivation in time-dependence experiments compared with the wild type peptide Eval418 (Figure 6d). In particular, Eval418-FH5 reduced the PFUs by more than 50% after only a 5-minute incubation.
The inhibitory activities of the Eval418 derivative peptides when added to cells during the HSV-1 attachment or entry steps were also evaluated (Figure 6e, 6f). The IC50 values of Eval418-FH2, Eval418-FH3, Eval418-FH4 and Eval418-FH5 during the viral attachment step were 1.43, 0.86, 0.63 and 0.67 μg/mL, respectively, which were also significantly decreased from 3.70 μg/mL of the wild type peptide. The SIs of the derivative peptides were 19.30, 31.20, 43.78 and 159.22, respectively (Table 1). The IC50 values of Eval418-FH2, Eval418-FH3, Eval418-FH4 and Eval418-FH5 during the viral entry step were 8.63, 4.23, 4.37 and 2.89 µg/mL, respectively. The inhibitory activities of the derivative peptides during the viral entry step were significantly improved when compared with that of the wild type peptide Eval418 (IC50 = 31.71 µg/mL) (Table 1).
The suppressive activities of Eval418 and its derivative peptides against HSV-1 proliferation in the post-entry assay were then determined (Figure 6g). The extracellular and intracellular infectivity of HSV-1 were assessed by plaque forming assay, and the intracellular DNA quantity was measured by real-time PCR. The wild type peptide Eval418 exerted no effect, whereas the extracellular and intracellular infectivity of HSV-1 were significantly decreased by all four derivative peptides. Among these peptides, Eval418-FH5 exhibited the strongest inhibition and reduced the infectivity of extracellular and intracellular HSV-1 by 81.90% and 77.65%, respectively.
Figure 6
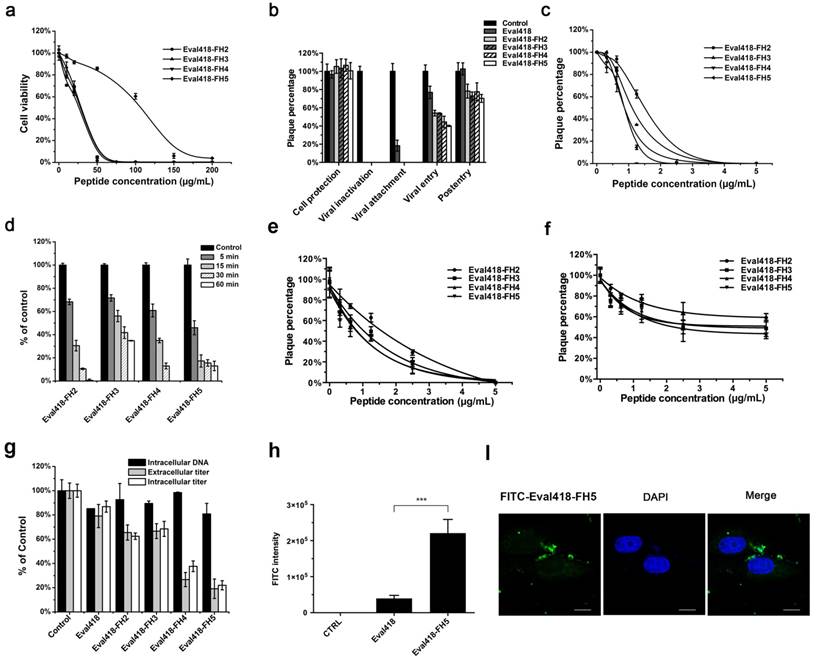
Cytotoxicities, anti-HSV-1 activities and intracellular distributions of the Eval418-derived peptides. (a) Cytotoxicities of Eval418-derived peptides. The cytotoxicities of Eval418-FH2, Eval418-FH3, Eval418-FH4 and Eval418-FH5 were measured on Vero cells by MTT assay. The concentrations ranged from 0 to 200 μg/mL, and the CC50 values were 27.60, 26.83, 27.58 and 106.68 μg/mL, respectively. (b) Anti-HSV-1 activity assay of the Eval418-derived peptides in the time of addition experiment. The inhibitory effects of the Eval418-derived peptides in each treatment mode were determined by plaque reduction assay. (c) Concentration-dependent inactivation activities of Eval418 derivative peptides. HSV-1 (60 PFU/well) was directly treated with Eval418-derived peptides. Vero cells were infected with the virus-peptide mixtures at a serial final concentration of the derivative peptides. (d) Time-dependent inactivation activities of Eval418 derivative peptides. Eval418-derived peptides were added to HSV-1 (60 PFU/well) and incubated over a series of indicated durations. The mixtures were then used to infect Vero cells, and the plaque reduction assay was completed. (e) Inhibitory activities of the Eval418-derived peptides when added to cells during the HSV-1 viral attachment step. Peptides were added to culture medium of Vero cells simultaneously with HSV-1 and