Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(17):4255-4275. doi:10.7150/thno.20270 This issue Cite
Research Paper
Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice
Chao Li1, Yiping Lu1, Sitong Du1, Siyi Li1, Yiting Zhang1, Fangwei Liu1, Ying Chen1, Dong Weng1, 2 ![]() , Jie Chen1
, Jie Chen1 ![]()
1. Division of Pneumoconiosis, School of Public Health, China Medical University, Shenyang, PR China;
2. Department of Respiratory Medicine, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, PR China.
Received 2017-3-26; Accepted 2017-8-29; Published 2017-9-26
Citation:
Li C, Lu Y, Du S, Li S, Zhang Y, Liu F, Chen Y, Weng D, Chen J. Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice. Theranostics 2017; 7(17):4255-4275. doi:10.7150/thno.20270. https://www.thno.org/v07p4255.htm
Other stylesAbstract

Inhalation of crystalline silica particles leads to pulmonary fibrosis, eventually resulting in respiratory failure and death. There are few effective drugs that can delay the progression of this disease; thus, patients with silicosis are usually only offered supportive care. Dioscin, a steroidal saponin, exhibits many biological activities and health benefits including its protective effects against hepatic fibrosis. However, the effect of dioscin on silicosis is unknown.
Methods: We employed experimental mouse mode of silicosis. Different doses of dioscin were gavaged to the animals 1 day after crystalline silica instillation to see the effect of dioscin on crystalline silica induced pulmonary fibrosis. Also, we used RAW264.7 and NIH-3T3 cell lines to explore dioscin effects on macrophages and fibroblasts. Dioscin was also oral treatment but 10 days after crystalline silica instillation to see its effect on established pulmonary fibrosis.
Results: Dioscin treatment reduced pro-inflammation and pro-fibrotic cytokine secretion by modulating innate and adaptive immune responses. It also reduced the recruitment of fibrocytes, protected epithelial cells from crystalline silica injury, inhibited transforming growth factor beta/Smad3 signaling and fibroblast activation. Together, these effects delayed the progression of crystalline silica-induced pulmonary fibrosis. The mechanism by which dioscin treatment alleviated CS-induced inflammation appeared to be via the reduction of macrophage, B lymphocyte, and T lymphocte infiltration into lung. Dioscin inhibits macrophages and fibroblasts from secreting pro-inflammatory cytokines and may also function as a modulator of T helper cells responses, concurrent with attenuated phosphorylation of the apoptosis signal-regulating kinase 1-p38/c-Jun N-terminal kinase pathway. Also, dioscin could block the phosphorylation of Smad3 in fibroblast. Oral treatment of dioscin could also effectively postpone the progression of established silicosis.
Conclusion: Oral treatment dioscin delays crystalline silica-induced pulmonary fibrosis and exerts pulmonary protective effects in mice. Dioscin may be a novel and potent candidate for protection against crystalline silica-induced pulmonary fibrosis.
Keywords: Crystalline silica, Pulmonary fibrosis, Dioscin, Immune response, Fibrocytes, Epithelial cells, Natural products therapy.
Introduction
There are estimated to be tens of millions of workers exposed to crystalline silica (CS) worldwide [1], many of whom are exposed to higher concentrations than the occupational limit in both developing and developed countries. Industrialization processes in developing countries have made this situation even worse [2]. Although an array of circumstances leading to occupational exposure has been existing for decades, such as mining, pottery, glass and concrete production, new occupational exposure circumstances continue to emerge [3, 4]. Inhalation of crystalline silica is associated with the development of pulmonary fibrosis, leading to silicosis [5], which is characterized by the progressive and irreversible destruction of normal lung architecture. This disease is incurable due to impaired particle clearance, resulting in persistent lung inflammation and fibrosis that may eventually lead to respiratory failure due to excessive extracellular matrix (ECM) deposition.
Crystalline silica-induced lung pulmonary fibrosis begins with a prominent acute inflammatory response that is caused by the recruitment and accumulation of inflammatory cells, including macrophages and lymphocytes, and the elevated secretion of pro-inflammatory and pro-fibrotic cytokines and chemokines [6]. The progression of silicosis involves a multi-factorial process that involves crystalline silica-induced persistent lung inflammation and ECM production. Cells responsible for the production of ECM proteins are myofibroblasts, which can be derived from three potential sources: (1) the expansion and activation of resident lung fibroblasts, (2) epithelial cells losing epithelial markers and gaining mesenchymal markers during the process of epithelial mesenchymal transition (EMT), or (3) the recruitment and differentiation of circulating mesenchymal precursors known as fibrocytes [6-9]. During the progression of fibrosis, immunomodulatory effects activate innate and adaptive immune responses, which create a milieu rich in pro-fibrotic growth factors, cytokines, and chemokines that foster the development of fibrosis [10] and are an integral part of the pathogenesis of silicosis.
At present, there are few effective therapies that can reverse or even delay the progression of crystalline silica-induced fibrosis; thus, patients with this disease are usually only offered supportive care [11]. Therefore, there is an urgent need to identify drugs that can prevent this disease or delay its progression. Natural products have recently been gaining more attention because of their many biological activities and desirable health benefits [12-16]. Dioscin (Dio), a steroidal saponin, is abundantly expressed in many medical plants such as Dioscorea nipponica Makino. Pharmacologists have performed extensive research studies that have demonstrated that this natural product has anti-tumor [17], anti-inflammatory [18], anti-virus [19], lipid-lowering [20], and hepatoprotective activities [21]. In addition, it has shown remarkable protective effects against several factors that induce hepatic fibrosis [22, 23]. However, to the best of our knowledge, there have been no studies on the effects of dioscin against pulmonary fibrosis, especially CS-induced fibrosis. Here, we explored the potential pulmonary protective effects of dioscin in a mouse model of silicosis and the possible mechanisms underlying its actions.
Materials and methods
Crystalline silica
Crystalline silica (CS) particulates were purchased from the U.S. Silica Company (Frederick, MD, USA). Particulate size distribution was as follows: 97% <5 μm in diameter, 80% <3 μm in diameter, median diameter of 1.4 μm. The equivalent spherical diameter is shown in Figure S1. Typical physical properties were: compacted bulk density, 656 kg/m³; uncompacted bulk density, 576 kg/m³; Mohs hardness, 7; Hegman fineness, 7; Yellowness index, 2.0; and Specific gravity, 2.65. Silicon dioxide accounted for 99.3% of its chemical composition. Particulates were ground in saline for 3 h, boiled in 1 N hydrochloric acid, washed, dried, and suspended in sterile saline. Suspensions were sonicated for 10 min before use.
Dioscin
Dioscin (Dio) was purchased from Spring & Autumn Biological Engineering Co. Ltd. (Nanjing, China) with a purity >98% that was determined by high-performance liquid chromatography (Figure S2). Dioscin was dissolved in 0.5% sodium carboxyl methyl cellulose (CMC-Na; Sigma, St. Louis, MO, USA) solution in distilled water for in vivo experiments. Dioscin was dissolved in 0.01% dimethyl sulfoxide (DMSO) for in vitro experiments.
Animal experiments
Female C57BL/6 mice 6-8 weeks of age were purchased from SLAC Laboratory Animal Co. Ltd. (Shanghai, China). All of the mice were housed in a pathogen-free facility at China Medical University (Shenyang, China). All of the animal experiments were approved by the Animal Care and Use Committee at China Medical University, and complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The silicosis model was performed as previously described [24]. Briefly, C57BL/6 mice were anesthetized with an injection of 10% chloral hydrate (0.4 mL/100 g) (Sangon Biotech, Shanghai, China) and placed on a platform. A 50 μL solution containing 2.5 mg CS was directly instilled by intratracheal instillation. Control mice were instilled with 50 μL sterile saline.
Program 1: The mice were randomly divided into the six groups (10 mice per group): Group I (Saline group), in which the mice were instilled with saline and orally administered vehicle only; Group II (dioscin control group), in which the mice were instilled with saline and orally administered dioscin (80 mg/kg); Group III (Crystalline silica group), in which the mice were instilled with crystalline silica as described before and orally administered vehicle only; Groups IV-VI (dioscin-treated groups), in which the mice were instilled with crystalline silica and orally administered dioscin at doses of 80, 40 or 20 mg/kg, respectively, once daily for consecutive 56 days. As Figure 1A describes, an experimental silicosis model was made on day 0. At the designated time points after crystalline silica instillation, mice were euthanized with chloral hydrate by intraperitoneal (i.p.) injection (0.8 mL/100 g). The eyeballs of mice were removed under anesthesia to get blood. Lungs were harvested for further analyses. For bronchoalveolar lavage fluid (BALF), the trachea was cannulated and lavaged twice with 1 mL sterile saline at room temperature. Samples were centrifuged at 1500 rpm for 5 min, after which the cell-free supernatant was saved for cytokine detection.
Program 2: Furthermore, we made another experimental mouse model to explore the effect of dioscin on established silicosis. As Figure 9A describes, after CS instillation, no treatment was given from day 1 to 10, during which silicotic acute inflammation was well underway. Dioscin (80 mg/kg) or vehicle were administered by gavage from day 11, and administered by daily intragastric gavage from then on. Mouse lungs were harvested 28 d, 42 d and 56 d after CS injury (8 mice per group per time point). Tissues were sectioned for Masson's trichrome to assess the degree of fibrosis (n=4-5). Collagen contents in the lung were measured with hydroxyproline assay.
In this in vivo experiment, the mouse primary alveolar macrophages were isolated and cultured. Briefly, the macrophages were obtained by lung lavage (n=3-4). Cells were suspended and cultured in RPMI-medium with 10% fetal bovine serum (FBS) and penicillin-streptomycin. Non-attached cells were removed after 2 h culture. The fresh RPIM-medium with FBS was added to the attached cells. LPS (25 ng/mL) was added in the culture medium for 6 h to induce cytokine secretion. Then the culture media were collected for cytokine measurement (IL-1β, IL-6, TNF-α and MCP-1).
Dioscin Toxicity Assay
The RAW 264.7 cells and NIH-3T3 cells were seeded into 96-well plates at a density of 5×104 cells/mL per well for 24 h before treatment and then incubated for another 24 h in the presence of different concentrations of dioscin (RAW264.7: 37.5, 75, 150, 300, 600, 1200, and 2400 ng/mL, NIH-3T3:125, 250, 500, 1000, 2000, 4000 8000 ng/mL). The cell viability was measured using the MTT method.
Cell culture and treatment
Mouse macrophage RAW264.7 cell line and NIH-3T3 mouse fibroblast cell line were purchased from National Infrastructure of Cell Line Resource (Beijing, China). RAW264.7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS). NIH-3T3 cells were maintained in DMEM with 10% FBS. Both medium were supplemented with 2 mM L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were grown at 37 °C in a humidified 95% air/5% CO2 incubator.
24 h prior to exposure, the RAW264.7 macrophages were plated into 12-well plates with a density of 3×105 per well, pre-treated with lipopolysaccharide (LPS, 25 ng/mL, Sigma-Aldrich Chemical Company) for 3 h, then exposed to crystalline silica particles (50 μg/cm2) in the presence of dioscin for 6 h [25, 26]. Dioscin was added with different concentrations of 300, 150, 75 ng/mL determined by MTT (Figure S5). Anisomycin (2 ng/mL, Cell Signaling Technology), activator of JNK and p38 MAPKs, was used to test the biological effect of dioscin on the MAPKs. The use of RAW264.7 cell is an established murine model in the literature for CS experiments [25-27]. After that, cultured medium was collected for ELISA analysis and cells were lysed for western blot analysis.
