Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(18):4577-4590. doi:10.7150/thno.20005 This issue Cite
Research Paper
Subcellular Localization of Survivin Determines Its Function in Cardiomyocytes
Tien-Jui Tsang1, 2, Ying-Chang Hsueh3, Erika I. Wei2, David J. Lundy2, Bill Cheng2, You-Tzung Chen1, 4, Shoei-Shen Wang5, Patrick C.H. Hsieh1, 2, 3, 4, 5 ![]()
1. Graduate Institute of Clinical Medicine, College of Medicine, National Taiwan University, Taipei, Taiwan;
2. Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan;
3. Institute of Clinical Medicine, National Cheng Kung University, Tainan, Taiwan;
4. Institute of Medical Genomics and Proteomics, College of Medicine, National Taiwan University, Taipei, Taiwan;
5. Department of Surgery, College of Medicine, National Taiwan University, Taipei, Taiwan.
Received 2017-3-9; Accepted 2017-8-10; Published 2017-10-13
Abstract

Rationale: Reducing cardiomyocyte death and enhancing their proliferation after myocardial infarction is perhaps the single largest challenge for cardiac tissue regeneration. Survivin (SVV) is the smallest member of the inhibitor of apoptosis (IAP) family but plays two important roles; inhibiting caspase-9 activation in the intrinsic apoptosis pathway, and regulating microtubule dynamics and chromosome segregation during cell division. Genetic depletion of cardiac SVV leads to incomplete cardiomyocyte division and abnormal heart development. However, the function of SVV in adult hearts after myocardial infarction remains unclear.
Methods: A homozygous inducible cardiomyocyte-specific SVV knockout transgenic mouse model was established through crossbreeding SVVflox/flox and αMHC-MCM transgenic mice. Adult mice received consecutive intraperitoneal injection of tamoxifen to induce genetic removal of SVV in cardiomyocytes. A SVV overexpressing model was established via local delivery of SVV in wild-type mouse hearts.
Results: We found that 30.82% of cardiomyocytes in the peri-infarct region of SVV knockout mice were apoptotic, significantly higher than the 22.18% in control mice. In addition, ejection fraction was 29.00±0.40% in knockout mice compared to 38.04±0.50% in control mice 21 days after myocardial infarction. On the contrary, locally overexpressing SVV in the heart improved cardiac functions. Unexpectedly, we found that altering the subcellular localization of SVV overexpression produced different outcomes. Overexpression of SVV in the cytoplasm decreased cardiomyocyte apoptosis, whereas overexpression of SVV in the nucleus enhanced cardiac regeneration. The ejection fraction of mice overexpressing SVV was 36.58±0.91%, significantly higher than 28.18±1.70% in the GFP control group. Apoptotic cardiomyocytes were only 4.63% in mouse overexpressing cytosolic SVV, compared to 9.31% in the GFP group, and activation of caspase-3 was also reduced. Moreover, mice overexpressing NLS-SVV exhibited a better ejection fraction (36.19±1.02%,) than GFP controls (26.69±0.75%). NLS-SVV enhanced H3P-positive cardiomyocytes in the border zone to 0.28%, compared to only 0.08% in GFP group, through interacting with Aurora B.
Conclusions: We demonstrate the importance of SVV subcellular localization in regulating post-MI cardiac repair and regeneration. We hope that this will open new translational approaches through targeted delivery of SVV.
Keywords: cardiac regeneration, myocardial infarction, subcellular localization, surviving.
Introduction
Acute myocardial infarction continues to be a major cause of mortality in many countries. Rapid re-establishment of blood flow following coronary artery occlusion is essential for reducing cardiomyocyte death, which reduces the degree of fibrosis and cardiac remodeling, and preserves cardiac function. However, since the adult human heart possesses only limited endogenous recovery ability, many patients nevertheless go on to develop heart failure. Therefore, there is great need for therapeutic strategies that can further reduce cardiomyocyte death and increase cardiomyocyte proliferation following injury, thus improving patient outcomes.
Several gene therapy trials have focused on administration of angiogenic factors for cardiovascular regeneration [1-4] or the targeted overexpression of anti-apoptotic proteins to reduce cell death and improve heart function [5], with varying degrees of success. Survivin (SVV) is the smallest member of the inhibitor of apoptosis family, and has been shown to play roles in both cell survival and cell proliferation. Therefore, manipulating SVV expression may be an ideal target for therapy post myocardial infarction (MI).
SVV performs its anti-apoptotic function by interfering with apoptosome complex recruitment and reducing apoptosis via inhibition of caspase-9 activation [6-9]. SVV also performs an essential role in cell division where it regulates spindle dynamics and stabilizes microtubule elongation [10]. SVV recognizes phosphorylation on T3 of histone 3 and recruits other members of the chromosomal passenger complex, including Aurora B [11-13]. Correct attachment of the microtubule-kinetochore enables cells to pass through the check point and move on to the next mitosis stage [14-16]. In cancer cells, SVV is over-expressed in almost all human malignancies and correlates to poor clinical outcome, therapeutic resistance, and tumor recurrence [17-20].
During development, SVV is expressed in fetal tissues and is essential for organ development [17]. Universal SVV knockout in mice is embryonically lethal [21], and therefore several conditional SVV depletion mice have been established. SVV deficiency in endothelial cells causes early embryonic lethality before E13.5, and heart development is impaired [22]. Cardiac-specific SVV knockout mice exhibit only a 50% survival rate 34 weeks after birth, and a large number of SVV-deficient cardiomyocytes display DNA polyploidy and reduced mitotic counts with incomplete cell division [23]. These data indicate that SVV plays an important role in heart development. In the human heart, SVV expression remains very low under normal conditions, but dramatically increases in patients with terminal heart failure [23].
Given that SVV can both reduce apoptosis and increase proliferation, we hypothesized that SVV may be involved in the response to cardiac injury, and be a viable target for future therapeutic applications. In this study, cardiac-specific, inducible SVV knockout mice were generated to investigate the role of SVV after heart injury. In particular, we focus on the differential functions of SVV in different subcellular localizations of cardiomyocytes after MI.
Materials and Methods
All experiments involving animals were completed following the Guide for the Use and Care of Laboratory Animals, and all the animal protocols have been proved by the Academia Sinica Institutional Animal Care and Utilization Committee.
Animals
The alpha myosin heavy chain (αMHC) promoter-driven MerCreMer (MCM), purchased from the Jackson Laboratory, were crossed with floxed SVV transgenic mice [24]. Wild-type C57BL/6 mice (WT) were purchased from National Applied Research Laboratories, National Laboratory Animal Center, Taiwan. Male animals aged 8-10 weeks were regarded as adult mice in all the experiments.
RT-PCR analysis
Heart tissues were harvested at different development time points and washed with phosphate-buffered saline (PBS) to remove excess blood. Ventricles, without aortas and atriums, were homogenized and total RNAs were extracted with Trizol reagent (Thermo Fisher Scientific). Total RNAs were reverse transcribed into cDNAs with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) and applied in the following PCR in GoTaq Green Master Mix (Promega). GAPDH primer sequences are ACCCAGAAGACTGTGGATGG and CACATTGGGGGTAGGAACAC, and SVV primer sequences are GCAAAGGAGACCAACAACAAGC and TGACGGGTAGTCTTTGCAGTCT.
