Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(18):4399-4409. doi:10.7150/thno.21403 This issue Cite
Research Paper
Theranostic Role of 32P-ATP as Radiopharmaceutical for the Induction of Massive Cell Death within Avascular Tumor Core
Mirco Galiè1, Federico Boschi2, Ilaria Scambi1, Flavia Merigo1, Pasquina Marzola2, Luisa Altabella3, Umberto Lavagnolo1, Andrea Sbarbati1, Antonello E Spinelli4 ![]()
1. Department of Neuroscience, Biomedicine and Movement, University of Verona, Piazzale L.A. Scuro 10, Verona 37134, Italy;
2. Department of Computer Science, University of Verona, Strada Le Grazie 15, 37134 Verona, Italy;
3. Medical Physics Department, San Raffaele Scientific Institute, Via Olgettina 60, 20182 Milan, Italy;
4. Experimental Imaging Centre, San Raffaele Scientific Institute, Via Olgettina 60, 20182 Milan, Italy.
Received 2017-6-9; Accepted 2017-8-1; Published 2017-9-30
Abstract

Drug inaccessibility to vast areas of the tumor parenchyma is amongst the major hurdles for conventional therapies. Treatment efficacy rapidly decreases with distance from vessels and most of the tumor cells survive therapy. Also, between subsequent cycles of treatment, spared cancer cells replace those killed near the vessels, improving their access to nutrients, boosting their proliferation rate, and thus enabling tumor repopulation. Because of their property of "acting at a distance," radioisotopes are believed to overcome the physical barrier of vascular inaccessibility.
Methods A novel molecular imaging tool called Cerenkov Luminescence Imaging (CLI) was employed for the detection of Cerenkov radiation emitted by beta particles, allowing in vivo tracking of beta-emitters. More precisely we investigated using a xenograft model of colon carcinoma the potential use of 32P-ATP as a novel theranostic radiopharmaceutical for tracing tumor lesions while simultaneously hampering their growth.
Results Our analyses demonstrated that 32P-ATP injected into tumor-bearing mice reaches tumor lesions and persists for days and weeks within the tumor parenchyma. Also, the high-penetrating beta particles of 32P-ATP exert a "cross-fire" effect that induces massive cell death throughout the entire tumor parenchyma including core regions.
Conclusion Our findings suggest 32P-ATP treatment as a potential approach to complement conventional therapies that fail to reach the tumor core and to prevent tumor repopulation.
Keywords: 32P-ATP radiopharmaceutical, Cerenkov luminescence imaging, magnetic resonance imaging, colorectal adenocarcinoma.
Introduction
Neoangiogenic programs in solid tumors generate convoluted and leaky blood vessels, which fail to uniformly perfuse the entire tumor parenchyma [1-4]. As new vessels stream from the peritumoral vascular network [1], vascularization is mainly restricted to the peripheral rim while the inner core is nearly avascular, hyper-dense, and subject to increased interstitial fluid pressure (IFP) [5]. The scarcity of vessels and the high IFP concur to form a physical barrier that limits drug penetration into the tumor core, reducing treatment efficacy [6]. Also, selective depletion of peripheral cells by radiotherapy or chemotherapy [7-10] shifts inner cells closer to vessels, improving their access to nutrients and oxygen [11], increasing their proliferation rate [9], and boosting tumor repopulation mechanisms thereby reducing efficacy during subsequent cycles of treatment [12, 13]. Hence, therapeutic approaches capable of killing tumor cells far from peripheral vasculature are of paramount medical importance.
Owing to their property of "acting at a distance," radioisotopes are believed to overcome the physical barrier of vascular inaccessibility. Beta emitters have been used in the past to kill cancer cells and some of them, such as 131I and 90Yttrium, have successfully entered clinical practice [14, 15]. Cheng et al. [16-18] have demonstrated that 32P bound to ATP molecules (32P-ATP) is effective in damaging tumor cells and reducing tumor growth rate in nude mice. The maximum beta energy of electrons emitted by 32P-ATP lies between that of 131Iodine and 90Yttrium with the particle range equal to 5 mm in tissues [19]. This property allows a “cross-fire effect” for cells located far away that cannot be physically reached through the bloodstream. Furthermore, the physical half-life of 32P is longer than 131Iodine and 90Yttrium, allowing the cell-damaging effect to persist for weeks after the first administration. This aspect is particularly relevant for avascular regions of tumor parenchyma where perfusion is delayed or inhibited [3].
The recent development of a novel molecular imaging tool based on the detection of Cerenkov radiation (CR) by beta particles allows tracking of the bio-distribution of beta-emitters and their uptake within tumor parenchyma in vivo [20-22]. It has been shown that Cerenkov luminescence imaging (CLI) can be used to measure the bio-distribution of beta plus [23, 24] and beta minus emitters [25, 26]. This suggests that 32P-ATP can be considered as a novel theranostic radiopharmaceutical providing measurements of physiological proprieties of tumors (on the basis of its tumor-specific uptake) while inhibiting their growth and progression.
To explore this hypothesis, we monitored the tumor-specific and whole-body distribution of 32P-ATP in vivo by using CLI and determined the effects of treatment with 32P-ATP on tumor growth and microregional architecture of tumor parenchyma. We found that 32P-ATP perfused scarcely within tumor parenchyma, but once penetrated, persisted for weeks. Consequently, while slightly reducing tumor volume, 32P-ATP treatment induced damaging effects throughout the entire tumor parenchyma and especially within the tumor core, which is usually spared by conventional therapies.
