Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(17):4229-4239. doi:10.7150/thno.21688 This issue Cite
Research Paper
Liposomal 64Cu-PET Imaging of Anti-VEGF Drug Effects on Liposomal Delivery to Colon Cancer Xenografts
Stephanie J. Blocker1, Kirk A. Douglas1, Lisa Anne Polin1, 3, Helen Lee2, Bart S. Hendriks2, Enxhi Lalo1, Wei Chen1, 3, Anthony F. Shields1, 3 ![]()
1. Department of Oncology, Wayne State University, Detroit, MI, USA;
2. Merrimack Pharmaceuticals, Cambridge, MA, USA;
3. Karmanos Cancer Institute, Detroit, MI, USA.
Received 2017-6-29; Accepted 2017-8-11; Published 2017-9-26
Abstract

Liposomes (LP) deliver drug to tumors due to enhanced permeability and retention (EPR). LP were labeled with 64Cu for positron emission tomography (PET) to image tumor localization. Bevacizumab (bev), a VEGF targeted antibody, may modify LP delivery by altering tumor EPR and this change can also be imaged.
Objective: Assess the utility of 64Cu-labeled LP for PET in measuring altered LP delivery early after treatment with bev.
Methods: HT-29 human colorectal adenocarcinoma tumors were grown subcutaneously in SCID mice. Empty LP MM-DX-929 (Merrimack Pharmaceuticals, Inc. Cambridge, MA) were labeled with 64CuCl2 chelated with 4-DEAP-ATSC. Tumor-bearing mice received ~200-300 μCi of 64Cu-MM-DX-929 and imaged with microPET. All mice were scanned before and after the treatment period, in which half of the mice received bev for one week. Scans were compared for changes in LP accumulation during this time. Initially, tissues were collected after the second PET for biodistribution measurements and histological analysis. Subsequent groups were divided for further treatment. Tumor growth following bev treatment, with or without LP-I, was assessed compared to untreated controls.
Results: PET scans of untreated mice showed increased uptake of 64Cu-MM-DX-929, with a mean change in tumor SUVmax of 43.9%±6.6% (n=10) after 7 days. Conversely, images of treated mice showed that liposome delivery did not increase, with changes in SUVmax of 7.6%±4.8% (n=12). Changes in tumor SUVmax were significantly different between both groups (p=0.0003). Histology of tumor tissues indicated that short-term bev was able to alter vessel size. Therapeutically, while bev monotherapy, LP-I monotherapy, and treatment with bev followed by LP-I all slowed HT-29 tumor growth compared to controls, combination provided no therapeutic benefit.
Conclusions: PET with tracer LP 64Cu-MM-DX-929 can detect significant differences in LP delivery to colon tumors treated with bev when compared to untreated controls. Imaging with 64Cu-MM-DX-929 is sensitive enough to measure drug-induced changes in LP localization which can have an effect on outcomes of treatment with LP.
Keywords: PET, liposomes, bevacizumab, EPR, colon cancer, irinotecan.
Introduction
Globally, colorectal cancer (CRC) is the third most common cancer in men (~746,000 cases) and the second most common in women (~614,000 cases) as of 2012 [1, 2]. The push for precision medicine has led to a greater understanding of the molecular and genetic subtypes of CRC among the population [3-6], and promoted the search for prognostic and predictive biomarkers. However, while multiple molecular markers have shown promise as prognostic indicators [7, 8], attempts to utilize them in the clinic have led to conflicting results [9-12]. Thus, tumor stage and supporting histological analysis remain the primary basis for therapeutic decision making in CRC [13, 14].
In addition to the search for prognostic markers for CRCs, research has also focused on uncovering better drug options. Standard cytotoxic agents for CRC include 5FU, often combined with irinotecan and/or oxaliplatin [14-22]. In patients with advanced disease almost all patients still develop resistance to treatment and succumb to tumor growth [23, 24]. Targeted antibodies are regularly used in treating mCRC, including agents that target vascular endothelial growth factor (VEGF) and its receptor (VEGFR) [25]. Targeting of VEGF pathways in CRC is designed to reduce tumor blood supply by disrupting tumor vessels, and has had some success in the clinic [26, 27]. One such therapy is bevacizumab (bev; Avastin™; Genentech, San Francisco, CA), a VEGF-targeted monoclonal antibody, which has been approved for CRC patients in combination with various chemotherapy regimens. Unfortunately, most therapeutic options in CRC have faced the problem of resistance in the clinic, often due to the heterogeneous nature of colon tumors [27-31].
Recently, there has been a growing interest in the development of nanoparticle-based therapies, such as liposomes (LP), for cancer as multiple preclinical studies have shown notable success in cellular and animal models [32-38]. Clinical trials utilizing LP for CRC treatment focus primarily on delivery of well-characterized drugs, including irinotecan and its metabolite, SN-38, or doxorubicin [13, 39, 40]. LP deposition in solid tumors is heavily influenced by enhanced permeability and retention (EPR), making the state of tumor blood vessels a key factor in delivery. Theranostic approaches for imaging delivery of LP could provide vital insight into the probability of success when treating with LP platforms for drug delivery [41-43]. In this study, we have utilized a 64Cu-loadable liposome formulation to image the effects of short-term bev treatment on LP delivery to colon tumor xenografts in mice. We chose to target tumor vasculature, as the state of vessels in solid tumors is critical in defining EPR, and thus macromolecular delivery [44-46]. Although the long-term effects of bev on tumor vasculature have been established, there is evidence that bev begins altering tumor vessels and affecting vascular permeability early into treatment [47]. Thus, we aimed to measure any early changes in LP localization induced by short-term bev with PET, and monitor subsequent therapy with liposomal irinotecan (LP-I; MM-398; Onivyde®; Merrimack Phamaceuticals Inc., Cambridge MA). In doing so, we generated a system to measure dynamic changes in LP deposition that could affect the efficacy of LP-based therapies on an individual basis. Furthermore, we were able to non-invasively measure significant differences in LP delivery between bev-treated tumors and control tumors early into bev treatment. Finally, the results seen with PET correlated with subsequent monitoring of treatment efficacies, suggesting that this platform could have utility in predicting and monitoring therapeutic LP success.
