Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results and Discussions
Conclusion
Experimental Section
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results and Discussions
Conclusion
Experimental Section
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(1):1-12. doi:10.7150/thno.21052 This issue Cite
Research Paper
Acid-degradable Dextran as an Image Guided siRNA Carrier for COX-2 Downregulation
Zhihang Chen ![]() , Balaji Krishnamachary, Marie-France Penet, Zaver M. Bhujwalla
, Balaji Krishnamachary, Marie-France Penet, Zaver M. Bhujwalla ![]()
Division of Cancer Imaging Research, Johns Hopkins University School of Medicine, Russell H. Morgan Dept. of Radiology & Radiological Science, Baltimore, MD 21205, USA
Received 2017-5-16; Accepted 2017-9-27; Published 2018-1-1
Citation:
Chen Z, Krishnamachary B, Penet MF, Bhujwalla ZM. Acid-degradable Dextran as an Image Guided siRNA Carrier for COX-2 Downregulation. Theranostics 2018; 8(1):1-12. doi:10.7150/thno.21052. https://www.thno.org/v08p0001.htm
Other stylesAbstract

Purpose: Effective in vivo delivery of siRNA to silence genes is a highly sought-after goal in the treatment of multiple diseases. Cyclooxygenase-2 (COX-2) is a major mediator of inflammation and its effective and specific downregulation has been of major interest to treat conditions ranging from auto-immune diseases to gastric inflammation and cancer. Here we developed a novel and efficient method to produce a multiple imaging reporter labeled cationic dextran nanopolymer with cleavable positive charge groups for COX-2 siRNA delivery.
Methods: Small molecules containing amine groups were conjugated to the dextran scaffold through acetal bonds that were cleaved in weak acid conditions. With multiple imaging reporters located on different regions of the nanopolymer, cleavage of acetal bonds was visualized and quantified by imaging, for the first time, in cancer cells and tumors.
Results: The biocompatibility of dextran and the rapid cleavage and release of amine groups minimized proinflammatory side effects and COX-2 induction observed with other siRNA carriers, to successfully achieve COX-2 downregulation in cancer cells and tumors. Imaging results confirmed that this nanoplex, consisting of the dextran nanopolymer with COX-2 siRNA, accumulated in tumors, and the amine functional groups were rapidly cleaved in cancer cells and tumors. Along with effective downregulation of COX-2, we also demonstrated, for the first time, effective downregulation of its major product prostaglandin E2 (PGE2).
Conclusions: We successfully developed an efficient method to produce an acid-degradable dextran nanopolymer containing cleavable amine groups as the siRNA carrier. Because of its biocompatibility, this degradable dextran delivered COX-2 siRNA within tumors and efficiently downregulated COX-2 expression.
Keywords: dextran, COX-2 siRNA therapy, biodegradable polymer, imaging, cancer
Introduction
As a molecular-based therapeutic strategy, specific gene knockdown using small interfering RNA (siRNA) is being actively investigated for the treatment of many diseases such as cancer, inflammation, diabetes, and neurodegenerative diseases.[1, 2] siRNA can enter the RNA-induced silencing complex (RISC) to induce enzyme-catalyzed degradation of the complementary mRNA in diseased cells, thus disrupting specific molecular pathways. siRNA-mediated silencing of target mRNAs has the potential to down-regulate pathways not accessible with pharmacological agents, without non-specific side-effects that are frequently associated with pharmacological agents.[3, 4] The down-regulation of cyclooxygenase-2 (COX-2) using siRNA is one example where the application of this technology would be of significant importance for a critical target that has not been fully utilized due to the side-effects of pharmacological inhibitors.[5] The two major isoforms of COX, COX-1 and COX-2, transform arachidonic acid into prostaglandins (PGs) that are important biological mediators of inflammation.[6, 7] COX-1 is constitutively expressed in normal tissues, whereas the inducible form, COX-2, shows high expression in inflammatory tissues and many cancers.[8] COX-2 has been found to play an important role in invasion and metastasis in prostate, colorectal, and breast cancer.[9-11] Consequently, COX-2 as a target has attracted significant pharmaceutical interest for multiple degenerative diseases and cancer.[12] Unlike pharmacological COX-2 inhibitors that have significant side-effects,[13] COX-2 siRNA can provide specific and effective down-regulation of COX-2.[14]
Although siRNA is a rapidly emerging class of new therapeutic molecules for the treatment of inherited and acquired diseases, there are some barriers in achieving efficient siRNA therapy. Because of the high molecular weight and negative charge in the form of phosphate, free siRNA cannot enter cells efficiently. Further, naked siRNA has a short lifetime in circulating blood due to nucleases and the innate immune system [15, 16], requiring the use of suitable carriers for successful siRNA therapy. The lack of suitable biocompatible carriers that can achieve effective siRNA delivery and downregulate target genes has, however, limited human translational applications of siRNA therapy. A large number of carriers such as viral vectors, cationic lipids, and polymers have been investigated for siRNA delivery.[17-23] Viral vectors raise safety concerns due to off-target immunogenicity, inflammatory response, and toxicity.[24] Therefore, non-viral gene delivery systems have been extensively explored as alternative gene carriers owing to lesser immunogenicity and toxicity than viral vectors. Among non-viral vectors, cationic polymers are increasingly investigated because of their versatility in delivering siRNA or cDNA, the ability to introduce specific targeting ligands, and ease of production.[25, 26] Compared with less stable cationic lipids, cationic polymers that trap molecular agents through electrostatic binding are the most promising due to their convenience and low cost of synthesis.
Effective COX-2 downregulation using carriers delivering COX-2 siRNA has remained a major challenge because COX-2 is easily induced by extrinsic chemicals, or by artificial cationic polymers with a high density of positive charges.[27] There are few, if any, reports about the use of artificial cationic polymers to downregulate COX-2 in cancer cells in vivo and in vitro. Because increase of COX-2 expression can increase aggressiveness,[11, 28] nanoparticles used in oncological applications should ideally not induce COX-2. Therefore, excellent biocompatibility of a cationic polymer as a carrier is vital for successful siRNA delivery in general, and COX-2 siRNA delivery in particular. Such a cationic polymer should be biodegradable and should rapidly release siRNA in cancer cells.[29] Small molecules that are products of polymer degradation are more rapidly metabolized than cationic polymers, resulting in a shorter residence time that minimizes side-effects. Biodegradation triggered within cells provides a strategy to resolve the coexistence of two conflicting requirements in the rational design of polymeric vectors: condensation and release of siRNA.[30, 31] Cationic polymers possess the ability to form a compact polymer complex with siRNA and to transfer siRNA into cells efficiently; however, such strong complexation impedes the release of siRNA for initiation of post-transcriptional silencing. Once the degradation of the polymer occurs in cells, it loses the ability to compact the siRNA resulting in rapid siRNA release. This makes rapid polymer degradation once inside cells, a highly desirable attribute for effective siRNA delivery.