Western blotting
Heart tissue, containing infarcted ventricle region only, was homogenized and proteins were extracted with RIPA buffer (150mM NaCl, 50mM Tris-base, 0.5% deoxycholate, 0.1% SDS, and 1% Triton X-100) for SDS-PAGE. Membranes were blocked with 5% non-fat milk and hybridized with the following primary antibodies: mouse anti-SVV (1:500, Novus NB500-238), mouse anti-GAPDH (1:2000, Millipore), mouse anti-phospho-SVV (Thr34) (1:100, Novus), mouse anti-caspase-3 (1:100, GeneTex), and mouse anti-caspase-9 (1:100, Cell Signaling). Secondary antibody, conjugated with HRP, was purchased from Invitrogen (1:2000).
Immunohistochemical staining
Heart tissues were harvested and then washed or perfused with PBS to remove excess blood. Clean tissues were fixed with 4% paraformaldehyde (PFA) at 4˚C overnight and embedded in paraffin. Immunostaining was performed on 5-μm sections, blocked with PBS containing serum (20% goat serum and 20% FBS), and hybridized with the following primary antibodies: rabbit anti-SVV (1:100, Cell Signaling), mouse-anti cardiac troponin I (cTnI) (1:100, Developmental Studies Hybridoma Bank), mouse anti-phospho-histone 3 (Ser10) (H3P) (1:100, Millipore), and wheat germ agglutinin (WGA) conjugated with Alexa-488 (Invitrogen). In immunofluorescence staining, the following secondary antibodies were applied: goat anti-rabbit conjugated with Alexa-586 (1:200, Invitrogen), and goat anti-mouse conjugated with Alexa-488 (1:200, Invitrogen). Nuclei were counterstained with 4',6-di-amidino-2-phenyliodole (DAPI, 1 μg/mL; Sigma-Aldrich). For DAB (3,3'-diaminobenzidine) staining, secondary antibody was applied with goat anti-rabbit conjugated with HRP (1:200, Invitrogen), and ABC kit and peroxidase substrate kit (Vector Laboratories) were used. Nuclei were counterstained with hematoxylin (Invitrogen). Neonatal mouse hearts were fixed with 4% PFA and incubated in PBS containing 0.25% Triton X-100. Following staining, the primary antibody, mouse anti-cTnI, and secondary antibody, goat anti-mouse conjugated with Alexa-586 (Invitrogen), were used. Nuclei were counterstained with DAPI.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
TUNEL staining was applied on heart tissue sections or fixed neonatal mouse hearts following the kit protocol. For the DAB system, DeadEnd Colorimetric TUNEL system (Promega) was used, and nuclei were counterstained with hematoxylin. For the fluorescence system, Apoptag Red In Situ Apoptosis Detection Kit (Millipore) was used, and nuclei were counterstained with DAPI.
Adenovirus establishment
PCR products of the full length of SVV, mutant SVV-T34A (T34A), SVV fused with nuclear localization signal (NLS) in C-terminal, and green fluorescence protein (GFP), as vehicle control, were cloned and purified. Cytomegalovirus (CMV) promoter and PCR product were ligated into pENRT vector (Invitrogen). Promoter and target gene on pENRT were homologously recombinated into pAd/PL-DEST vector following the protocol and reagents of Gateway LR Clonase II Enzyme Mix (Thermo Fisher). Plasmids were transfected into HEK-293A cells, and the adenovirus was amplified and purified followed the procedure reported previously [25].
Trichrome staining
Scar tissue was stained on heart tissue sections with Trichrome Stain (Masson) Kit (Sigma Aldrich), following the manufacturer's protocol. Ventricles below the coronary artery ligation point were equally divided into three parts, and three sections were collected from each heart. Scar area, length, and left ventricle wall thickness were evaluated and quantified with ImageJ. Scar area percentage was evaluated by the ratio of scar area to left ventricle area. Scar length was quantified with the mid-line length. Left ventricle wall thickness was measured in three points, with two end points and one in the middle, and reported as the mean.
Animal surgery and echocardiography
Adult transgenic mice were intraperitoneally injected with tamoxifen (Sigma Aldrich) 100 mg/kg for 14 consecutive days. After stopping the injections for 28 days, mice were anesthetized with isoflurane USP (Halocarbon) and received left anterior descending artery ligation with 6-0 polypropylene thread. Cardiac functions were recorded and analyzed by echocardiography.
Statistics
All statistical data were analyzed using Graphpad Prism and are shown as mean ± standard error of the mean (SEM). Unpaired Student's t-test and two-way ANOVA were applied for statistical comparisons and a P-value of less than 0.05 was considered significant.
Results
Cardiac SVV expression attenuates after birth but re-activates after MI
To determine the role that SVV plays in heart healing, the expression pattern of SVV in normal heart development and in post-MI was investigated. Murine hearts were isolated at different time points from embryonic to postnatal stages. SVV was found to be highly expressed throughout the embryonic stages but declined after birth (Figure 1A and Figure S1A), and the protein level was downregulated from postnatal day 4 onwards (Figure 1B). Immunostaining of heart tissue sections revealed that SVV was co-localized with the cardiomyocyte nucleus (Figure 1C). Additionally, the numbers of cells stained positively for cTnI and SVV also decreased from embryonic to postnatal stages (Figure 1D), suggesting that SVV played an important role during embryonic development of the heart, but not after birth, and that it mainly localized in the nucleus of cardiomyocytes.
Expression pattern and localization of Survivin (SVV) in heart development and after myocardial infarction (MI). A, RT-PCR of SVV mRNA expression level during heart development. B, Western blot of SVV protein expression level in different development stages. C, Immunofluorescence staining of SVV expression in cardiomyocytes during heart development. cTnI, cardiac troponin I; blue, nucleus; green, cardiomyocyte marker. White bar, 20 μm; yellow bar, 5 μm. D, Quantification of SVV positive cardiomyocytes during different heart development stages. E, Western blot showing SVV protein expression levels at different time points after MI. F, Immunofluorescence staining showing localization of SVV after injury. White bar, 20 μm; yellow bar, 5 μm.
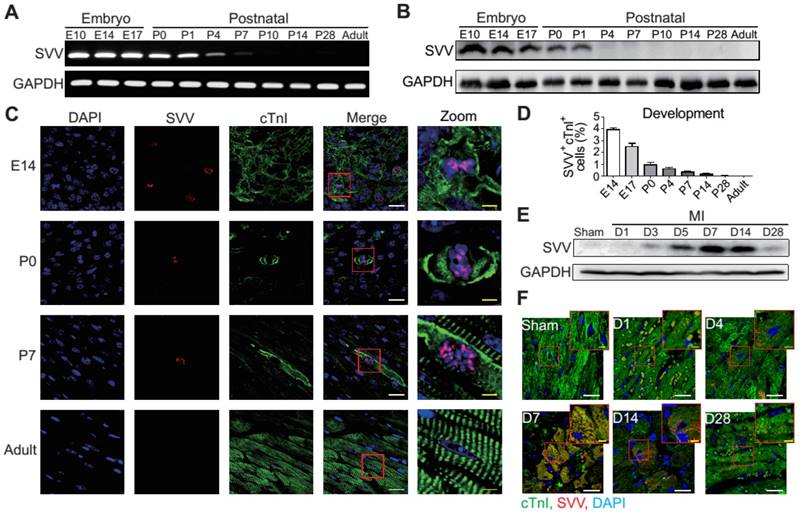
Although its expression was not detected in postnatal development, strong expression of SVV was detected in the heart after MI (Figure 1E and Figures S1B and S2). Within 7 days post-infarction, the expression level of SVV gradually increased, and started to decline after 14 days. Interestingly, unlike its localization in the nucleus of embryonic hearts, SVV was found to localize in the cytoplasm of the cardiomyocytes at the peri-infarct region (Figure 1F and Figure S3). Thus, the data suggest that SVV plays a different role during heart development and after injury, which may be attributed to the differences in subcellular localization.