NIH-3T3 is a murine fibroblast cell line that is widely used to investigate roles of fibroblast in fibrogenesis [28-30]. NIH-3T3 cells were seeded in 12-well plates (8×104 cells/ well) and cultured overnight. Then the cells were serum-starved for 24 h. 3T3 cells were incubated with or without TGF-β (5 ng/mL, Sino Biological Inc), with or without dioscin (1000, 500, 250 ng/mL) (Figure S5). After 30 min, cells were lysed for western blot analysis to test the effects of dioscin on TGF-β-smad3 signaling. In addition, 3T3 cells were incubated with or without TNF-α (25 ng/mL, Sino Biological Inc), with or without dioscin (1000, 500, 250 ng/mL). After 30 min, cells were lysed for western blot analysis to test the effects of dioscin on ASK-1-p38/JNK signaling. After 12 h, cells were lysed to get mRNA and detect Il-1β, Il6, and Tnf-α expression.
Co-culture experiment
This co-culture model was conducted as previously described [30]. Briefly, in our experiment, we used 24 mm Transwell® with a 0.4 μm Pore Polyester Membrane Insert from Corning Company. LPS-primed RAW264.7 cells seeded in the lower well were first treated with CS (50 μg/cm2) for 24 h together with different concentrations of dioscin (300, 150, 75 ng/mL), and then NIH 3T3 cells which had attached on the top of the insert for 24 h were co-cultured with RAW264.7 cells in the Transwell 6-well plate system for another 24 h. After that, NIH-3T3 cells were collected to detect the mRNA levels of collagen I and α-SMA.
Hydroxyproline assay
Lung collagen content was measured with a hydroxyproline (HYP) kit (Nanjing Jian Cheng Institute, Nanjing, China). The left lungs cleared of blood were used following the manufacturer's instruction. The results are expressed as microgram HYP per gram of wet lung weight by HYP standards.
Histological analysis and immunohistochemistry
The lung samples were fixed in 4% paraformaldehyde and embedded in paraffin. Samples were sectioned for staining with hematoxylin and eosin (H&E) or Masson's trichrome staining to assess the degree of fibrosis. Immunohistochemistry (IHC) and immunofluorescence (IF) were performed as previously described. Briefly, 5 µm sections were deparaffinized and rehydrated. After the antigen was recovered by microwave heating of histologic sections in citrate buffer (Beyotime). Then the tissues were incubated with the appropriate primary antibodies at 4°C overnight and with horseradish peroxidase (HRP) polymer secondary antibodies (Santa Cruz) at room temperature for 30 min, followed by DAB color development (Santa Cruz). Primary antibodies specific for type I collagen, CD45 (Abcam), cluster of differentiation 3 (CD3; Santa Cruz), B220 (eBioscience), Mac-2 binding protein (Cedarlane), E-cadherin (Cell Signaling Technology), and N-cadherin (Cell Signaling Technology) were used for staining. This was followed by incubation with Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies (Abcam). The nucleus was labeled with DAPI (Life Technology). The numbers of Mac-2 positive cells, B220 positive cells and fibrocytes were counted by a blind method. Image-Pro Plus 6.0 software was used for cell counting. Three different fields were selected from a lung section and three sections per animal were evaluated to obtain a mean value. 3-4 mice from each group were used to obtain an overall value for subsequent statistical analysis.
Quantitative PCR analysis
We performed RNA isolation and quantitative PCR (qPCR) as previously described. Briefly, total RNA was isolated from lungs cleared of blood using TRIzol Reagent (Invitrogen) and was reverse transcribed into cDNA with PrimeScript RT kit (Takara) based on the manufacturer's instructions. Target gene expression was determined using the SYBR Green Master Mix Kit (Takara). All of the RNA samples in the different groups were isolated and amplified in parallel. The sequences of specific primer pairs are described in the Supplementary Materials (Table S1). GAPDH was used as the internal control for determining ΔCT values. Fold increases in expression were normalized to the saline control group by determining the 2-ΔΔCT values.
Fluorescence-activated cell sorting analysis
Fluorescence-activated cell sorting (FACS) analysis was performed as previously described. Briefly, the hilar lymph node (HLN) was mechanically dissected and digested with 0.25% trypsin for 5 min at 37°C. The HLN cell pellet was washed and re-suspended in phosphate-buffered saline (PBS). The total population of HLN cells was stained with CD3, CD4, CD8, CD25, forkhead box P3 (Foxp3), interferon-gamma (IFN-γ), interleukin-4 (IL-4) and interleukin-17A (IL-17A) antibodies (all from BD Bioscience, San Jose, CA, USA). For lung cells, mouse lung was digested in buffer containing collagenase (15 mg), DNase I (250 KU), and complete media (RPMI+10% FBS) at 37°C for 30 min, and was then filtered through a 70 μm cell strainer. The total population of lung cells was analyzed by flow cytometry after staining with antibodies to CD45 (BD Bioscience), Type I collagen (Rockland Immunochemicals, Limerick, PA, USA), epithelial cell adhesion molecule (EpCAM; eBioscience), and fibroblast specific protein-1 (FSP-1; Abcam). FACS was performed on the BD Canto II FACS station, and data were analyzed using FlowJo V10 software.
Western blot analysis
The proteins were extracted from lung tissue and cells following standard protocols as previously described. RIPA buffer (Beyotime) containing protease inhibitors (Beyotime) and phosphatase inhibitors (Roche, Basel, Switerland) was used. All of the lung tissues used in the analysis were cleared of blood. Total protein was estimated using the Pierce BCA Protein Assay Kit (Thermo Scientific). Lung lysates from each mouse were diluted to a concentration of 3 μg/μL. Cell lysates were also diluted to the same concentrations. Then, 10 μL aliquots (i.e., 30 μg protein or equivalent) of each sample were resolved on sodium dodecyl sulfate polyacrylamide gels and electrophoretically transferred to PVDF membranes. After blocking in 5% defatted dried milk, membranes were washed and incubated with the following primary antibodies: anti-Fibronectin-1 (Abcam), anti-Collagen-1 (Abcam), anti-apoptosis signal-regulating kinase 1 (ASK1; Abcam), anti-transforming growth factor beta 1 (TGF-β1; Abcam), anti-phospho-ASK1 (Santa Cruz), anti-p38, anti-phospho-p38, anti-c-Jun N-terminal kinase (JNK), anti-phospho-JNK, anti-Vimentin, anti-Smad3, anti-phospho-Smad3, and anti-β-Actin (all from Cell Signaling Technology) overnight at 4°C. Then membranes were incubated with horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit or goat anti-rat; Cell Signaling Technology). Blots were developed with a high-performance luminol substrate solution (PexBio, Beijing, China). β-Actin was used as the loading control.
Cytokine analysis
BALF was obtained as previously described. The cytometric bead array (CBA) mouse inflammation kit (BD Biosciences) was used to determine the cytokine levels in BALF according to the manufacturer's instructions. Briefly, multiple capture beads were mixed together and co-incubated with 50 μL BALF supernatant and detection reagent for 2 h. Then beads were washed carefully and re-suspended. Samples were analyzed using a FACS Canto II station, and data were analyzed with FCAP Array software. In addition, levels of IL-1β, IL-2, IL-4, IL-13, and IL-17 (R&D Systems, Minneapolis, MN, USA) in BALF were measured with a commercial enzyme-linked immunoassay (ELISA) kit according to the manufacturer's instructions. Cytokines including IL-1β, IL-6, MCP-1, TNF-α (R&D Systems, Minneapolis, MN, USA) in the cell cultured medium were measured by ELISA following the instructions.
Statistical analysis
All of the quantitative experiments were repeated at least once with consistent results. The sample data from independent experiments were calculated to obtain the mean ± standard deviation. One-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test was used to compare treatment groups with the control. P values less than 0.05 were considered statistically significant. GraphPad Prism 7.0 software or SPSS 19.0 was used for the statistical analyses.
Results
Dioscin attenuates crystalline silica-induced pulmonary fibrosis
To investigate the biological effects of dioscin (chemical structure is shown in Figure 1B) on CS-induced pulmonary fibrosis, we used an experimental silicosis mouse model. The mice were treated with 20 mg/kg, 40 mg/kg, or 80mg/kg dioscin by intragastric gavage daily after CS instillation (Figure 1A). We evaluated collagen deposition in the lung tissues of dioscin or vehicle treated mice by analyzing the HYP content, and found that dioscin treatment reduced the amount of collagen in the lungs of CS-injured mice (Figure 1C). Accordingly, Masson's trichrome staining showed decreased collagen deposition in mice treated with dioscin compared with vehicle treated mice (Figure 1D). Furthermore, attenuated fibrosis was supported by decreased protein levels of type I collagen and fibronectin in the lungs at 56 d (Figure 1E, F). Moreover, qPCR analysis showed decreased mRNA type I collagen and fibronectin expression in the lungs (Figure 1G, H). We also showed by IHC staining that dioscin treatment reduced type 1 collagen deposition in the lungs (Figure 1I). What's more, we recorded the body weight of mice in each group during the intragastric administration and found that dioscin promoted body weight recovery during the first three weeks after CS instillation (Figure S3). Taken together, these data show that dioscin reduces collagen deposition in the lungs of CS-injured mice.
Dioscin targets the innate and adaptive immune systems
To investigate the mechanisms underlying the attenuation of CS-induced pulmonary fibrosis by dioscin, we first performed bioinformatics analysis utilizing STITCH, a database used to explore known and predicted interactions between chemicals and proteins. Several protein factors were involved, and predicted protein-protein interactions were demonstrated (Figure 2A). Gene Ontology (GO) analysis was used to determine how these protein factors are involved in dioscin's effects. GO analysis of the biological process showed that the inflammatory response may be an important biological effect related to dioscin (Figure 2B). It has also been reported that persistent inflammation drives fibrotic procession in some fibrosis animal models [31]. Thus, we analyzed the inflammatory responses in dioscin- or vehicle-treated mice 7 d after CS instillation. Notably, compared with saline-treated mice, CS-injured mice showed severe pulmonary inflammation, whereas the dioscin-treated mice had decreased lung inflammation in a dose-dependent manner (Figure 2C).
Figure 1
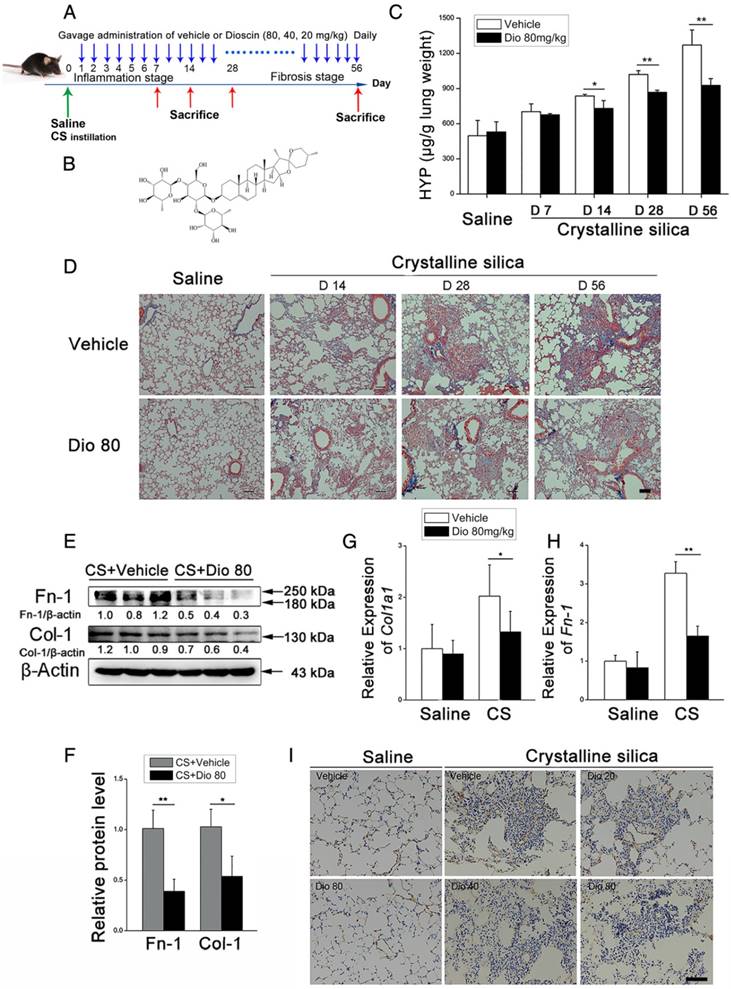
Pulmonary fibrosis caused by CS was attenuated by intragastric administration of dioscin. (A) Intervention dosing regimen of dioscin in experimental mouse model of silicosis. C57BL/6 mice were intragastric gavaged with different doses of dioscin or vehicle (n=10 per group) daily after CS instillation. Lungs were harvested at indicated time points for the following analyses. (B) Chemical structure of Dioscin. (C) Hydroxyproline (HYP) contents in lung tissues from dioscin or vehicle-treated mice were measured at 7, 14, 28, and 56 d (n = 3-4 per group; *, P<0.05; **, P<0.01). (D) Masson trichrome staining of collagen on lung sections taken from mice at 14, 28, and 56 d after CS instillation. Representative images are shown (n=4 per group). (E-H) Mice were treated with saline or CS, dioscin or vehicle for 56 d after which lung tissues were harvested for subsequent analyses. Western blot analysis of collagen I (Col-1, E) and fibronectin (Fn-1, E), β-Actin was used as a loading control (n=3). Quantification of Col-1 and Fn-1 protein levels relative to β-Actin is shown below each band. (F) Quantifications of the single protein. (G-H) qPCR analysis of Col-1a1 and fibronectin (Fn-1) mRNA levels in lung tissues (n=6) (*, P<0.05; **, P<0.01). (I) Immunohistochemical analysis of collagen I in lung sections. Representative staining images are shown (n=4 per group). (D and I) Bar, 100 μm. The experiments were performed three times. (C, F, G, H) Error bar indicates mean ± SD.
