Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(17):4071-4086. doi:10.7150/thno.20168 This issue Cite
Research Paper
Transforming doxorubicin into a cancer stem cell killer via EpCAM aptamer-mediated delivery
Dongxi Xiang1, Sarah Shigdar1, Andrew G Bean2, Matthew Bruce2, Wenrong Yang3, Motilal Mathesh3, Tao Wang1,4, Wang Yin1, Phuong Ha-Lien Tran1, Hadi Al Shamaileh1, Roberto A Barrero5, Pei-Zhuo Zhang6, Yong Li7, Lingxue Kong8, Ke Liu9, Shu-Feng Zhou10, Yingchun Hou11, Aina He12 ![]() , Wei Duan1
, Wei Duan1 ![]()
1. School of Medicine and Centre for Molecular and Medical Research, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria 3216, Australia;
2. CSIRO Australian Animal Health Laboratory, Private Bag 24, Geelong, Victoria 3220, Australia;
3. Centre for Chemistry and Biotechnology, School of Life and Environmental Sciences, Faculty of Science, Engineering and Built Environment, Deakin University, Waurn Ponds, Victoria 3216, Australia;
4. School of Nursing, Zhengzhou University, 100 Kexue Ave, Zhengzhou, P. R. China, 450001;
5. Centre for Comparative Genomics, Murdoch University, 90 South Street, Murdoch, WA 6150, Australia;
6. Suzhou GenePharma, 199 Dongping Street, Suzhou 215123, P. R. China;
7. Cancer Care Centre, St George Hospital and St George and Suthland Clinical School, University of New South Wales (UNSW), High Street, Kensington, NSW 2052, Australia;
8. Deakin University, Australia, Institute for Frontier Materials, 75 Pigdons Road, Waurn Ponds, Victoria, 3216;
9. College of Life Sciences, Sichuan University, No.24 South Section 1, Yihuan Road, Chengdu 610041, P. R. China;
10. Department of Bioengineering and Biotechnology, College of Chemical Engineering, Huaqiao University, 668 Jimei Avenue, Xiamen, Fujian 361021, P. R. China;
11. Center for Qinba Region's Sustainable Development, Shaanxi Normal University, No.199, South Chang'an Road, Xi'an, Shaanxi 710062, P. R. China;
12. Department of Oncology, Affiliated Sixth People's Hospital, Shanghai Jiaotong University, No. 600, Yishan Road, Shanghai 200233, P. R. China.
Received 2017-3-20; Accepted 2017-6-25; Published 2017-9-20
Abstract

Chemotherapy-resistant cancer stem cells (CSCs) are a major obstacle to the effective treatment of many forms of cancer. To overcome CSC chemo-resistance, we developed a novel system by conjugating a CSC-targeting EpCAM aptamer with doxorubicin (Apt-DOX) to eliminate CSCs. Incubation of Apt-DOX with colorectal cancer cells resulted in high concentration and prolonged retention of DOX in the nuclei. Treatment of tumour-bearing xenograft mice with Apt-DOX resulted in at least 3-fold more inhibition of tumour growth and longer survival as well as a 30-fold lower frequency of CSC and a prolonged longer tumourigenic latency compared with those receiving the same dose of free DOX. Our data demonstrate that a CSC-targeting aptamer is able to transform a conventional chemotherapeutic agent into a CSC-killer to overcome drug resistance in solid tumours.
Keywords: doxorubicin, cancer stem cell killer
Introduction
Current approaches including chemotherapy and radiotherapy are widely used for cancer treatment, but their lack of specificity to cancer cells often results in increased side effects and limited therapeutic efficacy1, 2. The dose-limiting toxicity of free drugs used in clinic limits the possibility to treat a patient at a planned dose or to timely start a new cycle of treatment, thereby underlying the failure of chemotherapy3, 4. The current failure of cancer treatment is largely attributed to our inability in eradicating cancer stem cells (CSCs)5, 6. In the CSC model, only a small population of cancer cells possess the ability to extensively proliferate, self-renew, differentiate to multiple lineages and generate a tumour5, 7. CSCs, as 'roots of cancer', are defined by their abilities to form new tumours that histologically resembles the original tumour when transplanted into immunodeficient mice; they are resistant to traditional therapies such as chemo- and/or radio-therapy8-10. As CSCs are more drug resistant, more aggressive, and more invasive with higher metastatic potential than their non-CSC counterparts, this population of cells must be effectively targeted in order to improve the survival of patients with cancer5.
To achieve this goal, tumour active targeting systems have been intensively explored as they have the tumour-targeting potential to deliver therapeutic agents more selectively to cancer cells, thereby enhancing the therapeutic efficacy while minimizing non-specific toxicity to healthy cells 11, 12. Active targeting entails the guidance of therapeutic agents, with the aid of targeting ligands, to tumour cells and promotes their subsequent cellular entry through receptor-mediated endocytosis 13-15. Aptamers, as chemical antibodies, are novel ligands for drug targeting and offer significant advantages over antibodies in terms of smaller size, low immunogenicity and high stability, as well as the high reproducibility and ease for chemical modification16-19.
Currently, there is no approved agent on the market that is able to effectively eliminate CSCs. Instead of developing new drugs to target CSCs, this study aimed to explore a novel strategy that utilizes a CSC-targeting moiety to deliver existing chemotherapeutic agents to target CSCs. Aptamers, also known as chemical antibodies, are single-stranded DNA or RNA molecules that could specifically bind their targets with high affinity and specificity 20, 21, which have been broadly applied for targeted cancer therapy 22-24. We have developed two RNA aptamers against CSC surface markers (EpCAM and CD133) 25, 26. Doxorubicin (DOX) is widely used for treating a spectrum of cancers including hematological malignancies and solid cancers of breast, ovarian, lung and others. DOX functions by intercalating double-stranded G-C sequences of DNA and inhibits the synthesis of nuclei acids, resulting in DNA damage, epigenome and transcriptome deregulation 27. However, the dose-limiting toxicity of DOX has limited its success in anticancer treatment due to non-specific targeting and increased emergence of drug resistance 27. By directly conjugating DOX to a CSC-targeting EpCAM aptamer without any chemical modification of the drug, we reasoned that this strategy may be able to effectively target EpCAM-expressing CSCs followed via enhanced cellular internalization and delivery the drug to the nuclei of CSCs. To test this hypothesis, a colorectal cancer mouse xenograft model using HT29 cells was established and a set of in vitro, ex vivo and in vivo studies were conducted to evaluate the potential of the CSC-targeted aptamer-mediated active targeting as a promising strategy for effective eradicating CSCs.