Results
Single dose injection of 32P-ATP inhibits tumor growth over two weeks
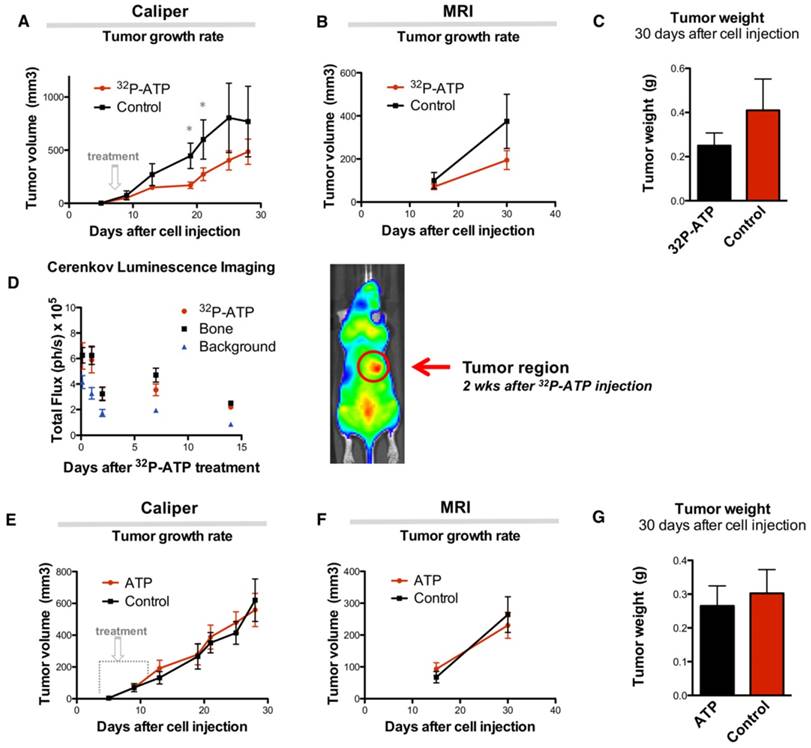
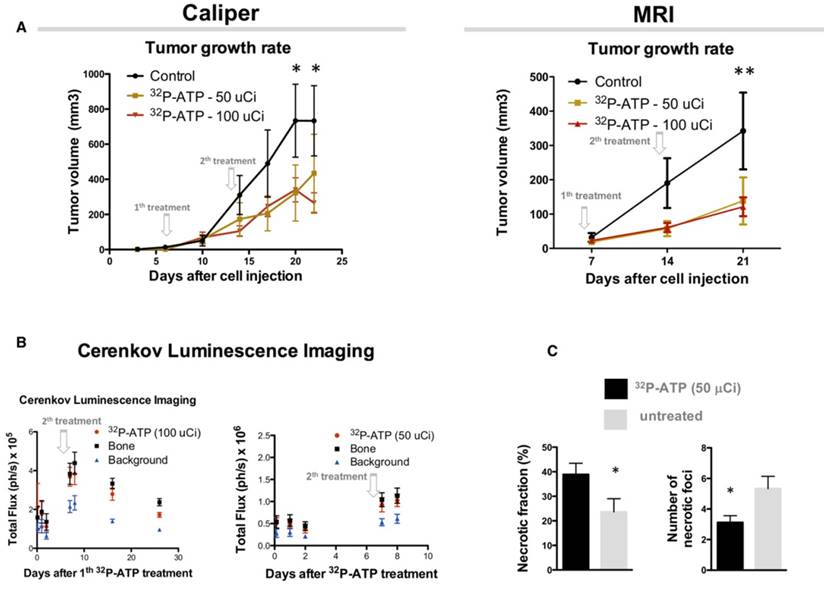
Sixteen nude mice were subcutaneously (s.c.) injected in the right rear flank with 2x106 HT-29 cells. At day 9 after injection, mice were randomized, and 8 of them received intravenous (i.v.) injection of a single dose of 100 μCi of 32P-ATP in the lateral tail vein. Treatment induced a significant reduction of tumor growth rate compared to untreated animals, which persisted over 2 weeks after treatment (Figure 1A and B). At the endpoint of the experiment (30 days after cell injection), animals were sacrificed, and tumor growth was examined. Tumors from treated mice were 60% in weight compared to those from control mice (0.25±0.05 g and 0.41±0.14 g, respectively), although the difference was not statistically significant (p=0.23) (Figure 1C).
The strong but transitory inhibition of tumor growth by a single dose injection of 32P-ATP is consistent with the half-life of 32P-ATP, which is equal to 14.3 days. In vivo monitoring of intratumor radioactivity by CLI confirmed that the beta particle emission within tumors shows a progressive decline after the initial i.v. injection but persists above the background level over 2 weeks (Figure 1D).
The antitumor effect does not rely on the intrinsic properties of ATP
It has been previously reported that extracellular ATP may provide per se a potent antitumor effect [27]. To further investigate this, two groups of mice were treated with Phosphate-Buffered Saline (PBS) or non-radioactive ATP using a dosage which is several orders of magnitude higher than that used in the 32P-ATP treatment. In this setting, we did not find any appreciable difference in tumor growth rate between ATP treated and control tumors (Figure 1E-G). This demonstrates that, at least in our model, the antitumor effect of the 32P-ATP treatment is mainly due to the radioactivity and not because of the intrinsic properties of ATP per se.