Cardiac-specific, inducible SVV knockout mice have lower cardiac recovery ability
A previous study has shown that cardiac-specific SVV depletion mice exhibited unhealthy cardiac function, and had only a 50% survival rate at 34 weeks after birth [23]. Thus, to allow a better understanding of the biological function of SVV in the heart, cardiac-specific but inducible SVV knockout (KO) mice were established (Figure 2A). The αMHC-MCM transgenic mice, expressing Cre fusion protein driven by αMHC, specifically in cardiomyocytes, were crossbred with SVVflox/flox mice that contained 2 LoxP sequences in the front and back of the exons 1 and 2 of SVV. To study the loss function of SVV, the homozygous αMHC-MCM+/SVVflox/flox transgenic mice and control MCM mice were all given daily tamoxifen for 14 days, and received MI surgery 28 days after tamoxifen injections ceased. The expression level of SVV at day 7 post-infarction was high in the MCM mice. In contrast, the expression level of SVV was not detected in the KO mice that received surgically-induced MI (Figure 2B and Figure S4). The data indicate that inducible knockout of cardiac-specific SVV was achieved through tamoxifen treatment.
Phenotype of SVV knockout (KO) mice after MI. A, Schematic diagram showing establishment of inducible, cardiac-specific SVV KO mouse. B, Western blot showing SVV KO efficiency after MI. C, Survival curve of MCM and KO mice over 28 days following MI surgery. D-G, Cardiac function of MCM and KO mice 1 day and 21 days after MI. EDV, end diastolic volume; ESV, end systolic volume; FS, fraction shortening; EF, ejection fraction.*, P<0.05; **, P<0.01; ***, P<0.005. H, TUNEL staining indicating apoptotic cardiomyocytes, marked with yellow arrows. White bar, 20 μm. I, Quantification of TUNEL positive cardiomyocyte (CM) at the border zone. ***, P<0.005. J, Trichrome staining of hearts 30 days after receiving MI surgery. K-M, Quantification of left ventricle (LV) scar area percentage, LV wall thickness, and scar length percentage of heart after MI. *** P<0.005.
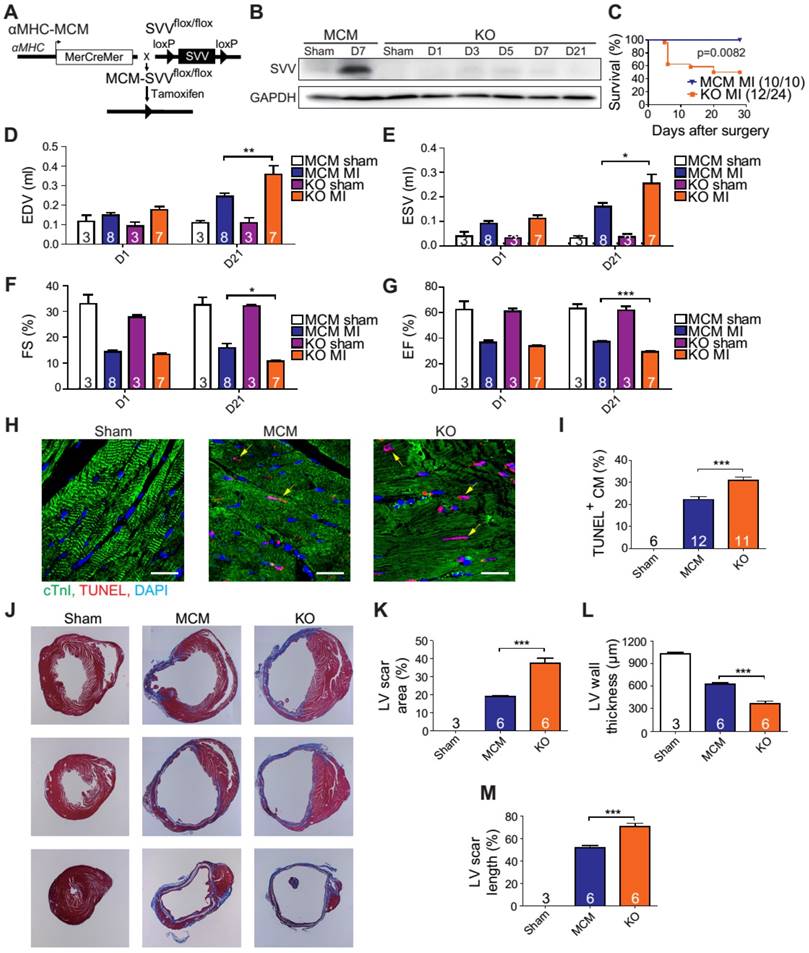
The KO mice were found to have a survival rate of only 50% at 28 days post-infarction as opposed to 100% survival rate for the MCM mice (Figure 2C). There were no significant differences in the overall cardiac function between survival MCM and KO mice at 1 day post-infarction. However, at 21 days post-infarction the KO mice showed deterioration in all of the measured echocardiographic parameters (Figure 2D-G). These data indicate that SVV depletion led to impaired cardiac recovery after myocardial injury. In order to further investigate the role that SVV plays in heart injury, apoptotic cardiomyocytes at the peri-infarct region were labeled by TUNEL staining (Figure 2H). More apoptotic cardiomyocytes were identified at the border zone of the KO murine heart 2 days after MI compared to the MCM group (Figure 2I). This supports previous findings that SVV plays an important role in anti-apoptotic function. However, we found no statistical difference between the percentage of proliferating cardiomyocyte in MCM and KO mice (Figure S5). Scar area, length, and left ventricle wall thickness were quantified based on trichrome staining to evaluate the scale of scar formation (Figure 2J). Compared to MCM mice, KO mouse hearts exhibited a larger scar area, longer scar length in circumference, and a thinner left ventricle wall (Figure 2K-M). This implies that SVV can attenuate cardiomyocyte apoptosis, thus leading to milder scar formation. Both MCM and KO mice exhibit enlarged cardiomyocytes, but there was no statistically significant difference between these two groups 30 days after MI (Figure S6).
Overexpressing SVV attenuates cardiomyocyte apoptosis
To further characterize the anti-apoptotic activity of cardiac SVV, adenovirus containing SVV fused with GFP were prepared. Subsequently, neonatal mouse cardiomyocytes were infected with SVV overexpressing or GFP control (adGFP) virus (Supplemental Figure S7). Cells were cultured in serum-free medium for 24 h and then treated under hypoxic conditions for 36 h to induce cell apoptosis. The control cardiomyocytes that were in normoxia did not show positive signals for TUNEL staining. Approximately 12% of cardiomyocytes, either with or without control virus infection, were TUNEL-positive after hypoxia treatment, and the percentage of the TUNEL-positive cardiomyocytes in the SVV overexpressing group was significantly lower (Figure 3A-B). Similar reductions in apoptosis could also be observed in doxorubicin-induced injury (Figure S8). These results indicated that SVV overexpression in cardiomyocytes could attenuate cell apoptosis.