Results
pH-dependent release of DOX from aptamer-drug complex
We have previously developed and optimized an 18-nt RNA aptamer (18-nucleotides) against a CSC marker EpCAM 25, 26. To target a traditional anticancer agent, doxorubicin (DOX), to CSCs, we developed a self-assembled and CSC-targeted drug conjugate. As our work previously demonstrated that it is the loop of this RNA aptamer that determines its target binding and the modifications made to the stem portion of the aptamer have no discernible impact on target interaction 25, 26, we engineered the stem of the EpCAM aptamer (Fig. 1A). As DOX binds to RNA poorly, the stem of the original RNA aptamer was replaced with a 10-bp DNA G-C stem. In addition, 5'-methyl-deoxycytidine (dC) was deployed in the newly engineered DNA stem to achieve increased duplex stability and reduced immunogenicity. Next, a self-assembled Apt-DOX conjugate was created by conjugation of DOX with chemically modified DNA-RNA hybrid EpCAM aptamer27. The images of atomic force microscopy (AFM) showed the morphologies of the corresponding nanostructures of aptamer and the conjugation of aptamer and DOX (Fig. 1B and Supplementary Fig. 1A). We observed the formation of two different adsorbed nanostructures, which indicated molecular interaction of DOX to the aptamer using atomic force microscopy (AFM). An aptamer, which has a 2'-O-methyl (2'-O-Me) instead of 2'-fluoropyrimidine modifications in the loop of the RNA was used as a negative control throughout the study as it does not bind to EpCAM 25, 26.
Characterization of EpCAM Apt-DOX conjugates. (A) Schematic illustration of the hybrid RNA-DNA EpCAM aptamer. (B) AFM analysis of intercalation between the aptamer and DOX. Based on cross section analysis, the size of aptamers were around 1.3 nm, while the conjugated structure has size of around 2.5 nm. (C) The fluorescence quenching of 10 μM DOX after 30 min incubation with an increasing molar ratio of aptamer-to-DOX (0.001, 0.002, 0.004, 0.006, 0.1, 0.6 and 1). (D) Time-dependent release of DOX from aptamer conjugates at pH 7.4 in vitro. The “Free DOX” sample was used to demonstrate the ability of the DOX dissociated from the aptamer can cross the membrane of the dialysis device. (E) pH-dependent release of DOX from aptamer conjugates at pH of 5.0, 7.4 and 8.0. Data shown are means ± SEM (n=3).
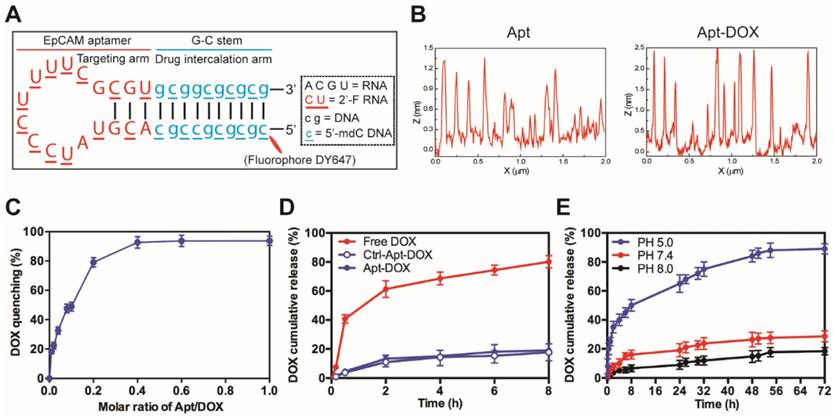
The optimal molar ratio for DOX loading into the aptamer was determined to be 0.4 (~2.5 molecules DOX assembled per aptamer) at which the quenching of the DOX fluorescence reached a plateau around 92% (Fig. 1C and Supplementary Fig. 1B). As shown in Fig. 1D, less than 18% of DOX was released from both Apt-DOX and control Apt-DOX (Ctrl-Apt-DOX) after 8 h in PBS buffer (pH 7.4), indicating a high stability of the Apt-DOX complex under physiological pH when transiting in the blood. We have previously shown that upon binding to cell surface EpCAM, the EpCAM aptamers are internalized via endocytosis 28. As the lysosomes are the end subcellular location for cargos entering the cells via endocytosis and they have a pH of approximately 5.0, we next studied the pH-dependent release of DOX from the engineered EpCAM aptamer at pH 5.0, pH 7.4 and pH 8.0 at 37 °C. As shown in Fig. 1E, there was an initial burst of DOX release at pH 5.0 followed by a steady and continued release at acidic conditions (pH 5.0), with a 3.1-fold to 4.8-fold increase of DOX release from Apt-DOX conjugates compared with that in the neutral or slightly basic conditions. Specifically, approximately 89.2 % of the intercalated DOX was released after 72 h at pH 5.0, while only 27.6 % and 18.4 % of the DOX were released from the engineered aptamer after 72 h at pH 7.4 and pH 8.0, respectively. Such a pH-dependent drug release is desirable as it could minimize the systemic exposure of DOX to sensitive organs under physiological conditions (pH 7.4), but allow a swift release of DOX from the Apt-DOX complex after endocytosis 29.
Time- and dose-dependent delivery leads to enhanced on-target retention
Next, we examined the effect of the conjugation of DOX to EpCAM aptamer on binding affinity and specificity of the aptamer to target EpCAM proteins. As shown in Fig. 2A and Supplementary Fig. 2A, the EpCAM Apt-DOX conjugate displayed negligible binding to the EpCAM-negative HEK293T cells (with an apparent K'd of >1000 nM); while it bound to EpCAM-positive HT29 cells with a K'd being 16.08 ± 4.83 nM (Supplementary Fig. 2B). The improved binding affinity of Apt-DOX conjugate over the free aptamer could be attributed to a more stable 3-D structure of the Apt-DOX conferred by the 10-bp GC stem and the conjugation of the DOX. The specificity of such interaction was further demonstrated by the lack of binding to target cells by a negative Ctrl-Apt-DOX that had an identical nucleotide sequence but an altered 3-D structure due to a different chemical modification at 2'-pyrimidines that abolishes its binding to EpCAM (Supplementary Fig. 2A). The ability of the Apt-DOX conjugate to successfully undergo endocytosis into EpCAM-positive but not in EpCAM-negative target cells was confirmed using a 3-D culture model via confocal microscopy (Fig. 2A).
Specific and enhanced delivery of DOX into target cells via aptamer-guided delivery. (A) Specificity of uptake of EpCAM Apt-DOX into EpCAM-positive HT29 tumourspheres but not the EpCAM-negative HEK293T tumourspheres at 37 °C for 30 min. Scale bar is 10 μm. Time-dependent (B) and dose-dependent (C) intracellular delivery of Apt-DOX into monolayer HT29 cells. (D) Time-dependent total cellular uptake and retention of Apt-DOX (1.5 μM of DOX equivalent) in HT29 cells. (E-F) Time-dependent uptake and retention of DOX by Apt-DOX in the nuclei of HT29 cells after incubating cells with 1.5 μM of DOX or equivalent Apt-DOX at 37 °C for 30 min or 2 h, followed by washing and further 2 h or 24 h incubation with fresh medium. (E) EpCAM-Apt-DOX; (F) free DOX. Scale bar is 5 μm. Data shown are means ± SEM. (n=3). **P < 0.01, ***P < 0.001 compared with free DOX treatment groups (two-tailed Student's t-test).