32P-ATP induces extensive cell death within the inner tumor parenchyma
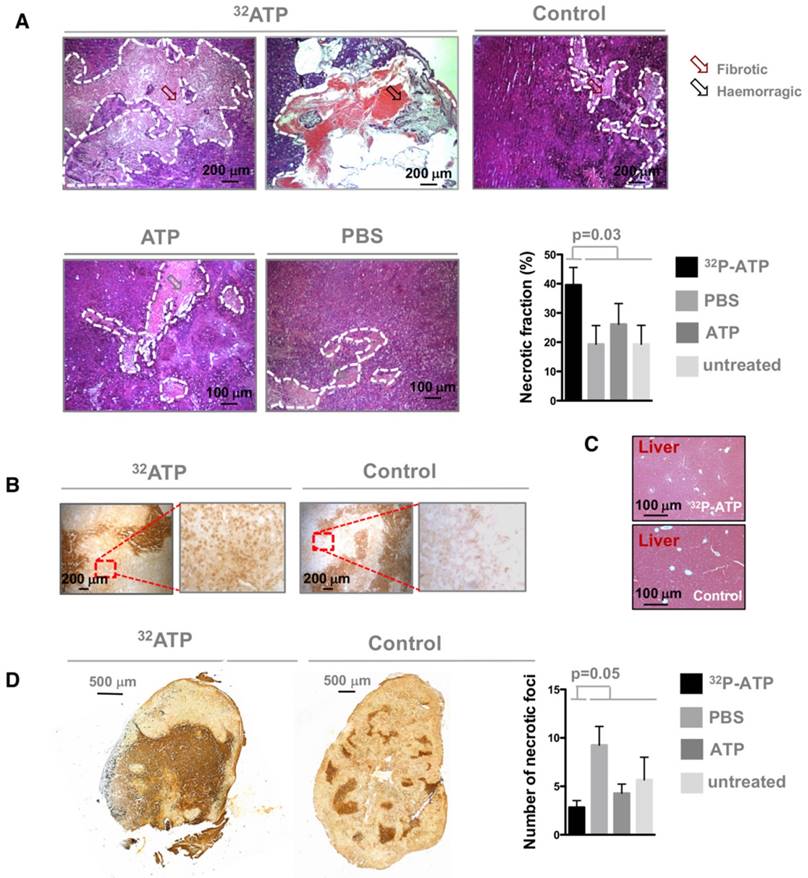
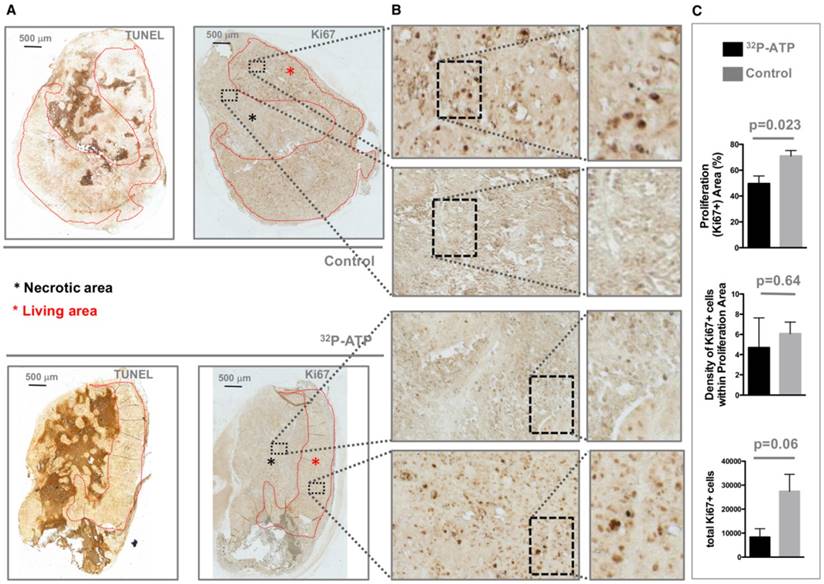
It is difficult to discern the microregional changes in the proliferation/death rate within the tumor parenchyma. Although the difference in tumor sizes between the 32P-ATP-treated and control mice at the end of the experiment was not statistically significant, histopathological analysis of tumor sections revealed that 32P-ATP caused a 2-fold increase in necrotic fraction compared to other treatments (Figure 2A). The extensive death areas in 32P-ATP tumors exhibited signs of either fibrotic or hemorrhagic necrosis. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay showed extensive DNA degradation in grossly necrotic areas. Also, in 32P-ATP tumors, but not in controls, TUNEL positive cells were still visible in close contiguity with necrotic areas, indicating an even larger spatial extension of death (Figure 2B). Necrosis was tumor-specific as it was absent in livers from the same animals (Figure 2C). Strikingly, while necrosis appeared fragmented in multiple small foci in control tumors, it appeared as a single patch or as few large necrotic foci often covering the entire tumor core in 32P-ATP-treated tumors (Figure 2D). However, outside necrotic areas, the density of Ki67-positive cells was unchanged in 32P-ATP-treated tumors compared to untreated tumors. This indicates that 32P-ATP treatment induces massive cell death but does not directly affect cell proliferation rate in these tumors (Figure 3).
Single dose injection of 100 μCi of 32P-ATP induces a temporary decline in tumor growth rate. Manual measurement by caliper (A); Magnetic Resonance Imaging (MRI) (B); At the endpoint of the experiment, tumor volume in 32P-ATP treated mice was reduced by 60% but did not reach statistical significance (C). In vivo monitoring 3 h to 14 days post injection) by CLI shows that after single dose injection, 32P-ATP emission decreases exponentially in the tumor as well as in other organs, but persists above the background value for over 2 weeks; (D). Treatment of HT29-tumor-bearing mice with non-radioactive ATP does not affect tumor growth rate, as measured either manually by caliper (E) or MRI (F), and tumor volume at the endpoint of the experiment.