incubated for 1 h at 4 ℃. The cells were then rinsed and replenished with a cover layer, and the plaque reduction assay was completed. (f) Inhibitory activities of the Eval418-derived peptides against viral entry of HSV-1. Cells were incubated with HSV-1 at 4 ℃ for 1 h. After HSV-1 was washed out, the derivative peptides were added and incubated at 37 ℃ for 1 h. The cells were then rinsed and replenished with a cover layer. The inhibitory rates were measured by plaque reduction assay. (g) Anti-HSV-1 activities of Eval418 and its derivative peptides during the post-entry step. Extracellular and intracellular HSV-1 infectivity levels were assessed by plaque forming assay, and the DNA content was assessed by real-time PCR. (h) Flow cytometry measurement of the cellular uptake of Eval418 and Eval418-FH5 peptides. Vero cells were incubated with FITC-labeled Eval418 or Eval418-FH5 for 24 h, and the average FITC intensity of each cell was measured by flow cytometry. ***P< 0.001. (i) Confocal microscopic examination of cellular localization of Eval418-FH5. Eval418-FH5 was labeled by FITC and used to treat Vero cells for 24 h. The cellular localization was determined using confocal microscopy. Scale bars: 10 µM.
To determine whether internalization of the Eval418 derivative peptides was promoted by the addition of histidine residues, Eval418 and Eval418-FH5 were labeled by FITC and incubated with Vero cells for 24 h. After that, cellular uptake and localization were measured by flow cytometry and confocal microscopy. As shown in Figure 6h and Figure 6i, Eval418-FH5 promoted a significant cellular uptake and a dispersed distribution compared with the wild type peptide Eval418.
Discussion
In the past decades, many antiviral peptides have been discovered or designed, and most have exerted potential virucidal activities. However, the greatest difficulty in treating viral diseases is the clearance of established infection in most natural and clinical conditions. However, the lipophilic muramyl peptide MTP-PE has been reported as a potent inhibitor of HIV replication in macrophages [23]. NP-1, a rabbit α-defensin, can simultaneously prevent the entry and intercellular spread of HSV-2 [24]. The derivative virucidal peptide, C5A, exhibits antiviral activities against HCV, HIV-1 and HSV-1 by disturbing viral membranes. In addition, C5A can block viral entry and suppress established infection [25-27]. Our group also has completed several studies of antiviral peptides that can inhibit established viral infection. Anti-HBV experiments demonstrated that a scorpion-derived short linear peptide, mucroporin-M1, can inhibit HBV replication in vitro and in vivo by activating the MAPK pathway and down-regulating HNF4α expression [14]. Designed histidine-rich peptides from the template of the scorpion venom peptide Ctry2459 showed considerable inhibition against HCV RNA when added at 4 h, 24 h and 48 h after infection [16]. The scorpion defensin BmKDfsin4 can inhibit HBV replication in vitro [18] and also showed ion-channel blocking activity and antibacterial activity in another study by us [28].