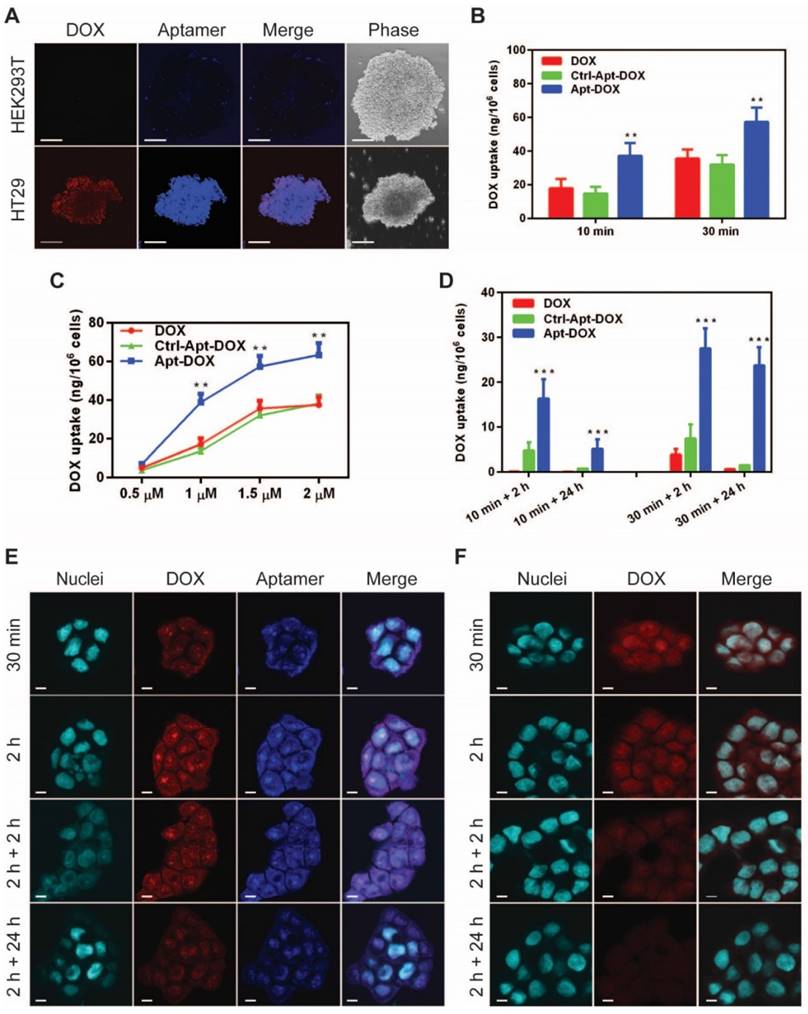
We next sought to evaluate whether EpCAM Apt-DOX enters target cells in a time- and dose-dependent manner. In time course studies over either a 10-min or 30 min period, the cumulative cellular uptake of the Apt-DOX was found to increase by 2-2.5 fold compared with that of free DOX and the Ctrl-Apt-DOX, respectively (P < 0.01) (Fig. 2B). Similarly, a dose-dependent uptake of the Apt-DOX was observed over a dose of up to 2 µM equivalent of DOX (Fig. 2C). Taken together, these results suggest that Apt-DOX is capable of efficiently targeting HT29 cells and enhancing the intracellular delivery of DOX both in a time- and dose-dependent manner. Since the persistence in on-target intracellular drug concentration is critical to its clinical efficacy, we further analyzed the intracellular retention of DOX as delivered by EpCAM aptamer. In studies involving a 10 min incubation with 1.5 µM DOX or equivalent Apt-DOX followed by a wash-out period of 2 h, the HT29 cells treated with Apt-DOX retained 16.34 ng DOX per 1 x 106 cells, while only 0.12 ng and 4.79 ng DOX per 1 x 106 cells were retained in cells treated with free DOX and Ctrl-Apt-DOX, respectively (Fig. 2D, left bars). Importantly, after a 24 h wash-out period, there was a limited residual amount of DOX left in cells treated with free DOX or Ctrl-Apt-DOX, while cells treated with Apt-DOX still retained 5.16 ng DOX per 1 x 106 cells. (P < 0.01) (Fig. 2D, left bars). Consistently, similar results were obtained in cells undergoing a 30 min incubation with 1.5 µM DOX or equivalent Apt-DOX followed by a wash-out period of 2 or 24 h (Fig. 2D, right bars). Next, we investigated if DOX was retained in the nuclei of EpCAM Apt-DOX treated HT29 cells using confocal microscopy. As shown in Fig. 2E, after incubation for 0.5 h or 2 h, a large amount of Apt-DOX was found in the nuclei, evident from the overlapping of red (for DOX) or blue (for EpCAM aptamer) with cyan (for nuclei) fluorescence (Fig. 2E). After 2 h or 24 h drug wash-out period, the red fluorescence signal for DOX in the cells treated with Apt-DOX was clearly detectable. In contrast, little red fluorescence in the nuclei of the cells incubated with free DOX were discernable after a 2 h wash-out period (Fig. 2F and Supplementary Fig. 2C). Thus, delivering DOX by EpCAM aptamer led to an enhanced time- and dose-dependent cellular uptake as well as a significantly improved on-target DOX accumulation in target cells.
Elimination of self-renewal cells by aptamer-guided DOX treatment both in vitro and ex vivo
Having established that Apt-DOX can deliver a large dose of DOX into colorectal cancer cells, it is important to evaluate if this translates into effective pharmacological outcome of eliminating CSCs. To this end, we used a tumoursphere assay to determine the impact of treatment on a key feature of CSC, namely the self-renewal potential. Using the limiting dilution assay (LDA), serial dilutions of HT29 cells were seeded in stem cell culture conditions and incubated with 1 μM of free DOX or equivalent concentration of Apt-DOX30. Salinomycin, an agent known to be able to robustly kill CSCs, was used as a positive control to validate the sensitivity of the tumoursphere assay system in detecting the elimination of cells with self-renewal capacity 31. Cells treated with free DOX showed moderate decrease in the frequency of tumoursphere formation, especially for those with a cell seeding dose of 5 cells/well or less (Fig. 3A, Supplementary Fig. 3A and Supplementary Table 1). In contrast, cells treated with, aptamer targeted delivery of DOX (Apt-DOX) resulted in a 16.7-fold decrease of CSC frequency compared to that of free DOX (Fig. 3A). After Apt-DOX treatment, the percentage of tumoursphere formation in the first passage was found to be 8.7 % which was a 10.4-fold decrease than that of free DOX treated tumour cells. This reduction in tumoursphere formation was more pronounced during the second (12.37-fold lower) and third round (170-fold lower) of tumoursphere culture during which cells without true self-renewal capability that managed to survive the first-round tumoursphere assay are more effectively eliminated (Fig. 3B). The efficacy of aptamer targeted delivery of DOX in eliminating putative CSCs in the tumoursphere assays was confirmed in two additional carcinoma models, namely the ovarian cancer cell line SKOV-3 and the breast cancer cell line T47D (Supplementary Fig. 3B and Supplementary Table 2). Thus, these results indicate that the Apt-DOX treatment could effectively impair the tumorigenicity of CSCs in at least three types of solid tumours.