Overexpression of SVV attenuates cardiomyocyte apoptosis. A, Effect of overexpressing SVV in hypoxia-induced cardiomyocyte apoptosis. B, Quantification of TUNEL positive CM. **, P<0.01. C, Sequence of mutant SVV-T34A (T34A). D, Western blotting showing phosphorylation level of SVV-T34. ad, adenovirus. E, Co-immunoprecipitation of CM overexpressing SVV with caspase-9. F, Co-immunoprecipitation of CM overexpressing T34A with caspase-9. G, Western blotting showing that overexpressing SVV attenuates caspase-3 activation but not T34A. Dox, doxorubicin. H, TUNEL staining indicating apoptotic cardiomyocytes in the hearts with overexpressing vehicle, SVV, T34A, or NLS-SVV after MI. NLS, nuclear localization signal. White bar, 50 μm; yellow bar, 10 μm. I, Quantification of apoptotic CM at the border zone. *, P<0.05. J, Western blot showing caspase-3 activation.
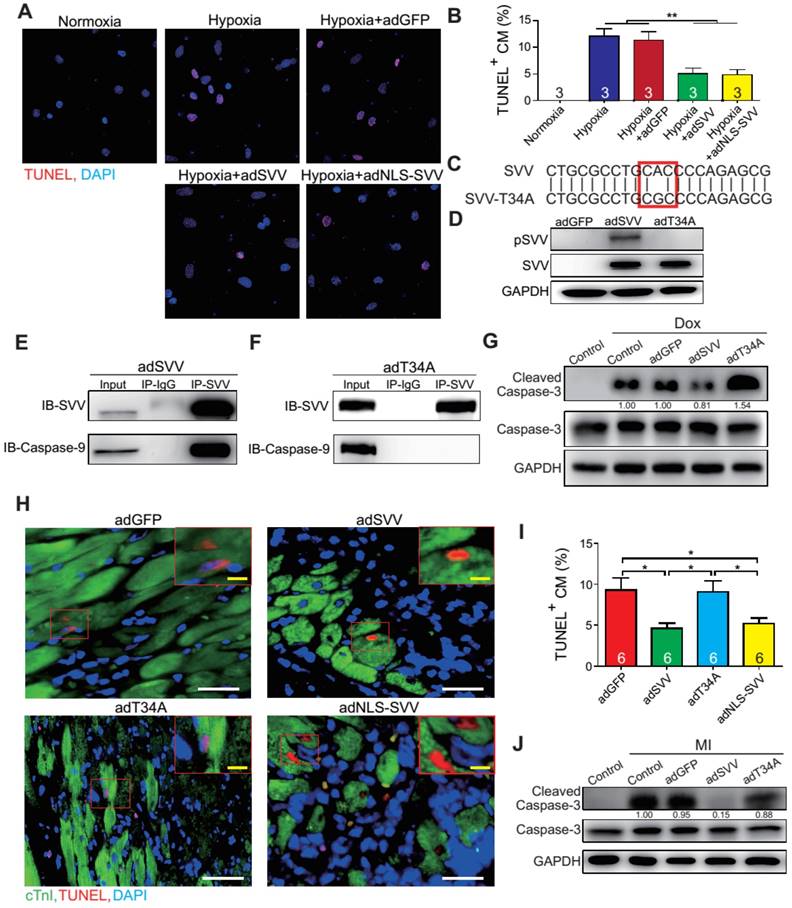
It is known that loss of phosphorylation on T34 of SVV abrogates interactions with caspase-9 and eliminates its function to inhibit caspase-9 activation [26]. To investigate how SVV attenuated cardiomyocyte apoptosis, a virus carrying mutant SVV T34A (adT34A) was generated, in which the mutant could not inhibit caspase-9 activation (Figure 3C). Unlike the overexpression of wildtype SVV in neonatal mouse cardiomyocytes, phosphorylation at Thr34 could no longer be detected on the SVV in the cardiomyocytes that were infected with mutant SVV T34A (Figure 3D). Moreover, a pull-down assay revealed that caspase-9 showed strong interactions with the wildtype SVV in cardiomyocytes (Figure 3E) but not with the mutant SVV T34A (Figure 3F). Compared to the GFP control and the SVV T34A mutant overexpressing cardiomyocytes, the expression level of active caspase-3 was lower in the wildtype SVV overexpressing cardiomyocytes (Figure 3G). These data suggest that the loss of interaction between SVV and caspase-9 eliminated the anti-apoptosis function of SVV, and that SVV attenuated cell apoptosis through the inhibition of caspase-9 activation.
To explore the function of SVV overexpression in vivo, 109 plaque-forming unit (pfu) of virus that contained either wild type SVV or mutant SVV T34A, both fused with GFP, were intramyocardially injected into adult C57BL/6JNarl mouse hearts at 2 days before the MI operation (Figure S9A). Mice that received the wildtype SVV gene had a significantly lower number of apoptotic cardiomyocytes at the peri-infarct region compared to GFP control and mutant SVV T34A groups (Figure 3H-I). Caspase-3 activation was also attenuated in the wildtype SVV treated group but not in the control and mutant SVV T34A treated groups (Figure 3J). These data demonstrate that overexpressing SVV could reduce cardiomyocyte apoptosis through interaction with caspase-9.
Adenoviral overexpression of SVV improves post-MI cardiac function
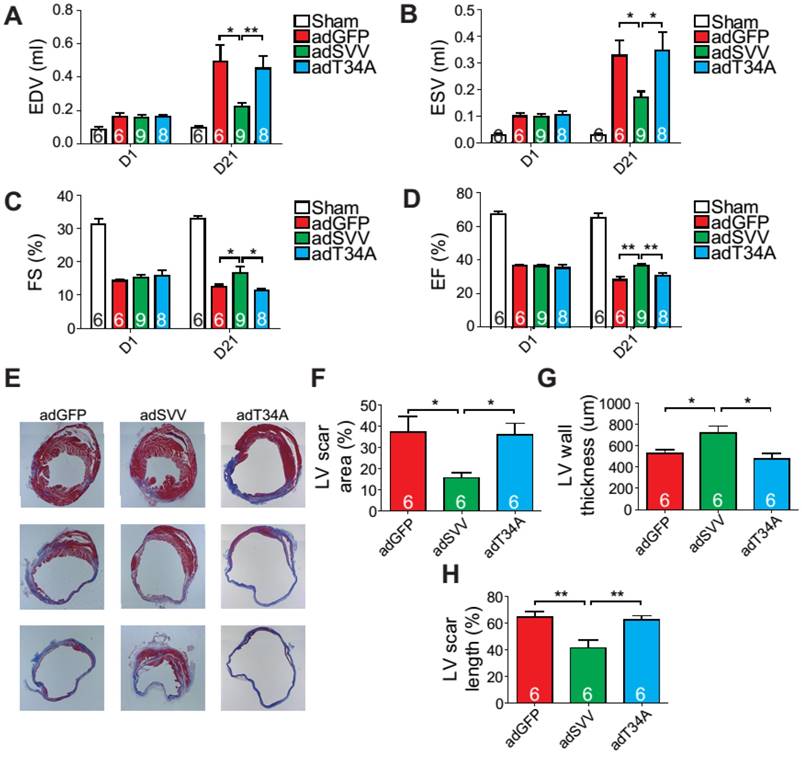
Since intramyocardial injection of adSVV was shown to inhibit caspase-3 activation in infarcted hearts, we decided to investigate the overall cardiac function of the heart after the administration of viral packaged SVV DNA. Compared to the adGFP control group and adT34A treated group, the mice that received adSVV treatments had better performance in all the measured echocardiographic parameters at 21 days after MI (Figure 4A-D). Furthermore, the ability of SVV to attenuate apoptosis in the peri-infarct region was also reflected in terms of scar formation. Mouse hearts overexpressing SVV showed smaller scar area, shorter scar length in circumference, and thicker LV wall at 30 days after MI (Figure 4E-H). These results indicate that overexpressing SVV in the border zone resulted in better cardiac recovery.
