Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(17):4340-4349. doi:10.7150/thno.18563 This issue Cite
Research Paper
Sustained Release Talazoparib Implants for Localized Treatment of BRCA1-deficient Breast Cancer
Jodi E. Belz1, 2, Rajiv Kumar2, 3, Paige Baldwin1, 2, Noelle Castilla Ojo2, Ana S. Leal4, 5, Darlene B. Royce4, Di Zhang4, Anne L. van de Ven2, 3, Karen T. Liby4, 5, Srinivas Sridhar2, 3 ![]()
1. Northeastern University, Department of Bioengineering, Boston, MA;
2. Nanomedicine Science and Technology Center, Northeastern University, Boston, MA;
3. Northeastern University, Department of Physics, Boston, MA;
4. Geisel School of Medicine at Dartmouth, Department of Pharmacology, Hanover, NH;
5. Michigan State University, Department of Pharmacology and Toxicology, East Lansing, MI.
Received 2016-11-30; Accepted 2017-6-17; Published 2017-9-26
Abstract

Talazoparib, a potent PARP inhibitor, has shown promising clinical and pre-clinical activity by inducing synthetic lethality in cancers with germline Brca1/2 mutations. Conventional oral delivery of Talazoparib is associated with significant off-target effects, therefore we sought to develop new delivery systems in the form of an implant loaded with Talazoparib for localized, slow and sustained release of the drug at the tumor site in Brca1-deficient breast cancer. Poly(lactic-co-glycolic acid) (PLGA) implants (0.8 mm diameter) loaded with subclinical dose (25 or 50 µg) Talazoparib were fabricated and characterized. In vitro studies with Brca1-deficient W780 and W0069 breast cancer cells were conducted to test sensitivity to PARP inhibition. The in vivo therapeutic efficacy of Talazoparib implants was assessed following a one-time intratumoral injection in Brca1Co/Co;MMTV-Cre;p53+/- mice and compared to drug-free implants and oral gavage. Immunohistochemistry studies were performed on tumor sections using PCNA and γ-H2AX staining. Sustained release of Talazoparib was observed over 28 days in vitro. Mice treated with Talazoparib implants showed statistically significant tumor growth inhibition compared to those receiving drug-free implants or free Talazoparib orally. Talazoparib implants were well-tolerated at both drug doses and resulted in less weight loss than oral gavage. PARP inhibition in mice treated with Talazoparib implants significantly increased double-stranded DNA damage and decreased tumor cell proliferation as shown by PCNA and γ-H2AX staining as compared to controls. These results demonstrate that localized and sustained delivery of Talazoparib via implants has potential to provide superior treatment outcomes at sub-clinical doses with minimal toxicity in patients with BRCA1 deficient tumors.
Keywords: Talazoparib, PARP inhibitor, BRCA-1, breast cancer, local therapy, sustained delivery.
Introduction
The breast cancer-associated gene 1 (BRCA1) is the most frequently mutated tumor suppressor gene found in familial breast cancers. Women with this mutation have a 50-80% risk of developing breast cancer in their lifetime. As many as 75% of these cases are diagnosed as triple-negative breast cancer (TNBC), a subtype known for its poor prognosis [1-3]. Mutations of the BRCA1 gene modulate many cellular functions including DNA damage and repair, homologous recombination, cell-cycle regulation, transcription, epigenetic modification and apoptosis. Typically, BRCA1 is required for homologous recombination, a commonly utilized double-stranded break DNA repair mechanism. In tumor cells which have a loss of both BRCA1 genes, homologous recombination is lost and cells must rely on non-homologous end joining, a more error prone pathway, for survival [4-7].
Poly-ADP-Ribose Polymerase (PARP) plays a key role in the repair of single-stranded DNA breaks and is recruited to sites of DNA damage to initiate repair [6, 8, 9]. PARP inhibition (PARPi) produces cell death via synthetic lethality in cancers with genetic predispositions for impaired DNA repair or impaired transcription, such as BRCA1 mutants [1, 10, 11]. Drugs that inhibit PARP1 activity prevent the repair of single-stranded DNA breaks, which propagate into double-stranded breaks after DNA replication. PARPi in cells deficient in homologous recombination results in genomic instability and ultimately cell death by forcing cells to repair double-stranded breaks using lower fidelity methods such as non-homologous end joining [12-16]. PARP inhibitors have shown promising results in clinical trials for breast cancers, particularly those due to either inherited mutation or somatic inactivation of BRCA1/2 genes which are present in ~15% of the patient population [17-19]. These drugs are also expected to have wide applicability beyond BRCA mutations in breast cancers that exhibit “BRCAness” such as sporadic tumors with homologous recombination defects [20-22].
Talazoparib (BMN-673) is known for high PARP1 and PARP2 inhibition with potency at much lower concentrations than earlier generations of PARP1/2 inhibitors like Olaparib or Veliparib [10, 23]. The cytotoxicity of Talazoparib at nanomolar concentrations is believed to be related to the trapping of PARP-DNA complexes based on studies in PARP1-/- mouse models [24-26]. However, the hydrophobic nature of Talazoparib, combined with its oral delivery route, limits drug bioavailability (~50% after first-pass metabolism), necessitating the use of high doses in patients and leading to off-site toxicity [10]. Thus, Talazoparib is an attractive drug candidate for breast cancer patients with Brca1 mutations, but its potency may limit long-term use [14, 27].