Impairment of CSC self-renewal function following Apt-DOX treatment in vitro and ex vivo. (A) The percent of CSC frequency of HT29 cells based on in vitro limiting dilution assay. (B) The percent of tumoursphere following 5-7 days incubation of HT29 cells (8 × 103) with various agents as indicated under stem cell culture conditions. (C) Tumour growth of colorectal tumours that were transplanted with 1 × 105 cells/mouse following treatment with various agents as indicated. (D) Kaplan-Meier survival curves of NOD/SCID mice bearing xenograft tumours treated with varies agents as described above. Data shown are means ± SEM. (n=3, unless indicated otherwise).
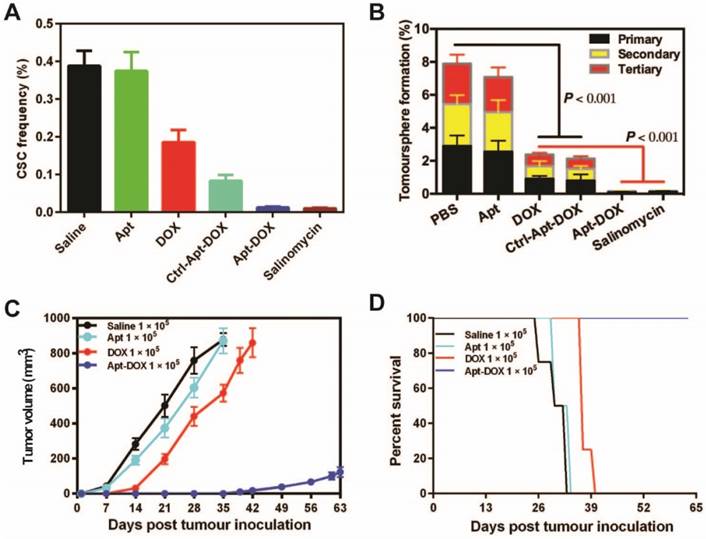
To further determine if the treatment of cancer cells with Apt-DOX would affect the ability of CSC to form tumours in immunocompromised mice, HT29 cells were treated in vitro as indicated in Fig. 3C for 5 days and then injected subcutaneously (s.c.) into NOD/SCID mice at two cell doses of 1 × 105 or 1 × 104 per site. In vitro treatment of Apt-DOX resulted in an approximately 3.9 × 105-fold lower CSC frequency than that of cells treated by free DOX in NOD/SCID mice (Supplementary 3C. and Supplementary Table 3). Remarkably, no tumour was formed in NOD/SCID mice when 1 × 104 cells treated with Apt-DOX were implanted, in sharp contrast to the mice receiving the same dose of cells treated with free DOX or the saline or aptamer-only controls (Supplementary Fig. 3D and 3E). As for the tumour latency, mice that received 1 × 105 cells treated by Apt-DOX exhibited a significantly longer tumour-free period of ~6 weeks, compared to a latent period of 2 weeks in mice that received cells treated with free DOX (Fig. 3C). Accordingly, the mice-bearing HT-29 cells treated with Apt-DOX survived more than 3 weeks longer than the mice-bearing cells treated with free DOX at the inoculating cell dose of 1 × 105 (Fig. 3D). These results strongly suggest that the EpCAM aptamer-guided delivery of DOX successfully targeted and eradicated CSCs in vitro, resulting in suppressed tumour growth, prolonged tumour-free tendency and extended survival.
PEGylation improves pharmaceutical profile and tumour delivery of Apt-DOX in vivo
To improve the pharmaceutical profile of Apt-DOX, the EpCAM aptamer was further engineered by conjugating a terminal 20 kDa polyethylene glycol (PEG) to increase its blood retention32 and the pharmacokinetics was evaluated in healthy Sprague Dawley rats. Following a single intravenous (i.v.) injection, the DOX concentration was measured as shown in Fig. 4A and Supplementary Table 4. In contrast to a short half-life (t1/2: 0.87 h) for free DOX, the PEGylated Apt-DOX displayed much prolonged half-life of approximately 7 h, a significantly increased the mean residence time (MRT) of 16.25 h compared to a MRT of 2.48 h for free DOX, as well as a 7-fold higher area under the curve (AUC) and a 10-fold lower clearance (CL). These results revealed that the DOX intercalated to PEGylated aptamer has a substantially improved pharmacokinetic profile than free DOX in vivo.
Pharmaceutical profile and biodistribution of PEGylated Apt-DOX. (A) Pharmacokinetic profiles of DOX after a single i.v. injection of free DOX, PEGylated Apt-DOX and control PEGylated Apt-DOX conjugate into SD rats at an equivalent dosage of 5 mg/kg DOX followed by quantification of DOX in blood plasma. (B) Biodistribution profiles of DOX accumulation in tumour and organs 24 h after i.v. injection of agents at an equivalent dosage of 5 mg/mL DOX. (C-D) Biodistribution of DOX in the tumour (C) and heart (D), 24 h after i.v. injection. Data shown are means ± SEM. (n=3, unless indicated otherwise). *P < 0.05, **P < 0.01 compared with free DOX administration groups (two-tailed Student's t-test).
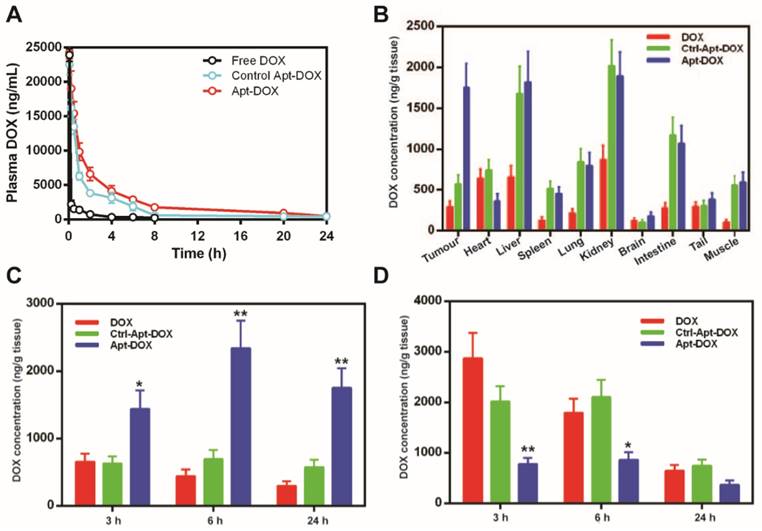
To assess if the improved serum pharmacokinetics translates into better in vivo uptake of Apt-DOX in the target tissue, the biodistribution of DOX in various tissues were studied after a single i.v. injection of various agents. DOX concentration in the tumour in mice receiving free DOX exhibited a low peak concentration at 3 h followed by a steady decrease of DOX over the 24 h period (Fig. 4B and C). In contrast, during the same period, the concentration of DOX in the tumour of mice receiving Apt-DOX was 100-200% higher than those receiving free DOX, reaching a peak concentration of DOX in tumours at 6 h followed by a slight decline, to around 73 % of the peak, at 24 h after administration (Fig. 4B and C). Remarkably, even 24 h after the administration of Apt-DOX, the DOX concentration in the tumour was still 22% higher than that at 3 h after administration (Fig. 4C). Interestingly, the levels of DOX in the heart, the site of principal dose-liming toxicity of DOX, of the tumour-bearing mice receiving Apt-DOX treatment were 39% to 70% lower than those treated with free DOX at all three time points studied (Fig. 4D). To investigate whether the PEGylated Apt-DOX could reduce side effects of DOX, changes in body weights of mice-bearing xenograft tumours were monitored after treatments. HT29 tumour-bearing mice receiving free DOX treatment lost 5.7-fold more weight than those treated with pegylated Apt-DOX (P < 0.01), while tumour-bearing mice receiving free aptamer and saline displayed an increase in body weight (Supplementary Fig. 4). Taken together, the aptamer-guided delivery of DOX not only improved DOX accumulation in tumours, but also enhanced the safety profile, with reduced cardiac exposure.