Materials and Methods
Materials
HT-29 cells and McCoy's 5a Modified Medium were purchased from ATCC (Manassas, VA) and kept below 15 passages following receipt. 4-DEAP-ATSC chelator, empty MM-DX-929 liposomes, and LP-I were provided by Merrimack Pharmaceuticals, Inc. (Cambridge, MA). 64CuCl2 was purchased from the Department of Radiology at Washington University (St. Louis, MO). Chelation efficiency was measured with iTLC-SG plates (Agilent Technologies, Santa Clara, CA). Loading efficiency was assessed with Sephadex G-50 DNA Grade Illustra Nick columns (GE Healthcare, Pittsburg, PA).
Gamma spectroscopy was performed on a Packard Cobra II gamma counter (Perkin-Elmer Inc., Waltham, MA). PET scans were acquired on an R4 microPET (Concorde Microsystems, Knoxville, TN). CT images were acquired using an Inveon microSPECT/CT (Siemens Preclinical Imaging Solutions, Malvern, PA). Images were registered and analyzed using PMOD Image Matching and Fusion Tool ver3.6 (PMOD group, Switzerland). Statistical analyses were performed using GraphPad Prism, v7 (GraphPad Software Inc., La Jolla, CA).
Labeling MM-DX-929 with chelated 64Cu
Upon receipt of 64CuCl2, 64Cu was chelated with 4-DEAP-ATSC (98±2% chelation efficiency), followed by loading into empty liposome (95±3% loading efficiency). Briefly, 64CuCl2 was vortexed with 4-DEAP-ATSC solution (0.06 mg/mL chelator in 0.1 M citrate buffer, pH 6) at room temperature for 10 s, then allowed to sit for one minute and vortexed again. Efficiency of 64Cu chelation was determined by diluting a sample in citrate buffer for instant thin layer chromatography as described previously [48]. Briefly, radioactivity of the solvent front (free 64Cu in solution) and at the sample origin (64Cu-DEAP-ATSC complex) was measured by gamma spectroscopy of the iTLC plates. Greater than 90% chelation efficiency was required to proceed to loading.
64Cu-MM-DX-929 was prepared by mixing 64Cu-DEAP-ATSC with empty MM-DX-929 liposomes (15mM phospholipid in 10 mM HEPES buffered saline, 150 mM sodium chloride, pH 6.5) and heated for 10 min at 65°C, followed by immediate cooling in an ice water bath for 1 min, as previously described [48]. Loading efficiency of 64Cu was assessed by performing size exclusion chromatography on an Illustra NICK column with a small sample of prepared 64Cu-MM-DX-929 in HEPES buffered saline (HBS). Radioactivity of the eluent containing labeled 64Cu-MM-DX-929 in HBS, and the column containing 64Cu-DEAP-ATSC was measured with gamma scintigraphy. Greater than 90% labeling efficiency was achieved before proceeding with animal imaging.
Cell Culture
HT-29 human colorectal adenocarcinoma cells were cultured in McCoy's 5a Modified Medium supplemented with 5% fetal bovine serum and 1% penicillin/streptomycin, as described by ATCC. Cells were kept at 37°C with 5% CO2 and were passaged with trypsin at approximately 80% confluence. Prior to inoculation in mice, cells were not passaged more than ten times in culture. Cell line identity was authenticated at time of all studies with the PowerPlex® 16 System from Promega (Madison, WI) in the Applied Genomics Technology Center at Wayne State University. Analyses were performed using ATCC and DSMZ reported karyotypes.
Animal Studies
Tumor model
Cultured HT-29 human colorectal adenocarcinoma cells were used to establish a subcutaneous tumor model in female SCID NCr mice (Charles River Labs; MA) and thereafter maintained in serial passage.
MicroPET studies
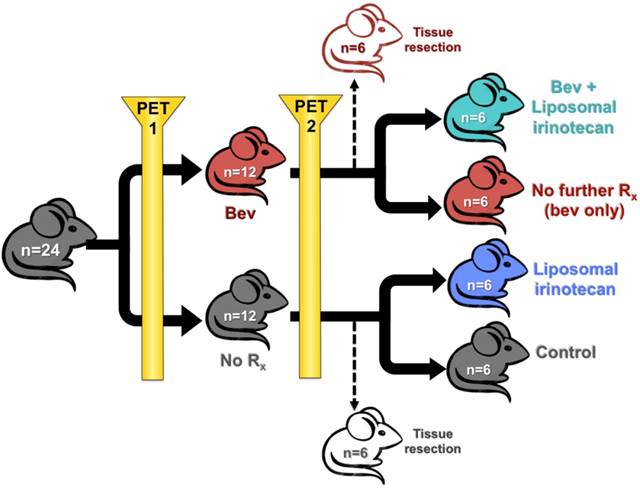
Schematic representation of mouse study design is presented in Figure 1. Tumor fragments were subcutaneously implanted into SCID NCr female mice on day 0 by trochar. Tumors were upstaged to 250 mg (range: 200-300 mg, day 12), and mice were non-selectively randomized into their respective control (No Rx) and treatment groups (bev). All mice were imaged with microPET before and after bev treatment (on days 13 and 20) 24 h after IV administration of 64CuCl2 MM-DX-929. Scans were compared for changes in LP accumulation during this time period. Mice were euthanized under anesthesia with whole blood and tissues collected after the second PET for biodistribution measurements and histological analysis. For subsequent studies, after the 2nd scan, mice were further divided into 4 groups of n=6 (No Rx, Bev, LP-I, and Bev + LP-I) to assess tumor progression post bev treatment, with or without LP-I, compared to untreated controls. All mice were weighed and observed daily for the duration of the study. Tumors were measured by caliper 2-3x/week with the formula [volume (mg) = length (mm) x width2 (mm2)/2] used to calculate tumor mass.
Animal study design. Schematic representation of treatment groups and timeline for mice treated with bev, LP-I, bev followed by LP-I, and controls.
Tracer preparation and injection
Empty LP MM-DX-929 (Merrimack Pharmaceuticals, Cambridge, MA) were labeled with 64CuCl2 chelated with 4-DEAP-ATSC). Mice were administered 200-300 μCi/injection intravenously (IV) within a 0.1-0.3 mL volume range; 22-24 h prior to each microPET scan.