Local, sustained delivery of drugs has been achievable in the past few decades with the use of biodegradable polymers. Poly (lactic-co-glycolic) acid or PLGA is an FDA approved biodegradable and biocompatible copolymer that has been widely studied for the use of drug delivery systems. PLGA has tunable mechanical and degradation properties and is widely characterized for its extensive use in developing devices for controlled delivery and release of many types of molecules, proteins, and drugs [28, 29]. Many groups have shown that PLGA can be injected subcutaneously or intratumorally for long-term controlled drug delivery without the need for surgery. The hydrophobicity of the polymer can be altered by adjusting the ratio of the two polymers PLA and PGA, so that a wide variety of payloads can be incorporated into nanoparticles, microspheres, or implants [30, 31]. PLGA as an implant has been used intratumorally to deliver molecules, magnetic particles, nanoparticles, drugs, and imaging agents to improve the bioavailability at the tumor site and minimize any leaking of particles from the tumor [32-36]. Substantial research has been conducted that supports the use of biodegradable PLGA implants in the use of cancer treatment for prolonged drug delivery with minimal systemic side effects.
Here, we report a novel, biodegradable implant for localized and sustained delivery of Talazoparib to treat Brca1-deficient breast cancer with minimal treatment associated toxicities. The in vitro efficacy of Talazoparib was compared to that of other PARP inhibitors Olaparib and Niraparib using two breast cancer cell lines derived from Brca1-deficient mice. For in vivo delivery, Talazoparib implants were fabricated using poly(lactic-co-glycolic) acid (PLGA) with tuned release of drug over several weeks. Implants containing 25 or 50 µg Talazoparib were administered intratumorally into Brca1Co/Co;MMTV-Cre;p53+/- mice and their treatment efficacy was compared to drug-free control implants and Talazoparib treatment by oral gavage. Mice were monitored for signs of gross toxicity and immunohistochemistry was performed to assess DNA damage and cell proliferation in the tumors. The results showed that localized delivery of Talazoparib significantly delays tumor growth and prolongs survival time at sub-clinical doses. This new approach of localized treatment using Talazoparib implants is an attractive strategy to treat the 75% of TNBC patients and 33% of overall breast cancer patients which would benefit from PARPi therapy [12, 37, 38].
Materials and Methods
All solvents used were HPLC grade and purchased from Sigma-Aldrich unless otherwise stated. Talazoparib, Olaparib and Niraparib were purchased from SelleckChem (Houston, TX). Poly(lactic-co-glycolic) acid (PLGA) (Mw 7-17 kDa) was purchased from Sigma-Aldrich. Syringes and silicon tubing (i.d. 0.8 mm) were purchased from Fisher Scientific. Unless otherwise stated, all cell culture products (nutrient media, trypsin, Penstrep, fetal bovine serum) were obtained from Life Technologies and were used without further purification. Antibodies were procured from Abcam and Calbiochem. For cell viability assay CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) reagent was procured from Promega.
Talazoparib Implant Fabrication
Talazoparib implants were fabricated using a polymer extrusion method as reported previously by our group with several modifications [36]. The Talazoparib implants are composed of a solid Poly(lactic-co-glycolic) acid (PLGA, 50:50 PLA:PGA and Mw 7-17kDa) matrix embedded with 4% (w/w) Talazoparib. Briefly, Talazoparib implants were fabricated by dissolving Talazoparib (9 mg) in chloroform and mixing with PLGA solution (190 mg) in chloroform. The polymer/drug mixture was mixed thoroughly and left at 4°C for 30 minutes or until the mixture became a viscous paste. The paste was transferred to a 1 mL syringe and extruded into sterile silicone tubing (i,d, 0.8 mm) at a rate of 2.5 µL/min using an infusion pump (Harvard Apparatus, Holliston, MA). After extrusion, tubes were dried overnight at 60 °C. Implants were then cooled to room temperature and cut to appropriate size (2±0.3 mm in length) for storage in a closed vial at -20 °C. Drug loading per unit length (mm) spacer was determined from randomly selected implants (n=3). Implants were measured for length, dissolved, and Talazoparib was quantified using high-performance liquid chromatography (HPLC).
Implant Characterization
The loading of the drug in the Talazoparib implants was quantified via HPLC method using an Agilent 1100 system and reverse phase C18 Zorbek column. A standard curve was prepared for free Talazoparib in both the mobile phase and PBS buffer at concentrations ranging from 1-100 µg/mL. Talazoparib implants (n=3) were dissolved in dichloromethane followed by extraction of Talazoparib using methanol. The drug was quantified using a 50:50 methanol (0.1% TFA):water (0.1%TFA) mobile phase with a flow rate of 0.8 mL/min. Absorbance was detected at 232 nm with tret=3.98 minutes. The standard curves were fit with a linear regression curve (R2=1) and the curve was used to quantify the concentration of Talazoparib in each implant.
Scanning Electron Microscopy
Talazoparib implants were flash frozen in liquid nitrogen and fractured using a cooled razor. Fractured and non-fractured implants were attached to a specimen mount using a conductive carbon adhesive tab and then sputter coated with 10 - 15 nm of platinum using a vacuum evaporator (DV502, Denton Vacuum, Moorestown, NJ). Spacer samples were imaged in both SE (secondary electrons) and BSE (back-scattered electrons) mode using aS-4800 field emission SEM (Hitachi, Chiyoda, Tokyo, Japan) at 5 kV.