Here we developed a novel method to efficiently synthesize a multiple imaging reporter labeled biodegradable dextran to use as an efficient cationic nanopolymer carrier for COX-2 siRNA delivery. As a homopolysaccharide of glucose, dextran has been used as a drug carrier in human applications due to its biodegradability, wide availability, and ease of modification.[32] The potential applications of dextran, such as in micelles and hydrogels for siRNA delivery, have recently been demonstrated.[33-35] In our study, amine groups that electrostatically bind siRNA were conjugated to the dextran platform through acetal bonds. Acetal bonds are attractive because they are cleaved under acidic conditions that occur within endocytotic compartments [36, 37], to rapidly release the amine groups. Rhodamine was conjugated to the amine groups to detect release of these groups from the dextran scaffold and their removal from cancer cells following cleavage of the acetal bonds, while the dextran scaffold was labeled with Cy5.5. With these imaging reporters, the distribution and degradation of this dextran carrier in vitro and in vivo were investigated and visualized by optical imaging, for the first time. The rapid release of amine groups minimized the proinflammatory side-effects of the positively charged amine group, making this cationic nanopolymer a useful carrier for siRNA delivery to downregulate COX-2 expression in cancer cells and tumors. Since increased COX-2 expression can result in an aggressive phenotype in cancer cells, identification of a nanopolymer that does not increase COX-2 expression provides a clear path for translational applications of siRNA delivery in cancer.
Results and Discussions
Synthesis and Characterization of Nanoplex
The synthesis of the dextran polymer is presented in Scheme 1. Dextran (70 kDa) was oxidized to form dextran with an aldehyde functional group. The actual oxidation degree determined by the trinitrobenzene sulfonic acid (TNBS) assay [38] was 3.1 ± 0.2%. Excess ethylenediamine (>20 eq) was added to the oxidized dextran PBS solution (pH 7.0) to form the Schiff base. This unstable Schiff base was reduced to a stable nitrogen-carbon bond compound by sodium cyanoborohydride. Cy5.5 was conjugated to the dextran platform (1.1 Cy5.5 molecule per dextran molecule), and the unreacted amine groups were blocked by N-succinimidyl acetate to prevent further side-reactions, such as the conjugation between rhodamine and amine groups, during further modifications. The Cy5.5 labeled dextran scaffold was reacted with an excess of compound 1 to attach the amine groups to the dextran polymer through the acetal bonds. 1H NMR spectra indicated that the functionalized degree of glucose residues was ~0.5. Finally, rhodamine was conjugated to the cleavable amine groups (1.2 rhodamine molecule per dextran molecule).
Scheme 1
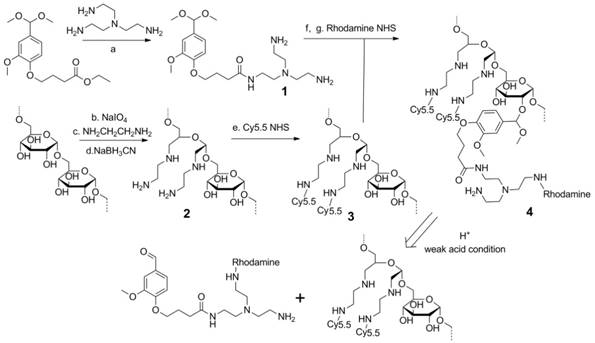
Synthesis procedure and degradation mechanism of the dextran siRNA carrier under acidic conditions. (a) 100°C, overnight. (b) pH 5.0, room temperature, 4 h. (c) PBS pH 7.0 buffer, room temperature, 2 h. (d) PBS pH 7.0 buffer, room temperature, overnight. (e) HEPES buffer, pH8.4, room temperature, 2 h. (f) p-toluenesulfonic acid, 4 Å molecular sieves, 65°C, overnight. (g) HEPES buffer, pH 8.4, room temperature, 2 h.
Although the use of acid degradable dextran with acetal bonds as a siRNA carrier has been previously reported, [39, 40] limitations in the synthesis prevented further modification of the dextran scaffold. In the previously reported method, the formation of acetal bonds preceded the introduction of amine functional groups, resulting in problems arising from the premature introduction of acetal bonds. Primarily, in the reported method, 20-40 equivalent amine molecules per acetal bond were reacted with the dextran platform at 50 °C for two weeks to conjugate the amine functional groups. With concentrated alkali, long reaction times, and high temperature, this reaction condition was not compatible with attaching several functional groups to the dextran scaffold. In fact, the cyanine and rhodamine dyes used as imaging probes in our nanopolymer were not stable under this reaction condition. As a result, this previously reported method could not be used to label imaging reporters to the dextran platform to investigate distribution and degradation in cells and in vivo. Additionally, attaching amines during the final step would cause crosslinking between the dextran molecules, affecting the size distribution of the dextran polymer. To achieve efficient binding with siRNA, a large number of small molecules with multi amine functional groups were conjugated to the dextran scaffold by reaction with ester. If the amine groups in one molecule reacted with different dextran scaffolds, this would induce crosslinking between the dextrans. Use of the previously reported method resulted in a wider size distribution of the dextran nanopolymer, and there were some particles with abnormally large sizes. Our novel cationic dextran polymer synthesis method overcame both limitations of the previous method, demonstrating the ability to modify the dextran scaffold with a variety of functional groups such as imaging reporters and targeting moieties, but avoiding crosslinking of the dextran scaffold. Using this novel synthesis method, we synthesized multi-imaging reporter labeled degradable dextran nanopolymer as an siRNA carrier; the imaging reporters allowed detection of degradation, distribution and metabolism of the nanocarrier in vitro and in vivo.
Dynamic light scattering (DLS) was performed to investigate the hydrodynamic radius of the dextran carriers (Figure 1A). The radius of dextran (70 kDa) was ~6 nm, and the radius increased to ~9.8 nm after modifications. This result was also confirmed by transmission electronic microscopy (TEM) (Figure 1B) that identified a diameter of ~20 nm. After binding with COX-2 siRNA, the size of the nanoplex did not change significantly, and remained at a radius of ~9.9 nm. With the attachment of cleavable amino groups, the zeta potential of compound 4 increased to 22.2±2.3 mV, compared to -12.3±0.4 mV of natural dextran. This positive charge was sufficient to achieve electrostatic binding with siRNA. We also examined the complexation between siRNA and the amino-dextran using gel electrophoresis. Compound 4 formed firm complexes with siRNA at nitrogen/phosphate (N/P) ratios of 15 within 20 min incubation in PBS solution (pH 7.4) (Figure S2). With this N/P ratio the COX-2 siRNA in this nanoplex was stable in mouse and human serum for up to 8 h of incubation (Figure S3). The zeta potential of compound 4/siRNA nanoplex at N/P ratio of 15 was 12.2±1.7 mV. Cytotoxicity of compound 4 evaluated using an MTT assay showed the absence of toxicity in MDA-MB-231 human breast cancer cells with concentrations as high as 100 µg/mL (Figure S4).
Figure 1
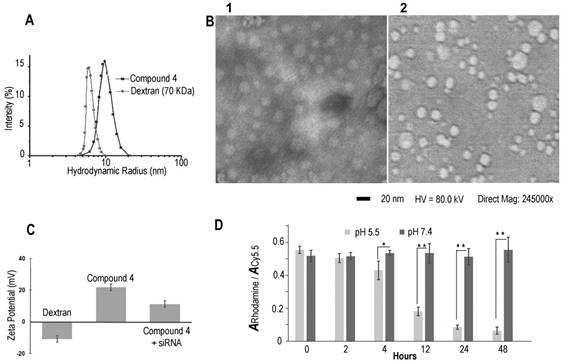
(A) Hydrodynamic radius of dextran and compound 4. (B) TEM image of compound 4 (B1) and COX-2 siRNA/compound 4 complex (B2). (Negative staining with phosphotungstic acid (PTA), scale bar is 20 nm). (C) Zeta potential of dextran and compound 4. (n=3, values represent mean ± SD.) (D) The ratio of rhodamine absorbance to Cy5.5 absorbance at pH 5.5 and pH 7.4 buffer at different time points. (n=3, values represent mean ± SD, **: P < 0.01, *: P < 0.05).