Overexpression of SVV recuses cardiac function after infarction. A-D, Cardiac function of hearts overexpressing SVV 1 and 21 days after MI. *, P<0.05; **, P<0.01. E, Trichrome staining indicating scar tissue in left ventricle (LV) 30 days after MI. F-H, Quantification of LV scar area, wall thickness, and scar length. *, P<0.05; **, P<0.01.
Nuclear SVV enhances cardiomyocyte proliferation
Since SVV was localized in the nucleus of cardiomyocytes during heart development but was found in the cytoplasm after injury, the effect of intracellular localization on the bioactivity of SVV was investigated. Adenovirus carrying SVV fused with nuclear localization signals (NLS) (adNLS-SVV) and GFP were established, and were transfected into neonatal mouse cardiomyocytes (Figure 5A). Overexpression of the SVV without NLS (adSVV) was identified in cardiomyocytes, whereas NLS-SVV was detected in both nucleus and cytoplasm (Figure 5A). Similar results were also supported by cell fractionation Western blot, as adSVV could only be detected in the cytoplasm, whereas NLS-SVV could be detected in both nuclear and cytoplasmic compartments of cardiomyocytes (Figure 5B). Since SVV was identified in the nucleus of cardiomyocytes during embryonic development of the heart, we suspected that the protein had a role in cell mitosis. Aurora kinase B (Aurora B) has been previously shown to complex with proteins such as SVV to facilitate chromosomal segregation during cell mitosis [14-16]. The pull-down assay of adNLS-SVV treated neonatal cardiomyocytes also showed strong detection of the complex formed between NLS-SVV and endogenous Aurora B (Figure 5C). Moreover, there were more cells in the cardiomyocyte sample that received adNLS-SVV treatment compared to those that received adSVV or adNLS-SVV + AZD1152, an Aurora B specific inhibitor (Figure 5D). Collectively, these results suggest that viral delivery of NLS-SVV could potentially initiate cardiomyocyte proliferation during postnatal development through interaction with Aurora B.
Overexpression of NLS-SVV enhances cardiomyocyte proliferation. A, Immunofluorescence staining revealing localization of SVV in neonatal mouse CM overexpressing SVV or NLS-SVV. NLS, nuclear localization signal; adNLS-SVV, adenovirus carrying NLS-SVV. B, Western blotting showing SVV expression in the cytoplasm or the nucleus fraction. C, Co-immunoprecipitation indicating the interaction of SVV and Aurora B in NLS-SVV-overexpressing CM. D, Quantification of CM number after overexpressing NLS-SVV. AZD1152, 10nM. *, P<0.05. E, Western blotting showing histone 3 phosphorylation (H3P) level in CM overexpressing NLS-SVV. F, Immunofluorescence staining indicating H3P positive cardiomyocytes at the border zone. Scale bar, 20 μm. G, Quantification of H3P-positive CM at the border zone. *, P<0.05. H, Western blotting showing H3P phosphorylation level after overexpressing NLS-SVV in vivo.
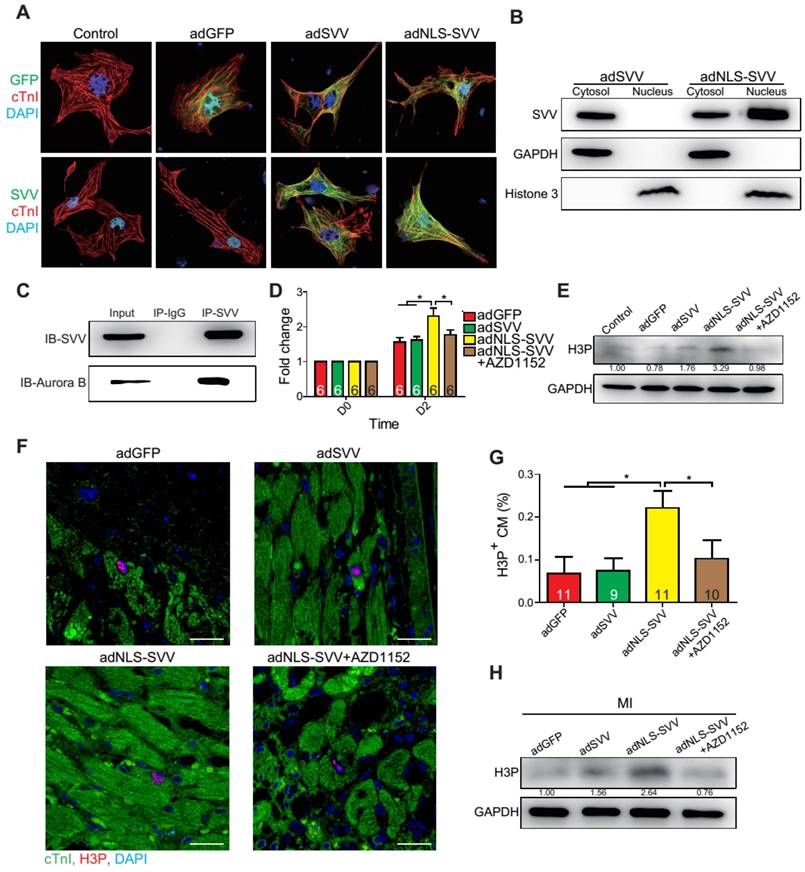
In addition to its interactions with Aurora B, NLS-SVV was also studied for its effect on the phosphorylation level of histone 3 (H3) (Figure 5E). During cell mitosis, Aurora B phosphorylates H3 on S10 [27-30]. Thus, it was suspected that the presence of Aurora B would increase the number of neonatal cardiomyocytes with phosphorylated H3 (H3P). The number of H3P+ cardiomyocytes was low in samples that were treated with either the GFP control or adSVV. In contrast, adNLS-SVV treatment increased the number of the H3P+ cardiomyocytes, but the addition of AZD1152 returned the expression to the basal level. Similarly, intramyocardial delivery of adNLS-SVV also increased the number of H3P+ cardiomyocytes in the peri-infarct region (Figure 5F-G). However, administration of adSVV did not increase cardiomyocyte proliferation in an EdU-pulse labeling assay (Figure S10). The elevated level of H3P in the infarcted hearts that received adNLS-SVV administration was confirmed by Western blot (Figure 5H). Accordingly, the delivery of adNLS-SVV increased the localization of the overexpressed SVV in the nucleus of neonatal cardiomyocytes, in which the proteins displayed strong interactions with the endogenous Aurora B.