Release Kinetics studies
The release profile of Talazoparib from the implants was studied using HPLC. To determine the release kinetics Talazoparib implants were cut to predetermined lengths, measured, and placed in 1 ml of PBS (pH 6.0 adjusted with 1 N HCl) at 37°C with gentle shaking to mimic biological tumor conditions for 28 days. At each predetermined time point, buffer was completely removed and replaced with fresh buffer to maintain the concentration gradient between drug depot and buffer. Each collected buffer fraction was subjected to HPLC analysis as described earlier and released Talazoparib was quantified. Care was taken as to not agitate and disrupt the integrity of the spacer during the buffer transfer. After 28 days, the spacer was dissolved and any remaining drug was quantified. The total drug released and remaining in the spacer was summed and compared to the estimated drug loading from HPLC analysis for the given length of spacer. The released drug from the implants was plotted as a function of time.
In vitro Therapeutic Efficacy
W0069 and W780 cell lines derived from mammary tumors from genetically engineered Brca1Co/Co;MMTV-Cre;p53+/- mice were used for all studies. All cells were grown in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% FBS and 1% Penn/Strep. To evaluate and compare the efficacy of various PARP inhibitors, W0069 and W780 were treated with 10 µM Olaparib, Talazoparib, or Niraparib for 24-72 hours. [3] Cell lysates were processed for Western blotting with antibodies against cleaved PARP (Upstate), cleaved caspase 3, γ-H2aX (Abcam), or α-tubulin (Calbiochem). IC50 values were calculated to determine the sensitivity of the cell lines to PARP inhibitors Talazoparib, Olaparib, and Niraparib. W0069 and W780 cells were seeded into 96 well plates (Corning) at 1000 cells per well. The following day cells were treated with Talazoparib concentrations ranging from 0 to 100 nM. One week after seeding, cell viability was measured with an MTS assay (Promega) to measure the metabolic activity of the cells. Dose response was plotted and fit using a variable slope four parameter logistic equation constrained at 0 to determine the IC50 value (Prism). All experiments were done in triplicate.
In vivo Therapeutic Efficacy
All animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Dartmouth College. For in vivo therapeutic efficacy evaluation, two different lengths of Talazoparib implants were used. Fabricated implants with the same unit loading of Talazoparib were cut into two different lengths of 1 mm and 2 mm. Since the drug loading in Talazoparib implants was 25 μg per mm, 1 and 2 mm long implant contain a total of 25 and 50 μg of Talazoparib respectively. The in vivo therapeutic efficacy of Talazoparib implants was carried out in a genetically engineered Brca1Co/Co;MMTV-Cre;p53+/- breast cancer mouse model. Treatment in mice was started when the tumor size was approximately 4-6 mm in diameter. The Talazoparib implant was administered intratumoraly using 18G brachytherapy applicator needles (Bard Medical, Covington GA). For intratumoral implantation, the Talazoparib implant was preloaded into the applicator needle and guided laterally into the center of the tumor. The implant was ejected by pressing the stylet simultaneously as the outer needle (cannula) was withdrawn from the tumor. Animal skin glue was used to seal the small hole in the skin to avoid any leakage of the implant out of the tumor. Four groups of animals (n=6-8 animals per group) were used in the study; a control arm received intratumoral implantation of blank PLGA implants (no Talazoparib), one additional control arm was treated with free Talazoparib via oral gavage and two Talazoparib implant groups which were treated with a single 1 mm or 2 mm long implant. Free Talazoparib was given orally for comparison to the current clinical administration of treatment. An equivalent dose of free Talazoparib (as in 2 mm long implant) was given a total of six times by oral gavage every other day over a period of 12 days (8.3 µg per gavage). Tumor size was monitored using calipers and mice were weighed twice weekly to evaluate weight loss. Tumor volumes were calculated using the formula: volume = length x width x height/2. Mice were sacrificed if the tumor diameter exceeded 1 cm, the tumor volume exceeded 10% of body weight, or if a tumor ulcerated or interfered with mobility. For immunohistochemistry, tumors were harvested, fixed in neutral-buffered formalin, embedded in paraffin and sectioned. Sections were stained with PCNA (Santa Cruz 1:200) or γ-H2aX (Abcam 1:100) antibodies as previously described [3]. To determine the percentage of PCNA- or γ-H2aX-positive cells, two sections on the periphery of each tumor (n=4-5/treatment, ~1000 cells each) were counted. Slides were randomized and group labels removed prior to counting.
Statistical Analysis
Results are expressed as mean ± standard error of mean (SEM) and were analyzed using the t test, χ2 test, or one-way ANOVA on ranks (Wilcoxon signed rank test) using Prism5 or SigmaStat3.5 software. All P values were two-sided with a P value of <0.05 considered statistically significant.