In molecular agent (cDNA, siRNA) delivery, release of the molecular agent from its carrier is critical to achieve successful transfection. Although the proton sponge hypothesis has been proposed as an advantage for effective endosomal escape of cationic polymer carriers, release of molecular agents from the carrier is equally important to achieve good transfection efficiency.[41] In our dextran carrier, the amine groups were linked to the dextran scaffold though acetal bonds that were cleaved under weak acid conditions. With the cleavage of acetal bonds, the amine groups broke away from the dextran scaffold to release the molecular agent cargo. Degradation studies of compound 4 were performed at low and neutral pH conditions (acetate buffer, pH 5.5 and PBS buffer, pH 7.4) using a colorimetric assay to detect Cy5.5 and rhodamine. The progress of degradation was monitored by comparing the ratio of the absorbance of rhodamine at 530 nm to the absorbance of Cy5.5 at 670 nm. The Cy5.5 probe was conjugated to the dextran platform through the amide bond that was stable to variations of pH. As a result, the absorbance of Cy5.5 showed very small variations at different pH buffers over time. Because rhodamine was conjugated to the amine group that was cleaved from the dextran scaffold under weak acid conditions, the absorbance of rhodamine decreased in pH 5.5 buffer over time. Since the compound was stable in pH 7.4 buffer, the ratio of the absorbance of rhodamine to the absorbance of Cy5.5 should not show significant change at this pH. In contrast, at pH 5.5 buffer this ratio should decrease after removing the released dye by molecular weight cut-off centrifugation. As shown in Figure 1D, in PBS buffer at pH 7.4 there was no obvious degradation and the absorbance ratios remained nearly constant up to 48 h of incubation. In contrast, amine groups were cleaved from the dextran scaffold at pH 5.5 starting at 2 h, and the absorbance of rhodamine decreased significantly as the incubation time progressed. These compounds were almost completely degraded after 48 h. Although aldehyde derivatives predominantly generate acyclic acetals that have a shorter half-life in water (pH 5.5 or pH 7.4) than their cyclic analogs, there were small amounts of more stable, difficult to cleave, cyclic acetal byproducts that caused weak absorbance of rhodamine after 48 h incubation in pH 5.5 acetate buffer. Results of the rhodamine to Cy5.5 absorbance ratio in different pH buffers demonstrated that compound 4 was stable at pH 7.4 up to 7 days of (168 h) incubation, and was cleaved effectively under weak acid conditions of pH 5.5. This cleavage at pH 5.5 buffer was also confirmed by NMR spectroscopy (Figure S4) and gel permeation chromatography (Figure S5).
Mechanism of Nanoplex Cellular Internalization
Although several reports have identified the acetal bond as an acid sensitive bond that can be cleaved in weak acidic conditions this has only been demonstrated in buffer systems.[42, 43] Here, for the first time, we used multiple fluorescence reporters combined with imaging to investigate the fate of our dextran/siRNA nanoplex, including the intracellular distribution and cleavage of acetal bonds, in cells. In general, non-viral carriers enter cells through endocytosis vesicles. The carriers are trapped in early endosomes where the pH drops from neutral to around pH 6. More often, the delivery systems are trafficked to late endosomes that are rapidly acidified to pH 5-6 by the action of the membrane-bound ATPase proton-pump. Subsequently, the late endosomes fuse with lysosomes that are at pH 4.5 and contain several degradative enzymes.[44] Endocytosis is therefore a key step for acid controlled degradation of our dextran/siRNA nanoplex because of the weak acid conditions that exist in endosomes. Evidence of the localization of our degradable dextran in endosomes would identify and confirm the degradation in cells in culture. We therefore incubated MDA-MB-231 human breast cancer cells pretreated with CellLight® Early Endosomes-GFP, a fusion construct between Rab5a and GFP that specifically binds to early endosomes, with the siRNA/compound 4 nanoplex for 1 h. In Figure 2A, the early endosomes-GFP can be observed in green, and most of the siRNA/compound 4 nanoplexes, detected by red fluorescence, were localized within these endosomes. These data indicated that this siRNA/compound 4 nanoplex localized in organelles that were weakly acidic in nature, a precondition required for rapid cleavage of acetal bonds.
Figure 2
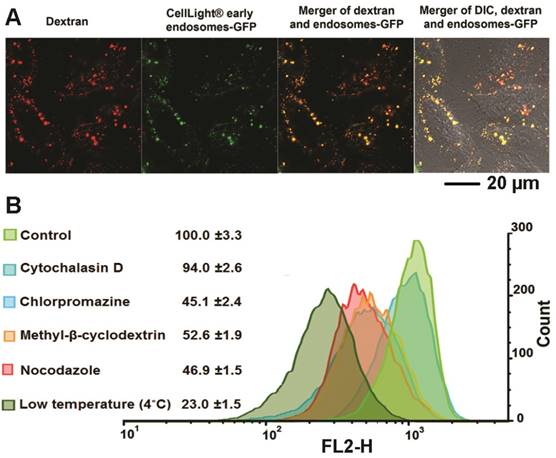
(A) Representative laser confocal fluorescence microscopy fields of view image of MDA-MB-231 cells incubated for 1 h with siRNA/compound 4 nanoplex (red, rhodamine) and endosome marker (green, CellLight® Early Endosomes-GFP) (siRNA concentration: 20 nM; N/P ratio: 15). (B) Quantification of relative fluorescence intensity of the siRNA/compound 4 nanoplex in MDA-MB-231 human breast cancer cells treated with different endocytosis inhibitors (values represent mean ± SD from three independent experiments).
We further investigated the endocytosis pathway of siRNA/compound 4 nanoplex through quantification of the fluorescence intensity of the internalized nanoplex in the presence of endocytosis inhibitors. There are several different pathways for the endocytosis of particles, with specific inhibitors available to block these pathways to determine the effect on the nanoplex cellular uptake. The relative intensity of red fluorescence obtained by flow cytometry (normalized to intensity without inhibitor treatment) was measured to determine the amount of nanoplex uptake (Figure 2B). Cytochalasin D was used to inhibit phagocytosis and micropinocytosis;[45] chlorpromazine hydrochloride was used to inhibit clathrin mediated endocytosis;[46] methyl-β-cyclodextrin was used to inhibit caveolae mediated endocytosis;[20] and nocodazole was used as a microtubule-disrupting agent. [47] Among these treatments, chlorpromazine hydrochloride, methyl-β-cyclodextrin and nocodazole demonstrated inhibition of the siRNA/compound 4 nanoplex uptake. A slight inhibition by cytochalasin D indicated that the uptake of the nanoplex was only mildly dependent on the phagocytosis and micropinocytosis pathways that are used to endocytose large particle (>100 nm). The significant reduction of fluorescence intensity at low temperature indicates an energy dependent uptake, and the absence of internalization through passive diffusion.
These data confirmed that our degradable nanoplex entered cells through endocytosis, and localized within the endosome under weak acid conditions after entering cells. Localization in weak acidic cell organelles supported the possibility of degradation once the nanoplex was within cells. In Figure 2A, we detected strong fluorescence from the nanoplex within 1 h of incubation, indicating that this dextran nanopolymer with suitable introduction of amine groups could rapidly deliver siRNA within cells. The endocytosis pathway study also provided evidence of the size distribution of our nanoplex. Due to the novel synthesis method, the size distribution of the dextran was well controlled, and there were no large nanopolymers induced by crosslinking. Inhibition of phagocytosis and micropinocytosis pathways that are only used to endocytose large particles, did not modify uptake of the nanoplex.