Improved antitumour efficacy afforded by Apt-DOX
Given that aptamer-guided DOX delivery could efficiently impair CSC's self-renewal function in vitro, we next studied the antitumour efficacy of Apt-DOX in HT-29 colorectal xenografts in NOD/SCID mice. Treatment of s.c. tumour-bearing mice with Apt-DOX resulted in a significantly reduced rate of tumour growth with an 8-fold and 3-fold smaller tumour size compared with the mice receiving saline or free DOX treatment, respectively (P < 0.01) (Fig. 5A). Mice receiving free DOX or negative control Apt-DOX succumbed to the tumours at day 51, while those treated with Apt-DOX had a remarkably prolonged survival beyond day 63 (Fig. 5B). Therefore, the aptamer-guided DOX delivery could effectively target CSCs in vivo and thus significantly inhibited the tumour growth and extended overall survival of the tumour-bearing mice.
Apt-DOX treatment enhanced apoptosis and inhibited cell proliferation in HT29 xenograft tumour. NOD/SCID mice bearing HT29 xenograft tumours with a volume of 50 mm3 were treated with agents as indicated. (A-B) Aptamer-guided DOX delivery inhibited tumour growth and extended survival rate of mice-bearing HT29 tumours. NOD/SCID mice-bearing HT29 xenograft tumour were randomized into six groups and treated as described in the legend. (A) The change of the tumour volume over 63 days (n=4). (B) Kaplan-Meier survival curves of mice (n=4) bearing HT29 tumour treated as indicated. (C) Quantification of apoptotic cells in the treated xenograft tumours using TUNEL assay. (D) Quantification of Ki-67 positive cells in HT29 xenograft tumours treated as indicated. Data shown are means ± SEM. (n=3, unless indicated otherwise). ** P < 0.01 compared with mice receiving free DOX (two-tailed Student's t-test).
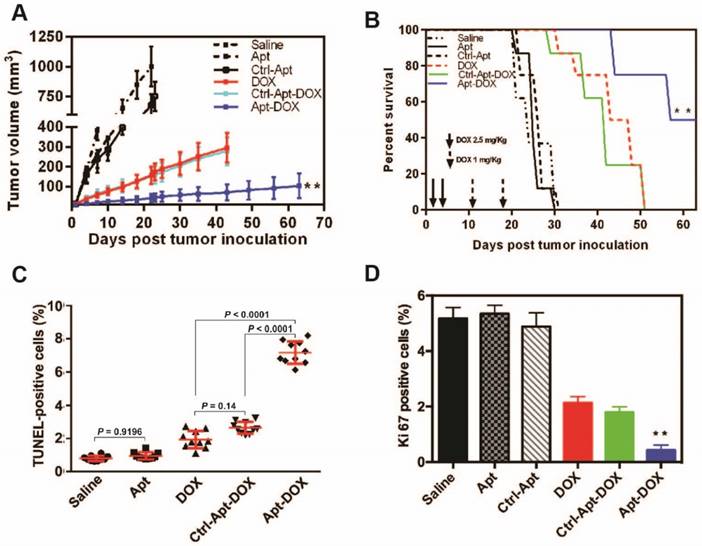
The progression of cancer is controlled by the rate of cancer cell proliferation and apoptosis 33. We next sought to evaluate the effects of Apt-DOX treatment on apoptosis and the proliferation of HT29 xenograft tumour cells. As shown in Fig. 5C, while single treatment with DOX alone induced limited apoptosis in tumours, Apt-DOX treatment elicited a 9.3-fold and 3.75-fold increase in apoptotic tumour cells compared with that in saline- and free DOX-treatment groups, respectively (Fig. 5C and Supplementary Fig. 5A). These data indicate that the major inhibition of tumour growth was at least partly due to the increased apoptosis effect induced by Apt-DOX. Similarly, a Ki-67 index observed in tumours treated with Apt-DOX was significantly reduced (~6-fold decrease) compared to tumours treated with free DOX or Ctrl-Apt-DOX (Fig. 5D and Supplementary Fig. 5B), indicating that the aptamer-guided DOX treatment not only strongly promoted apoptosis but also inhibited the proliferation of tumour cells.
Diminished tumorigenicity of colorectal CSCs treated with Apt-DOX
To assess the impact of Apt-DOX treatment on the self-renewal capacities of CSCs in vivo, an in vitro tumoursphere initiation with LDA was carried out on single cell suspensions prepared from xenograft tumours after in vivo treatment. As shown in Supplementary Table 5, there was a 207.8- and 18.1-fold decrease in the frequency of sphere re-initiating cells from tumour-bearing mice treated with Apt-DOX compared to those treated with saline and free DOX, respectively. To verify the in vivo efficacy of Apt-DOX in targeting CSCs, the self-renewal capacity of CSCs from the treated xenograft tumours was studied using the secondary in vivo tumour initiation assays at serial limiting cell doses34-36. To this end, single cell suspensions prepared from the tumours removed from the mice treated by various agents were re-implanted into naïve NOD/SCID mice s.c. at cell doses of 1 × 105, 1 × 104, 1 × 103 and 1 × 102/site. At the inoculating cell dose of 1 × 103, tumours formed in all mice receiving cells treated with free drug or a control (Supplementary Table 6). In contrast, no tumour was detected in mice receiving 1 × 103 cells dissociated from the mice treated with Apt-DOX after 3 months, suggesting that Apt-DOX conjugate treatment eliminated most, if not all, CSCs at this cell dose. Implantation of 1 × 104 cells treated by Apt-DOX exhibited a significantly longer tumour-free period than those receiving free DOX (7 weeks vs 4 weeks) (Supplementary Table 6). At higher inoculating cell doses of 1 × 105, there were a ~20 days longer tumour-free period in mice that received Apt-DOX than those treated with free DOX. There was a 30-fold reduction in the frequency of self-renewing CSCs in naïve mice receiving tumour cells from mice that had been treated with Apt-DOX compared with those that received tumour cells from mice treated with free DOX (P < 0.0001) (Supplementary Table 6). Thus, DOX, a classic anti-cancer drug, has been transformed into a new agent that effectively eliminates CSCs when it is delivered by the EpCAM aptamer.