Results
PLGA implants continuously release Talazoparib
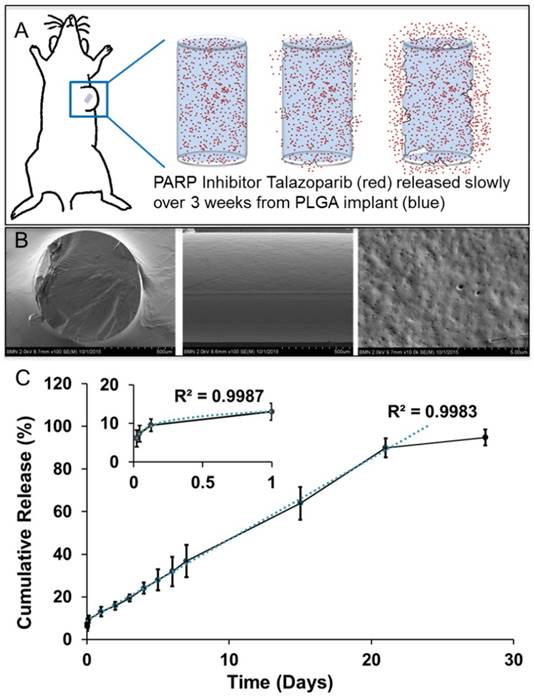
Poly(lactic-co-glycolic) acid implants (0.8 mm diameter) containing 25 µg/mm Talazoparib were fabricated by extrusion followed by curing. A single batch of fabrication with the above mentioned PLGA/Talazoparib mixture yields approximately 20 implants of 2 mm length. The implant was designed to allow direct placement into solid tumors using an 18G needle, after which Talazoparib was slowly released by PLGA degradation and allowed to diffuse into the tumor (Fig 1A). In these studies, we used implants of 1 and 2 mm length, equivalent to 25 µg and 50 µg of Talazoparib respectively, as measured by HPLC. Scanning electron microscopy images of flash-frozen and fractured PLGA implants showed a smooth surface intercalated with homogenously distributed nanoscale pores (Fig 1B). It is likely that these pores represent pockets of Talazoparib distributed throughout the implant. Talazoparib implants showed continuous drug release in phosphate buffered saline (pH 6.0) at 37°C. Drug release was characterized by an initial burst release (10% in day 1), followed by a linear release (R2 = 0.9983) until day 28 (Fig 1C). The initial burst release can be attributed to desorption of drug molecules from the implant surface. Release kinetics studies carried out with different length implants showed that the percentage of drug release was independent of implant length indicating that drug dosing to the tumor can be adjusted by changing the length of the implant without affecting the kinetics of the drug released.
Biodegradable PLGA implants for sustained intratumoral delivery of Talazoparib. A) Schematic depiction of PLGA implant degradation (blue) for localized delivery of Talazoparib (red). B) Scanning Electron Microscopy images of flash-frozen and fractured, drug-loaded PLGA implants a solid smooth surface with nanoscale pores homogenously spread throughout surface of implant. C) In vitro release profile of Talazoparib from 2 mm PLGA implants (n=3) in phosphate buffered saline (pH 6.0).
Sustained PARP inhibition produces apoptosis and DNA damage in vitro
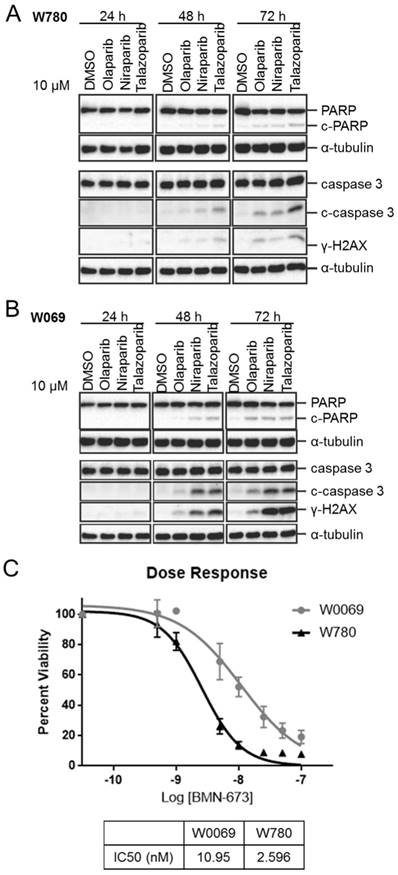
PARP inhibitors Olaparib, Niraparib, and Talazoparib were compared in BRCA1-mutant breast cancer cell lines W780 and W0069 derived from Brca1Co/Co;MMTV-Cre;p53+/- genetically engineered mice to evaluate the potency of three PARP inhibitors in these cell lines. Talazoparib was found to be the most potent inhibitor in the W780 cell line (Fig. 2A), as evidenced by an increase in cleaved PARP (c-PARP), cleaved caspase 3 (c-caspase 3), and γ-H2aX protein expression following 48-72 hours treatment. These proteins are markers of apoptosis and DNA damage, respectively. Similar results were obtained in W0069 cells (Fig. 2B), with both Talazoparib and Niraparib observed to be more potent than Olaparib. Both W0069 and W780 were highly sensitive to growth inhibition by Talazoparib, with IC50 values of 11 and 2.6 nM respectively (Fig. 2C).
In vitro analysis of different PARP inhibitors on W780 and W0069 breast cancer cell lines derived from Brca1Co/Co;MMTV-Cre;p53+/- mice. Western Blot data testing protein expression after treatment with Olaparib, Niraparib, and Talazoparib on Brca1-deficient cell line in A) W780 B) W0069 cells. C) Measured dose response curve and IC50 of Talazoparib in W780 and W0069 cells.
Sustained PARP inhibition produces tumor shrinkage
To determine the in vivo therapeutic efficacy of sustained Talazoparib delivery, drug-loaded PLGA implants were locally injected into Brca1Co/Co;MMTV-Cre;p53+/- spontaneous mammary gland tumors 4-6 mm in diameter. Mice received a one-time implantation of a single 1 or 2 mm length implant using an 18G brachytherapy needle. Untreated mice showed no significant difference in tumor growth rate or mouse weight to mice treated with empty implants (data not shown). For the entirety of the manuscript, controls refer to mice treated with control implants. As shown in Table 1, tumors treated with 1 mm control implants (0 µg Talazoparib) grew exponentially and had an average life span of 11.8 ± 1.9 days. These tumors increased in volume more than 7-fold, from an initial average of 67 ± 28 mm3 to a final average of 504 ± 67 mm3 (P=0.002). In contrast, tumors treated with 1 mm Talazoparib implants (25 µg drug) decreased in size by 50%, from an average initial tumor volume of 130 ± 42 mm3 to a final volume of 65 ± 14 mm3 (p = 0.002). The average lifespan of the mice treated with 1 mm drug-loaded implants was more than twice as long as mice in the control group (29 vs. 12 days; P < 0.001).