In this study, we screened the cDNA library from the scorpion venomous gland and identified six scorpion venom peptides, 12-14 amino acids in length, as antimicrobial candidates. The viral inactivation activities of these six scorpion venom peptides against HSV-1 were screened by PRA assay. Eval418, which contained 13 amino acid residues, cleared almost 100% of PFUs. The cytotoxicity and hemolysis activity of Eval418 were further tested, and the results showed that Eval418 is minimally toxic at a concentration of 10 μg/mL. Furthermore, Eval418 showed a significant dose-dependent inhibition when pre-incubated with HSV-1 or added during the HSV-1 viral attachment step with IC50 values of 2.48 and 3.70 μg/mL, respectively. Unfortunately, Eval418 scarcely exhibited any inhibition of plaque formation when pre-incubated with cells before infection or added to cells after infection. In addition, Eval418 only exerted a weak inhibition when added to cells during the viral entry step. The results of confocal microscopy and flow cytometry analyses showed that Eval418 exerted an extremely poor cellular uptake as no visible FITC-labeled Eval418 was detected in the incubated cells.
Various methods have been employed to overcome the limitations surrounding intracellular AMP antiviral activities. For example, addition of D-amino acids [29, 30] or fluorinated amino acids [31] may enhance peptide transmembrane delivery. The presence of lysosomotropic agents or introduction of histidine residues can enhance the transfection efficiencies of vehicle peptides as well as AMP cellular uptake and antiviral ability [16, 32-34]. In a previous study from our group, a glycine residue and a proline residue of the scorpion venom derived peptide mucroporin were replaced with a lysine residue and an arginine residue, respectively, to enhance the net positive charge of the hydrophilic side of the molecule. After the modifications, mucroporin-M1 exhibited a stronger virucidal activity and additional inhibition of HBV replication in vitro and in vivo [13, 14]. Ctry2459, another scorpion-derived peptide from the scorpion C. tryznai, has been modified into 2 histidine-rich mutant peptides to overcome the barriers of cellular uptake and endosome escape. These modifications significantly enhanced the anti-HCV activities and cellular uptake and introduced inhibition of established viral infection [16].
In our study, Eval418 was designed and modified into four histidine-rich derivative peptides by introducing 2-5 histidine residues. All four derivative peptides exhibited the same α-helix structures as the wild-type peptide Eval418, indicating that the addition of histidine residues did not affect the peptide structure but enhanced its helicity. In addition, all four derivative peptides showed improved virucidal effects. The IC50 values of Eval418-FH2, Eval418-FH3, Eval418-FH4 and Eval418-FH5 were 1.50, 1.01, 0.87 and 0.86 μg/mL, respectively. The blocking activities of the four derivative peptides against HSV-1 viral attachment were also improved. Their IC50 values were enhanced from 3.70 μg/mL to 1.43, 0.86, 0.63 and 0.67 μg/mL, respectively. The abilities of the derivative peptides to inhibit viral entry were also significantly improved compared with that of Eval418. In addition, the derivative peptides showed additional inhibition of established HSV-1 infection when added to cells 1 h after infection.
It is worth noting that Eval418-FH5, in which the most histidine residues were introduced, exhibited a significantly reduced cytotoxicity compared with the other derivative peptides. The CC50 of Eval418-FH5 was 106.68 μg/mL, but the CC50 of Eval418 was 68.50 μg/mL. Eval418-FH5 also showed a rapid action by clearing more than 50% of PFUs after incubation with HSV-1 for only 5 min. The IC50 of Eval418-FH5 for HSV-1 inactivation and attachment were 0.86 and 0.67 μg/mL, respectively. Therefore, the SIs of Eval418-FH5 were significantly enhanced to 124.05 and 159.22, respectively, whereas those of the wild type peptide Eval418 were 27.62 and 18.51, respectively. The flow cytometry and confocal microscopy results showed that Eval418-FH5 exerted a considerable cellular uptake and a dispersed distribution, which may explain its superior antiviral activity in the presence of an established HSV-1 infection. Thus, the introduction of histidine residues into the hydrophilic face of Eval418 molecules can overcome barriers in cellular uptake, improve antiviral activity and therefore enable the Eval418 derivatives to interact with intracellular HSV-1.
The successful modification of Eval418 and other AMPs or cell-penetrating peptides (CPPs) in previous studies may provide an avenue to design novel active peptide agents with excellent cellular uptake and activities or modify previously discovered peptides to improve their antiviral properties. However, the detailed mechanisms by which the cytotoxicity and antiviral properties are influenced by histidine modification still requires further exploration, which will accelerate the modification and optimization of such strategy. Detailed studies about the toxicity and the in vivo antiviral properties of Eval418 and its derivative peptides need to be conducted before further therapeutic application of these peptides.