Drug preparation
Bevacizumab was prepped fresh for each injection from 25 mg/mL stock diluted with 0.9% sterile saline, pH 6.0 and injected intraperitoneally (IP) at 5 mg/kg in a volume of 0.2 mL/20g mouse on days 14 and 17.
Liposomal Irinotecan (LP-I; MM-398; Onivyde®, Merrimack, Cambridge, MA) was prepped fresh for each injection from 5.05 mg/mL stock diluted with 0.9% sterile saline, pH 6.0 and injected IV at 10 mg/kg in a volume of 0.2 mL/20g mouse on days 21, 24, and 28.
All animal studies were approved by and performed in strict accordance with the policies of the Institutional Animal Care and Use Committee (IACUC) at Wayne State University.
Animal Imaging with 64Cu-MM-DX-929 PET
64Cu-MM-DX-929 (104 nm) was used to approximate the systemic distribution of LP-I (110 nm), as it has been shown to predict the accumulation of LP-I in solid tumors (Helen Lee, Personal communication). Following 64Cu-liposome preparation, mice received approximately 200-300 µCi of 64Cu-MM-DX-929 (20 μmol/kg lipid) intravenously via the tail vein. 64Cu-MM-DX-929 was imaged with PET 24±2 h post-injection, as liposomes remain in the blood pool for extended periods before depositing in tissues. Anesthesia was induced with 3% inhaled isoflurane, and maintained during scanning with 2% isoflurane. Mice were positioned prone on the scanner bed with heating pad to maintain body temperature. Fiducials labeled with 64Cu were fixed to the bed for subsequent alignment of PET and CT images. PET acquisition was performed for 10 min, followed by CT scanning for 10 min to obtain anatomical images.
Attenuation correction was performed on the whole body microPET images based on previously recorded transmission scans. Images were reconstructed by applying an iterative ordered-subsets expectation maximization 2-dimensional algorithm [49]. Together with scatter correction, these parameters yielded an isotropic spatial resolution of approximately 2 mm in full width at half maximum [50]. Prior to study, a phantom for 64Cu was scanned to calculate conversion from counts/pixel/min to kBq(μCi)/cm3.
PET/CT image registration and analysis
PET and CT images were registered and aligned using the PMOD Image Matching and Fusion Tool ver3.6 (PMOD group, Switzerland). Regions of interest (ROIs) were defined manually on individual planes of the PET, using the aligned CT images for anatomical reference. 3-dimensional volumes of interest (VOIs) were generated from the stacked ROIs of the tissue of interest. Activity in the VOIs, as detected by PET in kBq(μCi)/cm3, was converted to standardized uptake values based on injected dose and body weight. SUVmax values were calculated by averaging the max pixel value in the ROI of the three hottest consecutive planes in a tissue, and normalizing to injected dose and body weight.
64Cu-MM-DX-929 PET imaging of short-term bev effects
A baseline 64Cu-MM-DX-929 PET was performed on all mice at day 14 post-tumor implant, followed by half of the mice receiving 2 injections of bev over 7 days. Bev was administered IP at 5 mg/kg in a single injection performed on days 14 and 17 (two total injections). All mice received a second 64Cu-MM-DX-929 PET on day 20. Day 20 scans (post-treatment) were compared to scans from day 13 (baseline) and analyzed for changes in 64Cu-MM-DX-929 delivery to tumor. Results were compared between bev-treated and control mice.
Whole body tissue distribution of 64Cu-MM-DX-929
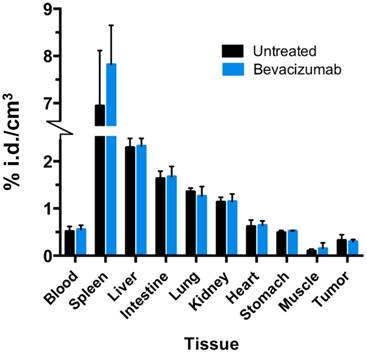
64Cu-MM-DX-929 retention in bulk tissues was assessed by gamma spectroscopy of resected tissues. Briefly, following the second PET scan (day 7), mice were sacrificed and tissues harvested (n=8). These included tumor, liver, heart, lung, intestine, stomach, kidney, spleen, and blood. Tissues were washed, weighed, and activity was measured for 1 min on a gamma counter. Activity in tissues was corrected for decay and normalized to tissue weight (kBq/cm3). Tissue biodistribution was compared between bev-treated and untreated mice to ensure that bev treatment was not affecting retention of 64Cu-MM-DX-929 in healthy tissues.
Immunohistochemistry and microvessel density analysis
Tumors resected after the second PET were fixed in formalin and paraffin embedded. Immunohistochemistry for CD34, and staining with hematoxylin (Sigma Aldrich, St. Louis, MO) was performed on 5 µm slices, and digital images of the entire cross section were captured. Sample identities were blinded, and images were analyzed with Pannoramic Viewer v1.15.4 (3DHISTECH Ltd., Budapest, Hungary). For each tissue, five distinct areas of 200 mm2 were utilized in assessing microvessel density. Briefly, tumor blood vessels (as identified by CD34 staining) were counted in each section, and distance measurements across the widest diameter of each vessel were used to determine vessel size. The average number of vessels per cm3 and the average vessel diameter were calculated.
Statistical Analysis
Tumor growth curves with mean ± standard error were plotted and growth rates were tested with linear mixed model. Tumor latency to 1 g total burden was tested with Kruskal-Wallis test, after normality assumption was evaluated with Shapiro-Wilk test. Statistical analyses were performed using R v3.2 (The R Foundation for Statistical Computing). All other statistical analyses were performed using GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA). Data were presented as the mean ± standard error. Comparisons between the bevacizumab and control were performed using two-sample Student's t-test. A p-value of <0.05 was considered statistically significant.
Results
64Cu-MM-DX-929 PET can detect significant differences in LP delivery between colon tumors treated with bev and untreated controls
Liposome distribution in mice bearing subcutaneous HT-29 colon tumor xenografts was measured with 64Cu-MM-DX-929 PET at baseline prior to any treatment. Due to the extended circulation times of liposomes in the body, images were acquired 24±2 h following tracer injection (approximately two half-lives of 64Cu, t1/2 = 12.7 h) to allow extravasation from the blood pool [48]. Tracer uptake was notable in liver (due to extensive vasculature) and spleen, and was still visible in the heart (residual blood pool). Tumors were easily detectable with 64Cu-MM-DX-929 PET, with relatively ubiquitous tracer distribution at baseline.