Talazoparib implants decrease tumor size and extend survival in BRCA1-deficient mice
| Initial Tumor Volume (mm3) | Final Tumor Volume (mm3) | # of mice/group | Lifespan after treatment (days) | |
|---|---|---|---|---|
| 0 µg control implant (1mm) | 66.7 ± 27.5 | 504 ± 67.4a | 5 | 11.8 ± 1.9 |
| 25 µg Talazoparib implant | 130.1 ± 42 | 65.4 ± 14.1b | 8 | 29 ± 3g |
| 0 µg control implant (2mm) | 72.1 ± 24.9 | 484.3 ± 68.8c | 7 | 11.9 ± 1.4 |
| 50 µg Talazoparib implant | 44.4 ± 5.2 | 14.9 ± 5.7c,d,e | 7 | 44.1 ± 1.8h |
| 50 µg Talazoparib gavage | 55.4 ± 16.2 | 537.1 ± 244.2f | 6 | 39.8 ± 6.4h |
Values are mean ± SEM.
a, P = 0.002 0 µg control implant (1mm) final volume vs. initial volume b, P = 0.002 25 µg Talazoparib implant final volume vs. 1mm control final volume c, P < 0.001 50 µg Talazoparib implant final volume vs. 2mm control final volume d, P < 0.05 50 µg Talazoparib implant final volume vs. 50 µg Talazoparib implant initial volume e, P < 0.001 50 µg Talazoparib implant final volume vs. 50 µg Talazoparib gavage final volume f, P = 0.026 50 µg Talazoparib gavage final volume vs. initial volume g, P < 0.001 25 µg Talazoparib implant vs. 0 µg control implant (1mm) h, P < 0.001 50 µg Talazoparib implant and 50 µg Talazoparib gavage vs. 0 µg control implant (2mm)
Since implants containing 25 µg Talazoparib were well-tolerated, we chose to test 2 mm implants containing 50 µg Talazoparib (Fig. 3). Measurements of tumor volume (Fig. 3A), tumor growth rates (Fig. 3B), and mouse survival (Table 1) demonstrated that the higher dose of 50 µg Talazoparib was more effective. Tumors treated with 50 µg Talazoparib implant decreased in volume by 67%, from 44 ± 5 mm3 at the beginning of treatment to 15 ± 6 mm3 after treatment (P < 0.05). The average lifespan of mice receiving Talazoparib implants was 44 days vs. 12 days (P < 0.001) for mice receiving control PLGA implants. An additional control arm, in which mice were treated with 50 µg Talazoparib by oral gavage over 12 days (8.3 µg x 6 doses), showed a slowed tumor growth rate compared to the 0 µg control implant, but was not as effective as the 50 µg implant group. As a result, the final tumor volume in the oral gavage group was no better than the control group but the average lifespan was increased (40 days vs. 12 days, P < 0.001).
Treatment with 50 µg Talazoparib implants decreases tumor size and slows tumor growth in Brca1-deficient mice. Implants (2 mm × 0.8 mm diameter) containing 0 µg (control) or 50 µg Talazoparib were injected into established mammary gland tumors in female BrcaCo/Co;MMTV-Cre;p53+/- mice. Mice treated by oral gavage received 6 doses of Talazoparib (50 µg Talazoparib total). A) Average tumor volumes before and after treatment (n=5-8/group). *, P < 0.05 vs. initial tumor volume; ǂ, P < 0.05 vs. all other groups. B) Change in tumor volume over a period of 28 days. C) Average body weight before and after treatment (* P < 0.05 vs. initial weight for the treatment group.
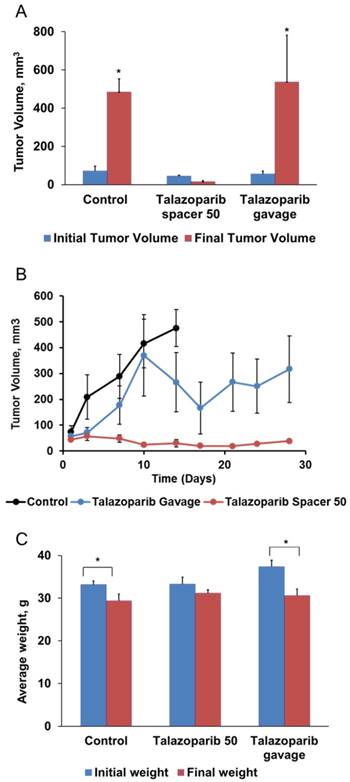
For evaluating the gross toxicity, mouse body weight was measured over the course of the survival studies. As shown in Fig. 3C, oral gavage of Talazoparib was accompanied by a statistically significant (P < 0.05) decrease of ~ 15% in mouse body weight that was not observed in mice treated with 50 µg Talazoparib implants. Talazoparib implants were well-tolerated and produced no significant change in initial vs. final body weight in either treatment group. Mice in the control group lost weight (P < 0.05), which was likely the result of ill health as they became moribund or perhaps a consequence of the relatively short lifespan (~12d) after treatment which did not allow for full recovery from the procedure. The absence of any adverse toxic effects in the Talazoparib implants groups also indicate that the Talazoparib implants have no residual solvents which can cause toxicity.