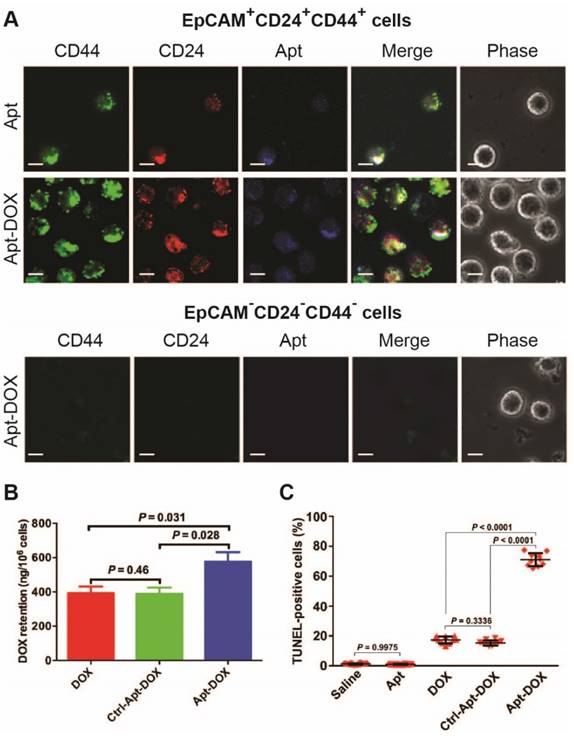
To evaluate the in vivo CSC-targeting capability of Apt-DOX, direct evidence on eliminating of CSCs was sought through experiments conducted directly on the putative colorectal CSCs isolated from xenograft HT29 tumours that had undergone various treatments in vivo (Fig. 6). It was previously shown by others that CSC population in HT29 tumours can be defined using a set of cell surface markers EpCAM+CD24+CD44+.37, 38 Thus, we first determined the binding and internalization of the Apt-DOX in FACS-sorted populations of EpCAM+CD24+CD44+ CSCs and EpCAM-CD44-CD24- non-CSCs from the xenograft tumours. The confocal microscopy confirmed that DY647-labelled Apt-DOX was indeed internalized into colorectal CSCs (Fig. 6A). No DY647-labelled Apt-DOX was found in EpCAM-CD44-CD24- cells. Second, approximately 24 h after the last treatment, the intracellular concentration of DOX delivered by Apt-DOX in EpCAM+CD24+CD44+ cells was found to be 1.5-fold higher than that in mice receiving free DOX (P = 0.031) (Fig. 6B). Third, 78% of EpCAM+CD24+CD44+ cells from mice treated with Apt-DOX were found to be apoptotic (TUNEL positive), in sharp contrast to ~19% apoptotic cells in the EpCAM+CD24+CD44+ cells isolated from tumours treated with free DOX s (P < 0.0001) (Fig. 6C and Supplementary 6A). Therefore, the Apt-DOX is able to target CSCs in HT29 xenograft tumour in vivo.
Impairment of CSC self-renewal capability in xenograft tumours following Apt-DOX treatment in vivo. (A-C) Apt-DOX was delivered into EpCAM+/CD44+/CD24+ cells in treated xenograft tumours and eliminated CSCs. (A) Representative micrographs showing efficient targeting of Apt-DOX to EpCAM+CD24+CD44+ cells in HT29 xenograft tumours. Scale bar is 10 μm. (B) Retention of DOX in FACS-sorted EpCAM+CD24+CD44+ cells in xenograft tumours after indicated treatments. The tumour-bearing animals were euthanized 3 h after the last treatment. (C) Quantification of apoptotic EpCAM+CD24+CD44+ cells after various treatments using TUNEL assay (n=4). Data shown are means ± SEM. (n=3, unless indicated otherwise). P value was obtained by the two-tailed Student's t-test.
Discussion
Current anticancer strategies that mainly focus on eliminating the bulk cancer cells fail to cure cancers as the remaining population of CSCs may cause tumour recurrence regarding their treatment resistance, aggressiveness and metastatic potential 39. EpCAM is expressed at low levels in normal epithelial cells, but its expression is 800~1000-fold higher in cancer cells 20, 21, 40. EpCAM is considered as a stem cell marker in multiple solid tumours, including colon, breast, pancreas, liver, and prostate tumours; and tumour cells with high EpCAM expression have been shown to correlate with CSC's phenotype and properties 25, 26, 41, 42. Therefore, EpCAM is a valid target for CSC-targeted therapy. Here the RNA EpCAM aptamer was utilized to develop a novel therapeutic strategy to transform a traditional chemotherapeutic drug into an agent for eradicating CSCs.
Despite DOX being a robust anticancer drug that is able to kill the majority of rapidly proliferating cells in cancer, abundant preclinical and clinical studies have revealed that DOX is largely ineffective in eliminating CSCs 43-46. Furthermore, the clinical applications of DOX are often associated with the emergence of drug resistance and enrichment of CSCs 47-51. Here an RNA aptamer was engineered with a binding loop specific to EpCAM and a docking DNA stem for loading DOX 52, 53. The retention of DOX with the aptamer during transit at neutral pH and the release of DOX at acidic pH are critical to the success of targeted cancer therapy. Indeed, the payload (DOX) is largely retained within the Apt-DOX conjugate at pH of 7.4, but released efficiently at pH 5.0 (Fig. 1D, E). These data suggest that DOX would remain stably conjugated with the EpCAM aptamer in the circulatory system and tissue interstitium, but be released from the aptamer following intracellular delivery in the endosome-lysosome compartment. This allows DOX to mainly exert its cytotoxic effect after entering the target cells, enhancing the therapeutic index of the Apt-DOX conjugate. Furthermore, this pH-dependent intracellular release of DOX from the Apt-DOX complex could circumvent the efflux by ABC transporters, allowing delivery of the DOX to the site of action, the nucleus. Indeed, confocal microscopy revealed that the treatment with Apt-DOX resulted in a more effective accumulation and retention of DOX in the nuclei compared with free DOX (Fig. 2E-F). Such data also suggest that free DOX is rapidly effluxed after entering cells via random diffusion, while DOX delivered by Apt-DOX could circumvent the highly elevated drug efflux systems in cancer cells.