Figure 2
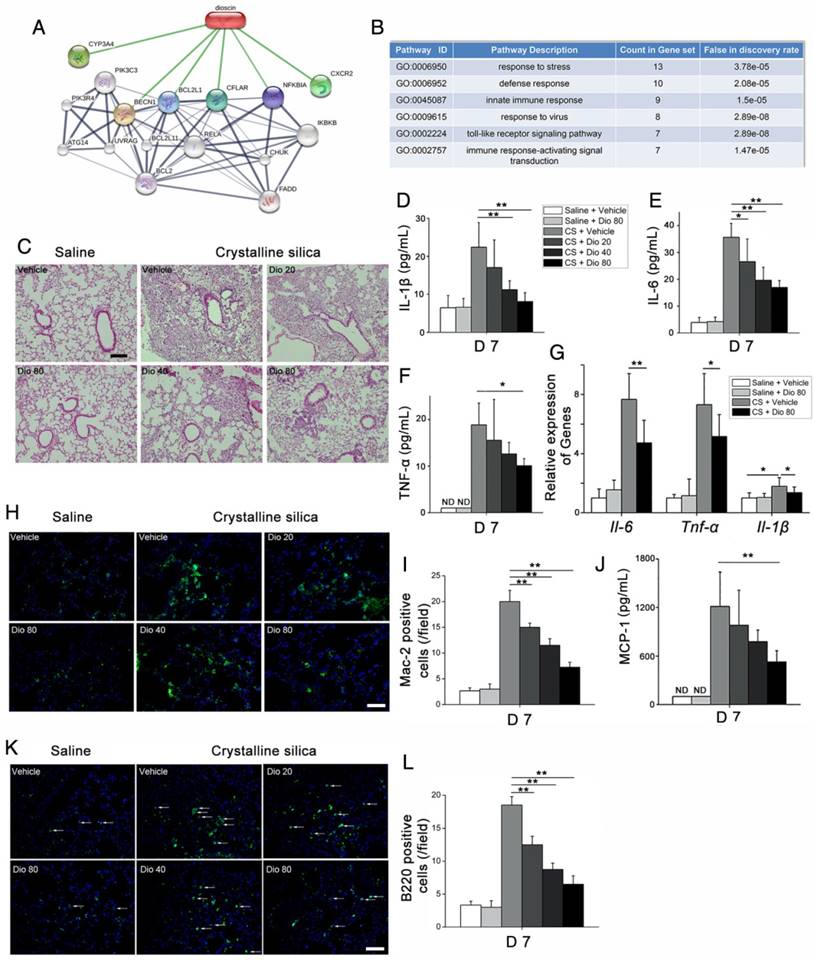
CS-induced pulmonary inflammatory responses were alleviated by dioscin. (A) Bioinformatics analysis: chemical-protein interaction network in STITCH v.5.0. A screenshot from STITCH shows a network associated with dioscin. (B) Gene Ontology (GO) analysis of biological processes. (C) Lung sections of dioscin-treated mice for 7 d were stained with H&E. Representative images are shown (n=4-5 per group) Scale bar indicates 200 μm. (D-F) ELISA analysis of IL-1β, IL-6, and TNF-α in BALF of dioscin-treated mice for 7 d (n=5-6 per group). ND, not detected. (G) qPCR analysis of Il-6, Il-1β, and Tnf-α mRNA levels in the lung tissues of dioscin-treated mice for 7 d (n=6 per group). (H) Mac-2-positive macrophages were detected by immunofluorescence analysis of the lung sections (green). Nuclei were visualized with DAPI (blue). (I) Quantification of Mac-2 positive cells in lung sections (n=3-4 per group). (J) CBA analysis of MCP-1 in BALF of dioscin-treated mice for 7 d (n=5 per group). (K) B220-positive B cells were detected by immunofluorescence analysis of the lung sections (green). Nuclei were visualized with DAPI (blue) (n=3-4 per group). (L) Quantification of B220 positive cells in lung sections (n=3-4 per group). White arrows indicate positive staining. The lung sections used in (H, K) were from different mice treated for 7 d. Scale bar indicates 100 μm in H, K. *, P<0.05; **, P<0.01; the experiments were performed three times. Error bar indicates mean ± SD.
Next, we measured levels of the pro-inflammation cytokines IL-6, IL-1β, and TNF-α, and found that their expression in BALF was altered in a dose-dependent manner upon dioscin treatment (Figure 2D-F). The qPCR analysis showed that mRNA levels of Il-6, Il-1β, and Tnf-α in the lungs of CS-injured mice were decreased by dioscin treatment (Figure 2G). There was also markedly increased cellularity near the perivascular and peribronchial regions of lungs exposed to CS, but not in control mice. In addition, cell infiltration was reduced by dioscin treatment (Figure S4A).
Acute inflammatory infiltration induced by CS particles is typified by macrophages. In saline-treated lungs, Mac-2-positive macrophages were barely detected, whereas in lungs exposed to CS, they were readily seen. In addition, the number of macrophages was clearly decreased in the lungs of dioscin-treated mice (Figure 2H, I). We also measured the expression of the classical macrophage marker CD68 in the lungs of dioscin-treated mice by qPCR analysis, and found that similarly to Mac-2, dioscin-treated mice showed reduced mRNA levels of CD68 (Figure S4B). Monocyte chemotactic protein-1 (MCP-1), a potent chemokine of macrophages, exhibits increased expression in silicosis patients [32]. We used CBA to measure MCP-1 levels in BALF, and found that the levels decreased in a dose-dependent manner with dioscin treatment (Figure 2J). Because both innate and adaptive immune mechanisms are operative during the inflammatory phase [33], we used IF and IHC with antibodies against B220 and CD3 to determine if dioscin treatment affects B and T cells, respectively. We observed an increased number of B lymphocytes (B220+) in the lungs of mice exposed to CS compared with control mice. The number of B cells was also decreased in the lungs of mice exposed to CS but treated with dioscin (Figure 2K, L). Similarly, there were fewer T cells in the lungs of dioscin-treated mice (Figure S4C). We also performed FACS analysis of HLN cells, and found that CD3-positive cell counts were lower in dioscin-treated mice compared with CS-injured mice (Figure S4D). These results indicate that dioscin can alleviate CS-induced pulmonary inflammation by decreasing macrophage and lymphocyte infiltration into lung tissues.
Th responses are influenced by dioscin treatment
T helper (Th) cells play a prominent role in the progression of some experimental models of fibrosis [34, 35], and a previous study showed that Th1/Th2 imbalance plays a role in fibrogenesis [36]. Specifically, the Th cell balance shifts from Th1 dominant in the stage of inflammation to Th2 dominant during fibrosis development. We detected the percentage of Th1 cells in the HLN by FACS analysis. As shown in Figure 3A and B, the percentage of CD3+CD8-(CD4+) cells expressing IFN-γ, regarded as Th1 cells, was significantly increased in CS-treated mice compared with control mates during the inflammatory phase. This effect was reversed with dioscin treatment. In addition, in the fibrosis stage, dioscin could reduce Th2 proportion in CD3+CD8-(CD4+) cells (Figure 3G, H). Th cells orchestrate immune responses involved in the host defense by producing cytokines [37]; thus, we used ELISA to measure the concentrations of typical Th1 cytokines (IL-2, IFN-γ) and typical Th2 cytokines (IL-4, IL-13) in BALF. We found that the expression of these cytokines in BALF was altered in a dose-dependent manner by dioscin treatment (Figure 3C, D, I, J). Next, we used qPCR to determine the expression of Th1 and Th2 transcription factors, T-bet and GATA3, respectively, in lung samples from treated and control mice. T-bet was decreased in the inflammatory stage (7 d), whereas GATA3 was decreased in the fibrosis stage (56 d) of dioscin-treated mice (Figure 3E, K). We measured the mRNA expression levels of Ifn-γ and Il-4 in mouse lungs and found that the results were consistent with those from the ELISA analyses (Figure 3F, L).
We also performed FACS analysis of Th17 cells (CD3+CD8-IL-17A+) in the HLN and found that CS exposure increased the number of Th17 cells in the HLN, which was decreased upon dioscin treatment at the inflammation (7 d) and fibrosis (56 d) stages (Figure 3M, N). Next, we measured IL-17 concentration in BALF, and found that it was reduced by dioscin treatment in a dose-dependent manner (Figure 3O). Together, these data demonstrate that Th responses are involved in CS-induced pulmonary inflammation and fibrosis. In addition, Th1, Th2 and Th17 immune responses are significantly attenuated in the inflammation and fibrosis stages, respectively, with treatment of dioscin.
Dioscin blocks ASK-1-p38/JNK signaling in CS-injured lungs
Mitogen-activated protein kinases (MAPKs) are thought to play key regulatory roles in the production of pro-inflammatory cytokines. Studies have also shown that MAPK signaling modulates Th responses in several animal models [38, 39]. ASK-1 is a member of the MAP3K family, and is a critical upstream activator of p38 and JNK [40]. ASK-1 phosphorylation is correlated with several inflammatory diseases [41-43], and regulates IL-17 production [44]. To determine the mechanism by which dioscin affects immune responses, we examined the effects of dioscin on ASK-1-p38/JNK phosphorylation. Our western blot results showed that dioscin decreased CS-induced phosphorylation of ASK-1 (Figure 4A, B) and the up-regulated expressions of phosphorylated p38 and JNK in mouse lung samples (Figure 4C-E). These data demonstrate that dioscin may exert its anti-inflammatory effects via the ASK-1-p38/JNK pathway.