Intramyocardial injection of adNLS-SVV promoted cardiomyocyte regeneration
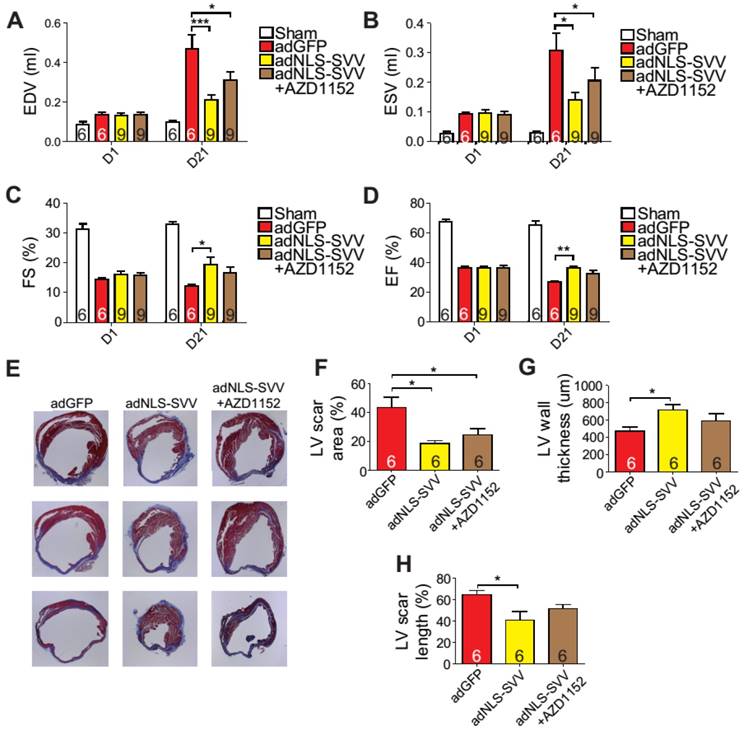
Given that the binding between NLS-SVV and Aurora B increased the number of H3P+ cardiomyocytes both in vitro and in vivo, and that the H3P+ level fell back to the basal level after the addition of AZD1152, we hypothesized that administration of adNLS-SVV to the infarcted hearts could improve the overall cardiac function by stimulating the mitotic activity of Aurora B. Therefore, we performed intramyocardial injection of adNLS-SVV following MI, and observed a significant improvement of cardiac function in the adNLS-SVV treated group compared to the adGFP treated group at 21 days after MI (Figure 6A-D). Although there was no significant difference in most of the measured echocardiographic parameters between the adNLS-SVV and adNLS-SVV + AZD1152 treated groups, the former group did show better overall cardiac function compared to the latter group. Trichrome staining also demonstrated that the adNLS-SVV treatments minimized the size of the damaged area, and that addition of AZD1152 abrogated the therapeutic effect (Figure 6E-H). All four key metrics of cardiac function were better in mice that received adNLS-SVV than those that received adSVV after MI (Table S1).
Overexpression of NLS-SVV reduces loss of cardiac function. A-D, Cardiac function of hearts overexpressing NLS-SVV 1 and 21 days after MI. *, P<0.05; **, P<0.01; ***, P<0.005. E, Trichrome staining revealing scar tissue in LV 30 days after MI. F-H, Quantification of LV scar area, wall thickness, and scar length. *, P<0.05.
Discussion
Cardiomyocyte loss following MI leads to pathological remodeling and heart failure, a leading cause of death in developed countries. Current standard-of-care treatments do not completely prevent cardiomyocyte death, nor do they induce new cardiomyocyte formation in or around the infarct. Hence, there is an urgent need for developing new therapeutic strategies to prevent cardiomyocyte loss. Here, we identify that SVV is a new therapeutic target for MI with a dual function of preventing cardiomyocyte apoptosis and increasing cardiomyocyte proliferation. We also demonstrate that this dual function is determined by the subcellular localization of SVV.
Previous studies have shown that the expression level of SVV in adult hearts was either low or undetectable, and that the expression of SVV is upregulated after MI in human patients [23, 31]. Similarly, the present study showed the protein expressions of SVV were highly present in the murine hearts during embryonic development but not after birth. Although mice have three different isoforms of SVV, both SVV-140 and SVV-121 were detected throughout the mouse embryonic heart development and after injury. SVV-40 was not detected in either embryonic or neonatal hearts at any stage of development or in the adult post-MI heart (Figure S1). Seven isoforms have been identified in humans so far, although only SVV-α (human analogue of murine SVV-140) has been reported in the heart [32]. Interestingly, expression of other human SVV isoforms such as SVV-2B and SVV-deltaEx3 were mostly reported in cancer patients, in which the expressions were upregulated at different stages of tumor development [33-36]. Under normal physiology, however, SVV-α is the main isoform identified in humans, which is known to regulate cell mitosis [32, 37]. This concurs with the finding of SVV in the nucleus but not in the cytosol of cardiomyocytes during embryonic heart development, which indicates that the presence of SVV in the nucleus was critical for cell mitosis to happen in embryonic hearts. A study on normal adult mice showed that the transcript of SVV-140 was detected in the spleen, testes, and thymus only, whereas SVV-121 mRNA was detected in the liver and lungs, and SVV-40 was not detected in any of the adult tissues [38]. Accordingly, SVV was not detected in normal adult murine heart; however, the expression was induced after MI.
The importance of SVV in overall cardiac output was clearly demonstrated in the SVV KO mice, in which cardiac SVV expression could only be downregulated after receiving tamoxifen delivery. The recombination efficiency of our αMHC-MCM strain was about 80%, and this lead to a variation in the SVV knockout efficiency between KO mice. Different knockout efficiencies might explain why 40% of KO mice died within 7 days after MI, and we speculate that the surviving mice were those with less severe loss of ejection fraction, thus resulting in the small difference between MCM and KO mice EF%. The KO mice had worse survival than MCM mice after MI, which clearly demonstrates the importance of SVV in preserving normal cardiac output in an adult murine model of MI. More importantly, the KO mouse model demonstrated that the physiological expression of SVV is required to minimize the extent of cardiomyocyte death resulting from the ischemic attack. It has also been found in human patients that the expression level of SVV in the heart was significantly upregulated after MI [23, 31]. The increase in the number of apoptotic cells at the peri-infarct region indicated that SVV alone could exert strong anti-apoptotic activity in reducing cell death after MI, as well as minimize the size of the resulting infarct. Similar findings have been described in cancer, where cancer cells reduce apoptosis by upregulating SVV expression [17].