The gold standard approach to study CSCs is to xenotransplant tumour cells into immunodeficient mice with LDA followed by evaluating functions of putative CSCs. Here, an in vitro surrogate of the gold standard CSC assay, tumoursphere formation assay, was employed to provide initial assessment of the CSC-targeting ability of Apt-DOX conjugates as it allows us to functionally test a key property of CSCs, self-renewal. Careful analysis at serial seeding doses of cells confirmed the ability of Apt-DOX conjugates in inhibiting sphere-initiating frequency (Fig. 3A and B, Supplementary Table 1). In addition to the colorectal cancer HT29 cells, two additional cancer cell lines from different types of solid tumour (ovarian and breast cancers) were also studied (Fig. Supplementary 3B, and Supplementary Table 2). The results indicated that the aptamer-guided DOX delivery to EpCAM-overexpressing cells could be a universal approach for CSC-targeted therapy. To functionally verify the CSC-targeting ability of Apt-DOX, an ex vivo LDA was conducted by xenotransplanting single cell suspensions from cells previously treated with Apt-DOX or controls in vitro into NOD/SCID mice. Consistent with the in vitro results from tumoursphere assays, the treatment of cells with Apt-DOX resulted in a significant reduction of tumour growth and a longer latency, compared to those treated with free DOX or other negative controls (Fig. 3C, Supplementary 3C and Supplementary Table 3). Most importantly, the Apt-DOX treatment remarkably prolonged the survival of mice-bearing HT29 tumours, indicating that the subpopulation of CSCs was effectively eliminated, at least partially, in vitro before they were xenotransplanted into mice (Fig. 3D, Supplementary 3D). Thus, targeted in vivo delivery of DOX leads to the elimination of cancer stem cells in the xenograft tumour mouse model studied.
Cytotoxic chemotherapy remains the corner stone of the first-line therapy for many advanced solid tumours. However, chemoresistance often develops and up to 90% of patients with metastatic cancer eventually succumb to the disease 54. Vast preclinical studies and clinical data suggest that when administrated in the current fashion, chemotherapeutic drugs, such as DOX, are unable to eliminate CSCs in most cases 55. A widely held view in the field is that CSCs are intrinsically resistant to chemotherapeutic agents. Therefore, heroic efforts have been devoted to develop next generation inhibitors to pathways leading to chemoresistance 56. However, the data presented in this study suggest that resistance to chemotherapeutic agents, such as DOX, might not be the consequence of the dysregulation of various molecular and cellular pathways currently being attributed to chemoresistance in CSC. Rather, the failure of chemotherapy in eliminating CSCs in the clinics could be due to our inability to fulfill a fundamental pharmacological and pharmaceutical principle: one needs to deliver a sufficient dose to the pharmacological subcellular location for sufficient time in order to kill a target cell. Our data demonstrate for the first time that as long as one can deliver sufficient amount of DOX to the nuclei of target cells for a sufficient period of time, the Aptamer-DOX, but not the free DOX administered at the same concentration/dose, can indeed eliminate CSCs both in vitro and in vivo. Furthermore, our results indicate that CSCs, as studied in our model, are actually intrinsically sensitive to DOX. Given the recent strong doubt casted on the feasibility, cost-effectiveness and the clinical success of precision-oncology 57, our findings suggest a new paradigm in cancer drug development in which one can utilize the current chemotherapeutic agents that we have extensive knowledge on their pharmacological and toxicological properties to develop novel delivery strategies in order to overcome chemoresistance. Considering the lengthy and costly road of developing a new drug, the smart delivery of existing anti-cancer drugs is indeed a very attractive alternative to combating cancer.
Over the past 2 decades, many drug delivery systems have been developed for doxorubicin58. In fact, the first US Food and Drug Administration approved nano-drug is Doxil®, a PEGylated nano-liposomal formulation of doxorubicin 59. More recently, efforts were devoted to develop targeted drug delivery system for doxorubicin 58. Some ingenious systems developed include a chemically crosslinked and functionalized biopolymer dextrin based nanogel 60; an anti-CD24 mAb-DOX conjugate via the non-cleavable GMBS (N-[gamma-maleic imide butyl acyl oxygen] succinimide ester) linker 61; a metal organic framework nanocarrier-based codelivery system functionalized with folic acid 62; a urokinase plasminogen activator receptor targeted magnetic iron oxide nanoparticles 63; a double-targeted nondrug delivery system via conjugating hyaluronic acid and grafting the doublecortin-like kinase 1 monoclonal antibody to the surface of PEG-PLGA nanoparticles 64; a nucleolin-binding F3 peptide functionalized liposome 65; a pH-sensitive triblock copolymer vesicles functionalized with EpCAM monoclonal antibodies 66; hyaluronic acid functional amphipathic and redox-responsive polymer particles with a redox-responsive drug release profile 67; EpCAM aptamer conjugated PEG-PLGA nanopolymersomes as well as direct aptamer-DOX conjugates 23, 68, 69. These innovations have significantly improved the pharmacokinetics and curtailed toxicity of doxorubicin in the preclinical experimental systems employed. In addition, these studies have also demonstrated markedly improve pharmacodynamics to the general population of cancer cells. However, it is unclear whether these DOX delivery systems can turn doxorubicin into a cancer stem cell killer. One cannot be ascertained if a DOX-delivery system is able to eliminate cancer stem cell that in the absence of the experimental data using the gold standard methodology for enumeration of cancer stem cell frequency via the limiting dilution transplantation analysis using immunodeficient mice 70, 71. In this contribution, we have provided evidence that our aptamer-DOX system can indeed physically deliver DOX into EpCAM+/CD44+/CD24+ colorectal cancer stem cells in the HT29 tumour xenograft mice model (Figure 6 A) and eliminate such cancer stem cells via apoptosis, which is a well know mode of action by DOX. Most critically, by employing the gold standard CSC functional assay via limiting dilution combined with xenotransplantation, we have provided unequivocal evidence that our EpCAM aptamer can indeed turn DOX into a robust cancer stem cell killer (Supplementary Figure 3, Supplementary Table 1, Supplementary Table 2, Supplementary Table 3, Supplementary Table 5 and Supplementary Table 6). Therefore, for the first time, we have demonstrated that one can indeed transform a traditional anti-cancer drug into a potent cancer stem cell killer via smart drug delivery without the use of nanoparticles. Such a CSC-busting system will facilitate the effective treatment of the disseminated cancer cells that are largely responsible for the death of patients with cancer 72.
In summary, an EpCAM-aptamer-based delivery system has been developed to target CSCs. The high concentration and prolonged retention of DOX delivered by Apt-DOX to the nuclei significantly improved the sensitivity of CSCs to DOX, leading to overcoming chemoresistance and elimination of CSCs both in vitro and in vivo. This novel strategy of drug delivery opens a new avenue for overcoming chemoresistance by transforming traditional chemotherapeutic agents into CSC killers. Further testing of this innovative approach in clinical trials will provide fresh insight into how to overcome chemoresistant cancers.