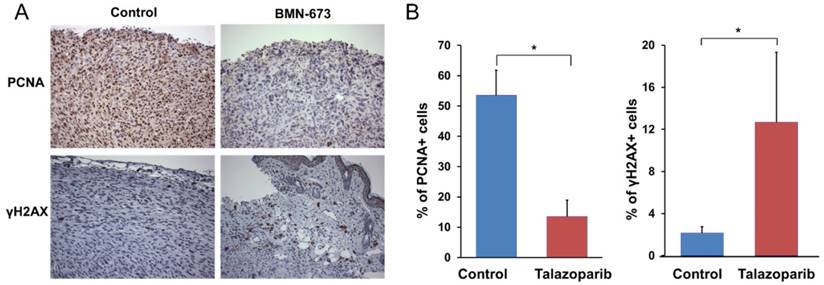
In order to verify tumor inhibition was a result of successful delivery of localized Talazoparib and its mechanism of action on tumor cells rather than a result of overt toxicity from the implant, tumor sections were analyzed by immunohistochemistry for assessment of cell proliferation and double stranded DNA breaks, two biomarkers indicative of successful treatment of PARP inhibitors. The results showed that the tumors treated with Talazoparib implants were not necrotic. Cell proliferation, as measured by PCNA staining (Fig. 4), was 4-fold lower than in control tumors (13.6 ± 5.3% vs. 53.5% ± 8.3%, P < 0.001). Talazoparib implants also showed significantly increased γ-H2aX expression, a marker of double-stranded DNA breaks. The percentage of γ-H2aX-positive cells in tumors treated with Talazoparib implants was 12.7 ± 6.6% compared to 2.2 ± 0.6% in control tumors treated with empty implants (P < 0.001). No overt signs of toxicity were observed.
Immunohistochemistry studies showing Talazoparib implants decrease PCNA staining and increase γ-H2aX staining in tumors from Brca1-deficient mice. A). Immunohistochemical staining for PCNA (top) and γ-H2aX (bottom) of tumors treated with 50 µg Talazoparib implants for 2 weeks. B). Quantification of the percentage of PCNA-positive or γ-H2aX-positive cells (mean ± SEM) from 4-5 tumors per group. (P < 0.001 versus control).
Discussion
Improved understanding of the genetic mutations prevalent in breast cancer has led to the development of new therapeutic strategies which target these mutations. Breast cancers with germline BRCA1 and BRCA2 mutations are promising candidates for DNA repair inhibition using Talazoparib. Talazoparib is a potent PARP1/2 inhibitor for the treatment of patients with deleterious germline BRCA1 or BRCA2 mutations and locally advanced and/or metastatic breast cancer. Currently, Talazoparib is administered orally, which not only limits its bioavailability but also results in treatment-associated toxicities in a majority of patients. Sustained, localized drug delivery systems that eliminate the need for daily oral dosing have the potential to improve patient outcomes and reduce systemic toxicity. Here we show that implant-based delivery of Talazoparib can address the limitations associated with oral Talazoparib.
Current brachytherapy procedures utilize inert spacers that are delivered intratumorally via an 18G needle. Our implants, of similar dimensions to brachytherapy spacers, can be easily and readily administered as an outpatient procedure using existing clinical protocols for a new breast cancer treatment. In this proof-of-concept study, we designed a degradable PLGA matrix containing embedded Talazoparib in order to slowly and continuously release this drug into tumors. Localized delivery of Talazoparib promises to not only reduce the amount of drug required to achieve therapeutic benefit but will also provide a continuous supply of Talazoparib to tumor cells over several weeks after a single administration. Thus, Talazoparib implants have the potential to bypass the concerns and challenges associated with oral delivery, metabolic stability, and pharmacokinetics. In the future, we envision that such implant could be used to deliver not just one drug but multiple therapies in order to produce synergistic therapeutic outcomes with reduced toxicity.
PLGA is an FDA-approved, biodegradable polymer with low glass transition temperature that provides an excellent matrix for implant fabrication. The hydrophobicity of Talazoparib ensures a uniform distribution of drug in the PLGA matrix, which was confirmed by scanning electron microscopy and release kinetic studies. Implant drug loading could be quantified by HPLC and the total available drug concentration could be tailored by changing implant length or administering multiple implants in the tumor. Talazoparib was selected for in vivo studies as it proved to be the most potent of the PARP inhibitors tested in the cell lines derived from the in vivo mouse model. The in vitro release kinetics showed a slow sustained release of the drug over 28 days, comparable to the 21 day time-frame in which Talazoparib is administered daily in clinical trials. While we expect these release kinetics to be altered in vivo due to enzymatic degradation of the PLGA polymer, we did observe a therapeutic benefit for as long as 28 days, suggesting that Talazoparib release in vivo is slow and sustained. Typically, PLGA is degraded via the hydrolysis of the ester bonds of the polymer backbone resulting in formation of acidic oligomers and we anticipate a similar degradation mechanism for Talazoparib implants. Since the release of the drug in buffer showed a diffusion based release, it is highly unlikely that there is any interaction between the drug molecules and PLGA matrix. The sustained release of the drug can be attributed to the diffusional constraints and slow degradation of the PLGA matrix. The competing dynamics with the continuous release of Talazoparib and simultaneous inhibitory response on the tumor cells will also alter the release profile of Talazoparib from the implants. However, the release profile in buffer indicates zero order kinetics suggesting a diffusion based release of Talazoparib. Thus, we predict that inside the tumor matrix, the release of Talazoparib will be a combination of diffusion as well as enzymatic degradation and surface erosion of the implants. Evidence of increased double-stranded DNA damage and decreased tumor cell proliferation was present at least 2 weeks after implantation in comparison to control tumors treated with empty implants. No gross toxicity was observed in any of the control mice, suggesting the method of fabrication completely absolves the implant of any trace amounts of toxic solvents. During fabrication, we dry the implants above the glass transition temperature of the polymer to induce the temperature dependent phase transition. This heating also allows for high vapor pressure and volatile solvents such as chloroform to completely evaporate during that drying time, which is evident in the lack of toxicity and biomarker changes in tumors treated with the implant. The tumor growth inhibition seen in our Talazoparib implant group weeks after treatment further attests to the long-term benefit of slow releasing Talazoparib implants and avoids the issue of low drug retention when injecting free drug into a tumor.