Figure 3
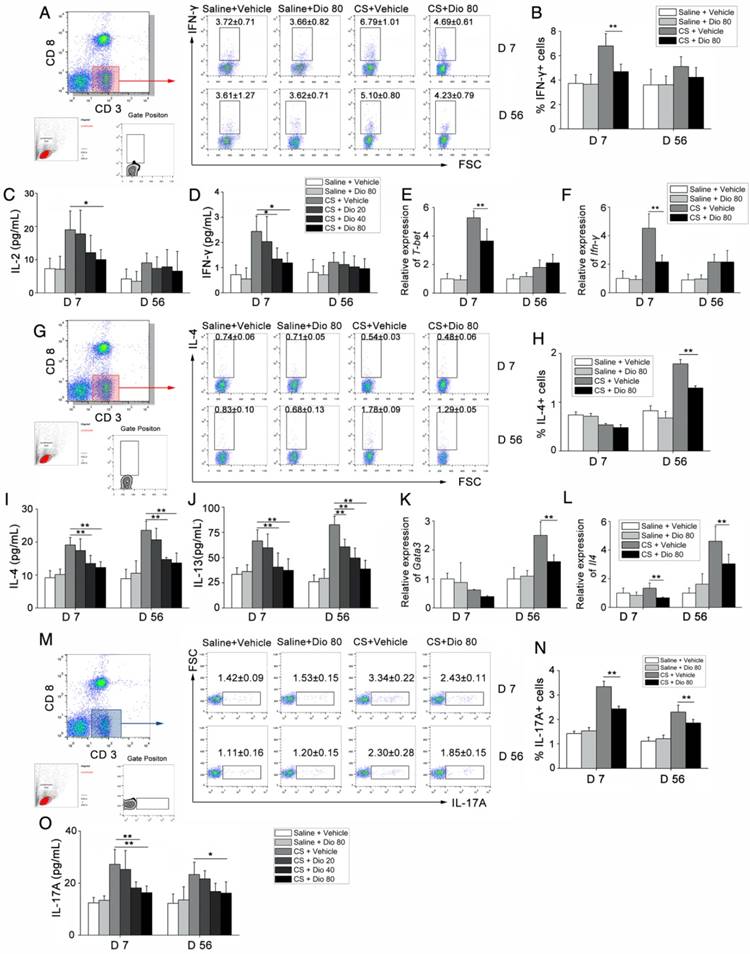
T helper cell immune responses were influenced by dioscin in the mouse model of CS-induced fibrosis. (A, B) Percentage of CD3+CD8-(CD4+) cells expressing IFN-γ (Th1 cells) in hilar lymph nodes was assayed by FACS analysis (n=5 per group). (C, D) ELISA analysis of IL-2 and IFN-γ in BALF (n=5 per group). (E, F) qPCR analysis of T-bet and IFN-γ in the lung tissues (n=5 per group). (G, H) Percentage of CD3+CD8-(CD4+) cells expressing IL-4 (Th2 cells) in hilar lymph nodes was assayed by FACS analysis (n=4-5 per group). (I, J) ELISA analysis of IL-4 and IL-13 in BALF (n=5 per group). (K, L) qPCR analysis of GATA3 and Il-4 in the lung tissues (n=5 per group). (M, N) Percentage of CD3+CD8-(CD4+) cells expressing IL-17 (Th17 cells) in hilar lymph nodes was assayed by FACS analysis. (n=5 per group). (O) ELISA analysis of IL-17 in BALF (n=5 per group). Gating strategy is shown in the figure. *, P<0.05; **, P<0.01; the experiments were performed twice with similar results. Error bar indicates mean ± SD.
Figure 4
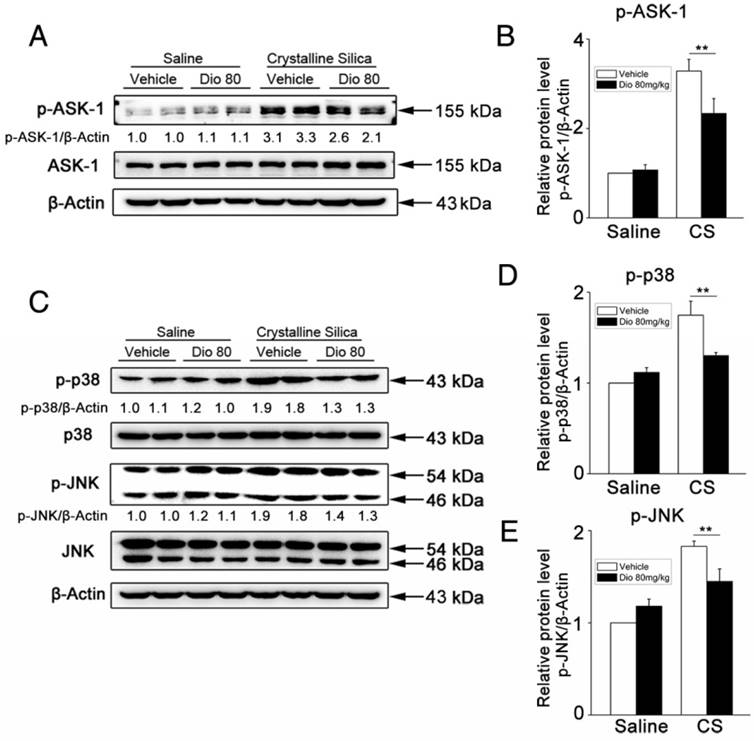
Phosphorylation of ASK-1 and downstream MAPK proteins in the lung was blocked by intragastric administration of dioscin. (A, C) Western blot analyses of ASK1 phosphorylation and downstream MAPK proteins (p38 and JNK/SAPK) at 7 d (n=3). Figure shows the representative images of three independent experiments. Quantification of each protein level relative to β-Actin is shown below each band. (B, D, E) The levels of phospho-ASK1, phosphor-p38 and phosphor-JNK were normalized to those of β-Actin. Data are the mean of three independent experiments. *, P<0.05; **, P<0.01. Error bars indicate the mean ± SD. The experiment was performed in triplicate.
Dioscin inhibits macrophages and fibroblasts from secreting pro-inflammatory cytokines through blocking ASK-1-p38/JNK signaling
To better illuminate the mechanism of dioscin's effects on individual cells of the lung, we did in vitro experiments, as well. Macrophages are the first line of defense against invading foreign bodies They can, in response to particles, produce pro-inflammatory and pro-fibrotic mediators that play important roles in inflammation and establishment of pulmonary fibrosis [45]. So, we used CS to stimulate macrophages while different concentrations of dioscin were added. We measured the cytokines in the culture medium. Results of ELISA showed that dioscin could effectively inhibit macrophages from secreting IL-1β, IL-6, TNF-α and MCP-1 (Figure 5E-H). We also did western blot analysis of the cell lysates. Results showed that dioscin effectively blocked the ASK-1-p38/JNK signaling pathway in macrophages (Figure 5A-D). To further illuminate the relationship between the cytokine reduction and blocked signaling pathway, we did a reverse experiment, in which we added dioscin together with anisomycin, a strong activator of JNK and p38 MAPK. Results showed that anisomycin could reverse the effect of dioscin on cytokines reduction (Figure 5E-H).
Figure 5
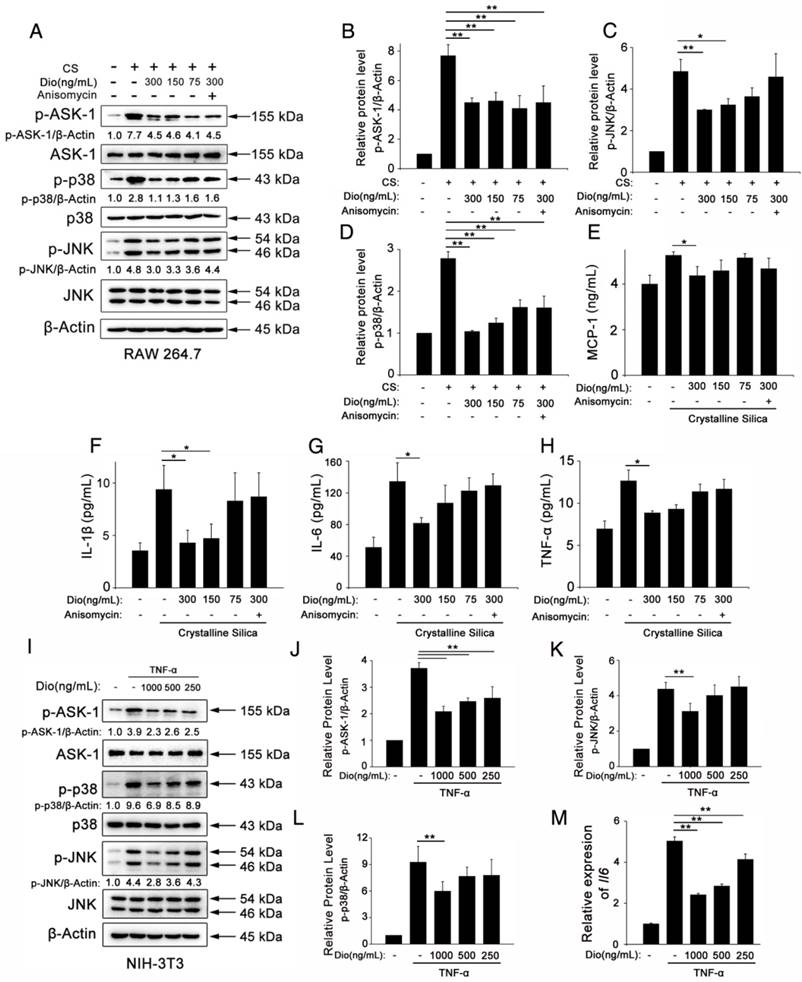
Dioscin inhibits macrophages and fibroblasts from secreting pro-inflammatory cytokines. (A) Western blot analyses of ASK1 phosphorylation and downstream MAPK proteins (p38 and JNK/SAPK) in LPS-primed RAW 264.7 macrophages treated with CS (50 μg/cm2) together with different does of dioscin for 6 h. Anisomycin was used to reverse dioscin's effect (2 ng/mL). Quantification of each protein level relative to β-Actin is shown below each band. (B-D) The levels of phospho-ASK1, phospho-p38 and phosphor-JNK were normalized to those of β-Actin (n=3). (E-H) ELISA analyses of pro-inflammation cytokines in the culture medium of different treated macrophages at 6 h. (E) MCP-1, (F) IL-1β, (G) IL-6, (H)TNF-α (n=3). Data are the mean of three independent experiments (n=3). (I) Western blot analyses of ASK1 phosphorylation and downstream MAPK proteins (p38 and JNK/SAPK) in TNF-α- (25 ng/mL) treated NIH-3T3 fibroblasts together with different does of dioscin for 30 min. Quantification of each protein level relative to β-Actin is shown below each band. (J-L) The levels of phospho-ASK1, phospho-p38 and phosphor-JNK were normalized to those of β-Actin (n=3). (M) qPCR analysis of Il-6 mRNA levels in TNF-α- (25 ng/mL) treated fibroblasts together with different concentrations of dioscin for 12 h (n=3). *, P<0.05; **, P<0.01. Error bars indicate the mean ± SD. The experiment was performed twice with similar results.
In addition, we did an in vitro experiment about fibroblasts. Fibroblasts are the effector cells of pulmonary fibrosis for the secretion of ECM proteins. But studies have shown that they could also secrete some pro-inflammation cytokines upon stimulation [46]. We treated NIH-3T3, a fibroblast cell line, with TNF-α together with various concentrations of dioscin. We did qPCR analysis to detect the mRNA levels of Il-6 Il-1β and Tnf-α in treated fibroblasts. Results showed that dioscin could reduce the mRNA level of Il-6 (Figure 5M). But we didn't detect the mRNA expressions of Il-1β and Tnf-α (Data not shown). We also did western blot analysis of the cell lysates of fibroblasts. As shown in Figure 5I-L, ASK-1-p38/JNK expression in fibroblasts was also blocked by dioscin in a dose-dependent manner. These results above showed that dioscin could inhibit macrophages and fibroblasts from secreting pro-inflammatory cytokines, which may be related to the alleviated pulmonary inflammation. The effects are related to dioscin's effect on the ASK-1-p38/JNK MAPK pathway.