Apoptosis lines the edge of necrotic areas in 32P-ATP-treated tumors
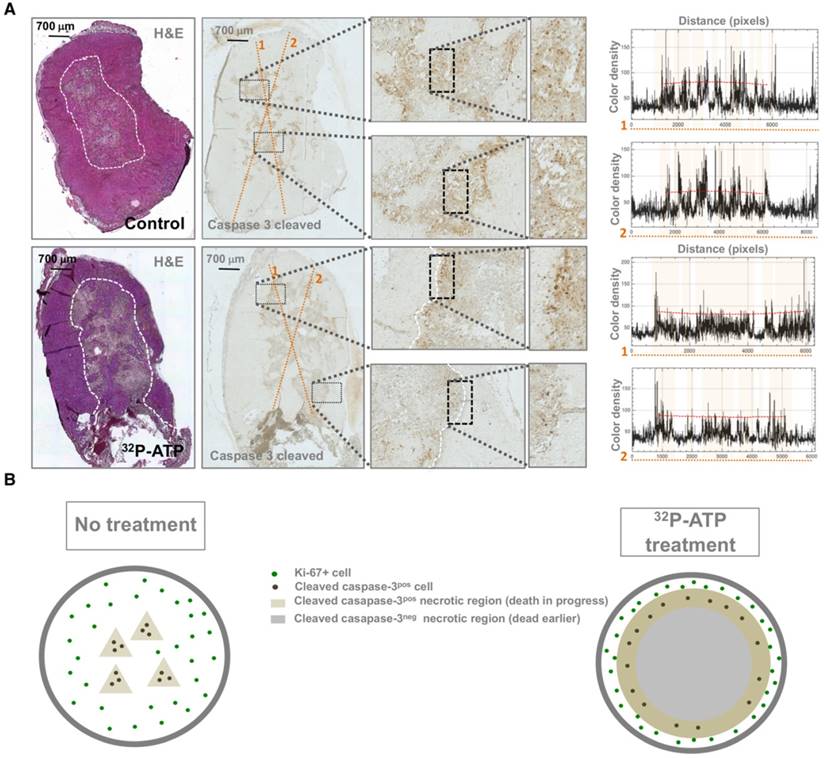
In general, extensive necrosis indicates that the death rate exceeds the ability of macrophages to clear dying cells, but does not provide information about timing and molecular pathways of cell death [28]. Molecular changes to tumor tissue following treatment precede changes in tumor morphology [29]. Activated caspase-3 is an early and specific marker of apoptotic cell death [30]. Spatial mapping of activated caspase-3 at the endpoint of the experiments might provide important clues about its dynamic distribution. Interestingly, while in control tumors, activated caspase-3 was homogeneously distributed throughout necrotic foci, in 32P-ATP-treated tumors, it was restricted to the peripheral edge of the extensive necrotic areas (Figure 4A). This pattern of activated-caspase-3 immunostaining in 32P-ATP-treated tumors indicated a centrifugal temporal gradient of cell death, where dying cells were located surrounding bulks of cells that had died earlier (Figure 4B).
Tumors from 32P-ATP-treated mice exhibited a significantly higher fraction of necrotic areas compared to those from either untreated, non-radioactive ATP-treated or PBS-treated mice (A). TUNEL assay on 32P-ATP-treated tumors revealed extensive contiguous DNA-fragmentation other than within grossly necrotic areas (B). Necrosis was restricted to tumors but was completely absent in other tissues such as liver (C). In 32P-ATP-treated tumors, necrosis extended over large regions often covering the entire tumor core while it appeared fragmented in multiple and small necrotic foci in control tumors (D).
Tumors from 32P-ATP-treated mice displayed a higher fraction of necrotic (TUNEL positive) areas (A, black asterisks) and consequently a much smaller proliferating (Ki67-positive) area (A, red asterisks) compared to those from control mice (A). The density of Ki67-positive cells within the Ki67-positive area appeared similar between the two experimental groups (B and C).
Necrotic regions exhibit a delayed but prolonged fluid perfusion
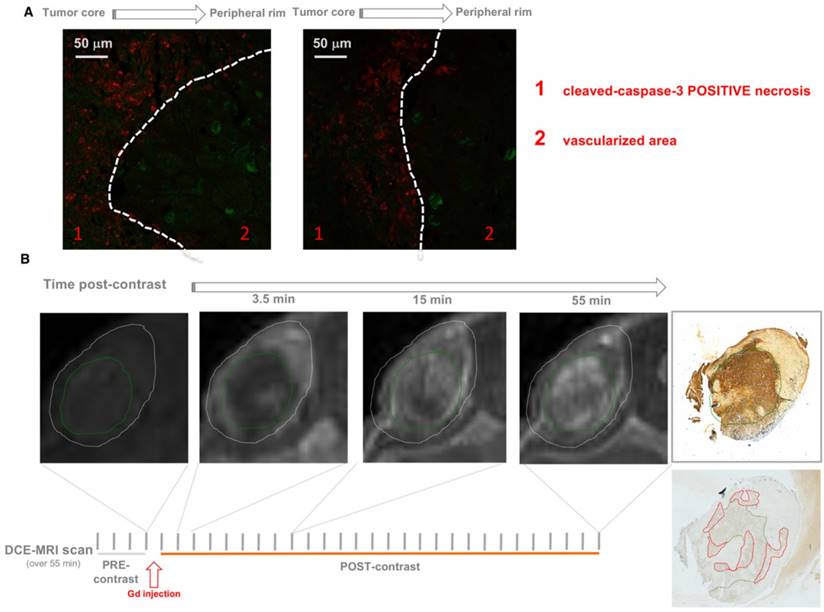
It has been previously shown that the effect of conventional chemotherapy decreases with increasing distance from tumor blood vessels [7]. In contrast with this notion, we found that caspase-3 positive cells were located in distal areas from vessels (Figure 5A).