For evaluating the therapeutic efficacy of the Talazoparib implants, two different lengths were prepared. The cumulative drug doses used this study are 5-10 times lower than those traditionally administered in preclinical oral dosing studies [10]. The 25 µg dose was well-tolerated and decreased the tumor size by 50%. When the treatment dose was doubled, the higher dose was found to significantly enhance tumor growth inhibition and survival time with no additional body weight loss. Taken together, this data suggests that treatment response is dose-dependent. Given that the implants do not appear to generate overt systemic toxicities, our approach opens new opportunities for safely treating patients that require drug concentrations higher than the conventional dosing scheme.
Localized drug delivery at the target site have several positive attributes which includes reduced systemic toxicity as well as minimal toxicity to surrounding tissues, increased control and decreased variability by eliminating reliance on patient circulation for distribution as in case of intravenous administration of drugs. Localized drug delivery formulations can also impart sustained release behavior of the entrapped drug and thus have the potential to increase the effectiveness of treatment at lower concentrations as opposed to systemic or oral administrations. These formulations also modulate the pharmacokinetics of the drug in systemic circulation with a high concentration of drug delivered directly to the tumor site. While delivering drugs directly at the tumor site can improve the bioavailability in diseased cells while bypassing administration and reducing toxicity to healthy organs, local delivery is a major limitation in the case of metastatic disease.
In this study, mice with spontaneous growing tumors were used to test the implant against the current clinical administration (oral), and in many instances mice grew multiple tumors. There was little difference between the average lifespan of mice treated with Talazoparib by either the intratumoral implant or by oral gavage. The primary tumor was greatly reduced in size in mice receiving implants, but the drug had no effect on the other spontaneous tumors growing concomitantly in this mouse model, necessitating mouse sacrifice due to tumor burden. Thus mice treated with Talazoparib implants did not show any enhanced survival compared to those treated by gavage. This limitation needs to be carefully considered to determine the appropriate cases for clinical application of Talazoparib implants (i.e. in unresectable tumors, or in combination with other inhibitors, chemotherapeutics, or radiation treatment). In the case of metastasis, our animal studies show that only the tumor treated with the intratumoral implant exhibit growth inhibition while other untreated tumors in the mouse have no therapeutic benefit from the implant. This result does suggest that local delivery of PARP inhibitors would not be useful for treating metastatic breast cancer, and future studies should compare surgical resection and radiation to local drug delivery and test the combination of Talazoparib implants in conjunction with standard of care radiation or chemotherapy. These implants can be administered prior to radiation therapy or chemotherapy to combat the metastatic conditions. Currently, our group has initiated in vivo experiments to evaluate the combination therapy with external beam radiation therapy (EBRT) and Talazoparib implants in lung cancer animal models as well as a combination therapy with cisplatin and Talazoparib implants in the above mentioned breast cancer animal model. Also, we did observe some evidence of primary tumor regrowth (resistance) in implant-treated mice that lived beyond 30 days (data not shown). At these later time-points, tumor drug levels were not detectable by HPLC/MS. In future studies, we will evaluate if a second administration of Talazoparib implants, or the placement of implants into concomitantly growing tumors, would further prolong the survival time.
The use of Talazoparib implants as a therapeutic intervention in unresectable tumors either as monotherapy or in combination with other standard of care therapies like radiation or chemotherapy have the potential to provide high therapeutic benefits with minimal systemic toxicities. Future studies will compare local delivery of Talazoparib to radiation and chemotherapy for this clinical issue. This approach of localized treatment has strong clinical relevance as the treatment using these implants is based on standard clinical brachytherapy procedure which is routinely followed in clinics and will not require additional procedures or protocols during treatment. The limitation associated with this localized treatment approach is mainly in the metastatic conditions but which can be addressed by careful selection of patient populations as well as combining the localized treatment with the standard of care therapy. Overall, a good therapeutic index, low systemic toxicity and the flexibility of the Talazoparib implants formulation to combine with other therapies provides a promising therapeutic platform which can be applied in treatment of different cancer types.
Conclusion
In conclusion, we fabricated a PLGA implant for localized and sustained intratumoral release of Talazoparib. This unique route of PARP inhibitor delivery significantly slows tumor growth and prolongs survival time in a spontaneous mouse model of Brca1-deficient breast cancer. Through optimization of the PLGA formulation, sustained drug release was achieved over a scale of ~28 days both in vitro and in vivo. PARP inhibition was associated with significantly increased double-stranded DNA damage and decreased tumor cell proliferation for several weeks after implantation, further supporting the long-term effectiveness of these implants. The treatment efficacy was dependent on drug dose of the implant, with 50 µg Talazoparib producing more tumor growth inhibition than a 25 µg Talazoparib dose. No acute or chronic toxicity was observed in mice treated with clinically relevant doses for 28 days, indicating that Talazoparib implants may present an easy and feasible way for controlling treatment outcomes with lower systemic toxicity.
Acknowledgements
We are grateful to the Breast Cancer Research Foundation (K. Liby) and NSF-DGE- 0965843 to Northeastern University (S. Sridhar) for funding to support these studies.