Anti-fibrotic effects of dioscin are associated with the decreased accumulation of fibrocytes
Fibrocytes are derived from the bone marrow, and they proliferate and differentiate into myofibroblasts, which release ECM components, leading to fibrosis [47]. The differentiation of fibrocytes is critically dependent upon CD4+ T cells, and the interaction between fibrocytes and Th cells promotes the pathogenesis of various diseases [48-51]. We hypothesized that dioscin affects fibrocyte accumulation in addition to its effects on T helper cells. To test this hypothesis, we performed double staining of fibrocytes with anti-CD45 (green) combined with anti-collagen I (red) on lung sections. Consistent with this idea, there was more fibrocyte accumulation in lungs exposed to CS compared with the control. This accumulation was reduced in a dose-dependent manner with dioscin (Figure 6A, B). FACS analysis also showed increased numbers of CD45+Collagen-1+fibrocytes in CS-injured lungs, and dioscin treatment consistently reduced fibrocyte accumulation (Figure 6C, D). To determine whether the reduced accumulation of fibrocytes was associated with a decrease of related chemokines, we performed qPCR analysis to evaluate the mRNA levels of Ccl12, Ccl19, Ccl21, and Cxcl12 in the lungs of dioscin-treated mice. As shown in Figure 6F, dioscin-treated mice had decreased mRNA levels of Ccl12 and Ccl19, but not Ccl21 or Cxcl12. These data indicate that the anti-fibrotic effects of dioscin are also associated with the decreased accumulation of fibrocytes, which appear to correlate with the down-regulation of related chemokines.
Dioscin protects epithelial cells from CS injury
To determine fibrocyte involvement in dioscin's anti-fibrotic effects, we performed FACS analysis. It is worth noting that CD45-negative Col-1-positive cells were also increased in CS-injured lungs. Interestingly, dioscin treatment also reduced this cell population, which we reasoned may be alveolar epithelium cells (AECs), as they account for most of the CD45-negative cells in the lung. Therefore, we re-examined the H&E-stained sections using a larger magnification for the alveolar areas. It appeared that mice treated with dioscin had a significant reduction of lung architectural damage 7 d after CS injury (Figure 7A). Next, we performed FACS analysis to determine whether the epithelium underwent a functional transition, and found an increased number of EpCAM+FSP-1+ cells in response to CS exposure. Dioscin treatment decreased this portion of double positive cells (Figure 7B, C). Then we performed qPCR analysis of the lung tissues isolated from treated mice and found decreased mRNA levels of epithelial markers, such as Occludin, E-cadherin, and Sftpc, and increased levels of mesenchymal markers such as vimentin and FSP-1. Dioscin treatment inhibited expression of the CS-induced mesenchymal markers and restored expression of the epithelial markers in the lungs of treated mice (Figure 7D, E). IF analysis of the lung sections showed that CS diminished E-cadherin expression in the lung, whereas dioscin treatment restored its expression in a dose-dependent manner. Conversely, CS-induced N-cadherin expression, which was inhibited upon dioscin treatment (Figure 7F). Western blot analysis was used to detect the protein levels of vimentin (a fibroblast marker) in the lung of treated mice. As shown in Figure 7G and H, increased levels of vimentin were induced by CS instillation, which were decreased by dioscin in a dose-dependent manner. These data demonstrate that dioscin can protect AECs from CS injury and functional transition.
Dioscin could block TGF-β-Smad3 signaling pathway in vivo and in vitro
TGF-β-Smad3 signaling plays central roles in lung fibrogenesis. It is tightly linked to fibroblast activation and fibrogenesis [52, 53]. Before, we detected that dioscin could alleviate CS-induced epithelial injury and reduce the expression of a fibroblast marker Vimentin. To illuminate dioscin's effect on TGF-β signaling, we first examined the expression of TGF-β in lung tissues at 56 d after CS instillation. Immunoblot and qPCR analyses of lung extracts showed higher TGF-β protein and mRNA expressions, respectively, which were reduced by dioscin treatment (Figure 8A, B and C). Next, we examined the effects of dioscin on the TGF-β downstream Smad signaling pathway. Dioscin treatment inhibited CS-induced phosphorylation of Smad3 in a does-dependent manner, but did not alter total Smad3 expression levels (Figure 8D, E). TGF-β-Smad3 signaling is tightly linked with the activation of fibroblast. So, we next performed an in vitro experiment, in which we treated the fibroblast cell line NIH-3T3 with TGF-β1 together with various concentrations of dioscin. Western blot analysis was used to determine dioscin's effects on the phosphorylation of Smad3. Results showed that dioscin could block phosphorylation of Smad3 in NIH-3T3 cells in a dose-dependent manner (Figure 8F, G). It is believed that TGF-β secreted by macrophages serves as a paracrine stimulus to fibroblasts to promote the transformation of fibroblasts into myofibroblasts [30]. Furthermore, we co-cultured NIH-3T3 with RAW 264.7 in a transwell assay (Figure 8H). After 24 h co-culturing, the mRNA expressions of collagen I and α-SMA in NIH-3T3 fibroblasts were detected by qPCR. As shown in Figure 8I and J, co-culturing with CS-treated RAW 264.7 increased the mRNA levels of Col-I and α-SMA in NIH-3T3 cells, while dioscin could reduce this increase. TGF-β is an immunosuppressive cytokine that is secreted by multiple cell types, including macrophages and epithelial cells. Regulatory T cells (Tregs) also produce TGF-β [54]. So, we performed FACS analysis using a single cell suspension of HLNs and found that dioscin treatment reduced the percentage of CD25+Foxp3+ in CD4+ cells at 7 d and 56 d (Figure 8K, L), which is in accordance with other studies that have demonstrated that Tregs promote fibrogenesis [55-57]. We also performed qPCR to determine the mRNA levels of Foxp3, the key transcription factor that controls Treg expression, in the lungs of treated mice, and found decreased levels of this protein as well upon dioscin treatment (Figure S6A). The mRNA levels of Il-10 were also inhibited upon dioscin treatment (Figure S6B). These data indicate that dioscin's pulmonary protective effect is via a reduction of TGF-β and inhibition of the TGF-β-Smad3 pathway in vivo and in vitro.
Dioscin could postpone the progression of established silicosis
Before, we demonstrated that dioscin treatment could attenuate CS-induced pulmonary fibrosis. In that in vivo experiment, dioscin was delivered at approximately the same time as CS instillation. We further asked whether dioscin could prevent or postpone the progression of established fibrosis. So, we performed another in vivo experiment, in which dioscin was instilled by intragastric gavage 10 days after CS instillation (Figure 9A). During the 10 days, acute inflammation caused by CS was diminished, and pulmonary fibrosis was established. We initially treated mice with dioscin (80 mg/kg) from day 11 after CS injury. During the experiment, we recorded the animal body weights weekly and found that during the first two weeks the two groups of animals weighed roughly the same. After that the mice treated with dioscin recovered faster than those treated with vehicle (Figure S7A). We isolated the lungs and did related experiments. As shown in Figure 9B, the lungs from CS-treated mice at 56 d had a granular surface and acquired a white round fleck, which were ameliorated by dioscin. HYP measurement showed that collagen content was significantly decreased in the mice treated with dioscin, compared with those treated with vehicle (Figure 9C). Masson's trichrome staining displayed a similar result (Figure 9D and E). In addition, we isolated alveoli macrophages of these treated mice and cultured the cells. We measured the cytokines in the cultured medium and found that these cytokines were significantly reduced by dioscin treatment (Figure S7B-E), which was analogous to the result of the in vitro experiments. These data indicated that dioscin could significantly postpone lung fibrosis even after fibrotic disease had been established.
Discussion
Silicosis, a form of pneumoconiosis, is a progressive occupational lung disease caused by the inhalation, deposition, and retention of CS particles [11]. The pathogenesis of silicosis has not been fully elucidated, and few treatments are available. Here, we show that oral treatment of dioscin in mice ameliorated CS-induced pulmonary fibrosis. To determine the mechanisms underlying its effects, we first conducted bioinformatics analysis to explore the biological function of dioscin, focusing on its effects on the inflammatory responses. We demonstrated that the anti-fibrotic effects of dioscin may be due to several mechanisms, including both immunoregulatory and direct pulmonary protective effects (Figure 10).
Immunity and inflammation are integral parts of the pathogenesis of silicosis. CS in the lungs induces recruitment of inflammatory cells and elevates the secretion of cytokines such as TNF-α, IL-1β, IL-6, MCP-1, and TGF-β1 during the innate immune phase. Macrophages, as typical representatives of innate immunity, engulf invading particles and participate in the fibrotic process [58]. They secrete multiple cytokines that regulate fibroblast recruitment, proliferation, and activation, among which TNF-α and IL-1β are recognized as the earliest factors [59-61]. We demonstrated that CS-injured mice given dioscin had reduced pulmonary inflammation, and decreased levels of TNF-α, IL-1β, and IL-6 in BALF (Figure 2). In addition, we did an in vitro study, in which we showed that dioscin could decrease CS-induced cytokines (IL-1β, IL-6, MCP-1 and TNF-α) released by LPS-primed macrophages (Figure 5). Studies on the role of macrophages in the fibrotic process have indicated that depleting macrophages during the early inflammatory phase leads to a reduction in scarred areas. In contrast, if macrophages are depleted during the late phase, fibrosis persists [62]. We found that macrophage infiltration into the lung was significantly reduced by dioscin treatment at 7 d, the early phase of the response to CS exposure, in accordance with the results of the aforementioned study. The immunoregulatory effects of dioscin on macrophages may be one reason for these anti-fibrotic effects.
We noticed massive cell infiltration near the perivascular region of the lungs of CS-injured mice. Thus, in addition to macrophages, we examined B and T lymphocytes as indicators of the adaptive immune response. B lymphocytes are involved in the progression of fibrosis and produce IL-6, a well-known fibroblast growth factor [63]. We found that dioscin treatment reduced IL-6 levels in BALF as well as B cell accumulation in the lung. Fibroblasts could also secrete cytokines upon stimulation. We did an in vitro experiment in which NIH-3T3 fibroblast was stimulated by TNF-α. Results showed that dioscin could block TNF-α-induced ASK-1-p38/JNK signaling and decrease IL-6 mRNA levels in NIH-3T3 fibroblasts. But, we did not detect IL-1β or TNF-α expressions in TNF-α-stimulated fibroblasts.