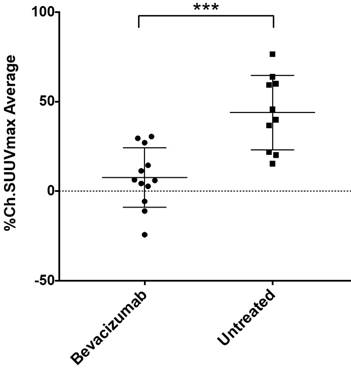
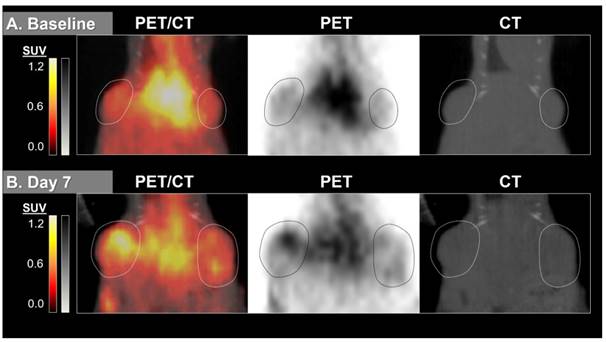
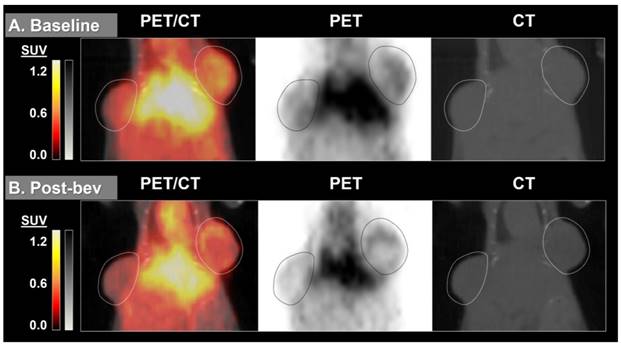
By measuring changes in tumor SUVmax between baseline and post-treatment scans (%ΔSUVmax) we found the difference in %ΔSUVmax of bev-treated tumors compared with the controls to be statistically significant (Figure 2). This trend was seen when comparing mice (2 tumors/mouse), but was also true when comparing individual tumors (Figure S1). Scans from two control mice were determined to be un-evaluable due to technical issues with one or both PET images for those mice. Changes in 64Cu-MM-DX-929 PET from baseline to post-treatment were noticeably different between tumors treated with bev and untreated controls. Tumors in control mice showed relative increases in 64Cu-MM-DX-929 retention after 7 days (Figure 3). Although these tumors often continued to grow between baseline and subsequent scans, increases in 64Cu-MM-DX-929 deposition was independent of individual tumor size or growth rate (data not shown). In mice treated with bev, however, 64Cu-MM-DX-929 delivery to tumor tissues appeared to remain stable between baseline and post-treatment scans (Figure 4). Again, these trends were independent of tumor size or growth rate (data not shown).
Changes in SUVmax of HT-29 colon tumors decreased after treatment with bev compared to untreated tumors. %ΔSUVmax of liposome accumulation in tumor tissues of mice that received no treatment (n=10), compared to mice treated with two doses of bev (n=12) as measured by 64Cu-MM-DX-929 PET. %ΔSUVmax values represent the average %ΔSUVmax of both tumors within an individual mouse. (***p=0.0002)
64Cu-MM-DX-929 delivery to HT-29 colon tumor xenografts increases after 7 days without therapeutic intervention. 64Cu-MM-DX-929 scans of a mouse bearing two subcutaneous HT-29 colon xenografts (outlined) at baseline (A) and after 7 days with no treatment (B). Images are coronal slices of the mouse midsection with fused PET/CT, PET alone, and CT alone. PET images were scaled from ½ background (kBq/cm3) to liver average (kBq/cm3) calculated based on average values from both scans.
64Cu-MM-DX-929 delivery to HT-29 colon tumor xenografts does not increase when treated with two doses of bevacizumab. 64Cu-MM-DX-929 scans of a mouse bearing two subcutaneous HT-29 colon xenografts (outlined) at baseline (A) and after 7 days of bev treatment (B). Images are coronal slices of the mouse midsection with fused PET/CT, PET alone, and CT alone. PET images were scaled from ½ background (kBq/cm3) to liver average (kBq/cm3) calculated based on average values from both scans.
Interestingly, 64Cu-MM-DX-929 PET scans visualized more frequent and dramatic shifts in the volumetric distribution of tracer across the mass of tumors treated with bev. This suggests that early into treatment, prior to measurable morphological differences, 64Cu-MM-DX-929 PET identified altered tumor vascularity in bev-treated tumors, as well as early effects of bev on LP distribution. 64Cu-MM-DX-929 PET images suggest that the early effects of bev may substantially alter or limit LP penetration into tumor tissues. Additionally, changes in tracer deposition in individual tumors were more highly variable in bev-treated tumors, while control tumors often exhibited similar increases in uptake over time (Figure S2). Taken together, 64Cu-MM-DX-929 PET was able to detect increased LP accumulation/delivery in colon tumor xenografts tended to increase as tumors progressed without intervention, but this trend was reduced or abolished with only two doses of bev. Thus, with 64Cu-MM-DX-929 PET we were able to measure the effects of bev therapy on LP delivery to solid tumors early into treatment.
Bevacizumab does not alter systemic distribution of 64Cu-MM-DX-929 in non-tumor tissues
Any treatment with the potential to alter systemic distribution of a PET tracer could confound image analysis and uptake quantitation. To verify that bev did not significantly alter 64Cu-MM-DX-929 global uptake in tissues, activity in resected normal tissues was measure by gamma spectroscopy and compared between treated and untreated mice. No significant differences were detected between normal tissues of bev-treated mice and control mice (Figure 5). This demonstrates that systemic distribution was not altered in a way which would be confounding for image analysis in tissues of interest.