Abbreviations
HSV-1: Herpes simplex virus type 1; AMPs: antimicrobial peptides; CD spectroscopy: circular dichroism spectroscopy; PNS: peripheral nervous system; MAPK pathway: mitogen-activated protein kinase pathway; HNF4α: hepatocyte nuclear factor 4α; TFA: trifluoroacetic acid; HPLC: high performance liquid chromatography; MEM: minimum essential medium; FBS: fetal bovine serum; CPE: cytopathic effect; PBS: phosphate buffered saline; DMSO: dimethyl sulfoxide; HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; PFU: plaque-forming units; PRA: plaque reduction assay; MOI: multiplicity of infection; DAPI: 4',6-diamidino-2-phenylindole; FITC: fluorescein isothiocyanate; CC50: 50% cytotoxicity concentration; IC50: 50% inhibition concentration; SI: selecting index.
Acknowledgements
This work was supported by grants from National Science Fund of China (Nos. 31422049, 31572289 and 81630091), International S&T Cooperation Program of China (No. S2016G3110), Hubei Science Fund (Nos. 2015CFA042 and 2016CFA018), China-Kazakhstan Cooperation Program (No. CK-07-09), and Fundamental Research Funds for the Central Universities in China (No. 2042017kf0242).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brandt C R. The role of viral and host genes in corneal infection with herpes simplex virus type 1. Experimental eye research. 2005;80:607-21
2. Liashkovich I, Hafezi W, Kuhn J M, Oberleithner H, Shahin V. Nuclear delivery mechanism of herpes simplex virus type 1 genome. Journal of molecular recognition: JMR. 2011;24:414-21
3. Schacker T. The role of HSV in the transmission and progression of HIV. Herpes: the journal of the IHMF. 2001;8:46-9
4. Schang L M. Herpes simplex viruses in antiviral drug discovery. Current pharmaceutical design. 2006;12:1357-70
5. Diamond G, Beckloff N, Weinberg A, Kisich K O. The roles of antimicrobial peptides in innate host defense. Current pharmaceutical design. 2009;15:2377-92
6. Baghian A, Jaynes J, Enright F, Kousoulas K G. An amphipathic alpha-helical synthetic peptide analogue of melittin inhibits herpes simplex virus-1 (HSV-1)-induced cell fusion and virus spread. Peptides. 1997;18:177-83
7. Baghian A, Kousoulas K G. Role of the Na+,K+ pump in herpes simplex type 1-induced cell fusion: melittin causes specific reversion of syncytial mutants with the syn1 mutation to Syn+ (wild-type) phenotype. Virology. 1993;196:548-56
8. Hancock R E, Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends in microbiology. 2000;8:402-10
9. Aboudy Y, Mendelson E, Shalit I, Bessalle R, Fridkin M. Activity of two synthetic amphiphilic peptides and magainin-2 against herpes simplex virus types 1 and 2. International journal of peptide and protein research. 1994;43:573-82
10. Gordon Y J, Huang L C, Romanowski E G, Yates K A, Proske R J, Mcdermott A M. Human cathelicidin (LL-37), a multifunctional peptide, is expressed by ocular surface epithelia and has potent antibacterial and antiviral activity. Current eye research. 2005;30:385-94
11. Carballar-Lejarazu R, Rodriguez M H, De La Cruz Hernandez-Hernandez F, Ramos-Castaneda J, Possani L D, Zurita-Ortega M, Reynaud-Garza E, Hernandez-Rivas R, Loukeris T, Lycett G, Lanz-Mendoza H. Recombinant scorpine: a multifunctional antimicrobial peptide with activity against different pathogens. Cellular and molecular life sciences: CMLS. 2008;65:3081-92