Figure 6
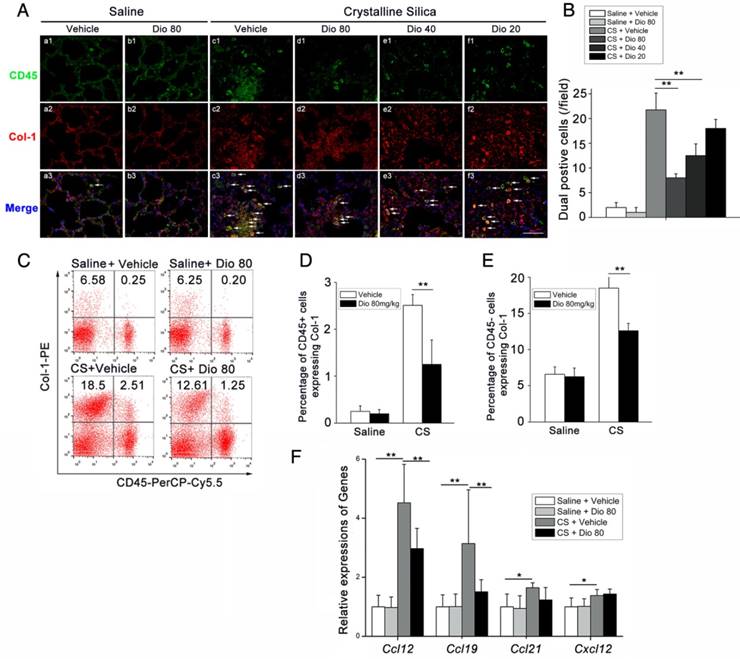
Fibrocyte recruitment into the lungs was reduced by dioscin treatment. (A) CD45 (green), Col-1 (red)-positive cells were detected by immunofluorescence analysis of the lung sections at 56 d. Nuclei were visualized with DAPI (blue) (n=3-4 per group). White arrow indicates fibrocytes. Scale bar indicates 100 μm. (B) Quantification of fibrocytes in lung sections (n=3-4 per group). (C) Representative flow cytometry plots of CD45 and Col-1-positive populations in lung single cell suspension from mice treated for 56 d (n=5). (D) Quantification of CD45+ Col-1+ cells. (E) Quantification of CD45- Col-1+ cells. (F) qPCR analysis of Ccl-12, Ccl19, Ccl-21, and Cxcl12 in the lung tissues at 56 d (n=5 per group). *, P<0.05; **, P<0.01. Error bars indicate the mean ± SD. The experiment was performed in triplicate
Figure 7
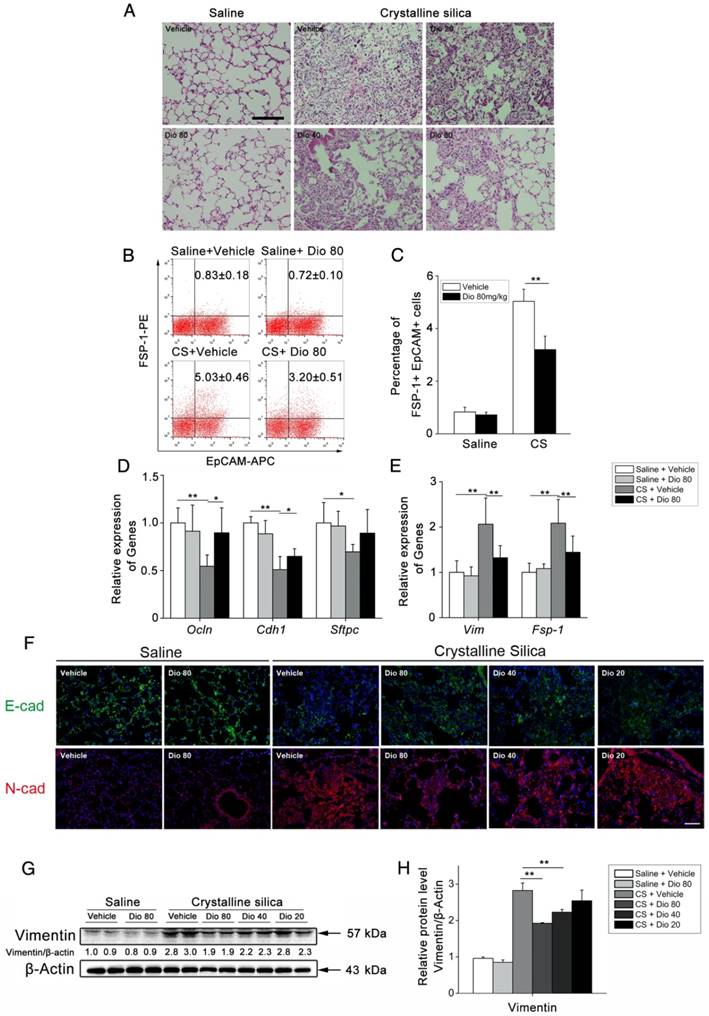
Dioscin protected alveolar epithelial cells from injury. (A) Alveolar areas of mice treated for 7 d were stained with H&E. Representative images of staining are shown (n=4-5 per group). (B C) Representative flow cytometry plots and quantification of EpCAM and FSP-1 double positive population in lung single cell suspension at 7 d. (n=5). (D) Relative mRNA levels of epithelial cell markers including Occuludin (Ocln), E-cadherin (Cdh1), Sftpc and (E) mesenchymal cell markers including vimentin (Vim) and FSP-1 in lungs from treated mice at 7 d (n=6 per group). The experiment was performed in triplicate. (F) E-cadherin- and N-cadherin-positive cells were detected by immunofluorescence analysis on the lung sections at 56 d. Nuclei were visualized with DAPI (blue). Scale bar indicates 100 μm. (G, H) Western blot analysis and quantification of vimentin in lungs of treated mice at 56 d (n=4). β-Actin was used as the loading control. Quantification of Vimentin levels relative to β-Actin is shown below the band. *, P<0.05; **, P<0.01. Error bars indicate the mean ± SD.
Figure 8
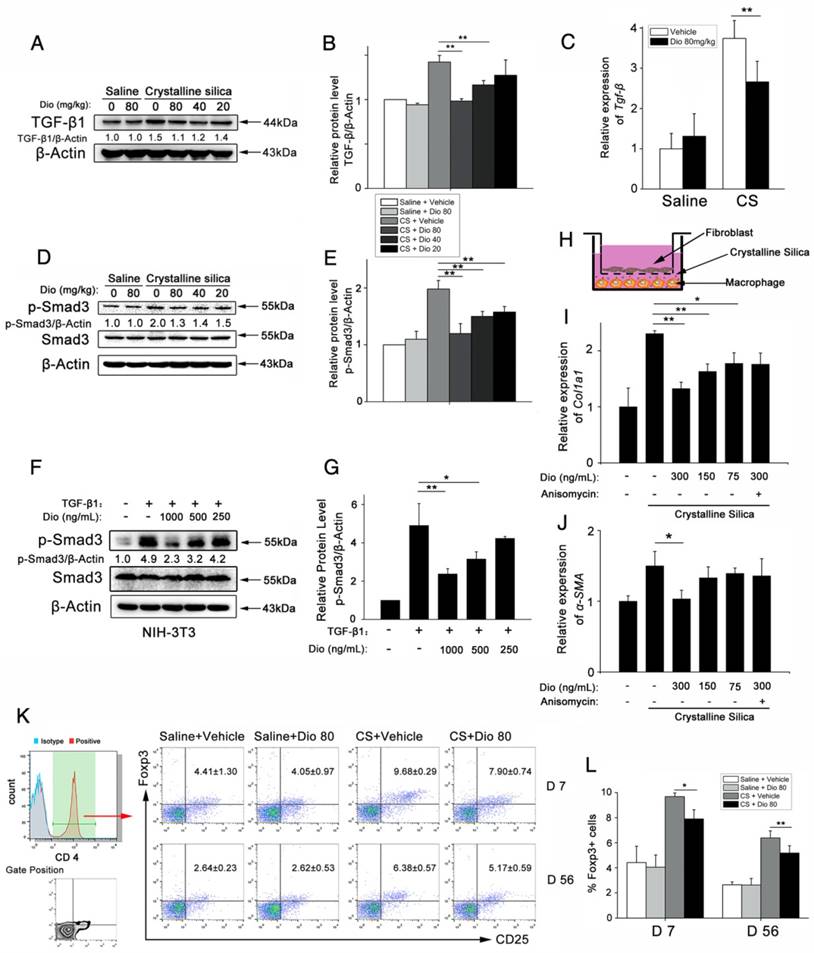
Dioscin regulated the TGF-β1-Samd3 signaling pathway and regulatory T cell functions. (A, B) Western blot analysis and quantification of TGF-β1 in lungs of dioscin-treated mice at 56 d (n=3). β-Actin was used as the loading control. Quantification of TGF-β1 levels relative to β-Actin is shown below the band. (C) qPCR analysis of Tgf-β1 in the lung tissues (n=6-8 per group). *, P<0.05; **, P<0.01. Error bars indicate the mean ± SD. The experiment was performed in triplicate. (D, E) Western blot analysis and quantification of Smad3 signaling in the lungs of treated mice at 56 d (n=3). β-Actin was used as the loading control. Quantification of p-Smad3 levels relative to β-Actin is shown below the band. (F, G) Western blot analysis of p-Smad3 and Smad3 in NIH-3T3 fibroblast, 30 min after TGF-β1 (5 ng/mL) and dioscin treatment. Quantification of p-Smad3 levels relative to β-Actin is shown below the band. (H) Schematic design of transwell experiment: RAW 264.7 macrophages were primed by LPS (25 ng/mL) for 3 h, and then treated with CS (50 μg/cm2) together with dioscin or anisomycin (2 ng/mL) for 24 h in the lower plate, while NIH-3T3 fibroblasts were attached in the upper chamber. After that, the upper chamber was inserted back to the lower plate, and macrophages and fibroblasts were co-cultured for another 24 h. Then, the mRNA expressions of Col1a1 (I) and α-SMA (J) in NIH-3T3 fibroblast were determined by qPCR (n=3). (K, L) Percentage of CD4+CD25+FoxP3+ cells (regulatory T cells) in hilar lymph nodes was assayed by FACS analysis (n=5). *, P<0.05; **, P<0.01. Error bars indicate the mean ± SD.
Figure 9
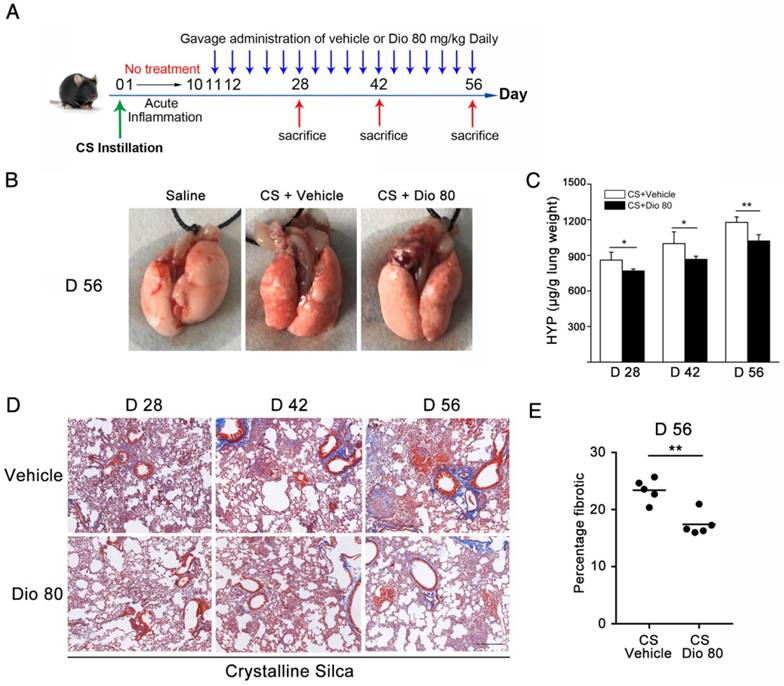
Dioscin could postpone the progression of established silicosis. (A) Intervention dosing regimen of dioscin in established silicosis. CS instillation was used to induce silicosis and no treatment was given during the first 10 d. Then, silicosis mice were intragastric gavaged with dioscin (80 mg/kg) or vehicle (n=8 per group) daily. Lungs were harvested at the indicated time points for HYP measurement and Masson trichrome staining. (B) Effects of dioscin on mice lungs. (C) HYP content in lung tissues from dioscin or vehicle-treated mice were measured at 28, 42, and 56 d (n = 4-5 per group; *, P<0.05; **, P<0.01). (D) Masson trichrome staining of collagen on lung sections taken from mice at 28, 42 and 56 d after CS instillation. Representative images are shown (n=4-5 per group). Scale bar indicates 200 μm. (E) Fibrotic score analysis of the lung sections of 56 d. The fibrotic area is presented as a percentage. **, P<0.01. Horizontal lines indicate mean.
Th1/Th2 cells play contrasting roles during the inflammatory phase of pulmonary fibrosis [31]. Th1 cells secrete IFN-γ and IL-2 to promote the clearance of CS, which may lead to lung injury, whereas IL-4- and IL-13-producing Th2 cells inhibit the Th1 immune response and have been causally associated with fibrogenesis. In our study, we observed that dioscin treatment inhibited Th1 responses and also limited production of the Th2-associated cytokines IL-4 and IL-13 throughout the development of fibrogenesis. We reasoned that dioscin treatment could attenuate the devastating Th1 immune response, leading to a relatively milder Th2 response; however, future studies are needed for confirmation. We noticed that the variation tendency of Th2 cells in FACS analysis was not the same as that of Th2 cytokines, but it was the same as GATA3 expression. Type 2 cytokines could be secreted by multiple cells, including mast cells, basophils, type 2 innate lymphoid cells (ILC2), IL-4-and/or IL-13-conditioned macrophages and T helper 2 (Th2) cells [64]. These cells may secrete IL-4 and IL-13 to restrain Th1 responses. IL-17 and Th17 have also been implicated in the pathogenesis of CS-induced pulmonary fibrosis [65]. In our study, we observed that dioscin treatment alleviated the Th17 response, which may be another way to reduce collagen deposition. IL-6 and TGF-β promote differentiation of Th17 cells, and IL-17-driven fibrosis is dependent on TGF-β. Dioscin treatment also reduced TGF-β production. These effects may account for the roles of Th17 and IL-17 in CS-induced fibrogenesis.
