Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental section
Results and Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(17):4289-4300. doi:10.7150/thno.21092 This issue Cite
Research Paper
Label-Free Biochips for Accurate Detection of Prostate Cancer in the Clinic: Dual Biomarkers and Circulating Tumor Cells
Lung-Hsuan Pan1*, See-Tong Pang2*, Po-Yu Fang1, Cheng-Keng Chuang2, Hung-Wei Yang1 ![]()
1. Institute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung 80424, Taiwan
2. Division of Urology, Department of Surgery, Linkou Chang Gung Memorial Hospital, Taoyuan 33305, Taiwan.
School of Medicine, Chang Gung University, Taoyuan 33302, Taiwan.
* These authors contributed equally.
Received 2017-5-19; Accepted 2017-8-10; Published 2017-9-26
Abstract

Purpose: Early diagnosis of prostate cancer (PCa) is essential for the prevention of metastasis and for early treatment; therefore, we aimed to develop a simple, accurate, and multi-analyte assay system for early PCa diagnosis in this study. Experimental design: We fabricated three kinds of biochips then integrated into microfluidic device for simultaneous detection of vascularendothelial growth factor (VEGF), prostate-specific antigen (PSA), and PCa circulating tumor cells (CTC) in human serum for accurate diagnosis of PCa. Then the integrated device can be put in the ELISA reader for signal analysis after sample incubation, no necessary of further fluorescence staining or microscopy counting. Result: The integrated device has wide liner detection ranges (0.05-25 ng/mL for both PSA and VEGF, and 5-300 cells/mL for PCa CTC), as well as high levels of sensitivity and selectivity, and demonstrated a high correlation with an enzyme-linked immunosorbent assay for sample detection in patients. Also, the presented biochips could maintain their stability when stored at 37°C for 49 days without significant differences in the red-shift (<5%). Conclusions: We have successfully developed a multi-analyte sensing system for rapid and easy detection of PSA, VEGF, and PC3 cells in PCa samples using label-free glass-based chips. This method presents the advantages of a broad working range, high specificity, label-free, high-speed, stability, and low cost detection method for point-of-care testing of PCa.
Keywords: label-free biochip, prostate cancer diagnosis, PSA, VEGF, circulating tumor cells.
Introduction
Prostate cancer (PCa) is the third most common cancer in men worldwide. In the United States (US), it is the most frequently diagnosed cancer (43%) and the second leading cause of death in men. Moreover, PCa is the second most common cancer in the world [1]. In 2016, over 180,000 new PCa cases were diagnosed in the US and approximately 15% have been estimated to be fatal [2]. Therefore, the development of simple assays for early and accurate detection of PCa is of utmost importance.
To date, prostatic specific antigen (PSA) is widely used for clinical PCa screening; the normal range of PSA values in serum is 0.5-2 ng/mL, and values above 4.0 ng/mL are considered abnormal [3]. However, its low accuracy across different cut-offs depending on factors such as age or other symptoms (i.e., benign prostatic hyperplasia, prostatitis, and cystitis) has warranted identification of other biomarkers that could further contribute in deciding whether the patient should be referred to prostatic biopsy for detailed examination. Vascular endothelial growth factor (VEGF) is a growth factor that can activate endothelial cell proliferation, angiogenesis, and induce blood vessel permeabilization, which are critical stages for tumor growth and development [4]. It has been shown to be expressed in many different types of tumors, including thyroid cancer, breast carcinoma, gliomas, lung cancer, and colorectal cancer [5-9]. VEGF overexpression has been demonstrated in normal, benign, and malignant prostate cells [10], and it plays a key role in microvascular remodeling in PCa to induce bone loss or osteolysis [11]. Of the four VEGF isoforms (VEGF-121, -165, -189, and -206) identified, VEGF-165 is predominantly implicated in physiological and pathological angiogenesis and has been repeatedly implicated in cancer growth and metastasis [12, 13]. In patients with PCa, the prostatic gland contributes considerably to circulating VEGF-165 levels [14]. Previous studies have shown that VEGF-165 concentrations increase with increasing PSA levels in patients' serum and a VEGF-165 concentration above 188.2 pg/mL is considered abnormal [15, 16]. Elevated plasma VEGF-165 levels could reflect prostatic VEGF-165 production, making VEGF-165 a potential tumor marker of PCa to increase the accuracy of PCa diagnosis with PSA detection.
To date, the typical enzyme-linked immunosorbent assay (ELISA) is the most common method used to analyze the concentration of cancer biomarkers such as PSA, VEGF, and carcinoembryonic antigen (CEA) in serum. However, ELISA has multiple disadvantages such as high cost for monoclonal antibodies, inaccuracy due to blocking failure, lengthy procedures, and the requirement for highly trained technicians. Therefore, novel sensing devices need to be developed that can easily, rapidly, and simultaneously detect different cancer biomarkers, including fluorescence-based, electrochemistry-based [17, 18], surface plasmon resonance (SPR)-based [19, 20], surface-enhanced Raman scattering-based [21, 22], and Bloch surface waves-based biosensors [23]. However, most of the above-mentioned methods have the main limitation of expensive equipment that renders them challenging to be used in point-of-care (POC) testing. Ideally, the new bioanalytical devices, which are urgently required, should be easy to manufacture, need simple equipment, and allow reagent-less and label-free detection.
Circulating tumor cells (CTC) are a group of rare cancer cells that have detached from a primary tumor and circulate in the bloodstream. Moreover, numerous clinical trials have shown that high CTC numbers correlate with aggressive disease, increased metastasis, and decreased time to relapse [24]. CTC have been used as a reliable marker to predict tumor response and patient survival and are detected even in the early stages of cancer [25, 26]. Therefore, CTC could be used as a potential biomarker for clinical decision making besides examining cancer-specific proteins. Moreover, the frequency of CTC in the peripheral blood of men with metastatic castration resistant PCa has been found to be 1-103 cells/mL. However, the normal total leukocyte count ranges from 4 × 106 to 10 × 106 cells/mL, and the normal erythrocyte count is up to 1 × 109 cells/mL [27]. Many methods have been developed for detection of CTC in patients with cancer [28-30]; however, only one platform, the CellSearch system (Veridex, USA), is authorized by the FDA as a diagnostic tool for monitoring CTC numbers in the blood samples. However, the CellSearch assay is expensive, time-consuming, and labor-intensive owing to multiple immunofluorescent staining procedures.
In this study, we aimed to develop a simple diagnostic platform by which PSA, VEGF, and CTC could be detected simultaneously in one sample for accurate PCa diagnosis. First, gold nanorods (GNR)-deposited chip (chipGNR), PSA antibody (PSAAb)-conjugated chipGNR (PSAAb-chipGNR), VEGF antibody (VEGFAb)-conjugated chipGNR (VEGFAb-chipGNR), and PC3 aptamer (AptPC3)-conjugated chip (AptPC3-GNR-chip) were constructed. Then, the diagnostic biochips were placed in the microfluidic device, which could integrate into the pedestal of a microtiter plate, for easy and fast signal analysis using a microplate spectrophotometer. Our results indicated that this label-free platform based on low-cost chipGNR could be developed as a powerful support for multi-analyte (PSA, VEGF, and PC3 cell) detection, and it would be beneficial for early, rapid, and simple diagnosis of PCa.