Degradation of Nanoplex in Cells
Multi labeled imaging provided a means to visualize the degradation of the nanoplex in cultured cells. In compound 4, Cy5.5 was conjugated to the dextran scaffold, rhodamine was labeled to the cleavable amine group, and the siRNA was labeled with fluorescein isothiocyanate F(ITC). Therefore, absence of co-localization of these dyes would indicate degradation of the siRNA nanoplex. Investigating the degradation of compound 4 in live cells was performed with laser scanning confocal microscopy (Figure 3). Cells were treated with FITC labeled siRNA/compound 4 nanoplexes for 2 h, and then were further incubated in fresh medium at the times shown in Figure 3. In Figure 3, the fluorescence from FITC is displayed in green, the fluorescence from Cy5.5 labeled on the dextran scaffold is displayed in cyan, and the fluorescence from rhodamine labeled on cleavable amines is displayed in red. After 1 h of incubation in fresh medium, strong fluorescence was observed from FITC, Cy5.5 and rhodamine. Co-localization of fluorescence from Cy5.5 and rhodamine was clearly evident. Delocalized fluorescence of FITC indicated partly released siRNA. After 4 h of incubation in fresh medium, strong fluorescent signals from FITC, Cy5.5 and rhodamine were still observed but the absence of co-localized fluorescence of rhodamine and Cy5.5 in the cytoplasm indicated that the acetal bonds had been cleaved, and free small molecules with amine groups were released into the cytoplasm. The changes of Mander's colocalization coefficients of Cy.5.5 and rhodamine (m1=0.901; m2=0.912 at 1 h, m1=0.921; m2=0.637 at 4 h) supported this absence of co-localization. At 12 h after treatment, a much weaker fluorescence of rhodamine, compared to fluorescence at 4 h, indicated that most acetal bonds had been cleaved. Because cleaved small molecules with amine groups are readily released from cells, the fluorescence intensity of rhodamine in the cytoplasm was very weak at 12 h. The amine groups were almost completely cleaved and removed from cells at 24 h after treatment, while the fluorescence of Cy5.5 that was conjugated to the dextran platform was still detected due to the slower metabolism of the larger sized dextran scaffold.
These imaging data indicated that the amine groups conjugated to the dextran scaffold through the acetal bonds were cleaved in endosomes under weak acidic condition. The increased delocalization of the siRNA (green color) and dextran scaffold (cyan color) proved that the cleavage of amine groups caused efficient release of siRNA from the nanoplex. The rapid release of cleaved small molecules with amine groups from cells was identified from the elimination of red fluorescence. Therefore, the side-effects of these small molecules were minimized. Although the imaging results showed the dextran scaffolds could be retained in the cells for a longer time compared with the small amine molecules, they did not induce inflammation due to their high biocompatibility.
Downregulation of COX-2 in Cells
Treatment with COX-2 siRNA should decrease COX-2 expression levels, but established artificial cationic polymer carriers enhanced COX-2 expression and were found to be proinflammatory. A cationic polymer carrier that induces minimal inflammation is critically important for delivering COX-2 siRNA to achieve effective downregulation. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed to measure COX-2 expression in MDA-MB-231 human breast cancer cells (Figure 4A). Cells were treated with COX-2 siRNA/compound 4 nanoplex for 8 h, after which the medium containing the released amines was replaced with fresh medium. After 16 h of further incubation, cells were collected and processed for qRT-PCR assay to determine messenger RNA (mRNA). In some experiments 12-O-tetradecanoylphorbol-13-acetate (TPA)[48] was added to the fresh medium to induce COX-2 expression at 6 h before cell harvest. As shown in Figure 4A, when polyethyleneimine-polyethylene glycol (PEI-PEG), an ordinary cationic polymer transfection agent,[49, 50] and LipofectamineTM 2000, which is a commercial transfection agent, were chosen to deliver COX-2 siRNA there was no decrease in COX-2 mRNA levels. Instead, following treatment with COX-2 siRNA with PEI-PEG, the level of COX-2 significantly increased to 139% (with TPA induction) or 109% (without TPA induction) compared to COX-2 mRNA levels without siRNA treatment. The commercial transfection agent LipofectamineTM 2000 showed similar results as PEI. In contrast, our degradable compound 4 with COX-2 siRNA demonstrated significant inhibition of COX-2 expression. After COX-2 siRNA/compound 4 nanoplex treatment, the level of COX-2 decreased to 58% (with TPA induction) or 42% (without TPA induction) of COX-2 level without siRNA treatment.
The cyclooxygenase activity of COX catalyzes the formation of prostaglandin G2 (PGG2), an unstable intermediate that is quickly converted to prostaglandin H2 (PGH2) by the peroxidase activity of the enzyme. From this precursor, PGE synthase initiates the production of PGE2. Its activity influences inflammation, fertility, immune modulation, and promotes cancer cell growth, survival, and angiogenesis.[51] Decreased expression of COX-2 should lead to a reduction of PGE2. We therefore investigated PGE2 concentration in cell culture medium following different treatments (Figure 4B). Compared with PGE2 concentration in the medium without treatment, we found that the dextran compound 4 did not affect PGE2 concentration. When we treated MDA-MB-231 human breast cancer cells with COX-2 siRNA/compound 4 nanoplex, PGE2 concentration in the medium significantly decreased to 126 pg/mL, which was 15% of the concentration with compound 4 treatment without siRNA. In contrast, following treatment with PEI-PEG, PGE2 concentration in medium increased significantly to approximately 5400 pg/mL. Because the PEI-PEG carrier increased PGE2 concentration, the COX-2 siRNA/PEI-PEG nanoplex was unable to reduce PGE2 concentration.
As mentioned earlier, there are few if any reports about cation polymers as carriers for COX-2 siRNA delivery. Here we used PEGylated PEI, a common cationic polymer applied in siRNA transfection [52, 53] as a control. PEI increased the expression of COX-2 to such an extent that it offset downregulation caused by siRNA. Our degradable compound 4 was cleaved to release amine groups with positive charge. Compared to macromolecules, the small molecules with amine groups were rapidly removed from cells. These detached amine groups were distributed more uniformly in the cytoplasm, which reduced positive charge density. Due to high biocompatibility, although the dextran scaffold was depolymerized to small molecules by α-1-glucosidases present in cells and various organs,[32] their retention in cells did not affect COX-2 expression. Consequently, our degradable compound 4 minimized inflammation, and was able to downregulate COX-2 successfully.
In Vivo and Ex Vivo Imaging of siRNA/compound 4 Nanoplex in Tumors
The in vivo distribution of COX-2 siRNA/compound 4 nanoplex was analyzed by fluorescence imaging of Cy5.5 (Figure 5A). Strong accumulation of the nanoplex in orthotopically implanted MDA-MB-231 tumors was observed at 24 h post injection, while fluorescence from Cy5.5 was observed in the liver and kidney. At 48 h post injection, accumulation of nanoplex in tumors slightly decreased compared to the 24 h time point. Accumulation of the nanoplex in tumors detected by imaging strongly indicated that the carrier could effectively deliver COX-2 siRNA within tumors. Because the fluorescence of rhodamine could not penetrate the tissue and skin, in vivo imaging could not confirm degradation of the dextran carrier in tumors in vivo. We therefore performed ex vivo imaging to identify degradation of compound 4 in tumors. In Figure 5B, fluorescence imaging at 24 h after injection detected strong signals from both Cy5.5 and rhodamine. At 48 h after injection, the fluorescence from Cy5.5 and rhodamine was weaker than at 24 h after injection. Decrease of the fluorescence intensity of rhodamine at 48 h was, however, more significant than Cy5.5. Figure 5C displays quantification of the ex vivo fluorescence intensity. The fluorescence intensity of Cy5.5 was 2.74 at 24 h, and decreased to 1.84 (67% of the value at 24 h) at 48 h. In contrast, the fluorescence of rhodamine was 4.33 at 24 h, and decreased to 0.79 (18% of the value at 24 h) at 48 h.