Contrast-enhanced magnetic resonance imaging (DCE-MRI) with gadolinium (Gd) as contrast agent allows monitoring of the dynamics of fluid perfusion within tumor parenchyma over time. Consistent with previous reports [3, 31], Gd perfusion is delayed in necrotic regions, but once penetrated, Gd is retained longer than it does in peripheral vascularized areas due to the inefficient wash-out. This may be because necrosis generated by the treatment might entrap 32P-ATP inside tumor parenchyma thus allowing a sustained cross-fire effect on surrounding cells over time. As demonstrated by Cheng et al., 32P was actively incorporated into the phosphate-ribose backbone of DNA of rapidly growing cells, which could be one possible mechanism of entrapment.
Repeated injection after 2 weeks enhances the inhibitory effect of 32P-ATP on tumor growth
As shown above, the effect of a single-dose of 32P-ATP on tumor growth is reduced 2 weeks after the injection, which is consistent with the physical half-life of 32P-ATP decay. We examined whether a second injection 7 days after the first one could prolong the effect of 32P-ATP and improve the efficacy of the treatment. As expected, the reduction in tumor growth rate that ensued after the first injection persisted over the entire duration of the experiment and at the endpoint (almost 20 days after cell injection), the tumor volumes of 32P-ATP-treated mice were significantly smaller than those from the controls (Figure 6A). There was no difference in the tumor growth rate between mice treated either with 50 μCi or 100 μCi of 32P-ATP. CLI signal immediately after the second injection was much higher than that after the first injection, indicating a cumulative effect of 32P-ATP within the tumor parenchyma (Figure 6B).
Apoptotic cells (caspase-3-cleaved -positive) were uniformly dispersed within necrotic foci in control tumors but were restricted to the edge of necrotic foci in 32P-ATP-treated tumors (A and B). Plots on the left of panel A display the histogram of color intensity as a function of distance along the yellow lines reported on the respective caspase-3-cleaved stained slide. Pink highlights on the plot indicate the tracts of the line passing through the grossly necrotic areas. Peaks appear to be uniformly spread along the line within the necrotic areas of control tumors and are mainly restricted to the edge of necrotic areas in the 32P-ATP treated tumors.
Histopathologic analysis of 32P-ATP-treated tumors showed a significantly reduced number of necrotic foci that covered a considerably larger total fraction of tumor parenchyma (Figure 6C).
Model of 32P-ATP antitumor effect
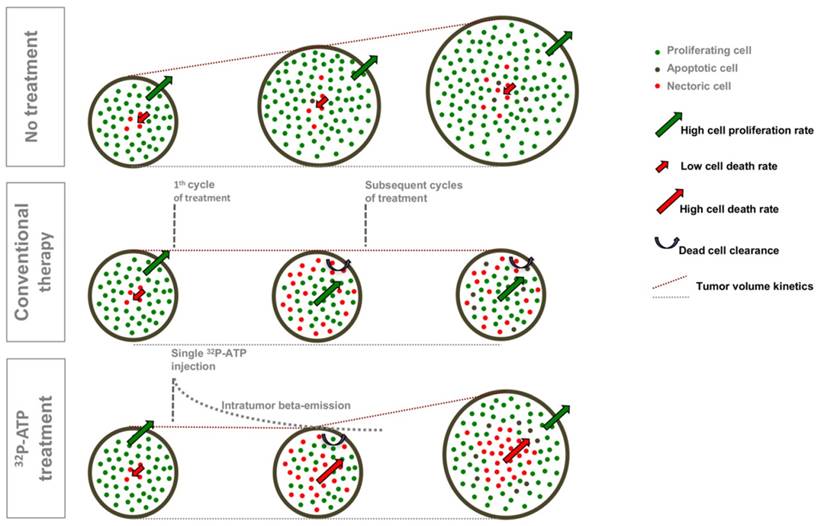
Our data suggest that the antitumor action of 32P-ATP proceeds through a dynamic process that is different from that of conventional chemotherapeutics (Figure 7). More precisely, in conventional therapies, drugs distribute mainly within the well-vascularized peripheral rim of the tumor and induce cell death locally. Depletion of peripheral cells improves vascularization of inner cells, which then proliferate faster and drive tumor repopulation between intermittent cycles of therapy. Sustained or multiple cycles of therapy allow a significant reduction of tumor volume, but inner cells become highly proliferative and more aggressive. The 32P-ATP treatment, on the other hand, reaches tumor core and its effect persists over days to weeks after a single dose injection. The proliferation of peripheral cells continues, but the extension of intratumor necrosis is large, and proliferating area at the peripheral rim is eroded. Although this results in limited reduction in tumor volume, the tumor core is destroyed and repopulation of inner cells is prevented.
Co-immunostaining of 32P-ATP-treated tumor slices for markers of endothelial cells (Von Willebrand Factor, VWF, Green) and apoptotic cells (caspase 3-cleaved, Red) revealed vessels and apoptosis in clearly distinct areas of tumor parenchyma (A). DCE-MRI with Gadolinium shows that necrotic regions (green circle) of 32P-ATP-treated tumors match with areas of tumor parenchyma where signal intensity appears later but persists longer (B).
Discussion
The limited accessibility of avascular tumor core to therapeutic drugs is amongst the major causes of therapy failure. Development of methods to avoid this limitation is an important challenge in medical oncology. Strategies that have been assessed to address this challenge in the last decades include: combination therapies aimed at improving tumor vascularization or reducing IFP [32, 33], development of drugs that selectively target hypoxic regions [34], and the use of devices that allow a controlled and continuous release of therapeutic drugs [35]. All these strategies, when successful, result in a significant increase of necrotic/apoptotic cell death throughout the entire tumor parenchyma, thus indicating the successful delivery of therapeutic drugs in regions that are normally inaccessible. Herein, we show that a single or double injection of 32P-ATP is sufficient to achieve the same result, inducing disruption of tumor structure throughout the entire tumor parenchyma and especially within the tumor core.