Bev treatment did not change overall biodistribution of 64Cu-MM-DX-929. %injected radioactive dose per g of resected tissues was measured directly after the second 64Cu-MM-DX-929 PET scan.
Colon tumor growth was delayed by short-term bev alone, liposomal irinotecan alone, or short-term bev followed by liposomal irinotecan
Following the second 64Cu-MM-DX-929 PET, bev-treated mice and untreated mice were further randomized into the following subsets: (1) untreated controls (n=6); (2) short-term bev (2q7d) only (n=6); (3) LP-I only (n=6); (4) short-term bev followed by LP-I (n=6) (Figure 1). One mouse assigned to receive LP-I only (group 3) was not included in data assessments due to lack of drug availability at the time of study. Mice were treated and tumor progression was monitored until tumor burden or weight loss warranted euthanasia. Tumor growth was considered individually, as well as by per-mouse analysis of total tumor burden.
As expected, HT-29 tumors in mice that received no treatment exhibited unrestrained growth (Figure 6A). Treatment with two doses of bev resulted in a measurable but modest delay in tumor progression compared to controls. Interestingly, tumors in mice treated with LP-I also exhibited delayed growth compared to controls, despite being administered later than bev, at advanced stage of disease. Succeeding short-term bev with LP-I demonstrated tumor inhibition compared to untreated controls, although there was no notable therapeutic advantage to this combination compared to bev or liposomal irinotecan alone with the specific doses and regimens tested.
HT-29 tumor growth is affected by bev, LP-I, and bev followed by LP-I compared to untreated controls. Tumor growth inhibition assessed by caliper measurements represented for each treatment group compared to control, represented as a mixed linear model (****p<0.0001) (A). Tumor latency to approximately 1 g total tumor burden was assessed for individual mice as a measure of growth delay due to treatment (B). Spaghetti plots of tumor growth in individual mice according to treatment with bev (C.b), LP-I (C.c), or bev followed by LP-I (C.d) compared to control tumors (C.a). Bev administration is indicated by red lines, LP-I administration is indicated by blue lines.
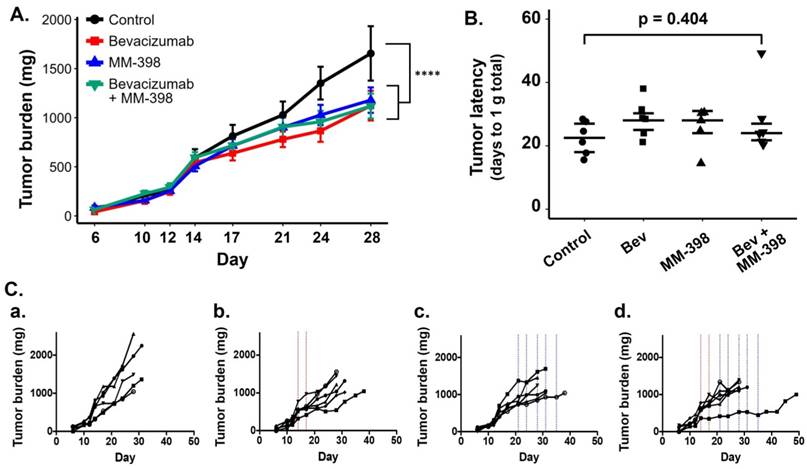
As was seen in the PET scans with bev, individual tumor and mouse responses varied in each of the treatment groups. Spaghetti plots of tumor burden in individual mice show that while untreated tumors progressed similarly quickly, each treatment regimen yielded variable rates of response in individuals (Figure 6Ca-d). Tumor growth rates and drug-induced growth inhibition were independent of tumor size at treatment initiation (data not shown). When measuring tumor latency to 1 g total burden per mouse, each treatment group shows increased latency compared to untreated controls, though the differences in group medians were not statistically significant (Figure 6B).
Bev treatment induced measurable changes in tumor blood vessels after two injections
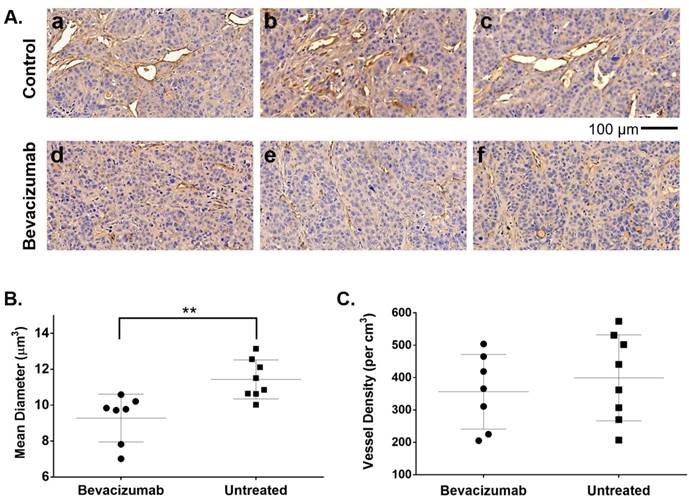
Although treated mice received only two injections of bev, HT-29 tumors resected after the second 64Cu-MM-DX-929/PET showed early evidence of bev action. Microvessel density (MVD) was assessed via immunohistochemical staining for CD34, followed by blinded analysis of tissues for vessel number and average diameter. CD34 staining revealed notable differences in vessel size between bev-treated and control tumors (Figure 7A). Short-term bev resulted in significantly smaller vessel diameters compared to untreated controls (Figure 7B). The total tissue area occupied by CD34+ vessels in treated tumors was 3.8% ± 1.5% compared to 5.7% ± 1.7% in control tumors (p=0.04, Figure S3). This indicates that 64Cu-MM-DX-929 in the blood pool has a very small contribution to the tumor tracer activity. While the size of the vessels was noticeably altered following bev, the vessel density (vessels/cm3) showed no measurable difference between bev-treated and untreated tumors (Figure 7C). These data would indicate that two injections of bev had begun eliciting an anti-vascular effect, and that the second 64Cu-MM-DX-929 PET was performed during the early stages of bev response. Taken together with trends seen in PET, these data show that 64Cu-MM-DX-929 PET was able to measure changes in LP delivery, which were likely due to the early effects of bev. Importantly, while bev-induced changes in vessel diameter were measurable at the time of the second PET, no difference was seen in tumor growth rates between treated and control mice (Figure 6A). Thus, 64Cu-MM-DX-929 PET was able to measure early fluctuations in LP delivery due to anti-vascular therapy, prior to any quantifiable changes to tumor morphology.