The enhanced permeability and retention (EPR) effect in solid tumors is a characteristic that facilitates tumor specific delivery of macromolecules. Because of high tumor vascular permeability, macromolecules accumulate in solid tumors.[54, 55] The size of the COX-2 siRNA/compound 4 nanoplex with a diameter of ~20 nm was ideal for the EPR effect, allowing significant accumulation of the nanoplex, as observed in Figure 5A. The prolonged retention time of the dextran scaffold caused a slow decrease of the fluorescence of Cy5.5 as shown in Figures 5B and 5C. The acidic environment within endosomes facilitated cleavage of the acetal bonds linking the amine groups to the dextran scaffold. Once the small amine groups were cleaved, they were rapidly metabolized compared to the dextran scaffold. Since rhodamine was only labeled on the small molecules with amine groups, rhodamine clearance was also more rapid than Cy5.5 that was attached to the dextran scaffold. A small amount of acyclic acetal bonds that were not cleaved resulted in some fluorescence from rhodamine, even 48 h after injection, in tumor tissues.
Figure 3
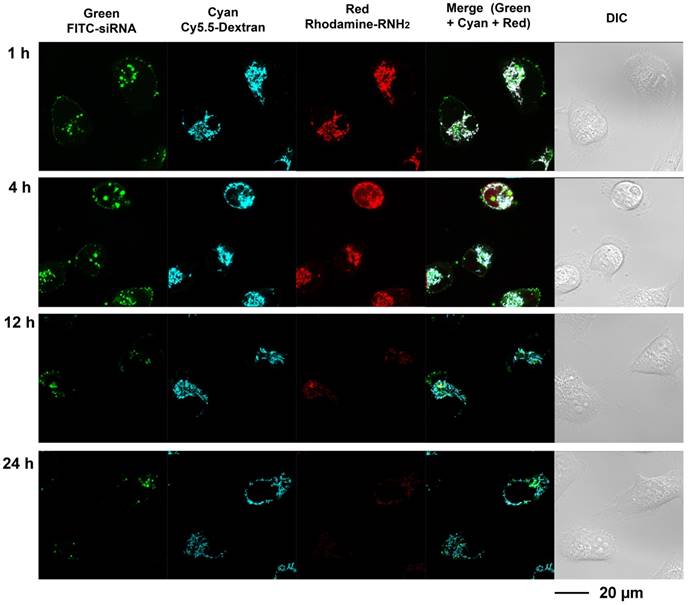
Live-cell laser confocal fluorescence microscopy of living MDA-MB-231 cells with siRNA/compound 4 nanoplex. Cells were treated with FITC-siRNA/compound 4 nanoplex (concentration of siRNA: 100 nM; N/P ratio: 15).
Downregulation of COX-2 in Tumors
Downregulation of COX-2 achieved by siRNA/compound 4 nanoplex in MDA-MB-231 tumors is presented in Figure 6. In the immunoblot results (Figure 6A), at 24 h after injection, all tumors treated with COX-2 siRNA/compound 4 nanoplex demonstrated significant downregulation of COX-2 expression. At 48 h post injection, a trend towards lower COX-2 expression continued. The difference in COX-2 expression between 24 and 48 h post injection could be due to the half-life of the unmodified siRNA in the nanoplex, presence of nucleases in the tumor microenvironment, and dilution of signal in the dividing tumor cells. To further validate this difference, qRT-PCR was performed to measure in vivo COX-2 mRNA levels in tumors (Figure 6B). The fold mRNA expression was normalized to the expression of 24 h treatment of compound 4 alone. With COX-2 siRNA treatment, mRNA expression decreased to 0.43-fold of expression without treatment at 24 h post treatment compared to 0.59-fold at 48 h. These results demonstrate that our degradable dextran compound 4 could effectively deliver COX-2 siRNA into tumors and downregulate the expression of COX-2. However continued dosing may be required to achieve sustained downregulation over longer periods of time. Because of the rapid degradation of the dextran nanopolymer, COX-2 siRNA was quickly released inducing downregulation of COX-2 protein and mRNA expression within 24 h.
Conclusion
In summary, we have developed an efficient method to produce an acid-degradable dextran nanopolymer containing cleavable amine groups as the siRNA carrier. The synthesis method allowed modifications of the dextran scaffold for conjugating multiple functional molecules, such as imaging reporters or targeting moieties. This degradable dextran compound is stable under neutral pH conditions, but under weak acid conditions, such as in endosomes, acid labile acetal bonds are cleaved, rapidly releasing the amine groups and siRNA. Cellular localization and degradation of this compound were observed through cell imaging. The labeled fluorescence probes also allowed identification of endocytosis as a pathway of the uptake of this dextran/siRNA nanoplex. The cleavage and release of amine groups did not increase COX-2 expression, unlike other cationic polymer carriers that induced COX-2 expression. This degradable dextran as a cationic polymer siRNA carrier delivered COX-2 siRNA within tumors and efficiently downregulated COX-2 expression. To the best of our knowledge, this is the first report describing the use of a cationic polymer as a successful COX-2 siRNA carrier to effectively downregulate COX-2 in cancer cells and in tumors. Because of its biocompatibility and synthesis reproducibility, this siRNA carrier has a clear path for translational applications to achieve effective COX-2 or other siRNA delivery in patients.
Figure 4
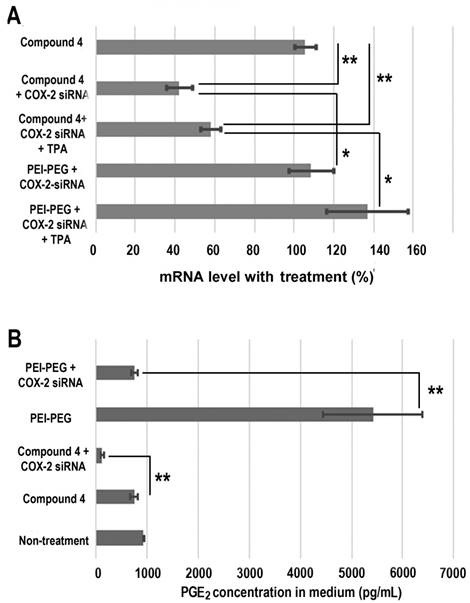
(A) Changes in mRNA levels in MDA-MB-231 cells following different siRNA treatments. (siRNA concentration: 100 nM; N/P ratio: 15. Cells were treated with siRNA-dextran for 8 h, following which the medium was replaced with fresh medium for a further incubation of 24 h before collection. 12-O-tetradecanoylphorbol-13-acetate (TPA) treated cells were incubated with 10 ng/mL TPA to induce COX-2 6 h before collection, and incubated for a further 24 h. Values represent mean ± SD (n = 3). **: P < 0.01, *: P < 0.05. mRNA levels were normalized to untreated cells. (B) PGE2 expression in MDA-MB-231cells: MDA-MB-231 cells were treated with COX-2 siRNA/compound 4 nanoplex (N/P = 15 for dextran; COX-2 siRNA concentration: 100nM). Values represent mean ± SD from three biological experiments. **p ≤ 0.01, n=3).