Notably, unlike during embryonic heart development, the MI-induced cardiac SVV was found to localize in the cytosol, not in the nucleus of cardiomyocytes, and had high binding affinity for the latent form of caspase-3. This binding was able to inhibit the activation of caspase-3, which explains how adSVV treatment was able to protect cell apoptosis from the exposure to doxorubicin. Studies have shown that HS-1 associated protein-1 (HAX-1) can protect from hypoxia/reoxygenation-induced cell death in adult rat cardiomyocytes, and HAX-1 directly bound to caspase-9 under cell-free conditions [39]. Caspase-9 is known as an element of the apoptosome, and that apoptosome formation autoprocesses caspase-9, further activating downstream caspase-3 [40, 41]. Although there was no direct evidence that adSVV disrupts apoptosome formation, we found that SVV bound to caspase-9 in neonatal mouse cardiomyocytes and attenuated the activation of caspase-3 after injury. Therefore, unlike its localization in the nucleus of cardiomyocytes during embryonic heart development, the MI-induced SVV was found to localize in the cytosol. Although the induction of cardiac SVV after MI has been reported before [23], we have demonstrated that the proteins were shown to localize in the cytosol of cardiomyocytes in adult mouse heart, and that its presence in the cytosol inhibited caspase-3 activation.
Since the localization of SVV was able to depict its bioactivity in cardiomyocytes after MI, we expected that the presence of nuclear localization signal on NLS-SVV would localize SVV exclusively in the nucleus of cardiomyocytes. However, as demonstrated in neonatal cardiomyocytes, only ~50% of NLS-SVV were localized in the nucleus and the other half were in the cytosol. Although the mechanism of SVV transport between the nucleus and the cytosol in cardiomyocytes remains to be elucidated, previous studies in cancer cells have shown that SVV may passively diffuse from the cytosol into the nucleus, but is actively transported from the nucleus to the cytosol under control of CRM1/exportin [42]. Acetylated SVV forms a homodimer in the nucleus, which is facilitated with CREB-binding protein (CBP), and deacetylated SVV forms a heterodimer with CRM1 to export from the nucleus [43]. We found that the expression level of CBP decreases in developing mouse heart and remains low in the adult stage (Figure S11). Thus, we suspect that the endogenous SVV is acetylated by CBP to regulate SVV's localization in the nucleus in the early development stage. Overexpression of NLS-SVV would also perform an anti-apoptotic function both in vitro and in vivo (Figure 3B,I). Nevertheless, we demonstrated that only the adNLS-SVV treated group, but not the adSVV treated group, exhibited cell proliferation, in which the overexpressed NLS-SVV was shown to bind to Aurora B to initiate cell mitosis. More importantly, the cell proliferative effect of NLS-SVV was also demonstrated in the murine model of MI, indicating that there is a strong correlation between the cellular location of SVV and its bioactivity. Although it is not clear why the MI-induced SVV only localized in the cytosol but not in the nucleus of cardiomyocytes, the human SVV does contain a nuclear exportation signal (NES) domain and mutant NES in SVV would facilitate its accumulation in the nucleus [44]. Thus, it is likely that there is another regulatory mechanism in the mature cardiomyocytes that regulates the transportation of SVV, in which the MI-induced SVV is restricted in the cytosol, thus carrying out its anti-apoptotic activity.
In the assessment of the therapeutic outcome of treating murine models of MI with either adSVV or adNLS-SVV, it was clear that both treatments displayed better outcomes than the adGFP control. Surprisingly, although the therapeutic actions between the two treatments are different, the overall therapeutic outcome was similar. Although the improvement seems minimal compared to the sham group, both treatments exhibited better cardiac output and were able to keep the size of infarct below 20% at 30 days post MI, as opposed to the >40% seen in the adGFP control group. Reduced LV scar size may be attributed to the inhibition of cardiomyocyte apoptosis [45-47].
In conclusion, the expression profile of SVV in heart development revealed that the proteins were only expressed in the embryonic developmental stages, and started to diminish after birth. More importantly, SVV was detected in the nucleus of cardiomyocytes during early heart development, and its expression was induced in the heart of murine models of MI. Although the precise mechanism that induces the expression of SVV in the heart after MI remains to be elucidated, it is clear that the MI-induced SVV was only found in the cytosol of cardiomyocytes. The biological functions of SVV were shown to depend on its localization, in which the SVV in the nucleus enables cell mitosis through interaction with Aurora B; whereas the SVV in the cytosol exerts its anti-apoptotic activity by downregulating caspase-3 activation. The improved therapeutic outcome in the infarcted heart after administrating either adSVV or adNLS-SVV, clearly shows the potential of SVV as a therapeutic target.
Abbreviations
Ad: adenovirus; αMHC: alpha myosin heavy chain; CBP: CREB-binding protein; CM: cardiomyocyte; CMV: cytomegalovirus; cTnI: cardiac troponin I; EDV: end diastolic volume; EF: ejection fraction; ESV: end systolic volume; FS: fraction shortening; H3: histone 3; H3P: phospho-histone 3 (Ser10); IAP: inhibitor of apoptosis; KO: knockout; LV: left ventricle; MCM: MerCreMer; MI: myocardial infarction; NES: nuclear exportation signal; NLS: nuclear localization signal; PFA: paraformaldehyde; Pfu: plaque-forming unit; SVV: survivin; T34A: mutant SVV-T34A; WGA: wheat germ agglutinin.
Acknowledgements
We gratefully acknowledge R.H. Lin and Y.P. Wong for their assistance in animal surgery, W.S. Lin and B.L. Lin for their help with echocardiography, Dr. Ed Conway (University of Leuven, Belgium) for sharing the floxed SVV transgenic mouse, and Dr. Tang K. Tang (IBMS, Academia Sinica) for helpful discussion and critical comments on the article.
Author contributions
TJT established the experiment design, participated in data collection, processing, and analysis, and wrote the manuscript; YCH established the inducible cardiac-specific SVV KO transgenic mouse; EIW constructed and generated adenovirus; DJL edited the manuscript; BC participated in manuscript writing; YTC and SSW supervised the work; PCHH established the experiment concept, supervised the work, and approved the final manuscript.
Sources of funding
This work was supported by the National Research Program for Biopharmaceuticals of the Ministry of Science and Technology, Taiwan (MOST 102-2321-B-001-069 and 104-2325-B-001-010) and the National Health Research Institutes grant EX10123SI.
Supplementary Material
Supplementary figures and tables.
Competing Interests
Patrick Hsieh received research grants from AstraZeneca, Sweden, and Takeda, Japan. The other authors declare no conflict of interest.