Figure 10
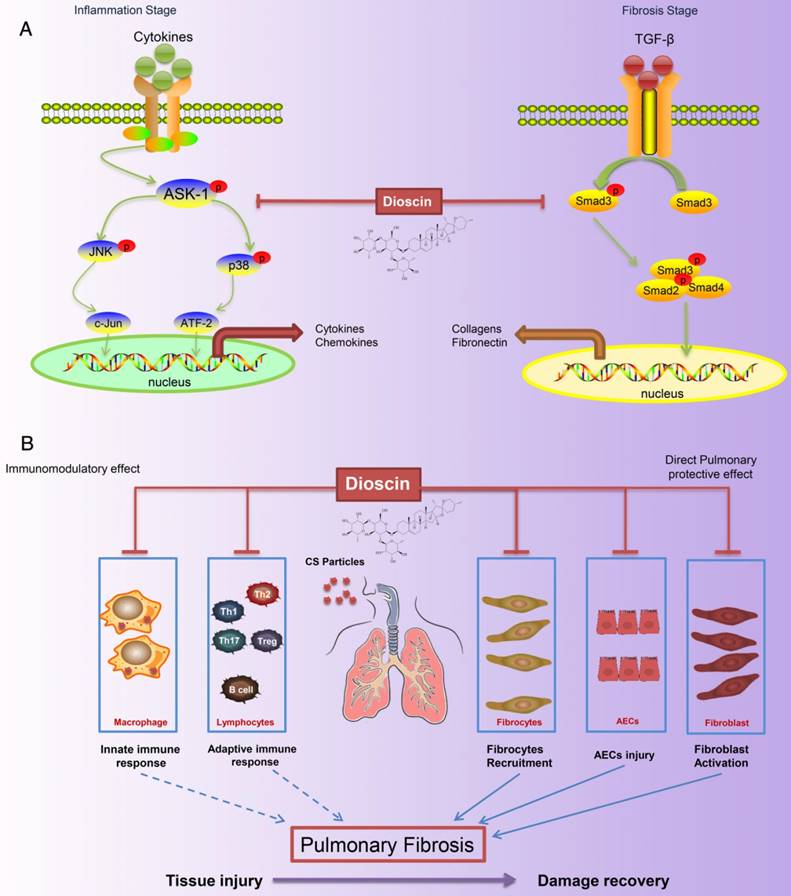
Schematic model of dioscin's anti-fibrotic effects. (A) Dioscin decreased the phosphorylation of ASK-1, leading to the attenuation of inflammation, and blocked TGF-β-Smad3 signaling, resulting in decreased collagen deposition. (B) Oral administration of dioscin alleviates CS-induced fibrosis by (i) Immunomodulatory effects: dioscin ameliorated CS-induced innate immune responses (typically macrophages), adaptive immune responses (lymphocytes) and Th immune responses (Th1/Th2/Th17, Tregs). (ii) Direct pulmonary protective effects: reducing the recruitment of fibrocytes (CD45+Col-1+), protecting alveolar epithelial cells (CD45-) from injury and decreasing fibroblast activation.
These data on macrophages, B lymphocytes, and T lymphocytes in vivo demonstrate that dioscin decreased CS-induced pulmonary inflammation via the modulation of innate and adaptive immune responses. To determine the signaling pathways by which dioscin mediated these effects, we evaluated several proteins involved in the inflammatory response, such as p38, JNK, and ASK-1, and found that dioscin inhibited their phosphorylation. In an in vitro study, we used anisomycin, an activator of p38 and JNK MAPKs, to reverse dioscin's effects. The combined results suggest that the immunomodulatory and immunoregulatory effects of dioscin were associated with inhibition of ASK-p38/JNK signaling.
Cytokines, chemokines, and growth factors produced by inflammatory cells provide the milieu for the activation, accumulation, and differentiation of myofibroblasts, which act as major effector cells to produce excessive amounts of collagen fibers and other ECM proteins [6]. Fibrocytes are a circulating bone marrow-derived population of fibroblast-like cells, which produce high levels of collagen matrix and low levels of pro-inflammatory cytokines when stimulated with pro-fibrotic cytokines. In this study, the recruitment of fibrocytes was significantly attenuated by dioscin treatment, most likely due (at least in part) to the decreased number of T lymphocytes. The differentiation of fibrocytes is dependent upon CD4+ T cells [49], and the interaction between fibrocytes and CD4+ T cells enhances their functions. In addition to directly secreting ECM, fibrocytes release other related factors through paracrine effects [66]. We believe that dioscin's effects on fibrocytes also contribute to its anti-fibrosis effects.
In addition to dioscin's effects on fibrocytes, we observed that dioscin treatment also decreased the amount of CD45-Col-1+ cells (Figure 6C, E). We first reasoned that this cell population may be AECs. Injured AECs may undergo the EMT process, losing epithelial markers and gaining mesenchymal markers [67, 68]. Although several studies have indicated that EMT is a source of fibroblasts, a recent tracing study argued against the role of EMT as a source of fibroblasts in pulmonary fibrosis [69]. EMT is identified by epithelial cells losing their markers and gaining mesenchymal markers; however, in our FACS analysis, we found a population of cells expressing both epithelial and mesenchymal markers in lungs, which is consist with data from another research study [70]. These cells may also contribute to aberrant epithelial-mesenchymal crosstalk that promotes fibrogenesis [68]. Dong et al. (2015) demonstrated that AECs from bleomycin-injured mice lungs have decreased expression of epithelial markers and increased expression of mesenchymal markers [70]. Thus, from this perspective, injured AECs could enhance a pro-fibrotic milieu and activate quiescent resident fibroblasts, rather than undergoing the EMT process that directly transitions them into fibroblasts. Yang et al. (2013) indicated that repetitive injuries to AECs subsequently provoke the activation of fibroblasts and differentiation of myofibroblasts [71]. We found that dioscin treatment caused retention of CS-induced loss of epithelial markers and decreased CS-induced gain of mesenchymal markers. We believe that dioscin may protect AECs from CS injury, resulting in declined accumulation and activation of fibroblasts, ultimately leading to reduced collagen deposition.
Considerable attention has been focused on the role of TGF-β in the pathogenesis of pulmonary fibrosis, as it is a potent stimulus of ECM secretion from fibroblasts. In addition, the pro-fibrotic activities of TGF-β are mediated by Smad3 [52]. Before, we showed that dioscin could alleviate CS-induced epithelial injury and reduced the expression of vimentin (a fibroblast marker). Further, we showed that treatment of dioscin in CS-injured mice reduced TGF-β production and inhibited downstream Smad3 signaling. These effects may lead to decreased differentiation and activation of resident fibroblasts and/or fibrocytes. To further testify dioscin's effects on TGF-β-Smad3 signaling, we did an in vitro experiment with NIH-3T3 fibroblasts. Our results showed that dioscin could block TGF-β-induced phosphorylation of Smad3. Researches about liver fibrosis showed that dioscin could block TGF-β/Smad signaling in vivo and reduce Smad 3 phosphorylation in HSC-T6 and LX2 cells, which is consistent with our results [72, 73]. We believe that dioscin could alleviate CS-induced epithelial injury while blocking fibroblast activation (Figure 10B).
TGF-β is a multi-source cytokine that can be secreted by a large array of cells including macrophages and injured epithelial cells. We proved that dioscin could reduce the accumulation of macrophages and protect epithelial cells from CS injury, both of which led to a decline in TGF-β production. Tregs, a subset of CD4+ T cells expressing Foxp3, functions as an immune regulator, and produces TGF-β [54]. Furthermore, studies on the relationship between Tregs and pulmonary fibrosis have suggested that CD4+CD25hiFoxp3+ Tregs exacerbate pulmonary fibrosis [55]. Our previous study indicated that Tregs could influence the Th response during experimental silicosis in mice, which promoted transition from the Th1 to Th2 response [74]. Here, we demonstrated that CD4+ T cells including Th1, Th2, Th17 and Tregs were all decreased by dioscin treatment, possibly due to inhibition of the inflammatory signaling pathway.
Our previous study indicated that a specific inhibitor of ASK-1 could ameliorate CS-induced pulmonary inflammation and fibrosis through blocking 4-1BB pathway, which functions as an immune accelerator [24]. We believe that mild inflammation protects lung tissue against fibrogenesis. Dioscin decreased ASK-1 phosphorylation in this study, similar to the effects of a specific inhibitor. Dioscin is a natural product that has advantages over artificial compounds such as fewer side effects. Notably, we demonstrated that dioscin could also inhibit TGF-β-Smad3 signaling, indicating that dioscin is a multifunctional product that can both alleviate inflammation and inhibit fibrogenesis. In addition, studies have also shown that dioscin has anti-virus, anti-tumor, lipid-lowering, and hepatoprotective activities. Silicosis patients have a higher risk of suffering from these diseases, especially lung cancer, as CS is a potential lung carcinogen [3]. The results of our study suggest that dioscin may postpone CS-induced fibrogenesis and may even have the potential to prevent or treat silica-induced diseases.
In summary, our study demonstrated that dioscin has protective effects against CS-induced pulmonary fibrosis. Its immunomodulatory effects on the innate and adaptive immune responses reduced tissue damage and decreased the secretion of pro-fibrotic cytokines, which are required for fibroblast differentiation and activation. In addition, dioscin decreased fibrocyte recruitment, which is a direct source of fibroblasts. These joint effects led to decreased collagen deposition. Additional studies are needed to determine if this natural product has a clinical application in CS-induced pulmonary fibrosis.