Experimental section
Fabrication of biochips
In this study, three kinds of biochips were fabricated for two kinds of cancer biomarker and PC3 cell detection. For fabrication of PSAAb-chipGNR and VEGFAb-chipGNR, the glass chips were chemically modified with thiol groups by immersing them into 10% (v/v) of (3-mercaptopropyl)trimethoxysilane ethanol solution for 1 h and were then annealed at 120°C for 3 h after washing three times with ethanol. To fabricate chipGNR, the glass chips were immersed into GNR solution with varying concentrations of NaCl (2, 4, 6, 8, 10, 12, and 14 mM) at room temperature for 6 h. The resulting chipGNR were deposited in each well of a 96-well plate after washing three times with deionized water (DI-water). Then, 50 µL of 0.05 mg/mL PSAAb or VEGFAb was added in the wells and incubated for 3 h at room temperature to allow conjugation of antibodies on chipGNR. After immobilization, the chips were washed three times with DI-water, and then air dried with nitrogen. Weakly adsorbed GNR were washed off to ensure a robust biochip.
For fabrication of AptPC3-GNR-chip, thiol-modified chips were reacted with 1 mg/mL of sulfosuccinimidyl sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) in 2-(N-morpholino)ethanesulfonic acid (MES; pH 8.5) at room temperature for 1.5 h in the dark. Then, excess NH2-mPEG-COOH was further conjugated on the chips via sulfo-SMCC linker to form carboxylated glass chips. The carboxylated glass chips were reacted with a mixture of 1-ethyl-3-(3-dimethylaminepropyl)carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide sodium salt (sulfo-NHS) 2-(N-morpholino)ethanesulfonic acid (MES) solution (pH 5.5) at 25°C and vortexed in dark for 40 min. After washing three times with 0.1 M MES buffer (pH 8.0), various concentrations (25, 50, 75, and 100 nM) of DNA AptPC3 (5′-NH2-(CH2)6-AAA-TGC-CAC-TAC-AGC-TGG-TTC-GGT-TTG-GTG-ACT-TCG-TTC-TTC-GTT-GTG-GTG-CTT-AGT-GGC-3′) solution were added into wells for conjugation at 37°C. The binding capacity of DNA AptPC3 immobilized on glass chips was measured by NanoDrop (NanoDrop One, Thermo Scientific, Wilmington, DE, USA). The resulting DNA AptPC3 monolayer functionalized chips were treated with excess thiol-terminated mismatched complementary ssDNA (ssDNAmis; 5′-SH-(CH2)6-GCC-ACT-AAG-CAC-CAC-3′) in an annealing buffer (10 mM Tris, pH 8.0; 1mM EDTA; 50 mM NaCl), hybridization was performed at room temperature for 2 h, and the chips were then mixed with GNR solution after washing three times with DI-water to form AptPC3-GNR-chip. The resulting AptPC3-GNR-chip was extensively rinsed with DI-water and dried under a stream of nitrogen prior to PC3 cell detection.
To determine the optimal hybridization time of ssDNAmis aptamer with AptPC3 on glass chip, 50 nM of ssDNAmis aptamer was incubated with AptPC3-chip for 15, 30, 45, or 60 min. The supernatant was then analyzed by electrophoresis at a constant voltage of 100 V (Bio-Rad Laboratories, Inc., Hercules, CA, USA) for 30 min in TAE buffer (40 mM Tris-base, 1 mM sodium ethylenediaminetetraacetic acid [EDTA], and 20 mM acetic acid [pH 8.5]), and then stained with GelStar™ Nucleic Acid Gel Stain (Lonza Rockland, Inc., Rockland, ME, USA). The gels were visualized under an ultraviolet transilluminator (Gel Doc 2000; Bio-Rad Laboratories, Inc.).
To prove successful hybridization between probe AptPC3 and ssDNAmis aptamer, annealing was performed with equal molar concentrations of probe AptPC3 and ssDNAmis aptamer in 10 mM Tris, pH 8.0, 1 mM EDTA, and 50 mM NaCl for 1 h. Aptamers were resolved on a 12% denaturing urea polyacrylamide gel electrophoresis (PAGE) gel and 12% native acrylamide gel.
Label-free biosensing system evaluation
To minimize non-specific binding, the chips were chemically treated with a blocking solution containing 1 mg/mL hexylamine in PBS buffer for 1 h. To perform label-free biosensing, the sample solution (50 μL) with respective PSA or VEGF concentration (upto 50 ng/mL in 10 mM PBS solution; pH = 7.4) was pipetted onto PSAAb-chipGNR or VEGFAb-chipGNR surface and incubated for 40 min until equilibrium. After washing three times with PBST, the chips were put on the holder in the UV-Vis-NIR spectrometer (Jasco V-770, USA) for optical measurement in the range of 400-1,000 nm with a step of 0.1 nm. Next, the UV-Vis-NIR absorption spectra were determined to observe red-shift (ΔRS) of longitudinal peak maxima in proportion to target binding on the GNR surface, the ΔRS would be increased with increasing the concentration of antigent (PSA or VEGF) captured on the chips. For PC3 cell detection, sample solution (50 μL) with a PC3 cell concentration upto 300 cells/mL, was pipetted onto the AptPC3-GNR-chip surface and incubated for 40 min until equilibrium. Afterwards, the UV-Vis-NIR absorption spectra were determined to observe absorbance intensity of longitudinal peak maxima in proportion to target binding on the AptPC3 aptamer, the absorbance intensity would be decreased with increasing the concentration of PC3 cells captured on the chips. For low CTC concentration detection, 100 μL of supernatant containing the dissociated ssDNAmis-GNR was reacted with 50 μL methyl orange (MO) dye (5 mM) and 50 μL NaBH4 (10 mM). The CTC concentration was determined by measurement of absorbance intensity of MO at 465 nm using UV-Vis-NIR absorption spectra, the absorbance intensity of MO would be decreased with increasing the concentration of PC3 cells captured on the chips.
To test the interference of the biochips, we incubated the PSAAb-chipGNR, VEGFAb-chipGNR, or AptPC3-GNR-chip with typical interfering species that exist in human plasma, including IgG (1000 ng/dL), IgM (192.5 mg/dL), glucose (5 mM), AA (4.3 μg/mL), UA (0.295 mM), 10% FBS, human serum protein (1 mg/mL), HEK293 cells (100 cells/mL), MBR614-2 cells (100 cells/mL), 25 ng/mL positive target proteins (PSA or VEGF), and positive target cells (PC3 cells; 50 cells/mL). The interference levels were evaluated on the basis of the change in ΔRS for PSAAb-chipGNR or VEGFAb-chipGNR and the change in absorbance intensity (ΔA) for AptPC3-GNR-chip at longitudinal peak wavelength by incubating different species, and additional washing with DI-water was performed after incubation prior to measurement.