The 32P-ATP-induced destruction of tumor parenchyma is due to two physical properties of 32P-ATP, the spatial extension and the temporal duration of its beta-emission. With regard to the spatial extension, 32P-ATP acts at a distance since the range of electrons emitted by 32P-ATP has a length of 5 mm in tissues19. Therefore, the intratumor spatial distribution of 32P-ATP with its "cross-fire" effect is expected to extend 5 mm beyond the edge of the distribution area. This means that electrons emitted by 32P-ATP located at the peripheral rim would reach the core of tumors. As for the temporal duration, once 32P-ATP enters the tumor, it emits beta particles for two weeks of its half-life and beyond unless it is cleared by the outward fluid flow. The lack of an efficient lymphatic drainage in the avascular tumor core limits 32P-ATP washout. This would enhance the efficacy of 32P-ATP therapy by allowing it to be entrapped, resulting in a persistent therapeutic action over days or weeks after the first injection. Consistent with this notion, necrosis exhibited a delayed but durable vascular perfusion by DCE-MRI [3]. Also, microscopic co-mapping of cleaved caspase-3 and the endothelial marker von Willebrand Factor showed accumulation of apoptotic cells distal from blood vessels, which is contrary to the reports showing a progressive decrease in the therapeutic effects of conventional drugs with increasing distance from vessels.
Although the dynamic distribution of 32P-ATP within the tumor parenchyma and its uptake by tumor cells remain to be further investigated, our results provide, for the first time, evidence that 32P-ATP can damage the architecture of tumor parenchyma to induce massive cell death that extends over the entire inner core. This suggests that 32P-ATP might be exploited alone, or in combination with other drugs, to overcome the physical barrier posed by the vascular inaccessibility of the tumor core. This can become a novel potential tool to remarkably improve the efficacy of treatments currently used in clinical settings.
Due to the intrinsic tendency of 32P to concentrate in the bone, liver, and spleen, 32P-based drugs have been successfully used for decades in the treatment of myeloproliferative diseases [36-38]. Our results extend the potential use of 32P for the treatment of solid tumors and, more specifically, in combination with other therapies whose efficacy might be limited by the scarce intratumor accessibility. Nevertheless, the therapeutic use of 32P-based drugs is discouraged as it may induce side effects in long-term, such as an increased risk to develop leukemia [36-38].
Our findings indicated clear benefits of 32P-ATP treatment in terms of massive cell death within the tumor core as well as a reduction in tumor volume. However, as for every new therapeutic approach, specific cost/benefit ratio needs to be determined in relation to a specific clinical context. By using CLI, we were also able to image and quantify the Cerenkov light emission of 32P-ATP in the whole animal and particularly in the tumor region. We can thus consider 32P-ATP as a theranostic agent, at least for preclinical studies involving small animals.
Injection of 2 subsequent doses of 32P-ATP at 1-week interval confirms and amplifies the anti-tumor effect reducing the tumor growth rate. Tumor volume in 32P-ATP-treated mice was significantly below the control group at the endpoint of experiments (A), as measured manually or by MRI. CLI displays an increase after the second injection indicating a cumulative effect (B). Necrosis covered a significantly larger total fractional area in 32P-ATCP treated tumors compared to controls but was concentrated in a reduced number of foci (C).
A comparative diagram of the dynamic models of tumor progression in untreated tumors or those treated with conventional therapies or with 32P-ATP. Untreated tumors grow fast with a minimal frequency of spontaneous apoptotic cell death within the tumor core; tumor treated with conventional therapies exhibit massive cell death along the peripheral rim where efficient vessels allow drug penetration. Tumor volume does not increase during the treatment, but inner cells proliferate faster and drive tumor repopulation. In 32P-ATP-treated tumors, the tumor volume is less reduced than it is in conventional therapies, but the tumor core is massively destroyed and tumor repopulation is prevented.
Materials and methods
Animals
All animal experiments were approved by Verona University Ethical Committee and authorized by Ministerial Decree (Aut. n.29/2014-B). Animal experiments were in accordance with the Amsterdam Protocol on animal protection and welfare and conducted according to the guidelines of Federation of European Laboratory Animal Science Associations (FELASA).
Treatment
A pilot experiment was performed aimed at comparing the anti-tumor effect of 32P-ATP with non-radiolabeled ATP. 24 nu/nu mice, inoculated subcutaneously with 2x106 HT-29 cancer cells, were subdivided into the following groups and received: 1) 32P-ATP-treated (n = 8): a single intra venous (i.v.) injection of 32P-ATP (3.7 MBq) at day 9 after HT-29 cell inoculation; 2) ATP-treated (n = 8): i.p injection of 1 ml of 25 mM ATP (pH 6.2) in PBS solution for 10 days starting from day 9 after HT-29 cell injection; 3) PBS-treated (n = 4): daily i.p. injection of 1 ml of PBS for 10 days; 4) non-treated (n = 4).
32P-ATP (Perkin Elmer) 3000 Ci/mmol was injected into the tail vein when the highest diameter of their tumors reached the size of 8-10 mm.
Once the beneficial effect of 32P-ATP treatment was demonstrated in this cancer model, we designed a new experiment aimed at assessing the effect of a double injection of 32P-ATP. 24 nu/nu mice were inoculated subcutaneously with 2x106 HT-29 cancer cells and subsequently randomized into 3 groups: 1) 32P-ATP - 100 µCi - treated (n = 9): received 2 injections of 32P-ATP (100 µCi) at day 7 and 14; 2) 32P-ATP - 50 µCi - treated (n = 9): received 2 injections of 32P-ATP (50 µCi) at day 7 and 14; 3) non-treated (n = 8).