Abbreviations
ASK-1: Apoptosis signal regulating kinase 1; AECs: Alveolar epithelial cells; BALF: bronchoalveolar lavage fluid; CD: Cluster of Differentiation; ECM : Excetral Cellular Matrix; FSP-1: Fibroblast specific protein-1; HLN: hilar lymph nodes; IL: interleukine; JNK: C-Jun N-terminal kinase; MCP-1: monocyte chemoattractant protein-1; Th: T helper cell; TNF-α: Tumour necrosis factor-α.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81573117) and the Program for Liaoning Innovative Research Team in University (LT2015028).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Leung CC, Yu ITS, Chen W. Silicosis. The Lancet. 2012;379:2008-18
2. Sharma N, Kundu D, Dhaked S, Das A. Silicosis and silicotuberculosis in India. Bull World Health Organ. 2016;94:777-8
3. Steenland K, Ward E. Silica: a lung carcinogen. CA Cancer J Clin. 2014;64:63-9
4. Barmania S. Deadly denim: sandblasting-induced silicosis in the jeans industry. Lancet Respir Med. 2016;4:543
5. Sayan M, Mossman BT. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol. 2016;13:51
6. Dong J, Ma Q. Myofibroblasts and lung fibrosis induced by carbon nanotube exposure. Part Fibre Toxicol. 2016;13:60
7. Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103-21
8. Lama VN, Phan SH. The extrapulmonary origin of fibroblasts: stem/progenitor cells and beyond. Proc Am Thorac Soc. 2006;3:373-6
9. Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leukoc Biol. 2009;86:1111-8
10. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028-40
11. Schleiff PL, Mazurek JM, Reilly MJ, Rosenman KD, Yoder MB, Lumia ME. et al. Surveillance for Silicosis - Michigan and New Jersey, 2003-2011. MMWR Morb Mortal Wkly Rep. 2016;63:73-8
12. Tao X, Sun X, Yin L, Han X, Xu L, Qi Y. et al. Dioscin ameliorates cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via HMGB-1 inhibition. Free Radic Biol Med. 2015;84:103-15
13. Chang CJ, Lin CS, Lu CC, Martel J, Ko YF, Ojcius DM. et al. Ganoderma lucidum reduces obesity in mice by modulating the composition of the gut microbiota. Nat Commun. 2015;6:7489
14. Tyagi A, Raina K, Gangar S, Kaur M, Agarwal R, Agarwal C. Differential effect of grape seed extract against human non-small-cell lung cancer cells: the role of reactive oxygen species and apoptosis induction. Nutr Cancer. 2013;65(Suppl 1):44-53
15. Subramani R, Gonzalez E, Nandy SB, Arumugam A, Camacho F, Medel J. et al. Gedunin inhibits pancreatic cancer by altering sonic hedgehog signaling pathway. Oncotarget. 2017;8:10891-10904
16. Subramani R, Gonzalez E, Arumugam A, Nandy S, Gonzalez V, Medel J. et al. Nimbolide inhibits pancreatic cancer growth and metastasis through ROS-mediated apoptosis and inhibition of epithelial-to-mesenchymal transition. Sci Rep. 2016;6:19819
17. Hsieh MJ, Tsai TL, Hsieh YS, Wang CJ, Chiou HL. Dioscin-induced autophagy mitigates cell apoptosis through modulation of PI3K/Akt and ERK and JNK signaling pathways in human lung cancer cell lines. Arch Toxicol. 2013;87:1927-37
18. Zhang Y, Xu Y, Qi Y, Xu L, Song S, Yin L. et al. Protective effects of dioscin against doxorubicin-induced nephrotoxicity via adjusting FXR-mediated oxidative stress and inflammation. Toxicology. 2017;378:53-64
19. Xu T, Zhang S, Zheng L, Yin L, Xu L, Peng J. A 90-day subchronic toxicological assessment of dioscin, a natural steroid saponin, in Sprague-Dawley rats. Food Chem Toxicol. 2012;50:1279-87
20. Li H, Huang W, Wen Y, Gong G, Zhao Q, Yu G. Anti-thrombotic activity and chemical characterization of steroidal saponins from Dioscorea zingiberensis C.H. Wright. Fitoterapia. 2010;81:1147-56
21. Zhao X, Cong X, Zheng L, Xu L, Yin L, Peng J. Dioscin, a natural steroid saponin, shows remarkable protective effect against acetaminophen-induced liver damage in vitro and in vivo. Toxicol Lett. 2012;214:69-80
22. Gu L, Tao X, Xu Y, Han X, Qi Y, Xu L. et al. Dioscin alleviates BDL- and DMN-induced hepatic fibrosis via Sirt1/Nrf2-mediated inhibition of p38 MAPK pathway. Toxicol Appl Pharmacol. 2016;292:19-29
23. Xu T, Zheng L, Xu L, Yin L, Qi Y, Xu Y. et al. Protective effects of dioscin against alcohol-induced liver injury. Arch Toxicol. 2014;88:739-53
24. Li C, Du S, Lu Y, Lu X, Liu F, Chen Y. et al. Blocking the 4-1BB Pathway Ameliorates Crystalline Silica-induced Lung Inflammation and Fibrosis in Mice. Theranostics. 2016;6:2052-67
25. Mischler SE, Cauda EG, Di Giuseppe M, McWilliams LJ, St Croix C, Sun M. et al. Differential activation of RAW 264.7 macrophages by size-segregated crystalline silica. J Occup Med Toxicol. 2016;11:57
26. Sandberg WJ, Lag M, Holme JA, Friede B, Gualtieri M, Kruszewski M. et al. Comparison of non-crystalline silica nanoparticles in IL-1beta release from macrophages. Part Fibre Toxicol. 2012;9:32
27. Gozal E, Ortiz LA, Zou X, Burow ME, Lasky JA, Friedman M. Silica-induced apoptosis in murine macrophage: involvement of tumor necrosis factor-alpha and nuclear factor-kappaB activation. Am J Respir Cell Mol Biol. 2002;27:91-8
28. Han R, Ji X, Rong R, Li Y, Yao W, Yuan J. et al. MiR-449a regulates autophagy to inhibit silica-induced pulmonary fibrosis through targeting Bcl2. J Mol Med (Berl). 2016;94:1267-79
29. Ozturk Akcora B, Storm G, Prakash J, Bansal R. Tyrosine kinase inhibitor BIBF1120 ameliorates inflammation, angiogenesis and fibrosis in CCl4-induced liver fibrogenesis mouse model. Sci Rep. 2017;7:44545
30. Wang P, Nie X, Wang Y, Li Y, Ge C, Zhang L. et al. Multiwall carbon nanotubes mediate macrophage activation and promote pulmonary fibrosis through TGF-beta/Smad signaling pathway. Small. 2013;9:3799-811
31. Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210
32. Zhang W, Wang R, Zhang H, Yu Y, Wang H, Zhou X. Observation of the inflammatory factors level and the correlation between induced sputum and whole lung lavage fluid for silicosis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2015;33:201-3
33. Wick G, Grundtman C, Mayerl C, Wimpissinger TF, Feichtinger J, Zelger B. et al. The immunology of fibrosis. Annu Rev Immunol. 2013;31:107-35
34. Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777-85
35. Misson P, Brombacher F, Delos M, Lison D, Huaux F. Type 2 immune response associated with silicosis is not instrumental in the development of the disease. Am J Physiol Lung Cell Mol Physiol. 2007;292:L107-13
36. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583-94
37. Re SL, Lison D, Huaux F. CD4+ T lymphocytes in lung fibrosis: diverse subsets, diverse functions. J Leukoc Biol. 2013;93:499-510
38. Noubade R, Krementsov DN, Del Rio R, Thornton T, Nagaleekar V, Saligrama N. et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011;118:3290-300
39. Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR, Ashwell JD. Lack of the T cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood. 2011;118:3280-9
40. Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol. 2008;48:199-225
41. Katome T, Namekata K, Guo X, Semba K, Kittaka D, Kawamura K. et al. Inhibition of ASK1-p38 pathway prevents neural cell death following optic nerve injury. Cell Death Differ. 2013;20:270-80
42. Ma FY, Tesch GH, Nikolic-Paterson DJ. ASK1/p38 signaling in renal tubular epithelial cells promotes renal fibrosis in the mouse obstructed kidney. Am J Physiol Renal Physiol. 2014;307:F1263-73
43. Eaton GJ, Zhang QS, Diallo C, Matsuzawa A, Ichijo H, Steinbeck MJ. et al. Inhibition of apoptosis signal-regulating kinase 1 enhances endochondral bone formation by increasing chondrocyte survival. Cell Death Dis. 2014;5:e1522
44. Mizukami J, Sato T, Camps M, Ji H, Rueckle T, Swinnen D. et al. ASK1 promotes the contact hypersensitivity response through IL-17 production. Sci Rep. 2014;4:4714
45. Vietti G, Lison D, van den Brule S. Mechanisms of lung fibrosis induced by carbon nanotubes: towards an Adverse Outcome Pathway (AOP). Part Fibre Toxicol. 2016;13:11
46. Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164-76
47. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339-50
48. Garibaldi BT, D'Alessio FR, Mock JR, Files DC, Chau E, Eto Y. et al. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. 2013;48:35-43
49. Galligan CL, Keystone EC, Fish EN. Fibrocyte and T cell interactions promote disease pathogenesis in rheumatoid arthritis. J Autoimmun. 2016;69:38-50
50. Garcia de Alba C, Buendia-Roldan I, Salgado A, Becerril C, Ramirez R, Gonzalez Y. et al. Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am J Respir Crit Care Med. 2015;191:427-36
51. Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Gobel N. et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A. 2009;106:17892-7
52. Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47-64
53. Lakos G, Takagawa S, Chen S-J, Ferreira AM, Han G, Masuda K. et al. Targeted Disruption of TGF-β/Smad3 Signaling Modulates Skin Fibrosis in a Mouse Model of Scleroderma. Am J Pathol. 2004;165:203-17
54. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775-87
55. Birjandi SZ, Palchevskiy V, Xue YY, Nunez S, Kern R, Weigt SS. et al. CD4(+)CD25(hi)Foxp3(+) Cells Exacerbate Bleomycin-Induced Pulmonary Fibrosis. Am J Pathol. 2016;186:2008-20
56. Thakur C, Wolfarth M, Sun J, Zhang Y, Lu Y, Battelli L. et al. Oncoprotein mdig contributes to silica-induced pulmonary fibrosis by altering balance between Th17 and Treg T cells. Oncotarget. 2015;6:3722-36
57. Liu F, Dai W, Li C, Lu X, Chen Y, Weng D. et al. Role of IL-10-producing regulatory B cells in modulating T-helper cell immune responses during silica-induced lung inflammation and fibrosis. Sci Rep. 2016;6:28911
58. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245-57
59. Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA. et al. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest. 1995;96:250-9
60. Guo J, Gu N, Chen J, Shi T, Zhou Y, Rong Y. et al. Neutralization of interleukin-1 beta attenuates silica-induced lung inflammation and fibrosis in C57BL/6 mice. Arch Toxicol. 2013;87:1963-73
61. Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529-36
62. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S. et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56-65
63. Hasegawa M, Fujimoto M, Takehara K, Sato S. Pathogenesis of systemic sclerosis: altered B cell function is the key linking systemic autoimmunity and tissue fibrosis. J Dermatol Sci. 2005;39:1-7
64. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15:271-82
65. Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG. et al. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol. 2011;187:3003-14
66. García de Alba C, Buendia-Roldán I, Salgado A, Becerril C, Ramírez R, González Y. et al. Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am J Respir Crit Care Med. 2015;191:427-36
67. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949-61
68. Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med. 2012;18:517-23
69. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J. et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475-83
70. Dong Y, Geng Y, Li L, Li X, Yan X, Fang Y. et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J Exp Med. 2015;212:235-52
71. Yang J, Wheeler SE, Velikoff M, Kleaveland KR, LaFemina MJ, Frank JA. et al. Activated alveolar epithelial cells initiate fibrosis through secretion of mesenchymal proteins. Am J Pathol. 2013;183:1559-70
72. Liu M, Xu Y, Han X, Yin L, Xu L, Qi Y. et al. Dioscin alleviates alcoholic liver fibrosis by attenuating hepatic stellate cell activation via the TLR4/MyD88/NF-kappaB signaling pathway. Sci Rep. 2015;5:18038
73. Zhang X, Han X, Yin L, Xu L, Qi Y, Xu Y. et al. Potent effects of dioscin against liver fibrosis. Sci Rep. 2015;5:9713
74. Liu F, Liu J, Weng D, Chen Y, Song L, He Q. et al. CD4+ CD25+ Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS One. 2010;5:e15404
Author contact
![]() Corresponding authors: Jie Chen e-mail: Jchenedu.cn, Tel: 86 24 31939079 and Dong Weng e-mail: wengdongedu.cn
Corresponding authors: Jie Chen e-mail: Jchenedu.cn, Tel: 86 24 31939079 and Dong Weng e-mail: wengdongedu.cn
Citation styles
APA
Li, C., Lu, Y., Du, S., Li, S., Zhang, Y., Liu, F., Chen, Y., Weng, D., Chen, J. (2017). Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice. Theranostics, 7(17), 4255-4275. https://doi.org/10.7150/thno.20270.
ACS
Li, C.; Lu, Y.; Du, S.; Li, S.; Zhang, Y.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice. Theranostics 2017, 7 (17), 4255-4275. DOI: 10.7150/thno.20270.
NLM
Li C, Lu Y, Du S, Li S, Zhang Y, Liu F, Chen Y, Weng D, Chen J. Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice. Theranostics 2017; 7(17):4255-4275. doi:10.7150/thno.20270. https://www.thno.org/v07p4255.htm
CSE
Li C, Lu Y, Du S, Li S, Zhang Y, Liu F, Chen Y, Weng D, Chen J. 2017. Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice. Theranostics. 7(17):4255-4275.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.