Experimental Section
Degradation of Compound 4 in Different pH Buffers
Compound 4 (2 mg) was dissolved in 2 mL buffer (pH 5.5 or pH 7.4) and incubated for the desired time. After incubation, the free cleaved amine molecules with rhodamine were removed by molecular weight cutoff centrifugation (Amicon ultra-15, 10,000 MW cutoff). Three further centrifugations in PBS (pH 7.4) buffer were performed to remove free rhodamine completely. Absorbances at 530 nm and 670 nm were recorded.
Cell Culture
MDA-MB-231 human breast cancer cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA). Fetal bovine serum, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). Cells were maintained in RPMI 1640 (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum in a humidified incubator at 37°C/5% CO2. Cells were seeded at a density of 400,000 cells per dish in a 6 cm dish (for RT-PCR experiments) or 20,000 cells per well in a 4-well slide chamber (for confocal laser scanning fluorescence microscopy studies) 24 h prior to the transfection experiment.
Localization of Compound 4/siRNA Nanoplex in Endosomes
MDA-MB-231 cells seeded in a 4-well coverglass chamber were treated with CellLight® Early Endosomes-GFP (Thermo Fisher Scientific, Waltham, MA) for 24 h. Then compound 4/siRNA nanoplex (concentration of compound 4: 1 µg/mL; N/P = 15) was added for 1 h of incubation. After incubation, the transfection mixture was removed, and cells were washed twice with fresh medium. Fluorescence microscopy images of MDA-MB-231 cells were generated on a Zeiss LSM 700 META confocal laser-scanning microscope (Carl Zeiss, Inc. Oberkochen, Germany).
Figure 5
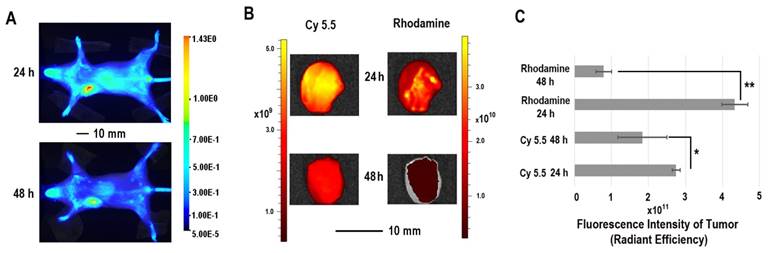
(A) Representative longitudinal in vivo Cy5.5 NIR fluorescence optical images of a SCID mouse bearing an MDA-MB-231 tumor. Mice were injected with nanoplex (compound 4: 2.0 mg/mouse, N/P=15; COX-2 siRNA: 4 nmol/mouse) through the tail vein. (B) Representative ex vivo Cy5.5 and rhodamine fluorescence optical images of MDA-MB-231 tumors at 24 h and 48 h. (C) Quantification of Cy5.5 and rhodamine fluorescence intensity from ex vivo tumors. (** P < 0.01, * P < 0.05, values represent mean ± SD, n = 3).
Quantification of Relative Fluorescence Intensity of Compound 4 Nanoplex in MDA-MB-231 Cells Treated with Different Endocytosis Inhibitors
MDA-MB-231 cells seeded in 6 cm dishes at 60% confluence were treated with endocytosis inhibitors (cytochalasin D: 5 µg/mL; chlorpromazine hydrochloride: 5 µg/mL; methyl-β-cyclodextrin: 6 mg/mL; nocodazole: 6 mg/mL), or incubated at 4 ˚C, for 45 min. Then siRNA/compound 4 nanoplex (siRNA concentration: 100 nM) was added to the solution for a further 2 h incubation under the same conditions. After incubation, cells were harvested, washed, and fixed for quantification of fluorescence intensity. Quantification of fluorescence intensity of dextran uptake was performed by BD FACSCalibur flow cytometry (BD Biosciences, San Jose, CA).
In Vitro Imaging of Degradation of siRNA Nanoplex
MDA-MB-231 cells seeded in 4-well coverglass chambers were treated with siRNA/compound 4 nanoplex (siRNA concentration: 100 nM) for 2 h. After treatment, this medium was replaced by fresh medium for further incubation and investigation. Fluorescence microscopy images of MDA-MB-231 cells were generated on a Zeiss LSM 700 META confocal laser-scanning microscope.
COX-2 qRT-PCR Assay and PGE2 ELISA of MDA-MB-231 Cells
The siRNA/compound 4 nanoplex in RPMI 1640 medium solution (concentration of siRNA: 100 nM, N/P = 15) was added to each dish for 8 h incubation. After incubation, cells were incubated in fresh medium for a further 16 h. In TPA treatment experiments, TPA was added to medium at 6 h before harvest. Before harvesting the cells, the supernatant of cell culture medium was collected, and PGE2 ELISA was performed following instructions provided by the manufacturer (Cayman Chemical, Ann Arbor, MI). ELISA was performed in triplicates. Total RNA was isolated from cells by using QIA shredder and RNeasy Mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. The expression of target RNA relative to the housekeeping gene hypoxanthine phosphoribosyl transferase 1 (HPRT1) was calculated based on the threshold cycle (Ct) as R = 2 - Δ(ΔCt), where ΔCt = Ct of target - Ct of HPRT1. Primer for COX-2 was purchased from Qiagen (Cat. No. QT00040586). The following primers against HPRT1-the house keeping gene-Fwd-5'-CCTGGCGTCGTGATTAGTGATG-3' and Rev-5'-CAGAGGGCTACAATGTGATGGC-3' were designed using either Beacon designer software 7.8 (Premier Biosoft, Palo Alto, CA, USA) or a free web-based software Primer3Plus.
Mouse Model and Tumor Implantation
All in vivo studies were done in compliance with guidelines established by the Institutional Animal Care and Use Committee of the Johns Hopkins University. MDA-MB-231 human breast cancer cells (2×106 cells/mouse) were inoculated orthotopically in the mammary fat pad of female severe combined immunodeficient (SCID) mice. Tumors were palpable within two weeks after implantation and reached a volume of ~300-400 mm3 within four to five weeks, at which time they were used for the studies.
In Vivo and Ex Vivo Optical Imaging Studies
In vivo optical images were acquired with a Pearl® Trilogy Small Animal Imaging System (LI-COR, Lincoln, NE), ex vivo optical images were acquired with an IVIS Lumina Series III Spectrum scanner (Perkin-Elmer, Waltham, MA), and fluorescence intensities in regions of interest (ROIs) were quantified by using Living Image 4.5 software (Caliper, Hopkinton, MA). For in vivo optical imaging of the distribution of COX-2 siRNA/compound 4 nanoplex, MDA-MB-231 tumor bearing mice were injected intravenously with 100 µL of dextran conjugated COX-2 siRNA nanoplex (dextran 2.0 mg/mouse, N/P=20. COX-2 siRNA, 4 nmol/mouse) through the tail vein. Delivery of the nanoplex was confirmed by imaging the mice at 24 h and 48 h. Subgroups of mice were sacrificed either at 24 h or at 48 h after nanoplex injection for ex vivo imaging studies, and tumors and muscle were excised to obtain the optical images.