Tumor growth was measured by the width (W) and length (L) of the tumor twice a week using calipers. Tumor volume was calculated using the formula (W2xL)/2.
Magnetic resonance imaging
All animals underwent MRI examination the day before injection of 32P-ATP and the first and second week post injection. Mice were anesthetized by inhalation of a mixture of air and O2 containing 0.5-1% isoflurane and were placed in a prone position inside a 3.5 cm i.d. transmitter-receiver birdcage coil. Images were acquired using a Biospec tomograph (Bruker, Karlsruhe, Germany) equipped with a 4.7-T, 33 cm bore horizontal magnet (Oxford Ltd., Oxford, UK). Turbo RARE-3D images were acquired for tumor localization and volume measurements using the following parameters: TR = 1200 ms, TE = 47 ms, slice thickness = 25 mm, field of view (FoV) = 5 cm × 2.5 cm × 2.5 cm, matrix size = 256 × 128 × 32.
DCE-MRI was performed with Gd-DTPA (Magnevist®; molecular weight 0.57 kDa) as a contrast agent. A dynamic series of transversal RARE t1-weighted images were acquired with the following parameters: repetition time/echo time 300/10.7 ms, matrix size 256 × 128, FoV = 6 cm × 3 cm, the number of averages = 8. The acquisition time for a single image was 57 s; dynamic scans of 30 images were acquired at 5 s intervals (total acquisition time approximately 31 min).
The contrast agent at 300 μmol/kg dosage was injected in a bolus during the interval between the fifth and the sixth scans. A phantom containing 1 mM Gd-DTPA in saline was inserted in the FoV and used as an external reference standard.
Cerenkov luminescence imaging acquisitions
All the CLI acquisitions were performed using the IVIS Spectrum optical imager (Perkin Elmer, Massachusetts, USA), equipped by a -90 ˚C cooled camera sensor back-thinned, back-illuminated grade 1 CCD 2.7 cm × 2.7 cm, 2048 pixel × 2048 pixel. During image acquisition, mice were kept under gaseous anesthesia (2% of isoflurane and 1 L/min of oxygen).
The images were acquired in bioluminescence modality with FoV = 13 cm and with the following parameters: exposure time = 300 s, f = 1, binning = 8. Dark measurements were obtained before image acquisition and subtracted from the luminescence images. Images were acquired and analyzed with Living Image 4.4 (Perkin Elmer, Massachusetts, USA). Measurements of the total flux (photons/s) were done on regions of interest (ROIs) drawn on the optical images corresponding to the entire body or the tumor region.
Post-mortem analyses
Quantification of the necrotic fraction
At the endpoint of the experiment, tumors were dissected, fixed overnight in 4% paraformaldehyde, and embedded in paraffin for subsequent histopathological analysis. Necrotic areas were quantified on 5 µm thick sections stained with Hematoxylin & Eosin. Multiple images were acquired covering the whole section and the percentage of necrotic area (i.e. necrotic area/ total area ratio) was calculated on each image using ImageJ software.
Immunohistochemistry
Proliferating cells were identified in tumor sections by immunohistochemical staining with an antibody specific for Ki67 antigen (Abcam #ab15580). Apoptotic cells were identified and quantified on tumor sections by immunohistochemical staining with an antibody specific for caspase-3-cleaved (Asp175, Cell Signaling #9661S). Ki67 and caspase-3-cleaved-positive cells were mapped and quantified. Pictures were obtained by the automated acquisition and assembling of multiple images covering the whole sections.
Death-associated DNA fragmentation was assessed by TUNEL assay (Roche Applied Sciences #11684817910) on tumor sections according to the manufacturer's protocol.
Abbreviations
CLI: Cerenkov Luminescence Imaging; IFP: Interstitial fluid pressure; CR: Cerenkov radiation; PBS: Phosphate-Buffered Saline; TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling; DCE-MRI: Contrast-enhanced magnetic resonance imaging; FoV: field of view; ROIs: regions of interest.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-57
2. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-6
3. Galiè M, Farace P, Merigo F, Fiorini S, Tambalo S, Nicolato E, Sbarbati A, Marzola P. Washout of small molecular contrast agent in carcinoma-derived experimental tumors. Microvasc Res. 2009;78:370-8
4. Marzola P, Farace P, Calderan L, Crescimanno C, Lunati E, Nicolato E, Benati D, Degrassi A, Terron A, Klapwijk J, Pesenti E, Sbarbati A, Osculati F. In vivo mapping of fractional plasma volume (fpv) and endothelial transfer coefficient (Kps) in solid tumors using a macromolecular contrast agent: correlation with histology and ultrastructure. Int J Cancer. 2003;104:462-8
5. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popović Z, Huang P, Bawendi MG, Boucher Y, Jain RK. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516
6. Fuso Nerini I, Morosi L, Zucchetti M, Ballerini A, Giavazzi R, D'Incalci M. Intratumor heterogeneity and its impact on drug distribution and sensitivity. Clin Pharmacol Ther. 2014;96:224-38
7. Saggar JK, Fung AS, Patel KJ, Tannock IF. Use of molecular biomarkers to quantify the spatial distribution of effects of anticancer drugs in solid tumors. Mol Cancer Ther. 2013;12:542-52