Bev induced significant changes in blood vessel diameter early into treatment. 20x images of HT-29 tumor tissues stained with CD34 to identify blood vessels (brown) and hematoxylin to denote cell nuclei (blue) show significant differences in vessel size between untreated controls (A. a-c) and tumors treated with bevacizumab (A. d-f). Microvessel density analysis of blood vessel diameter in bev-treated tumors compared to untreated controls (B). Vessel density (vessels per cm3) was compared between treated and untreated tumors (C). Data are presented as mean ± SD. **p=0.0042
Discussion
The urgent need for precision medicine for CRC is not limited to the development of more sophisticated therapies, but also techniques to predict and monitor therapeutic efficacy. Here we have demonstrated the utility of a dynamic system using 64Cu-labeled liposomes for PET to non-invasively measure the early effects of bev therapy on LP delivery to colon tumor xenografts in mice. Furthermore, significant differences measured with 64Cu-MM-DX-929 PET between bev-treated and control tumors provided early insight into therapeutic outcomes in mice subsequently treated with liposomal irinotecan.
Clinically, the potential advantages of LP are twofold: (1) sustained and/or local delivery of drugs or drug combinations to tumor tissues, and (2) reduced toxicity profiles as normal tissues are shielded from toxic drugs [51-55]. However, heterogeneous, inconsistent, or obstructed delivery of these nanoparticles to tumor tissues can hamper their effectiveness, and is hypothesized to be a contributor to the lack of clinical success seen with many LP. Passive targeting through EPR relies on specific properties of tumor blood vessels, which are constantly changing in response to the tumor environment and therapeutic intervention, particularly with antivascular agents. While antivascular agents are expected to disrupt tumor vessels, there is some evidence suggesting that early effects of bev treatment may transiently “normalize” tumor vessels, though these effects are not consistent [47, 56-58]. Thus, a non-invasive means of measuring LP delivery to tumor tissues could provide individualized information on the effect of drugs like bev on LP delivery and subsequent efficacy [48, 59-61].
In these studies, we found that we could use PET to quantify changes in LP accumulation in colon tumor xenografts very early into bev treatment. In mice that received no treatment in between PET scans, 64Cu-MM-DX-929 accumulation in tumors increased, indicating that LP-I were still able to reach and penetrate HT-29 tumor tissues and elicit an effect. While previous studies of LP-I in HT-29 tumors demonstrated significant anti-tumor effects when administered earlier into tumor progression [62, 63], the modest effects seen here were likely attributed to treatment initiation at late-stage disease. Because of this, we anticipated no significant difference in survival among the treatment groups. After two injections over the course of one week, bev had already begun eliciting anti-vascular effects, which were quantifiable with 64Cu-MM-DX-929 PET. While bev treatment conferred therapeutic advantage in HT-29 tumors, 64Cu-MM-DX-929 PET showed that even short-term bev treatment began to impede liposome delivery and penetration. This observation is consistent with the lack of therapeutic benefit seen in treating mice with LP-I that had already received bev.
In the clinic, bev and other antivascular agents, such as ziv-aflibercept (Zaltrap®; Regenron Pharmaceuticals Inc., Tarrytown, NY), are approved to treat patients with CRC. As liposome-based therapies are introduced for this population, understanding the effects of antivascular agents on LP delivery could reduce the probability of employing incompatible drug combinations. Furthermore, when designing clinical trials of LP-drug platforms for colon cancer, imaging techniques could be used to non-invasively monitor changes in LP delivery over time, or as a result of various therapies.
Along with the clinical implications of bev and LP-therapy in CRC patients, we have been able to employ a powerful model for dynamically assessing modulation of LP delivery. 64Cu-MM-DX-929 PET was able to non-invasively quantify the effects of bev on LP delivery, which likely affected subsequent therapy with liposomal irinotecan injection. This would suggest that 64Cu-MM-DX-929 PET may be sensitive enough to detect and monitor changes in LP delivery to solid tumors, which may directly influence therapeutic LP efficacy. Aspects of the tumor environment that affect LP distribution are dynamic, and are certain to vary among patient populations. Thus, predicting and monitoring LP delivery with non-invasive theranostic imaging is an invaluable tool in achieving precision medicine with LP for CRC patients.
Finally, the mission of individualized treatment plans for patients with cancer is one that requires a significant preclinical effort to identify diagnostic and therapeutic strategies. In this study, we have demonstrated a practical system for measuring therapeutic modulation of LP delivery that predicted and described subsequent therapeutic results. 64Cu-MM-DX-929 PET may be used in preclinical studies of therapeutic LP to efficiently measure the effect of combination therapies, treatment timelines/conditions, etc. on LP delivery. Utilizing imaging protocols with tracer LP like 64Cu-MM-DX-929 can quickly and non-invasively identify treatment conditions that improve or hinder LP delivery. In CRC, this could mean creating more avenues towards precision medicine with liposomes to improve outcomes for patients.
Abbreviations
ATCC: American Type Culture Collection; bev: bevacizumab; CRC: colorectal cancer; CT: computerized tomography; DSMZ: Deutsche Sammlung von Mikroorganismen und Zellkulturen; EPR: enhanced permeability and retention; HEPES: hydroxyethyl piperazineethanesulfonic acid; IP: intraperitoneal; iTLC-SG: instant thin-layer chromatography silica gel; iTLC: instant thin-layer chromatography; IV: intravenous; LP-I: liposomal irinotecan; LP: liposome; mCRC: metastatic colorectal cancer; MVD: microvessel density; PET: positron emission tomography; ROI: region of interest; SCID: severe combined immune deficiency; SPECT: single photon emission computed tomography; SUV: standardized uptake value; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; VOI: volume of interest; %i.d./cm3: percent injected dose per gram; 2q7d: twice weekly; 5FU: fluorouracil
Supplementary Material
Supplementary figures.