To evaluate the storage stability of biochips, PSAAb-chipGNR, VEGFAb-chipGNR, and AptPC3-GNR-chip were placed in a storage container and were then maintained in an electronic cabinet at room temperature. The stability of the biochips after varying storage time periods (1, 3, 5, 7, 9, 12, 14, 21, 28, 35, 42, 49, and 56 days) were evaluated by measuring ΔRS or ΔA after incubating with target proteins or cells compared to freshly prepared biochips.
Determination of PSA and VEGF concentrations using an ELISA assay
We used a human PSA (ab188388) and VEGF ELISA kits (ab100663), and followed the manufacturer's protocols to determine PSA and VEGF concentrations in human serum samples.
Preparation of serum samples for quantitative detection
The human plasma samples were prepared as follows. Plasma was isolated from whole blood sample (10 mL) treated with the anticoagulant heparin and centrifuged at 3500 rpm at 4°C for 15 min. The supernatant (serum) was collected for PSA and VEGF detection. This study was approved by the Institutional Review Board of the Chang Gung Memorial Hospital, Taiwan (IRB: 104-7457A3).
Biochips-integrated microfluidic device for detection of biomarkers and PC3 cells in serum samples of the patients
The three kinds of biochips prepared were placed in the detection wells of the microfluidic device (with 1 × 1 mm microfluidic channel) that could integrate into a pedestal of microtiter plate for easy and fast signal analysis using microplate spectrophotometer. Two milliliter of serum samples [containing varying concentrations of PC3 cells (10, 20, 50, and 100 cells/mL) and constant concentration of PZ cells (10,000 cells/mL) and HUVEC cells (10,000 cells/mL)] obtained from patients with PCa were streamed for 30 min in the microfluidic device for sample circulation using a circulating propeller pump (flow rate: 200 µL/min). The microfluidic device was then integrated into a pedestal for signal measurement after washing with PBS for 3 min, with the flow rate being 400 µL/min.
To determine the cell type captured on the AptPC3-GNR-chip, the cells on AptPC3-GNR-chip were fixed with ice-cold methanol for 5 min and rinsed twice with PBS. The cell nuclei were then stained with Hoechst stain for 20 min, and specific protein expressed on the PC3 cell membrane were stained with FITC-labeled cadherin-11 antibody for 1 h. Cells were washed twice with PBS and visualized with an inverted fluorescence microscope (Eclipse Ti-S, Nikon, Melville, NY, USA).
Results and Discussion
In our simulated cancer biomarkers and CTC measuring device, the optical output signals of gold-based materials varied with the increasing concentration of cancer biomarkers and CTC. The proposed ideas were executed in the present study following the scheme given in Figure 1. To determine the efficiency of the multiple-analyte system with a single test using patient samples for rapid PCa diagnosis, we constructed a simple microfluidic device with three detection wells for PSAAb-chipGNR, VEGFAb-chipGNR, and AptPC3-GNR-chip biochips to be placed in each well for simultaneous detection of PSA, VEGF, and CTC, respectively, for allowing low cost, simple, and label-free analysis. The presented multiple-analyte sensing system represents a novel tool for direct and parallel UV-Vis-NIR spectrophotometer analysis in three fluidically independent microfluidic wells. We first investigated the synthesis parameters of GNR that would significantly affect the shape and size to optimize the sensitivity of chipGNR. We found that the concentration of AgNO3 affects the GNR shape significantly but the seed solution does affect the absorption peak (Figure S1A). When the AgNO3 concentration in the growth solution increases, the aspect ratio (AR) of GNR increases gradually (Figure S1B). Thus, the peak red-shifted to 532 nm from 768 nm when the concentration of AgNO3 was increased to 0.1 mM from 0 mM in the growth solution (Figure S1C). However, the peak wavelength did not red-shift when the amount of AgNO3 (10 mM) in the growth solution was increased to more than 40 μL, which was most likely due to the ionic strength effect and/or depletion of Au3+ in the growth solution [31]. Centrifugation also plays an important role in determining the dispersion stability of GNR in water and the ability of ligand exchange because excess cetyltrimethylammonium bromide (CTAB) can adversely affect substrate assembly because of the electrostatic shielding effect, which stabilizes the colloidal solution so that attraction of nanoparticles to the surface becomes secondary [32, 33]. Concentration of the surface capping agent CTAB and zeta-potential decrease by increasing the wash times. The amount of CTAB decreased six-fold (determined by the absorbance at 205 nm) and the zeta-potential decreased to 18.7 ± 2.5 mV from 54.1 ± 3.3 mV after washing four times, but no aggregation occurred; however, significant aggregation was observed when the solution was washed one more time (Figure S1D). Thus, 20 µL of seed solution, 30 µL of AgNO3 (10 mM), and washing four times were finally used as the optimal parameters for GNR synthesis and purification.
Illustration of biochip (PSAAb-ChipGNR, VEGFAb-ChipGNR, AptPC3-GNR-chip) fabrication, and the biochip-integrated microfluidic device.
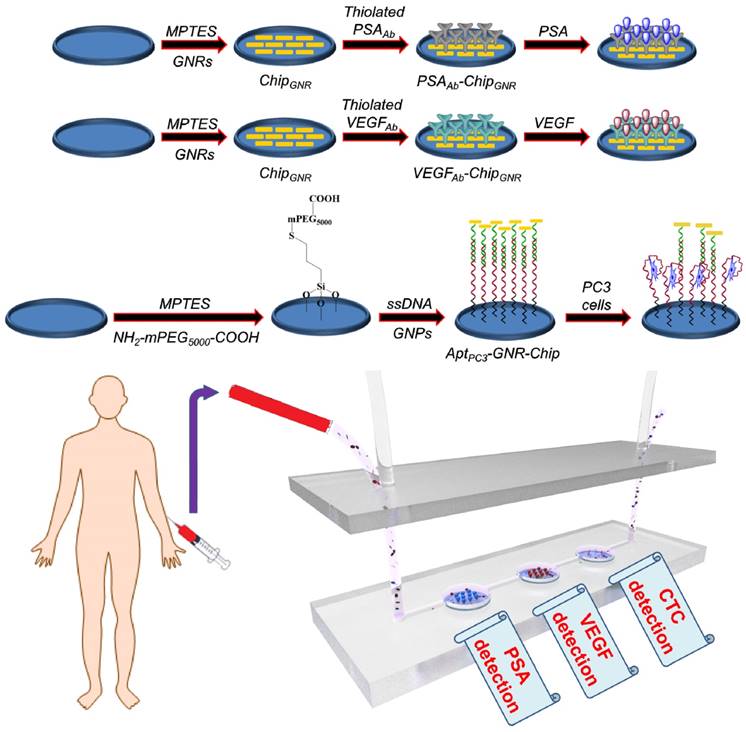
To construct high-quality sensing biochips, high density of GNR must be assembled on the thiolated chips to produce stronger signals, which can be performed by managing NaCl concentration through electrostatic attraction (the details of results and discussion are shown in Supplementary Information; Figure S2) [32].