Figure 6
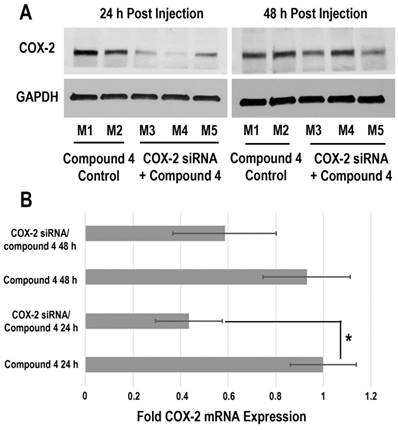
(A) Representative immunoblot assays of COX-2 protein expression in MDA-MB-231 tumors obtained from different mice (M). Mice were injected with siRNA/compound 4 nanoplex (compound 4: 2.0 mg/mouse, N/P=15; COX-2 siRNA: 4 nmol/mouse) through the tail vein. (B) Fold mRNA levels in MDA-MB-231 tumors following different treatments. Concentration of siRNA/ compound 4 nanoplex: compound 4, 2.0 mg/mouse, N/P=15; COX-2 siRNA, 4 nmol/mouse; values represent mean ± SD, n = 3. *: P < 0.05.
In Vivo Downregulation of COX-2 Expression
After imaging, tumors were excised at each time point and freeze clamped for molecular analysis. cDNA was synthesized from RNA isolated from the frozen samples. Total RNA was isolated from tumor tissues by using QIA shredder and RNeasy Mini kit according to the manufacturer's protocol. HPRT1 was used as a housekeeping gene for internal control. In the immunoblot analysis of tumor tissues, proteins were extracted using RIPA buffer with protease inhibitor cocktail (1/500, Sigma, St. Louis, MO), dithiothreitol (1/1,000, 1 M stock), phenylmethylsulfonyl fluoride (1/200, 0.2 M stock), sodium orthovanadate (1/500, 0.5 M stock) and sodium fluoride (1/500, 0.5 M stock). Protein was isolated and quantified from each tumor. 60 µg of protein was resolved on 4-15% gradient SDS gel. Proteins were transferred to a nitrocellulose membrane overnight at 40oC. The membrane was immunoblotted against goat polyclonal anti-COX-2 antibody. Antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control.
Abbreviations
COX-2: Cyclooxygenase-2; siRNA: small interfering RNA; RISC: RNA-induced silencing complex; TNBS: trinitrobenzene sulfonic acid; DLS: dynamic light scattering; TEM: transmission electronic microscopy; N/P ratio: nitrogen/phosphate ratio; FITC: fluorescein isothiocyanate; qRT-PCR: quantitative reverse transcription polymerase chain reaction; TPA: 12-O-tetradecanoylphorbol-13-acetate; PGH2: prostaglandin H2; PGG2: prostaglandin G2; PGE2: prostaglandin E2; PEI: polyethylenimine; EPR effect: enhanced permeability and retention effect; SCID mice: severe combined immunodeficient mice; HPRT1: hypoxanthine phosphoribosyl transferase 1; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; DIC: differential interference contrast.
Supplementary Material
Supplementary figures; detailed synthesis procedure, characterization of compounds and nanoplexes, stability of encapsulated siRNA in serum, degradation assays of compound 4, cytotoxicity assays of compound 4 and ex vivo images of nanoplex in mouse organ.
Acknowledgements
This work was supported by NIH R01 CA82337, R01 CA136576, R01 CA193365, R01 CA73850 and R35 CA209960. We thank Mr. Cromwell for inoculating the tumors. We gratefully acknowledge the support of Dr. K. M. Horton.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kurreck J. RNA Interference: From Basic Research to Therapeutic Applications. Angew Chem Int Edit. 2009;48:1378-98
2. Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11:59-67
3. Davidson BL, McCray PB. Current prospects for RNA interference-based therapies. Nat Rev Genet. 2011;12:329-40
4. Kozielski KL, Tzeng SY, Green JJ. siRNA nanomedicine: the promise of bioreducible materials. Expert Rev Med Devic. 2013;10:7-10
5. Patrignani P, Patrono C. Cyclooxygenase inhibitors: From pharmacology to clinical read-outs. Bba-Mol Cell Biol L. 2015;1851:422-32
6. Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LBA. et al. Cyclooxygenase in biology and disease. Faseb J. 1998;12:1063-73
7. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol. 1998;38:97-120
8. Khan Z, Khan N, Tiwari RP, Sah NK, Prasad GBKS, Bisen PS. Biology of Cox-2: An Application in Cancer Therapeutics. Curr Drug Targets. 2011;12:1082-93
9. Gupta RA, DuBois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11-21
10. Hsu AL, Ching TT, Wang DS, Song XQ, Rangnekar VM, Chen CS. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J Biol Chem. 2000;275:11397-403
11. Stasinopoulos I, O'Brien DR, Wildes F, Glunde K, Bhujwalla ZM. Silencing of cyclooxygenase-2 inhibits metastasis and delays tumor onset of poorly differentiated metastatic breast cancer cells. Molecular cancer research: MCR. 2007;5:435-42
12. Subbaramaiah K, Dannenberg AJ. Cyclooxygenase 2: a molecular target for cancer prevention and treatment. Trends Pharmacol Sci. 2003;24:96-102
13. Blobaum AL, Uddin MJ, Felts AS, Crews BC, Rouzer CA, Marnett LJ. The 2 '-Trifluoromethyl Analogue of Indomethacin Is a Potent and Selective COX-2 Inhibitor. Acs Med Chem Lett. 2013;4:486-90
14. Strillacci A, Griffoni C, Valerii MC, Lazzarini G, Tomasi V, Spisni E. RNAi-Based Strategies for Cyclooxygenase-2 Inhibition in Cancer. J Biomed Biotechnol. 2010
15. Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12:967-77
16. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129-38
17. Baigude H, Rana TM. Delivery of Therapeutic RNAi by Nanovehicles. Chembiochem. 2009;10:2449-54
18. Lee EJ, Lee SJ, Kang YS, Ryu JH, Kwon KC, Jo E. et al. Engineered Proteinticles for Targeted Delivery of siRNA to Cancer Cells. Adv Funct Mater. 2015;25:1279-86
19. Li C, Penet MF, Wildes F, Takagi T, Chen ZH, Winnard PT. et al. Nanoplex Delivery of siRNA and Prodrug Enzyme for Multimodality Image-Guided Molecular Pathway Targeted Cancer Therapy. Acs Nano. 2010;4:6707-16
20. Lin QY, Chen J, Zhang ZH, Zheng G. Lipid-based nanoparticles in the systemic delivery of siRNA. Nanomedicine-Uk. 2014;9:105-20
21. Xu JB, Li JM, Lin S, Wu TY, Huang HQ, Zhang KY. et al. Nanocarrier-Mediated Codelivery of Small Molecular Drugs and siRNA to Enhance Chondrogenic Differentiation and Suppress Hypertrophy of Human Mesenchymal Stem Cells. Adv Funct Mater. 2016;26:2463-72
22. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
23. Zhao J, Feng SS. Nanocarriers for delivery of siRNA and co-delivery of siRNA and other therapeutic agents. Nanomedicine-Uk. 2015;10:2199-228
24. Somia N, Verma IM. Gene therapy: Trials and tribulations. Nat Rev Genet. 2000;1:91-9
25. Nimesh S, Gupta N, Chandra R. Cationic Polymer Based Nanocarriers for Delivery of Therapeutic Nucleic Acids. J Biomed Nanotechnol. 2011;7:504-20
26. Zhang SB, Zhao B, Jiang HM, Wang B, Ma BC. Cationic lipids and polymers mediated vectors for delivery of siRNA. J Control Release. 2007;123:1-10
27. Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomed. 2012;7:5577-91