Acknowledgements
We would like to thank the Animal Model and Therapeutics Evaluation Core, the Biostatistics Core, and the Microscopy, Imaging and Cytometry Core at Wayne State University for their input and technical support. We thank Xin Lu for his assistance during imaging studies. We also thank the Biobanking and Correlative Sciences Core at Wayne State for performing cell line identification. This work is supported by the National Cancer Institute T32-CA009531 and 5P30-CA22453 grants, the Colon Cancer Alliance Chris4Life Research Fund, and the Thomas C. Rumble Fellowship.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
2. Ferlay J SI, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer. 2013 Available from: http://globocan.iarc.fr, accessed on 05/08/2017
3. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113-30
4. Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L. et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453
5. Sinicrope FA, Okamoto K, Kasi PM, Kawakami H. Molecular Biomarkers in the Personalized Treatment of Colorectal Cancer. Clin Gastroenterol Hepatol. 2016;14:651-8
6. De Rosa M, Rega D, Costabile V, Duraturo F, Niglio A, Izzo P. et al. The biological complexity of colorectal cancer: insights into biomarkers for early detection and personalized care. Therap Adv Gastroenterol. 2016;9:861-86
7. De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594-603
8. Douillard JY, Rong A, Sidhu R. RAS mutations in colorectal cancer. N Engl J Med. 2013;369:2159-60
9. Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J. et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261-70
10. Mouradov D, Domingo E, Gibbs P, Jorissen RN, Li S, Soo PY. et al. Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am J Gastroenterol. 2013;108:1785-93
11. Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D. et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466-74
12. Cushman-Vokoun AM, Stover DG, Zhao Z, Koehler EA, Berlin JD, Vnencak-Jones CL. Clinical utility of KRAS and BRAF mutations in a cohort of patients with colorectal neoplasms submitted for microsatellite instability testing. Clin Colorectal Cancer. 2013;12:168-78
13. Kotelevets L, Chastre E, Desmaele D, Couvreur P. Nanotechnologies for the treatment of colon cancer: From old drugs to new hope. Int J Pharm. 2016;514:24-40
14. Lee JK, Chan AT. Molecular Prognostic and Predictive Markers in Colorectal Cancer: Current Status. Curr Colorectal Cancer Rep. 2011;7:136-44
15. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
16. Poon MA, O'Connell MJ, Moertel CG, Wieand HS, Cullinan SA, Everson LK. et al. Biochemical modulation of fluorouracil: evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J Clin Oncol. 1989;7:1407-18
17. Brandi G, De Lorenzo S, Nannini M, Curti S, Ottone M, Dall'Olio FG. et al. Adjuvant chemotherapy for resected colorectal cancer metastases: Literature review and meta-analysis. World J Gastroenterol. 2016;22:519-33
18. Andre T, Louvet C, Maindrault-Goebel F, Couteau C, Mabro M, Lotz JP. et al. CPT-11 (irinotecan) addition to bimonthly, high-dose leucovorin and bolus and continuous-infusion 5-fluorouracil (FOLFIRI) for pretreated metastatic colorectal cancer. GERCOR. Eur J Cancer. 1999;35:1343-7
19. Ychou M, Rivoire M, Thezenas S, Quenet F, Delpero JR, Rebischung C. et al. A randomized phase II trial of three intensified chemotherapy regimens in first-line treatment of colorectal cancer patients with initially unresectable or not optimally resectable liver metastases. The METHEP trial. Ann Surg Oncol. 2013;20:4289-97
20. Souglakos J, Androulakis N, Syrigos K, Polyzos A, Ziras N, Athanasiadis A. et al. FOLFOXIRI (folinic acid, 5-fluorouracil, oxaliplatin and irinotecan) vs FOLFIRI (folinic acid, 5-fluorouracil and irinotecan) as first-line treatment in metastatic colorectal cancer (MCC): a multicentre randomised phase III trial from the Hellenic Oncology Research Group (HORG). Br J Cancer. 2006;94:798-805
21. Twelves C. Can capecitabine replace 5-FU/leucovorin in combination with oxaliplatin for the treatment of advanced colorectal cancer? Oncology (Williston Park). 2002;16:23-6
22. Akiyama Y, Fujita K, Ishida H, Sunakawa Y, Yamashita K, Kawara K. et al. Association of ABCC2 genotype with efficacy of first-line FOLFIRI in Japanese patients with advanced colorectal cancer. Drug Metab Pharmacokinet. 2012;27:325-35
23. Giacchetti S, Perpoint B, Zidani R, Le Bail N, Faggiuolo R, Focan C. et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2000;18:136-47
24. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330-8
25. Seow HF, Yip WK, Fifis T. Advances in targeted and immunobased therapies for colorectal cancer in the genomic era. Onco Targets Ther. 2016;9:1899-920
26. Prenen H, Vecchione L, Van Cutsem E. Role of targeted agents in metastatic colorectal cancer. Target Oncol. 2013;8:83-96
27. Nandikolla AG, Rajdev L. Targeting angiogenesis in gastrointestinal tumors: current challenges. Transl Gastroenterol Hepatol. 2016;1:67
28. Axelrod DE, Vedula S, Obaniyi J. Effective chemotherapy of heterogeneous and drug-resistant early colon cancers by intermittent dose schedules: a computer simulation study. Cancer Chemother Pharmacol. 2017;79:889-98
29. Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR. et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219-26
30. Leguisamo NM, Gloria HC, Kalil AN, Martins TV, Azambuja DB, Meira LB. et al. Base excision repair imbalance in colorectal cancer has prognostic value and modulates response to chemotherapy. Oncotarget. 2017;8:54199-214
31. Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA. Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer. 2010;126:2457-68
32. Maksimenko A, Alami M, Zouhiri F, Brion JD, Pruvost A, Mougin J. et al. Therapeutic modalities of squalenoyl nanocomposites in colon cancer: an ongoing search for improved efficacy. ACS Nano. 2014;8:2018-32