For further detection of PSA and VEGF, PSAAb or VEGFAb was reacted with Traut's reagent to transform the amine groups on the antibody into thiol groups, which could directly react with GNR via Au-S chemisorption to form PSAAb-chipGNR or VEGFAb-chipGNR. Then the localized surface plasmon resonance (LSPR) was used to detect the capture efficiency of chips for PSA and VEGF. The LSPR position is highly sensitive to any change in the refractive index, and therefore adsorption of molecules on the surface of metallic nanoparticles induces a shift in their LSPR [34, 35]. Upon antibody immobilization, a clear plasmon red-shift in LP was recorded in the spectrum, a 21-nm or 20-nm spectral red-shift caused after PSAAb or VEGFAb immobilization (Figure 2A, B), because the absorption band is sensitive to the localized refractive index change caused by deposition of biomolecules [36]. Fluorescence staining with tetramethylrhodamine (TRITC)-labeled secondary antibody revealed that a strong red fluorescence signal was exhibited on PSAAb-chipGNR and VEGFAb-chipGNR compared to that exhibited by only chipGNR (Figure 2C), indicating the success of antibody immobilization on chipGNR, which was confirmed by X-ray photoelectron spectroscopy (XPS) analysis (Figure S3). An N 1s signal was observed at 399 eV for chipGNR (from CTAB), but the signal became more significant after VEGFAb immobilization (from CTAB and VEGFAb) and both showed Au signals at 82.8 eV (Au 4f7/2) and 86.5 eV (Au 4f5/2). These results confirmed the successful fabrication of PSAAb-chipGNR and VEGFAb-chipGNR for rapid and accurate PSA and VEGF detection.
The UV-vis absorbance spectra of chipGNR after (A) PSAAb conjugation and (B) VEGFAb conjugation. (C) Chips were stained with TRITC-labeled secondary antibody to confirm the successful conjugation of antibodies on the chips (red color: TRITC-labeled secondary antibody). (D) Detection of PSA targets with varying concentration using PSAAb-chipGNR. Lower left, enlarged view at longitudinal peak region; Lower right, calibration curve of longitudinal plasmon red-shift vs. PSA concentration. (E) Detection of VEGF targets with varying concentration using VEGFAb-chipGNR. Lower left, enlarged view at longitudinal peak region; Lower right, calibration curve of longitudinal plasmon red-shift vs. VEGF concentration. Each datum point is presented as mean ± SD.
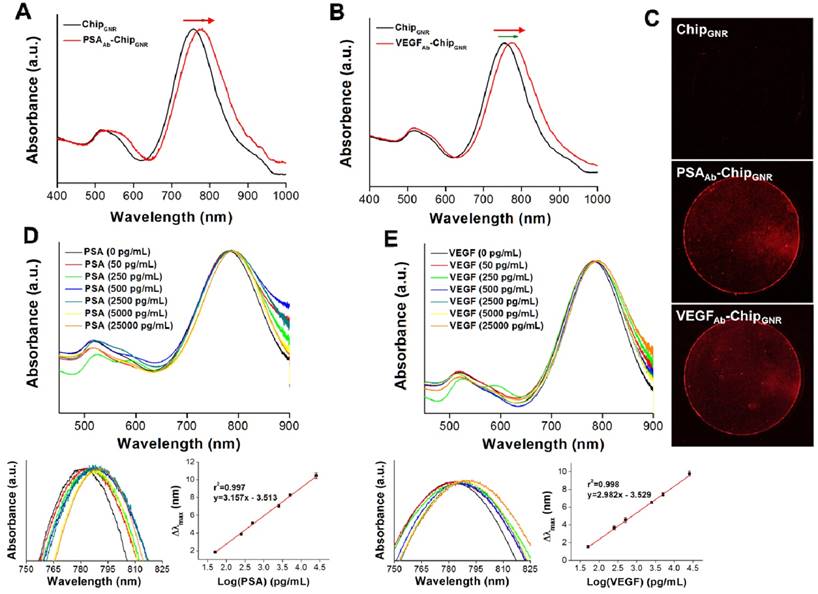
Next, we evaluated sensing performance of PSAAb-chipGNR and VEGFAb-chipGNR for human PSA and VEGF detection. Marked red-shifts in LP were observed with progressively larger shifts in response to increased concentration of the analyte. Standard curves were linear and in the range of 50 pg/mL to 25 ng/mL for both PSA (r2 = 0.997; Figure 2D) and VEGF (r2 = 0.998; Figure 2E) and clear differentiation of the target amount in this region was observed. The wide detection range is suitable for ascertaining the cut-off between a healthy person and a patient with PCa. The specificity of PSAAb-chipGNR and VEGFAb-chipGNR was also evaluated (Figure S4). In addition to the target proteins (PSA and VEGF), the biochips were also probed with other non-specific substances that are always present in the human blood, including ascorbic acid (AA; 4.3 μg/mL), uric acid (UA; 0.295 mM), IgG (1000 ng/dL), IgM (192.5 μg/dL), fetal bovine serum (FBS; 10%), bovine serum albumin (BSA; 1 mg/mL), and human serum albumin (HSA; 1 mg/mL). Red-shifts of the above-mentioned proteins were compared with those of PSA or VEGF (25 ng/mL; positive control). As expected, these interfering proteins resulted in <0.4 nm spectral red-shift because they presented with no specific affinity with PSAAb or VEGFAb immobilized on chipGNR. In contrast, PSA or VEGF caused a significant longitudinal resonant red-shift of approximately 10.2 nm for PSA and approximately 9.5 nm for VEGF, which was in consistence with the calibration curve, indicating that PSAAb-chipGNR and VEGFAb-chipGNR resisted interference well and exhibited excellent selectivity. Readings of the longitudinal resonant red-shifts were noted to investigate the storage stability of PSAAb-chipGNR and VEGFAb-chipGNR at 37°C (Figure S5). The results showed that our presented biochips could maintain their stability when stored at 37°C for 49 days without significant differences in the red-shift (<5%) because of the reduction in freedom of antibodies caused by immobilization on chipGNR that prevents morphological change in their surroundings. However, the biochips lost their activity on storing at room temperature for 56 days, and the red-shift reduced by approximately 47.6% for PSAAb-chipGNR and by 40.2% for VEGFAb-chipGNR compared with new biochips that were stored at room temperature for 56 days.