28. Krishnamachary B, Stasinopoulos I, Kakkad S, Penet MF, Jacob D, Wildes F. et al. Breast cancer cell cyclooxygenase-2 expression alters extracellular matrix structure and function and numbers of cancer associated fibroblasts. Oncotarget. 2017;8:17981-94
29. Son S, Namgung R, Kim J, Singha K, Kim WJ. Bioreducible Polymers for Gene Silencing and Delivery. Accounts Chem Res. 2012;45:1100-12
30. Islam MA, Park TE, Singh B, Maharjan S, Firdous J, Cho MH. et al. Major degradable polycations as carriers for DNA and siRNA. J Control Release. 2014;193:74-89
31. Jere D, Arote R, Jiang HL, Kim YK, Cho MH, Cho CS. Bioreducible polymers for efficient gene and siRNA delivery. Biomed Mater. 2009:4
32. Mehvar R. Dextrans for targeted and sustained delivery of therapeutic and imaging agents. J Control Release. 2000;69:1-25
33. Cohen JL, Schubert S, Wich PR, Cui L, Cohen JA, Mynar JL. et al. Acid-Degradable Cationic Dextran Particles for the Delivery of siRNA Therapeutics. Bioconjugate Chem. 2011;22:1056-65
34. Kim JS, Oh MH, Park JY, Park TG, Nam YS. Protein-resistant, reductively dissociable polyplexes for in vivo systemic delivery and tumor-targeting of siRNA. Biomaterials. 2013;34:2370-9
35. Raemdonck K, Naeye B, Buyens K, Vandenbroucke RE, Hogset A, Demeester J. et al. Biodegradable Dextran Nanogels for RNA Interference: Focusing on Endosomal Escape and Intracellular siRNA Delivery. Adv Funct Mater. 2009;19:1406-15
36. Gillies ER, Goodwin AP, Frechet JMJ. Acetals as pH-sensitive linkages for drug delivery. Bioconjugate Chem. 2004;15:1254-63
37. Knorr V, Russ V, Allmendinger L, Ogris M, Wagner E. Acetal linked oligoethylenimines for use as pH-sensitive gene carriers. Bioconjugate Chem. 2008;19:1625-34
38. Maia J, Ferreira L, Carvalho R, Ramos MA, Gil MH. Synthesis and characterization of new injectable and degradable dextran-based hydrogels. Polymer. 2005;46:9604-14
39. Chen Z, Krishnamachary B, Bhujwalla ZM. Degradable Dextran Nanopolymer as a Carrier for Choline Kinase (ChoK) siRNA Cancer Therapy. Nanomaterials. 2016;6:34
40. Cui LN, Cohen JL, Chu CK, Wich PR, Kierstead PH, Frechet JMJ. Conjugation Chemistry through Acetals toward a Dextran-Based Delivery System for Controlled Release of siRNA. J Am Chem Soc. 2012;134:15840-8
41. Thomas M, Klibanov AM. Enhancing polyethylenimine's delivery of plasmid DNA into mammalian cells. P Natl Acad Sci USA. 2002;99:14640-5
42. Kauffman KJ, Do C, Sharma S, Gallovic MD, Bachelder EM, Ainslie KM. Synthesis and Characterization of Acetalated Dextran Polymer and Microparticles with Ethanol as a Degradation Product. Acs Appl Mater Inter. 2012;4:4149-55
43. Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JMJ. Acetal-derivatized dextran: An acid-responsive biodegradable material for therapeutic applications. J Am Chem Soc. 2008;130:10494 -+
44. Hernandez A, Serrano-Bueno G, Perez-Castineira JR, Serrano A. Intracellular Proton Pumps as Targets in Chemotherapy: V-ATPases and Cancer. Curr Pharm Design. 2012;18:1383-94
45. Kuhn DA, Vanhecke D, Michen B, Blank F, Gehr P, Petri-Fink A. et al. Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J Nanotech. 2014;5:1625-36
46. McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Bio. 2011;12:517-33
47. dos Santos T, Varela J, Lynch I, Salvati A, Dawson KA. Effects of Transport Inhibitors on the Cellular Uptake of Carboxylated Polystyrene Nanoparticles in Different Cell Lines. Plos One. 2011:6
48. Zhang DY, Li JX, Song L, Ouyang WM, Gao JM, Huang CS. A JNK1/AP-1-dependent, COX-2 induction is implicated in 12-o-tetradecanoylphorbol-13-acetate-induced cell transformation through regulating cell cycle progression. Molecular Cancer Research. 2008;6:165-74
49. Malek A, Czubayk F, Aigner A. PEG grafting of polyethylenimine (PEI) exerts different effects on DNA transfection and siRNA-induced gene targeting efficacy. J Drug Target. 2008;16:124-39
50. Chen Z, Penet MF, Krishnamachary B, Banerjee SR, Pomper MG, Bhujwalla ZM. PSMA-specific theranostic nanoplex for combination of TRAIL gene and 5-FC prodrug therapy of prostate cancer. Biomaterials. 2016;80:57-67
51. Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C. et al. The COX-2/PGE(2) pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377-86
52. Chen Z, Penet MF, Nimmagadda S, Li C, Banerjee SR, Winnard PT. et al. PSMA-Targeted Theranostic Nanoplex for Prostate Cancer Therapy. Acs Nano. 2012;6:7752-62
53. Malek A, Merkel O, Fink L, Czubayko F, Kissel T, Aigner A. In vivo pharmacokinetics, tissue distribution and underlying mechanisms of various PEI(-PEG)/siRNA complexes. Toxicol Appl Pharm. 2009;236:97-108
54. Maeda H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. P Jpn Acad B-Phys. 2012;88:53-71
55. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271-84
Author contact
![]() Corresponding authors: Zhihang Chen, PhD, Johns Hopkins University School of Medicine, Russell H. Morgan Dept. of Radiology & Radiological Science, Baltimore MD 21205 Tel: 410 955 6873 Email: zchen19edu and Zaver M. Bhujwalla, PhD, Johns Hopkins University School of Medicine, Russell H. Morgan Dept. of Radiology & Radiological Science, Baltimore MD 21205 Tel: 410 955 9698 Fax: 410 614 1948 Email: zaverjhu.edu
Corresponding authors: Zhihang Chen, PhD, Johns Hopkins University School of Medicine, Russell H. Morgan Dept. of Radiology & Radiological Science, Baltimore MD 21205 Tel: 410 955 6873 Email: zchen19edu and Zaver M. Bhujwalla, PhD, Johns Hopkins University School of Medicine, Russell H. Morgan Dept. of Radiology & Radiological Science, Baltimore MD 21205 Tel: 410 955 9698 Fax: 410 614 1948 Email: zaverjhu.edu
Citation styles
APA
Chen, Z., Krishnamachary, B., Penet, M.F., Bhujwalla, Z.M. (2018). Acid-degradable Dextran as an Image Guided siRNA Carrier for COX-2 Downregulation. Theranostics, 8(1), 1-12. https://doi.org/10.7150/thno.21052.
ACS
Chen, Z.; Krishnamachary, B.; Penet, M.F.; Bhujwalla, Z.M. Acid-degradable Dextran as an Image Guided siRNA Carrier for COX-2 Downregulation. Theranostics 2018, 8 (1), 1-12. DOI: 10.7150/thno.21052.
NLM
Chen Z, Krishnamachary B, Penet MF, Bhujwalla ZM. Acid-degradable Dextran as an Image Guided siRNA Carrier for COX-2 Downregulation. Theranostics 2018; 8(1):1-12. doi:10.7150/thno.21052. https://www.thno.org/v08p0001.htm
CSE
Chen Z, Krishnamachary B, Penet MF, Bhujwalla ZM. 2018. Acid-degradable Dextran as an Image Guided siRNA Carrier for COX-2 Downregulation. Theranostics. 8(1):1-12.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.