8. Tan Q, Saggar JK, Yu M, Wang M, Tannock IF. Mechanisms of Drug Resistance Related to the Microenvironment of Solid Tumors and Possible Strategies to Inhibit Them. Cancer J. 2015;21:254-62
9. Lankelma J, Dekker H, Luque FR, Luykx S, Hoekman K, van der Valk P, van Diest PJ, Pinedo HM. Doxorubicin gradients in human breast cancer. Clin Cancer Res. 1999;5:1703-7
10. Huxham LA, Kyle AH, Baker JH, Nykilchuk LK, Minchinton AI. Microregional effects of gemcitabine in HCT-116 xenografts. Cancer Res. 2004;64:6537-41
11. Naumov GN, Folkman J, Straume O. Tumor dormancy due to failure of angiogenesis: role of the microenvironment. Clin Exp Metastasis. 2009;26:51-60
12. Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer. 2005;5:516-25
13. Vassileva V, Allen CJ, Piquette-Miller M. Effects of sustained and intermittent paclitaxel therapy on tumor repopulation in ovarian cancer. Mol Cancer Ther. 2008;7:630-7
14. Goldsmith SJ. To ablate or not to ablate: issues and evidence involved in 131I ablation of residual thyroid tissue in patients with differentiated thyroid carcinoma. Semin Nucl Med. 2011;41:96-104
15. Nisa L, Savelli G, Giubbini R. Yttrium-90 DOTATOC therapy in GEP-NET and other SST2 expressing tumors: a selected review. Ann Nucl Med. 2011;25:75-85
16. Cheng Y, Yang J, Agarwal R, Green GM, Mease RC, Pomper MG, Meltzer SJ, Abraham JM. Strong inhibition of xenografted tumor growth by low-level doses of [(32)P]ATP. Oncotarget. 2011;2:461-6
17. Cheng Y, Senthamizhchelvan S, Agarwal R, Green GM, Mease RC, Sgouros G, Huso DL, Pomper MG, Meltzer SJ, Abraham JM. [32P]ATP inhibits the growth of xenografted tumors in nude mice. Cell Cycle. 2012;11:1878-82
18. Cheng Y, Kiess AP, Herman JM, Pomper MG, Meltzer SJ, Abraham JM. Phosphorus-32, a clinically available drug, inhibits cancer growth by inducing DNA double-strand breakage. PLoS One. 2015;10:e0128152
19. Lechner A, Blaickner M, Gianolini S, Poljanc K, Aiginger H, Georg D. Targeted radionuclide therapy: theoretical study of the relationship between tumour control probability and tumour radius for a 32P/33P radionuclide cocktail. Phys Med Biol. 2008;53:1961-74
20. Spinelli AE, Marengo M, Calandrino R, Sbarbati A, Boschi F. Optical imaging of radioisotopes: a novel multimodal approach to molecular imaging. Q J Nucl Med Mol Imaging. 2012;56:280-90
21. Spinelli AE, Boschi F. Novel biomedical applications of Cerenkov radiation and radioluminescence imaging. Phys Med. 2015;31:120-9
22. Boschi F, Spinelli AE. Cerenkov luminescence imaging at a glance. Current Molecular Imaging. 2014;3:106-117
23. Spinelli AE, D'Ambrosio D, Calderan L, Marengo M, Sbarbati A, Boschi F. Cerenkov radiation allows in vivo optical imaging of positron emitting radiotracers. Phys Med Biol. 2010;55:483-95
24. Boschi F, Calderan L, D'Ambrosio D, Marengo M, Fenzi A, Calandrino R, Sbarbati A, Spinelli AE. In vivo ¹⁸F-FDG tumour uptake measurements in small animals using Cerenkov radiation. Eur J Nucl Med Mol Imaging. 2011;38:120-7
25. Spinelli AE, Kuo C, Rice BW, Calandrino R, Marzola P, Sbarbati A, Boschi F. Multispectral Cerenkov luminescence tomography for small animal optical imaging. Opt Express. 2011;19:12605-18
26. Spinelli AE, Boschi, D'Ambrosio D, Calderan L, Marengo M, Fenzi A, Menegazzi M, Sbarbati A, Del Vecchio A, Calandrino R. Cherenkov radiation imaging of beta emitters: in vitro and in vivo results. Nucl Inst Meth Sect A. 2011;648:S310-S312
27. Rapaport E, Fontaine J. Anticancer activities of adenine nucleotides in mice are mediated through expansion of erythrocyte ATP pools. Proc Natl Acad Sci. 1989;86:1662-6
28. Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271-9
29. Shuhendler AJ, Ye D, Brewer KD, Bazalova-Carter M, Lee KH, Kempen P, Dane Wittrup K, Graves EE, Rutt B, Rao J. Molecular Magnetic Resonance Imaging of Tumor Response to Therapy. Sci Rep. 2015;5:14759
30. Duan WR, Garner DS, Williams SD, Funckes-Shippy CL, Spath IS, Blomme EA. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol. 2003;199:221-8
31. Dadiani M, Margalit R, Sela N, Degani H. High-resolution magnetic resonance imaging of disparities in the transcapillary transfer rates in orthotopically inoculated invasive breast tumors. Cancer Res. 2004;64:3155-61
32. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418-29
33. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grützmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457-61
34. Sun JD, Liu Q, Wang J, Ahluwalia D, Ferraro D, Wang Y, Duan JX, Ammons WS, Curd JG, Matteucci MD, Hart CP. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res. 2012;18:758-70
35. Shemi A, Khvalevsky EZ, Gabai RM, Domb A, Barenholz Y. Multistep effective drug distribution within solid tumors. Oncotarget. 2015;6:39564-77
36. Lawrence JH. Preliminary Report on a New Method of Treatment of Leukemia and Polycythemia. Radiology. 1940;35:51-60
37. Najean Y, Rain JD. Treatment of polycythemia vera: use of 32P alone or in combination with maintenance therapy using hydroxyurea in 461 patients greater than 65 years of age. The French Polycythemia Study Group. Blood. 1997;89:2319-27
38. Berlin NI. Treatment of the myeloproliferative disorders with 32P. Eur J Haematol. 2000;65:1-7
Author contact
![]() Corresponding author: spinelli.antonelloit
Corresponding author: spinelli.antonelloit