One way in which PCa diagnosis could be improved is by detecting CTC that are shed by primary tumors, travel through the bloodstream, and seed the growth of metastatic tumors [37]. In this study, we developed a new platform to capture and quantify CTC without additional staining or labeling. A specific single stranded DNA (ssDNA) probe, was selected using systematic evolution of ligands by exponential enrichment (SELEX) method [38] to specifically capture the PC3 cells immobilized on the glass chip via poly(ethylene glycol) 2-aminoethyl ether acetic acid (NH2-PEG-COOH; Molecular weight, 3500 Da) as a linker to increase the flexibility of ssDNA on the chip. Then, a thiol-terminated mismatched complementary ssDNA (ssDNAmis) was hybridized to the probe (AptPC3) forming AptPC3-chip and GNR was attached as a detection signal on the ssDNAmis to form AptPC3-GNR-chip. Peaks were observed at 400 eV (N 1s) and 132.6 eV (P2p), and the peak (C 1s) at 285 eV was stronger after immobilization of AptPC3 on the chip, indicating that AptPC3 was indeed immobilized on the glass chip (Figure 3A and Figure S6). To determine CTC capture and quantification, ssDNAmis must hybridize to AptPC3 on AptPC3-GNR-chip by complementary bonding for further attachment of GNR. The results of gel mobility support our hypothesis of the binding interaction between the two aptamers. The interaction of the AptPC3 aptamer with the ssDNAmis aptamer was evaluated by gel mobility assay. In the presence of the ssDNAmis aptamer, the migration of the AptPC3 aptamer band is slower on the gel (Figure 3B left, lane 1). The ssDNAmis aptamer per se cannot be visualized on the native gel (Figure 3B left, lane 2). In absence of the ssDNAmis aptamer, the AptPC3 aptamer appears as one predominant band and exhibits faster mobility in the gel. (Figure 3B left, lane 3). These results indicate that the AptPC3 aptamer forms complexes with ssDNAmis aptamer after co-incubation. The bands on native gel do not represent various sizes of the DNA aptamer because the probe DNA aptamer (AptPC3) migrates as one major band on the denaturing gel (Figure 3B right, lane 3). Small fragments or unincorporated nucleotides are found in the gel when the ssDNAmis aptamer is subjected to electrophoresis (Figure 3B right, lane 2). We also observed that the position of the AptPC3 aptamer band shifted slightly above under denaturing conditions after co-incubation with the ssDNAmis aptamer (Figure 3B right, lane 1), indicating a specific high-affinity interaction between these two aptamers.
(A) XPS surface survey spectra of chip and AptPC3-chip. (B) Gel shift analyses of interaction between probe aptamer and complementary aptamer. (Left) Quality of DNA aptamers visualized on a denaturing gel. Lane 1, total DNA aptamers from annealing reaction; Lane 2, the 15-nt ssDNAmis aptamer; Lane 3, the 60-nt DNA probe AptPC3; M, the 21-nt single-stranded oligomer as a size marker. The upper black arrow indicates probe AptPC3 and the lower black arrow indicates ssDNAmis aptamer. (Right) Annealing of two aptamers visualized by native acrylamide gel. The gel mobility of the DNA aptamers suggests annealing of the probe AptPC3 and ssDNAmis aptamers. Lane 1, total DNA aptamers from annealing reaction; Lane 2, the 15-nt ssDNAmis aptamer; Lane 3, the 60-nt DNA AptPC3. The upper red arrow indicates the annealing of probe AptPC3 and ssDNAmis aptamers. The ssDNAmis aptamer retards the mobility of the probe AptPC3. The lower red arrow indicates free DNA probe AptPC3, which migrates faster than the AptPC3-ssDNAmis complex. (C) SEM image of GNR assembled on AptPC3-chip. (D) Investigating the maximum conjugation concentration of AptPC3 on chip by agarose gel retardation analysis. Left lane, initial concentration of AptPC3 added for reaction; Right lane, the concentration of AptPC3 in the supernatant. (E) Investigating the optimal hybridization time of ssDNAmis aptamer with AptPC3 on chip by agarose gel retardation analysis. Left lane, initial concentration of ssDNAmis added for hybridization; Right lane, the concentration of ssDNAmis in the supernatant.
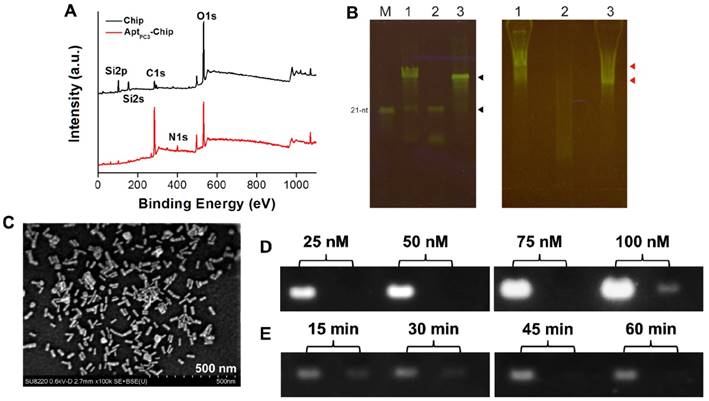
A LP at 760 nm appeared after GNR was attached to ssDNAmis, indicating successful formation of AptPC3-GNR-chip (Figure S7) that can be used to capture and quantify PC3 cells; SEM showed a randomly distributed monolayer of GNR on the AptPC3-chip, confirming DNA hybridization and GNR signal attachment (Figure 3C). The maximum concentration of AptPC3 immobilization onto the chip was determined by gel electrophoresis with GelStar™ Nucleic Acid Gel Stain. Total AptPC3 was successfully immobilized on the chip (with no fraction remaining in the supernatant) at 25, 50, and 75 nM. When the AptPC3 aptamer concentration was increased to 100 nM, a weak signal was observed for the supernatant after conjugation, indicating that the maximum concentration of immobilized AptPC3 on the chip was approximately 90 nM (Figure 3D). Also, we investigated the optimal hybridization time of the ssDNAmis aptamer with AptPC3 on the biochips. Reduction of the ssDNAmis aptamer in the supernatant was observed with increasing reaction time, and complete hybridization of ssDNAmis aptamer (50 nM) was observed on the chip after 45 min of reaction. Thus, 45 min was chosen as the optimal reaction time (Figure 3E).