12. Yan R, Zhao Z, He Y, Wu L, Cai D, Hong W, Wu Y, Cao Z, Zheng C, Li W. A new natural alpha-helical peptide from the venom of the scorpion Heterometrus petersii kills HCV. Peptides. 2011;32:11-9
13. Li Q, Zhao Z, Zhou D, Chen Y, Hong W, Cao L, Yang J, Zhang Y, Shi W, Cao Z, Wu Y, Yan H, Li W. Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides. 2011;32:1518-25
14. Zhao Z, Hong W, Zeng Z, Wu Y, Hu K, Tian X, Li W, Cao Z. Mucroporin-M1 inhibits hepatitis B virus replication by activating the mitogen-activated protein kinase (MAPK) pathway and down-regulating HNF4alpha in vitro and in vivo. The Journal of biological chemistry. 2012;287:30181-90
15. Chen Y, Cao L, Zhong M, Zhang Y, Han C, Li Q, Yang J, Zhou D, Shi W, He B, Liu F, Yu J, Sun Y, Cao Y, Li Y, Li W, Guo D, Cao Z, Yan H. Anti-HIV-1 activity of a new scorpion venom peptide derivative Kn2-7. PloS one. 2012;7:e34947
16. Hong W, Zhang R, Di Z, He Y, Zhao Z, Hu J, Wu Y, Li W, Cao Z. Design of histidine-rich peptides with enhanced bioavailability and inhibitory activity against hepatitis C virus. Biomaterials. 2013;34:3511-22
17. Hong W, Li T, Song Y, Zhang R, Zeng Z, Han S, Zhang X, Wu Y, Li W, Cao Z. Inhibitory activity and mechanism of two scorpion venom peptides against herpes simplex virus type 1. Antiviral research. 2014;102:1-10
18. Zeng Z, Zhang Q, Hong W, Xie Y, Liu Y, Li W, Wu Y, Cao Z. A Scorpion Defensin BmKDfsin4 Inhibits Hepatitis B Virus Replication in Vitro. Toxins. 2016:8
19. Zeng X C, Li W X, Zhu S Y, Peng F, Jiang D H, Yang F H, Wu K L. Cloning and characterization of the cDNA sequences of two venom peptides from Chinese scorpion Buthus martensii Karsch (BmK). Toxicon: official journal of the International Society on Toxinology. 2000;38:893-9
20. Ma Y, He Y, Zhao R, Wu Y, Li W, Cao Z. Extreme diversity of scorpion venom peptides and proteins revealed by transcriptomic analysis: implication for proteome evolution of scorpion venom arsenal. Journal of proteomics. 2012;75:1563-76
21. Li Z, Xu X, Meng L, Zhang Q, Cao L, Li W, Wu Y, Cao Z. Hp1404, a new antimicrobial peptide from the scorpion Heterometrus petersii. PloS one. 2014;9:e97539
22. Kichler A, Leborgne C, Marz J, Danos O, Bechinger B. Histidine-rich amphipathic peptide antibiotics promote efficient delivery of DNA into mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1564-8
23. Lazdins J K, Woods-Cook K, Walker M, Alteri E. The lipophilic muramyl peptide MTP-PE is a potent inhibitor of HIV replication in macrophages. AIDS research and human retroviruses. 1990;6:1157-61
24. Sinha S, Cheshenko N, Lehrer R I, Herold B C. NP-1, a rabbit alpha-defensin, prevents the entry and intercellular spread of herpes simplex virus type 2. Antimicrobial agents and chemotherapy. 2003;47:494-500
25. Bobardt M D, Cheng G, De Witte L, Selvarajah S, Chatterji U, Sanders-Beer B E, Geijtenbeek T B, Chisari F V, Gallay P A. Hepatitis C virus NS5A anchor peptide disrupts human immunodeficiency virus. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5525-30
26. Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland S F, Isogawa M, Fredericksen B, Selvarajah S, Gallay P A, Ghadiri M R, Chisari F V. A virocidal amphipathic {alpha}-helical peptide that inhibits hepatitis C virus infection in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3088-93
27. De Witte L, Bobardt M D, Chatterji U, Van Loenen F B, Verjans G M, Geijtenbeek T B, Gallay P A. HSV neutralization by the microbicidal candidate C5A. PloS one. 2011;6:e18917