33. Anitha A, Sreeranganathan M, Chennazhi KP, Lakshmanan VK, Jayakumar R. In vitro combinatorial anticancer effects of 5-fluorouracil and curcumin loaded N,O-carboxymethyl chitosan nanoparticles toward colon cancer and in vivo pharmacokinetic studies. Eur J Pharm Biopharm. 2014;88:238-51
34. Wang K, Liu L, Zhang T, Zhu YL, Qiu F, Wu XG. et al. Oxaliplatin-incorporated micelles eliminate both cancer stem-like and bulk cell populations in colorectal cancer. Int J Nanomedicine. 2011;6:3207-18
35. Judge AD, Robbins M, Tavakoli I, Levi J, Hu L, Fronda A. et al. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J Clin Invest. 2009;119:661-73
36. Jain A, Jain SK. In vitro and cell uptake studies for targeting of ligand anchored nanoparticles for colon tumors. Eur J Pharm Sci. 2008;35:404-16
37. Jain KK. Advances in the field of nanooncology. BMC Med. 2010;8:83
38. Urbanska AM, Karagiannis ED, Guajardo G, Langer RS, Anderson DG. Therapeutic effect of orally administered microencapsulated oxaliplatin for colorectal cancer. Biomaterials. 2012;33:4752-61
39. Chibaudel B, Maindrault-Goebel F, Bachet JB, Louvet C, Khalil A, Dupuis O. et al. PEPCOL: a GERCOR randomized phase II study of nanoliposomal irinotecan PEP02 (MM-398) or irinotecan with leucovorin/5-fluorouracil as second-line therapy in metastatic colorectal cancer. Cancer Med. 2016;5:676-83
40. Batist G, Gelmon KA, Chi KN, Miller WH Jr, Chia SK, Mayer LD. et al. Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clin Cancer Res. 2009;15:692-700
41. Vizirianakis IS. Nanomedicine and personalized medicine toward the application of pharmacotyping in clinical practice to improve drug-delivery outcomes. Nanomedicine. 2011;7:11-7
42. Frank D, Tyagi C, Tomar L, Choonara YE, du Toit LC, Kumar P. et al. Overview of the role of nanotechnological innovations in the detection and treatment of solid tumors. Int J Nanomedicine. 2014;9:589-613
43. Miller MA, Gadde S, Pfirschke C, Engblom C, Sprachman MM, Kohler RH. et al. Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci Transl Med. 2015;7:314ra183
44. Yamamoto S, Kato A, Sakurai Y, Hada T, Harashima H. Modality of tumor endothelial VEGFR2 silencing-mediated improvement in intratumoral distribution of lipid nanoparticles. J Control Release. 2017;251:1-10
45. Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172:782-94
46. Daldrup-Link HE, Okuhata Y, Wolfe A, Srivastav S, Oie S, Ferrara N. et al. Decrease in tumor apparent permeability-surface area product to a MRI macromolecular contrast medium following angiogenesis inhibition with correlations to cytotoxic drug accumulation. Microcirculation. 2004;11:387-96
47. Lassau N, Coiffier B, Kind M, Vilgrain V, Lacroix J, Cuinet M. et al. Selection of an early biomarker for vascular normalization using dynamic contrast-enhanced ultrasonography to predict outcomes of metastatic patients treated with bevacizumab. Ann Oncol. 2016;27:1922-8
48. Lee H, Zheng J, Gaddy D, Orcutt KD, Leonard S, Geretti E. et al. A gradient-loadable (64)Cu-chelator for quantifying tumor deposition kinetics of nanoliposomal therapeutics by positron emission tomography. Nanomedicine. 2015;11:155-65
49. Hudson HM, Larkin RS. Accelerated image reconstruction using ordered subsets of projection data. IEEE Trans Med Imaging. 1994;13:601-9
50. Peng F, Lu X, Janisse J, Muzik O, Shields AF. PET of human prostate cancer xenografts in mice with increased uptake of 64CuCl2. J Nucl Med. 2006;47:1649-52
51. Fonseca NA, Gregorio AC, Valerio-Fernandes A, Simoes S, Moreira JN. Bridging cancer biology and the patients' needs with nanotechnology-based approaches. Cancer Treat Rev. 2014;40:626-35
52. Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC. et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412-7
53. Ranganathan R, Madanmohan S, Kesavan A, Baskar G, Krishnamoorthy YR, Santosham R. et al. Nanomedicine: towards development of patient-friendly drug-delivery systems for oncological applications. Int J Nanomedicine. 2012;7:1043-60
54. Shapira A, Livney YD, Broxterman HJ, Assaraf YG. Nanomedicine for targeted cancer therapy: towards the overcoming of drug resistance. Drug Resist Updat. 2011;14:150-63
55. Reynolds JG, Geretti E, Hendriks BS, Lee H, Leonard SC, Klinz SG. et al. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol Appl Pharmacol. 2012;262:1-10
56. Mollard S, Ciccolini J, Imbs DC, El Cheikh R, Barbolosi D, Benzekry S. Model driven optimization of antiangiogenics + cytotoxics combination: application to breast cancer mice treated with bevacizumab + paclitaxel doublet leads to reduced tumor growth and fewer metastasis. Oncotarget. 2017;8:23087-98
57. Hutchinson LG, Mueller HJ, Gaffney EA, Maini PK, Wagg J, Phipps A. et al. Modeling Longitudinal Preclinical Tumor Size Data to Identify Transient Dynamics in Tumor Response to Antiangiogenic Drugs. CPT Pharmacometrics Syst Pharmacol. 2016;5:636-45
58. Wong PP, Bodrug N, Hodivala-Dilke KM. Exploring Novel Methods for Modulating Tumor Blood Vessels in Cancer Treatment. Curr Biol. 2016;26:R1161-R6
59. Lee H, Shields AF, Siegel BA, Miller KD, Krop I, Ma CX. et al. 64Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin Cancer Res. 2017;23:4190-202
60. Roy Chowdhury M, Schumann C, Bhakta-Guha D, Guha G. Cancer nanotheranostics: Strategies, promises and impediments. Biomed Pharmacother. 2016;84:291-304
61. Babu A, Templeton AK, Munshi A, Ramesh R. Nanodrug delivery systems: a promising technology for detection, diagnosis, and treatment of cancer. AAPS PharmSciTech. 2014;15:709-21
62. Kalra AV, Kim J, Klinz SG, Paz N, Cain J, Drummond DC. et al. Preclinical activity of nanoliposomal irinotecan is governed by tumor deposition and intratumor prodrug conversion. Cancer Res. 2014;74:7003-13
63. Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66:3271-7
Author contact
![]() Corresponding author: E-mail: shieldsaorg
Corresponding author: E-mail: shieldsaorg