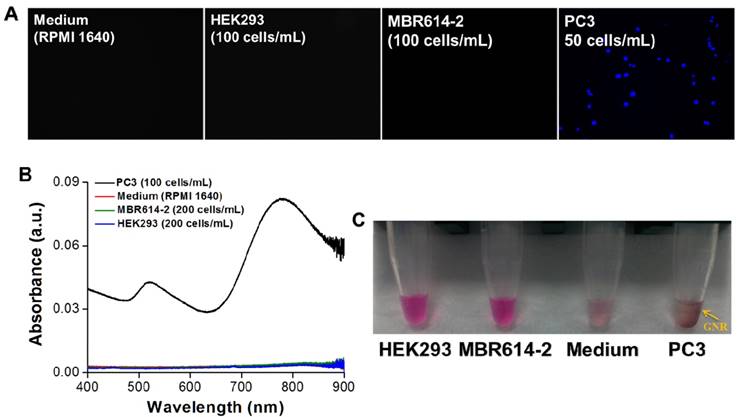
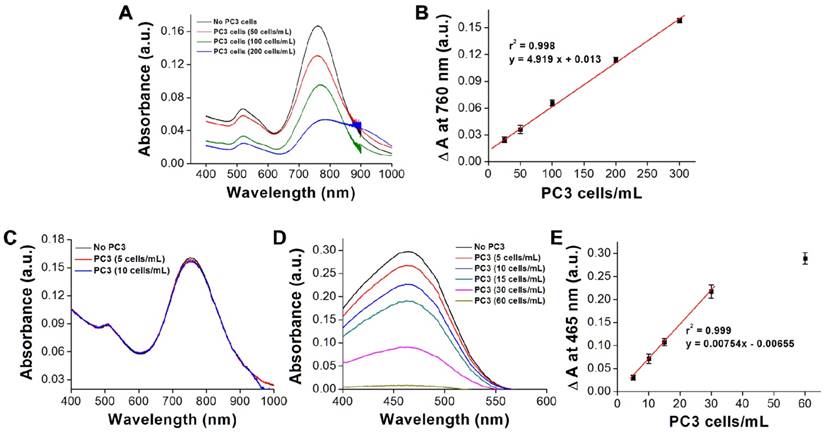
Next, we investigated the specificity of AptPC3-GNR-chip for PC3 cell capture by incubating with HEK293 (100 cells/mL), MBR614-2 (100 cells/mL), or PC3 (50 cells/mL). It was observed that HEK293 or MBR614-2 cells were not captured on AptPC3-GNR-chip after staining with Hoechst stain, with the results being the same as the control (medium only). Only PC3 cells were captured and stained and emitted blue fluorescence on AptPC3-GNR-chip (Figure 4A), confirming its excellent ability for detecting and quantifying CTC. In this study, we demonstrated a new strategy to easily and rapidly quantify PC3 cell counts. In the presence of PC3 cells, the AptPC3 aptamer bound to PC3 cells and folded into a complex structure, driving the equilibrium and denaturing the duplex due to PC3 cell binding, altering the configuration of the AptPC3 aptamer, and allowing the dissociation of the ssDNAmis strand-GNR complex (ssDNAmis-GNR) from AptPC3-GNR-chip to reduce the LP signal at 780 nm. UV-Vis-NIR spectroscopy was used to prove the detection mechanism (Figure 4B). Subsequently, the PC3 cells were incubated with AptPC3-GNR-chip to bind to AptPC3, which removed a large fraction of the ssDNAmis-GNR from AptPC3 in the supernatant, to demonstrate significant GNR absorption signal and precipitation after centrifugation (Figure 4C). However, GNR signal and precipitation were not observed after incubating with other cells (HEK293 or MBR614-2 cells). Next, we evaluated the performance of AptPC3-GNR-chip for PC3 cells detection by UV-Vis-NIR spectroscopy. The absorbance of LP at 760 nm decreased as more PC3 cells bound to the AptPC3 aptamer on AptPC3-GNR-chip causing increased dissociation of ssDNAmis-GNR in the supernatant (Figure 5A). The change in absorbance (ΔA) increased linearly (r2 = 0.998) with increasing cell concentration from 25 to 300 cells/mL (Figure 5B). However, no significant difference of ΔA observed when the concentration of incubated PC3 cells was lower than 10 cells/mL (Figure 5C) as only a few ssDNAmis-GNR dissociated from the chip. Therefore, we used the method of enhanced reduction of MO with NaBH4 in the presence of metallic nanoparticles as catalyst [39]. In this study, we amplified the signal by detecting the dissociated ssDNAmis-GNR in the supernatant after capture of PC3 cells on AptPC3-GNR-chip using the method of catalytic degradation of MO with rapid reaction time (<3 min). The absorption spectrum showed decreasing peak intensity at 465 nm for MO by NaBH4 in the presence of catalytically active GNR, as shown in Figure 5D. More ssDNAmis-GNR released from AptPC3-GNR-chip to the supernatant upon incubation with PC3 cells, which caused significant degradation of MO and a decrease in absorbance intensity at 465 nm. The linear correlation between ΔA versus incubated PC3 cells (5 to 30 cells/mL) indicated that the detection limit can be further reduced to 5 cells/mL from 25 cells/mL by detection of MO degradation (Figure 5E). The wide detection range reported here indicated that this biochip could be used for accurate quantitation of CTC in a 10 mL blood sample collected from a patient with PCa, which typically contains >50 CTC/mL [40]. The results indicated that this sensing system could be used for simple, rapid, and accurate quantification of CTC concentration without further staining and signal labeling.
(A) Fluorescence images of AptPC3-GNR-chip stained with Hoechst after incubating with various cell types. (B) UV-vis absorbance spectra of the supernatant after incubating the AptPC3-GNR-chip with solutions of various cell types. (C) Image of the supernatant after incubating the AptPC3-GNR-chip with solutions of various cell types.
(A) UV-vis absorbance spectra showing the decrease in absorbance signal as varying amounts of PC3 cells are captured. (B) Visual cell count compared to the change in signal on the chip surface. Each datum point is presented as mean ± SD. (C) UV-vis absorbance spectra showing the decrease in absorbance signal as varying amounts of PC3 cells are captured (low CTC concentration). (D) UV-vis absorbance spectra for the catalytic reduction of MO as varying amounts of PC3 cells are captured. (E) Visual cell count compared to the change in signal of MO in the supernatant. Each datum point is presented as mean ± SD.