28. Meng L, Xie Z, Zhang Q, Li Y, Yang F, Chen Z, Li W, Cao Z, Wu Y. Scorpion potassium channel-blocking defensin highlights a functional link with neurotoxin. The Journal of biological chemistry. 2016
29. Papo N, Oren Z, Pag U, Sahl H G, Shai Y. The consequence of sequence alteration of an amphipathic alpha-helical antimicrobial peptide and its diastereomers. The Journal of biological chemistry. 2002;277:33913-21
30. Shai Y, Oren Z. Diastereoisomers of cytolysins, a novel class of potent antibacterial peptides. The Journal of biological chemistry. 1996;271:7305-8
31. Cortesi R, Romagnoli R, Drechsler M, Menegatti E, Zaid A N, Ravani L, Esposito E. Liposomes- and ethosomes-associated distamycins: a comparative study. Journal of liposome research. 2010;20:277-85
32. Sandhu A P, Lam A M, Fenske D B, Palmer L R, Johnston M, Cullis P R. Calcium enhances the transfection potency of stabilized plasmid-lipid particles. Analytical biochemistry. 2005;341:156-64
33. Shiraishi T, Pankratova S, Nielsen P E. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chemistry & biology. 2005;12:923-9
34. Tanaka K, Kanazawa T, Ogawa T, Suda Y, Takashima Y, Fukuda T, Okada H. A novel, bio-reducible gene vector containing arginine and histidine enhances gene transfection and expression of plasmid DNA. Chemical & pharmaceutical bulletin. 2011;59:202-7
Author contact
![]() Corresponding author: Zhijian Cao (State Key Laboratory of Virology, College of Life Sciences, Wuhan University, Wuhan, Hubei 430072, PR China. E-mail: zjcaoedu.cn. Tel: +86-27-68752831; Fax: +86-27-68756746) and Youli Xie (Department of General Surgery, Zhongnan Hospital of Wuhan University, Wuhan, Hubei 430071, PR China. E-mail: xyl222com. Tel: +86-27-87325743; Fax: +86-27-87325743).
Corresponding author: Zhijian Cao (State Key Laboratory of Virology, College of Life Sciences, Wuhan University, Wuhan, Hubei 430072, PR China. E-mail: zjcaoedu.cn. Tel: +86-27-68752831; Fax: +86-27-68756746) and Youli Xie (Department of General Surgery, Zhongnan Hospital of Wuhan University, Wuhan, Hubei 430071, PR China. E-mail: xyl222com. Tel: +86-27-87325743; Fax: +86-27-87325743).
Citation styles
APA
Zeng, Z., Zhang, R., Hong, W., Cheng, Y., Wang, H., Lang, Y., Ji, Z., Wu, Y., Li, W., Xie, Y., Cao, Z. (2018). Histidine-rich Modification of a Scorpion-derived Peptide Improves Bioavailability and Inhibitory Activity against HSV-1. Theranostics, 8(1), 199-211. https://doi.org/10.7150/thno.21425.
ACS
Zeng, Z.; Zhang, R.; Hong, W.; Cheng, Y.; Wang, H.; Lang, Y.; Ji, Z.; Wu, Y.; Li, W.; Xie, Y.; Cao, Z. Histidine-rich Modification of a Scorpion-derived Peptide Improves Bioavailability and Inhibitory Activity against HSV-1. Theranostics 2018, 8 (1), 199-211. DOI: 10.7150/thno.21425.
NLM
Zeng Z, Zhang R, Hong W, Cheng Y, Wang H, Lang Y, Ji Z, Wu Y, Li W, Xie Y, Cao Z. Histidine-rich Modification of a Scorpion-derived Peptide Improves Bioavailability and Inhibitory Activity against HSV-1. Theranostics 2018; 8(1):199-211. doi:10.7150/thno.21425. https://www.thno.org/v08p0199.htm
CSE
Zeng Z, Zhang R, Hong W, Cheng Y, Wang H, Lang Y, Ji Z, Wu Y, Li W, Xie Y, Cao Z. 2018. Histidine-rich Modification of a Scorpion-derived Peptide Improves Bioavailability and Inhibitory Activity against HSV-1. Theranostics. 8(1):199-211.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.