Author's Contribution
JB, RK, KL, and SS designed the research and experimental plan. JB designed and fabricated the implants. JB, PB, and NC performed in vitro experiments. AL and DR carried out in vivo experiments. DZ completed the immunohistochemistry experiments. KL monitored and summarized the in vivo experiments. JB, KL, RK, and SS analyzed data. JB, KL, and RK and helped write/edit the manuscript, and AV and SS contributed to editing the manuscript. All authors read/approved the final manuscript.
Ethical Approval
All animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Dartmouth College.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ. et al. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol. Med. 2012;4:1087-96
2. Petrucelli N, Daly MB, Feldman GL. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010;12:245-59
3. To C, Kim E-H, Royce DB, Williams CR, Collins RM, Risingsong R. et al. The PARP Inhibitors, Veliparib and Olaparib, Are Effective Chemopreventive Agents for Delaying Mammary Tumor Development in BRCA1-deficient Mice. Cancer Prev. Res. 2014;7:698-707
4. Comen EA, Robson M. Poly(ADP-ribose) polymerase inhibitors in triple-negative breast cancer. Cancer J. 2010;16:48-52
5. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer. 2011;12:68-78
6. Deng C-X, Brodie SG. Roles of BRCA1 and its interacting proteins. Bioessays. 2000;22:728-37
7. Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell cycle. 2008;7:2902-6
8. Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer. 2011;105:1114-22
9. Hilton JF, Hadfield MJ, Tran M-T, Shapiro GI. Poly(ADP-ribose) polymerase inhibitors as cancer therapy. Front. Biosci. (Landmark Ed.). 2013;18:1392-406
10. Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R. et al. BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 2013;19:5003-15
11. Nijman SMB. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585:1-6
12. Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW. et al. Poly(ADP-Ribose) polymerase inhibition: targeted therapy for triple-negative breast cancer. Clin. Cancer Res. 2010;16:4702-10
13. Weil MK, Chen AP. PARP inhibitor treatment in ovarian and breast cancer. Curr. Probl. Cancer. 2011;35:7-50
14. Livraghi L, Garber JE, Gonzalez-Angulo A, Timms K, Liu S, Chen H. et al. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med. 2015;13:188
15. Goldberg MS, Xing D, Ren Y, Orsulic S, Bhatia SN, Sharp PA. Nanoparticle-mediated delivery of siRNA targeting Parp1 extends survival of mice bearing tumors derived from Brca1-deficient ovarian cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2011;108:745-50
16. Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: A new era of targeted therapy. Maturitas. 2015;81:5-9
17. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FBL, Imholz ALT. et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer. 2013;108:d2172-2177
18. De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Front. Oncol. 2013;3:1-12
19. Plummer R. Poly(ADP-ribose) polymerase inhibition: a new direction for BRCA and triple-negative breast cancer? Breast Cancer Res. 2011;13:218-23
20. Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011;10:1192-9
21. Turner N, Tutt A, Ashworth A. Opinion: Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer. 2004;4:814-9
22. Le Du F, Eckhardt BL, Lim B, Litton JK, Moulder S, Meric-Bernstam F. et al. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget. 2015;6:12890-908
23. Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br. J. Cancer. 2016;114:713-5
24. Huang J, Wang L, Cong Z, Amoozgar Z, Kiner E, Xing D. et al. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1-/- murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015;463:5551-556
25. Murai J, Huang SYN, Das BB, Renaud A, Zhang Y, Doroshow JH. et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588-99
26. Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S. et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014;13:433-43
27. Basu B, Sandhu SK, de Bono JS. PARP Inhibitors. Drugs. 2012;72:1579-90
28. Uhrich KE, And SMC, Langer* RS, Shakesheff KM. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999;99:3181-98
29. Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475-90
30. Shuwisitkul D. Biodegradable Implants with Different Drug Release Profiles. 2011;166.
31. Bret UD, Lakshmi NS, Laurencin CT. Biomedical Applications of Biodegradable Polymers. J. Polym. Sci. Part B Polym. Phys. 2011;3:832-64
32. Gad HA, El-Nabarawi MA, Abd El-Hady SS. Formulation and Evaluation of PLA and PLGA In Situ Implants Containing Secnidazole and/or Doxycycline for Treatment of Periodontitis. AAPS PharmSciTech. 2008;9:878-84
33. Chen Y, Jiang L, Wang R, Lu M, Zhang Q, Zhou Y. et al. Injectable smart phase-transformation implants for highly efficient in vivo magnetic-hyperthermia regression of tumors. Adv. Mater. 2014;26:7468-73
34. Ortiz R, Au JL-S, Lu Z, Gan Y, Wientjes MG. Biodegradable intraprostatic doxorubicin implants. AAPS J. 2007;9:E241-50
35. Wang S, Chen Y, Li X, Gao W, Zhang L, Liu J. et al. Injectable 2D MoS2-Integrated Drug Delivering Implant for Highly Efficient NIR-Triggered Synergistic Tumor Hyperthermia. Adv. Mater. 2015;27:7117-22
36. Kumar R, Belz J, Markovic S, Jadhav T, Fowle W, Niedre M. et al. Nanoparticle-Based Brachytherapy Spacers for Delivery of Localized Combined Chemoradiation Therapy. Int. J. Radiat. Oncol. 2015;91:393-400
37. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FBL, Imholz ALT. et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer. 2013;108:d2172-2177
38. Plummer R. Poly(ADP-ribose) polymerase inhibition: a new direction for BRCA and triple-negative breast cancer? Breast Cancer Res. 2011;13:218-23
Author contact
![]() Corresponding author: Srinivas Sridhar s.sridharedu
Corresponding author: Srinivas Sridhar s.sridharedu