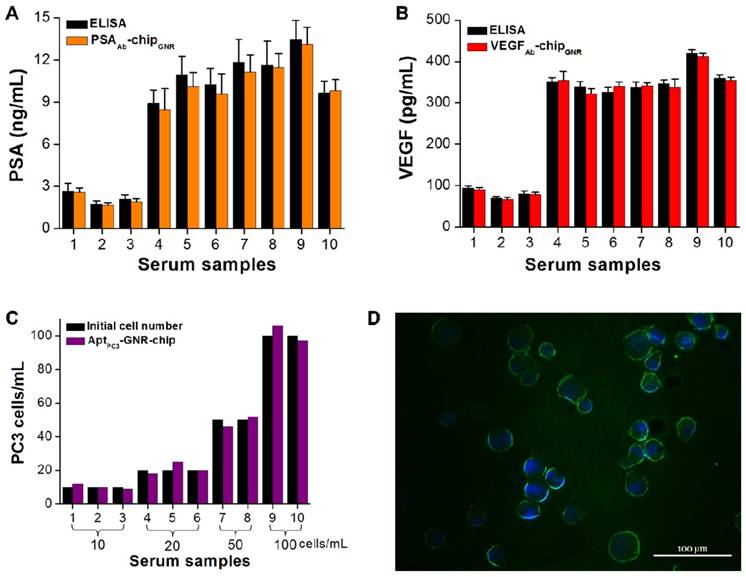
Some drawbacks of PSA are also related to its false-negative results. It has been demonstrated that PCa may be detected in about 15% of men with normal or very low levels of total PSA; thus, it is possible that a combination of PSA and a proper cut-off point for VEGF may result in overcoming such drawbacks. In the tests, ten serum samples were collected from different patients diagnosed with early stages of PCa to assess the utility and accuracy of this multi-detection system. The measured results by this multi-detection system were also compared with the results obtained from a conventional detection method (i.e., ELISA). The absorption spectra of PSA and VEGF showed the same trend, with both demonstrating a significant red-shift after incubating with varying concentration of PSA or VEGF. PSA and VEGF concentrations were calculated from calibration curves for both methodologies. The PSA and VEGF concentrations detected by the multi-analyte sensing system in the sera of healthy controls (samples 1-3; 1.7-2.6 ng/mL for PSA and 66.8-90.4 pg/mL for VEGF) were lower than those of patients with PCa (samples 4-10; 8.5-13.1 ng/mL for PSA and 321.8-412.8 pg/mL for VEGF; Figure 6A, B). Previous studies have shown that values above 4.0 ng/mL of PSA and 188.2 pg/mL of VEGF are considered abnormal [15, 16]. No significant differences were observed in terms of PSA and VEGF levels measured by the multi-analyte sensing system compared with ELISA. For proof-of-concept of rapid CTC detection and quantification, the cultured PC3 cells with other cell types (PZ and HUVEC cells) were added into the serum samples to test them together in the circulating microfluidic system. The results showed that the ssDNAmis-GNR aptamer gradually departed from AptPC3-GNR-chip, with PC3 cells captured by the AptPC3 aptamer, which decreased the absorbance intensity at 760 nm. This can be used to rapidly and easily quantify the concentration of PC3 cells in serum samples without any further signal labeling or staining. We also collected the supernatant for double-checking low CTC concentration in serum samples by reacting with MO. No significant difference was observed in the cell concentration counted by AptPC3-GNR-chip and the initial cell concentration added into the serum samples (Figure 6C), indicating that the chip can accurately quantify PC3 cells. However, the captured cells must be affirmed as PC3 cells and not other cell types by staining the cadherin-11 protein, which is overexpressed on the cell membrane of PC3 cells, using fluorescein isothiocyanate (FITC)-labeled cadherin-11 antibody. Interestingly, all captured cells on AptPC3-GNR-chip were stained with green fluorescence to sufficiently affirm that the captured cells were PC3 cells (Figure 6D). The results indicated that the present multi-analyte sensing system can rapidly and easily quantify PSA, VEGF, and CTC (PC3) concentrations in one sample without further signal labeling or staining.
Serum biomarker and CTC concentrations measured using the biochip-integrated microfluidic device and ELISA kits. (A) PSA, (B) VEGF, and (C) PC3 cells. Each datum point is presented as mean ± SD. (D) Immunofluorescence staining demonstrating that the isolated cells are cadherin-11 positive (green) for PC3 cells and Hoechst positive (blue).
Conclusion
In conclusion, we have designed a multi-analyte sensing system for rapid and easy detection of PSA, VEGF, and PC3 cells in PCa samples using label-free glass-based chips. The chips incorporated with a microfluidic device with multi-wells can then immediately measure the signal change by UV-vis spectroscopy after 30 min of sample circulation to quantify the concentrations of PSA, VEGF, and PC3 cells in one PCa sample. The detection limit for PSA and VEGF was 50 pg/mL and 5 cells/mL, respectively, in PC3 cells from PCa sample, and the measured concentrations of PSA and VEGF were not significantly different compared with those measured by ELISA. Our multi-analyte sensing system presents the advantages of a broad working range, high specificity, label-free, high-speed, stability, and low cost detection method. In addition, the simultaneous and combined detection of specific antigens and cancer cells results in higher accuracy of PCa diagnosis.
Supplementary Material
Supplementary experimental methods and figures (Figures S1-S7).
Abbreviations
PCa: prostate cancer; VEGF: vascularendothelial growth factor; PSA: prostate-specific antigen; CTC: circulating tumor cells; GNR: gold nanorods; ELISA: enzyme-linked immunosorbent assay; CEA: carcinoembryonic antigen; SPR: surface plasmon resonance; POC: point-of-care; sulfo-SMCC: sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate; EDC: 1-ethyl-3-(3-dimethylaminepropyl)carbodiimide hydrochloride; sulfo-NHS: N-hydroxysulfosuccinimide sodium salt; MES: 2-(N-morpholino)ethanesulfonic acid; EDTA: ethylenediaminetetraacetic acid; TRITC: fluorescence staining with tetramethylrhodamine; XPS: X-ray photoelectron spectroscopy; ssDNA: single stranded DNA; SELEX: systematic evolution of ligands by exponential enrichment.
Acknowledgements
We thank the Ministry of Science and Technology, and Chang Gung Memorial Hospital, Taiwan (R.O.C.), for the financial assistance provided (MOST105-2314-B-110-001, MOST105-2314-B-182A-086-MY3, MOST106-2628-E-110-001-MY3, CMRPG3F0221, CMRPG3F0701-02). We would also like to thank the Chang Gung Memorial Hospital Microscopy Core Laboratory for assistance with SEM.
Competing Interests
The authors have declared that no competing interest exists.