References
1. Sarkar N, Ruck A, Kallner G, S YH, Blomberg P, Islam KB. et al. Effects of intramyocardial injection of phvegf-a165 as sole therapy in patients with refractory coronary artery disease-12-month follow-up: Angiogenic gene therapy. J Intern Med. 2001;250:373-81
2. Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N. et al. Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation. 2002;105:2012-8
3. Grines CL, Watkins MW, Mahmarian JJ, Iskandrian AE, Rade JJ, Marrott P. et al. A randomized, double-blind, placebo-controlled trial of ad5fgf-4 gene therapy and its effect on myocardial perfusion in patients with stable angina. J Am Coll Cardiol. 2003;42:1339-47
4. Henry TD, Grines CL, Watkins MW, Dib N, Barbeau G, Moreadith R. et al. Effects of ad5fgf-4 in patients with angina: An analysis of pooled data from the agent-3 and agent-4 trials. J Am Coll Cardiol. 2007;50:1038-46
5. Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M. et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330-5
6. Dohi T, Beltrami E, Wall NR, Plescia J, Altieri DC. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J Clin Invest. 2004;114:1117-27
7. Altieri DC. Targeted therapy by disabling crossroad signaling networks: The survivin paradigm. Mol Cancer Ther. 2006;5:478-82
8. Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I. et al. Hbxip functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729-40
9. Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K. et al. An iap-iap complex inhibits apoptosis. J Biol Chem. 2004;279:34087-90
10. Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC. et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580-4
11. Yamagishi Y, Honda T, Tanno Y, Watanabe Y. Two histone marks establish the inner centromere and chromosome bi-orientation. Science. 2010;330:239-43
12. Kelly AE, Ghenoiu C, Xue JZ, Zierhut C, Kimura H, Funabiki H. Survivin reads phosphorylated histone h3 threonine 3 to activate the mitotic kinase aurora b. Science. 2010;330:235-9
13. Wang F, Dai J, Daum JR, Niedzialkowska E, Banerjee B, Stukenberg PT. et al. Histone h3 thr-3 phosphorylation by haspin positions aurora b at centromeres in mitosis. Science. 2010;330:231-5
14. Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (cpc): From easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol. 2012;13:789-803
15. Jeyaprakash AA, Klein UR, Lindner D, Ebert J, Nigg EA, Conti E. Structure of a survivin-borealin-incenp core complex reveals how chromosomal passengers travel together. Cell. 2007;131:271-85
16. Bourhis E, Hymowitz SG, Cochran AG. The mitotic regulator survivin binds as a monomer to its functional interactor borealin. J Biol Chem. 2007;282:35018-23
17. Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917-21
18. Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006;5:1087-98
19. Zaffaroni N, Daidone MG. Survivin expression and resistance to anticancer treatments: Perspectives for new therapeutic interventions. Drug Resist Updat. 2002;5:65-72
20. Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CH. Treat cancers by targeting survivin: Just a dream or future reality? Cancer Treat Rev. 2013;39:802-11
21. Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL. et al. Survivin and the inner centromere protein incenp show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319-28
22. Zwerts F, Lupu F, De Vriese A, Pollefeyt S, Moons L, Altura RA. et al. Lack of endothelial cell survivin causes embryonic defects in angiogenesis, cardiogenesis, and neural tube closure. Blood. 2007;109:4742-52
23. Levkau B, Schafers M, Wohlschlaeger J, von Wnuck Lipinski K, Keul P, Hermann S. et al. Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation. 2008;117:1583-93
24. Xing Z, Conway EM, Kang C, Winoto A. Essential role of survivin, an inhibitor of apoptosis protein, in t cell development, maturation, and homeostasis. J Exp Med. 2004;199:69-80
25. Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J. et al. A protocol for rapid generation of recombinant adenoviruses using the adeasy system. Nat Protoc. 2007;2:1236-47
26. O'Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A. et al. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103-7
27. Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK. et al. Mitotic phosphorylation of histone h3 is governed by ipl1/aurora kinase and glc7/pp1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279-91
28. Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R. et al. The small molecule hesperadin reveals a role for aurora b in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281-94
29. Hirota T, Lipp JJ, Toh BH, Peters JM. Histone h3 serine 10 phosphorylation by aurora b causes hp1 dissociation from heterochromatin. Nature. 2005;438:1176-80
30. Sabbattini P, Canzonetta C, Sjoberg M, Nikic S, Georgiou A, Kemball-Cook G. et al. A novel role for the aurora b kinase in epigenetic marking of silent chromatin in differentiated postmitotic cells. EMBO J. 2007;26:4657-69
31. Santini D, Abbate A, Scarpa S, Vasaturo F, Biondi-Zoccai GG, Bussani R. et al. Surviving acute myocardial infarction: Survivin expression in viable cardiomyocytes after infarction. J Clin Pathol. 2004;57:1321-4
32. Hirohashi Y, Torigoe T, Maeda A, Nabeta Y, Kamiguchi K, Sato T. et al. An hla-a24-restricted cytotoxic t lymphocyte epitope of a tumor-associated protein, survivin. Clin Cancer Res. 2002;8:1731-9
33. Caldas H, Honsey LE, Altura RA. Survivin 2alpha: A novel survivin splice variant expressed in human malignancies. Mol Cancer. 2005;4:11
34. Suga K, Yamamoto T, Yamada Y, Miyatake S, Nakagawa T, Tanigawa N. Correlation between transcriptional expression of survivin isoforms and clinicopathological findings in human colorectal carcinomas. Oncol Rep. 2005;13:891-7
35. Pavlidou A, Dalamaga M, Kroupis C, Konstantoudakis G, Belimezi M, Athanasas G. et al. Survivin isoforms and clinicopathological characteristics in colorectal adenocarcinomas using real-time qpcr. World J Gastroenterol. 2011;17:1614-21
36. Espinosa M, Ceballos-Cancino G, Callaghan R, Maldonado V, Patino N, Ruiz V. et al. Survivin isoform delta ex3 regulates tumor spheroid formation. Cancer Lett. 2012;318:61-7
37. Yamada Y, Kuroiwa T, Nakagawa T, Kajimoto Y, Dohi T, Azuma H. et al. Transcriptional expression of survivin and its splice variants in brain tumors in humans. J Neurosurg. 2003;99:738-45
38. Conway EM, Pollefeyt S, Cornelissen J, DeBaere I, Steiner-Mosonyi M, Ong K. et al. Three differentially expressed survivin cdna variants encode proteins with distinct antiapoptotic functions. Blood. 2000;95:1435-42
39. Han Y, Chen YS, Liu Z, Bodyak N, Rigor D, Bisping E. et al. Overexpression of hax-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ Res. 2006;99:415-23
40. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES. et al. Cytochrome c and datp-dependent formation of apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479-89
41. Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD. et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281-92
42. Rodriguez JA, Lens SM, Span SW, Vader G, Medema RH, Kruyt FA. et al. Subcellular localization and nucleocytoplasmic transport of the chromosomal passenger proteins before nuclear envelope breakdown. Oncogene. 2006;25:4867-79
43. Wang H, Holloway MP, Ma L, Cooper ZA, Riolo M, Samkari A. et al. Acetylation directs survivin nuclear localization to repress stat3 oncogenic activity. J Biol Chem. 2010;285:36129-37
44. Colnaghi R, Connell CM, Barrett RM, Wheatley SP. Separating the anti-apoptotic and mitotic roles of survivin. J Biol Chem. 2006;281:33450-6
45. Barile L, Lionetti V, Cervio E, Matteucci M, Gherghiceanu M, Popescu LM. et al. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res. 2014;103:530-41
46. Guo C, Deng Y, Liu J, Qian L. Cardiomyocyte-specific role of mir-24 in promoting cell survival. J Cell Mol Med. 2015;19:103-12
47. Yang B, Yan P, Gong H, Zuo L, Shi Y, Guo J. et al. Tweak protects cardiomyocyte against apoptosis in a pi3k/akt pathway dependent manner. Am J Transl Res. 2016;8:3848-60
Author contact
![]() Corresponding author: Patrick C.H. Hsieh, MD, PhD, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan; IBMS Rm.417, 128 Academia Road, Section 2, Nankang District, Taipei 115, Taiwan Phone: 886-2-27899170; fax: 886-2-27858594; email: phsiehsinica.edu.tw
Corresponding author: Patrick C.H. Hsieh, MD, PhD, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan; IBMS Rm.417, 128 Academia Road, Section 2, Nankang District, Taipei 115, Taiwan Phone: 886-2-27899170; fax: 886-2-27858594; email: phsiehsinica.edu.tw