References
1. DeSantis CE, Lin CC, Mariotto AB. et al. Cancer treatment and survivorship statistics, 2014. CA. Cancer J Clin. 2014;64:252-71
2. Wang Z, Xu L, Hu Y. et al. miRNA let-7b modulates macrophage polarization and enhances tumor-associated macrophages to promote angiogenesis and mobility in prostate cancer. Sci Rep. 2016;6:25602
3. Yang HW, Lin CW, Hua MY. et al. Combined detection of cancer cells and a tumor biomarker using an immunomagnetic sensor for the improvement of prostate-cancer diagnosis. Adv Mater. 2014;26:3662-6
4. Botelho F, Pina F, Silva P. et al. Vascular endothelial growth factor (VEGF) and prostate pathology. Int Braz J Urol. 2010;36:430-7
5. Zhu X, Zhou W. The emerging regulation of VEGFR-2 in triple-negative breast cancer. Front Endocrinol. 2015;6:159
6. Bunone G, Vigneri P, Mariani L. et al. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am J Pathol. 1999;155:1967-76
7. Zhang C, Hao L, Wang L. et al. Elevated IGFIR expression regulating VEGF and VEGF-C predicts lymph node metastasis in human colorectal cancer. BMC Cancer. 2010;10:184
8. Zhang Y, Meng X, Zeng H. et al. Serum vascular endothelial growth factor-C levels: A possible diagnostic marker for lymph node metastasis in patients with primary non-small cell lung cancer. Oncol Lett. 2013;6:545-9
9. Qin LJ, Jia YS, Zhang YB. et al. Interleukin-1β induces the upregulation of caveolin-1 expression in a rat brain tumor model. Biomed Rep. 2016;4:433-6
10. Wu TLT, Wang JS, Jiann BP. et al. Expression of vascular endothelial growth factor in Taiwanese benign and malignant prostate tissues. J Chin Med Assoc. 2007;70:380-4
11. Bruni-Cardoso A, Johnson LC, Vessella RL. et al. Osteoclast-derived matrix metalloproteinase-9 directly affects angiogenesis in the prostate tumor-bone microenvironment. Mol Cancer Res. 2010;8:459-70
12. Muhlhauser J, Merrill MJ, Pili R. et al. VEGF165 expressed by a replication-deficient recombinant adenovirus vector induces angiogenesis in vivo. Circ Res. 1995;77:1077-86
13. Gupta A, Zhou CQ, Chellaiah MA. Osteopontin and MMP9: Associations with VEGF expression/secretion and angiogenesis in PC3 prostate cancer cells. Cancers. 2013;5:617-38
14. George DJ, Regan MM, Oh WK. et al. Radical prostatectomy lowers plasma vascular endothelial growth factor levels in patients with prostate cancer. Urology. 2004;63:327-32
15. Botelho F, Pina F, Lunet N. VEGF and prostatic cancer: A systematic review. Eur J Cancer Prev. 2010;19:385-92
16. Sharif MR, Shaabani A, Mahmoudi H. et al. Association of the serum vascular endothelial growth factor levels with benign prostate hyperplasia and prostate malignancies. Nephro Urol Mon. 2014;6:e14778
17. Sarkar P, Pal PS, Ghosh D. et al. Amperometric biosensors for detection of the prostate cancer marker (PSA). Int J Pharm. 2002;238:1-9
18. Yang Z, Kasprzyk-Hordern B, Goggins S. et al. A novel immobilization strategy for electrochemical detection of cancer biomarkers: DNA-directed immobilization of aptamer sensors for sensitive detection of prostate specific antigens. Analyst. 2015;140:2628-33
19. Uludag Y, Tothill IE. Cancer biomarker detection in serum samples using surface plasmon resonance and quartz crystal microbalance sensors with nanoparticle signal amplification. Anal Chem. 2012;84:5898-904
20. Jana D, Matti C, He J. et al. Capping agent-free gold nanostars show greatly increased versatility and sensitivity for biosensing. Anal Chem. 2015;87:3964-72
21. Cho H, Yeh EC, Sinha R. et al. Single-step nanoplasmonic VEGF165 aptasensor for early cancer diagnosis. ACS Nano. 2012;6:7607-14
22. Mohammad JK, Ahmadi MT, Elnaz A. et al. Analytical modeling of bilayer graphene based biosensor. J Biosens Bioelectron. 2013;4:1000131
23. Sinibaldi A, Sampaoli C, Danz N. et al. Detection of soluble ERBB2 in breast cancer cell lysates using a combined label-free/fluorescence platform based on Bloch surface waves. Biosens Bioelectron. 2017;92:125-30
24. Plaks V, Koopman CD, Werb Z. Circulating tumor cells. Science. 2013;341:1186-8
25. Jiang ZF, Cristofanilli M, Shao ZM. et al. Circulating tumor cells predict progression-free and overall survival in Chinese patients with metastatic breast cancer, HER2-positive or triple-negative (CBCSG004): A multicenter, double-blind, prospective trial. Ann Oncol. 2013;24:2766-72
26. Goldkorn A, Ely B, Quinn DI. et al. Circulating tumor cell counts are prognostic of overall survival in SWOG S0421: A phase III trial of docetaxel with or without atrasentan for metastatic castration-resistant prostate cancer. J Clin Oncol. 2014;32:1136-42
27. Friedlander TW, Ngo VT, Dong H. et al. Detection and characterization of invasive circulating tumor cells derived from men with metastatic castration-resistant prostate cancer. Int J Cancer. 2014;134:2284-93
28. Song Y, Zhu Z, An Y. et al. Selection of DNA aptamers against epithelial cell adhesion molecule for cancer cell imaging and circulating tumor cell capture. Anal Chem. 2013;85:4141-9
29. Lee J, Kang HJ, Jang H. et al. Simultaneous imaging of two different cancer biomarkers using aptamer-conjugated quantum dots. Sensors. 2015;15:8595-604
30. Kang WJ, Chae JR, Cho YL. et al. Multiplex imaging of single tumor cells using quantum-dot-conjugated aptamers. Small. 2009;5:2519-22
31. Nikoobakht B, El-Sayed MA. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem Mater. 2013;15:1957-62
32. Ferhan AR, Guo LH, Kim DH. Influence of ionic strength and surfactant concentration on electrostatic surfacial assembly of cetyltrimethylammonium bromide-capped gold nanorods on fully immersed glass. Langmuir. 2010;26:12433-42
33. Vallery RS, Liu M, Gidley DW. et al. Characterization of fatigue-induced free volume changes in a bulk metallic glass using positron annihilation spectroscopy. Appl Phys Lett. 2007;91:261908
34. Gabudean AM, Biro D, Astilean S. Localized surface plasmon resonance (LSPR) and surface enhanced Raman scattering (SERS) studies of 4-aminothiophenol adsorption on gold nanorods. J Mol Struct. 2011;993:420-4
35. Huang H, He C, Zeng Y. et al. A novel label-free multi-throughput optical biosensor based on localized surface plasmon resonance. Biosens Bioelectron. 2009;24:2255-9
36. Tian L, Chen E, Gandra N. et al. Gold nanorods as plasmonic nanotransducers: Distance-dependent refractive index sensitivity. Langmuir. 2012;28:17435-42
37. Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011;11:512-22
38. Wang Y, Luo Y, Bing T. et al. DNA aptamer evolved by cell-SELEX for recognition of prostate cancer. PLOS ONE. 2014;9:e100243
39. Gupta N, Singh HP, Sharma RK. Metal nanoparticles with high catalytic activity in degradation of methyl orange:An electron relay effect. J Mol Catal A- Chem. 2011;335:248-52
40. Nagrath S, Sequist LV, Maheswaran S. et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235-9
Author contact
![]() Corresponding author: Tel.: +886-7-5252000x5842; fax: +886-7-5250151. E-mail address: howardyangnsysu.edu.tw
Corresponding author: Tel.: +886-7-5252000x5842; fax: +886-7-5250151. E-mail address: howardyangnsysu.edu.tw