Materials and Methods
Cell lines and cell culture
Human colorectal adenocarcinoma cell line HT-29 (ATCC®, HTB38™), human ovarian adenocarcinoma cell lines SKOV-3 (ATCC®, HTB77™), and human embryonic kidney cell HEK-293T (ATCC®, CRL-11268™), and human hepatocellular carcinoma cell lines Huh-7 and PLC/PRF/7 (gift from Dr. Liang Qiao, University of Sydney, Australia) were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 10,000 I.U./mL Penicillin, 50 µg/ml streptomycin (Invitrogen, Australia) and 1× Glutamax (Life Technologies, Australia) in a humidified atmosphere containing 5% CO2 at 37 °C.
Animals used in this study
All animals were purchased from The Animal Resources Centre (Perth, Australia). Six to eight weeks old NOD-SCID female mice were used for HT29 tumour xenograft establishment. The mice were housed in TECNIPLAST SealsafeTM individually ventilated cages. Both mice and Male Sprague-Dawley rats (200 to 250 g) were placed in a temperature-controlled room (25 ± 1 °C) with a 12-h light-dark cycle and fed ad libitum with a standard diet. Rats were fasted overnight before treatments administration. Tumour progression was monitored using digital calipers. All experiments were performed in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and Guidelines to Promote the Wellbeing of Animals Used for Scientific Purposes from Australian Government's National Health and Medical Research Council. The Deakin University Animal Ethics Committee approved all experimental protocols.
Aptamers used in this study
Aptamers were synthesized by IBA GmbH (Göttingen, Germany) followed by HPLC purification. The aptamers used in this study were, RNA EpCAM aptamer [5'- (DY647) - A (2'-F-C) G (2'-F-U) A (2'-F-U) (2'-F-C) (2'-F-C) (2'-F-C) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-C) G (2'-F-C) G (2'-F-U) -3'], negative control RNA EpCAM aptamer [5'- (DY647) - A (2'-O-Me-C) G (2'-O-Me-U) A (2'-O-Me-U) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-C) G (2'-O-Me-C) G (2'-O-Me-U) -3'], hybrid DNA-RNA EpCAM aptamer [5'- (DY647) - c g c g c g c c g c A (2'-F-C) G (2'-F-U) A (2'-F-U) (2'-F-C) (2'-F-C) (2'-F-C) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-C) G (2'-F-C) G (2'-F-U) c g g c g c g c g -3'], negative control Hybrid DNA-RNA EpCAM aptamer [5'- (DY647) - c g c g c g c c g c A (2'-O-Me-C) G (2'-O-Me-U) A (2'-O-Me-U) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-C) G (2'-O-Me-C) G (2'-O-Me-U) c g g c g c g c g -3'], PEGylated RNA EpCAM aptamer [5'-(20 kDa PEG-FITC)- c g c g c g c c g c A (2'-F-C) G (2'-F-U) A (2'-F-U) (2'-F-C) (2'-F-C) (2'-F-C) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-C) G (2'-F-C) G (2'-F-U) c g g c g c g c g - (Biotin or DY647) -3'], negative control PEGylated RNA EpCAM aptamer: 5'-(20 kDa PEG-FITC)- c g c g c g c c g c A (2'-O-Me-C) G (2'-O-Me-U) A (2'-O-Me-U) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-C) G (2'-O-Me-C) G (2'-O-Me-U) c g g c g c g c g - (Biotin or DY647) -3'].
In the above sequences, 2'-F represents 2'-fluoropyrimidine, 2'-O-Me indicates 2'-O-methyl modification. The lowercase letters indicate DNAs in which deoxycytidines were modified with 5'-methyl-deoxycytidine (5-Methyl-dC), which increases the Tm by as much as 0.5°C per limits unwanted immune responses 73, 74. The negative control aptamer is an aptamer of the same sequence as the EpCAM targeting aptamer but with a 2'-O-methyl modification at the pyrimidines that changes the 3-dimensional structure of aptamer and thus abolishing the binding of this control aptamer to EpCAM. The aptamers were folded in PBS containing 5 mM MgCl2, by denaturation at 85˚C for 5 min, followed by 10 min incubation at room temperature and refolding at 37˚C for at least 15 min.
Development of aptamer-drug conjugate
EpCAM aptamer was designed for conjugation with doxorubicin (DOX) (SIGMA-ALDRICH). DOX was mixed with folded aptamer in conjugation buffer containing 0.1 M sodium acetate, 0.05 M NaCl, and 5 mM MgCl2, pH 7.4 and incubated in orbital mixer/incubator at 37 ºC for 1 hour at 75 r.p.m. The conjugate mixture was then passed through a Sephadex®G-10 medium column (SIGMA-ALDRICH) to separate the Apt-DOX from free DOX. DOX was quantified as follows. Apt-DOX conjugate (30 µL) was added into 90 µL of acetonitrile and vortex for 1 min, followed by centrifugation for 5 min at 21,000 x g. The supernatant (50 μL) was diluted in 150 µL PBS and mixed well prior to another centrifugation for 5 min at 21,000 g. Sixty to one hundred micro-liters of supernatant was analyzed in a fluorescence VICTORTM X5 Plate Reader (PerkinElmer Life, Australia) at an excitation and emission wavelength of 470 nm and 585 nm, respectively. A standard curve of free DOX was also prepared in parallel.
Evaluation of DOX conjugation with aptamer
The natural fluorescence of DOX and its subsequent quenching after intercalating into the aptamer allows efficient quantification of DOX. Fluorescence intensity in solutions with different aptamer to DOX molar ratios (0, 0.01, 0.02, 0.04, 0.08, 0.1, 0.2, 0.4, 0.6, and 1) were measured using a VICTORTM X5 plate reader (PerkinElmer Life) at ex. 480 nm and em. 575 nm or by HPLC,) and compared with a standard curve of free DOX solution prepared in parallel. The efficient loading of DOX into aptamers was determined with a fixed concentration of DOX incubated with increasing amount of aptamer at a wavelength from 520 nm to 700 nm.
Determination of the stability of Apt-DOX conjugates
The release of DOX from Apt-DOX conjugates in vitro were studied by monitoring the release of DOX from the conjugates using a dialysis method with a Slide-A-Lyzer Dialysis Cassette (ThermoFisher Scientific). For determination of the stability, aptamer-DOX conjugates at an equivalent DOX concentration of 1µg/mL were dialysed against conjugation buffer at 37 °C. At various time points, (10 min, 0.5 h, 1 h, 2h, 4 h, 6 h, 8 h, 24 h, 26 h, 30 h, 32 h, 48 h, 50 h, 54 h and 72 h), 400 µL of each sample outside the dialysis cassette were collected the concentration of free DOX in the dialysis buffer was determined. Accumulative release of DOX from Apt-DOX was expressed as a percentage of the released DOX vs time.
Determination of binding affinity
The equilibrium dissociation constant (K'd) of 2'-F RNA aptamer species to EpCAM proteins expressed on the cell surface was determined using flow cytometry as described in our previous publication75.
Cellular uptake and retention of Apt-DOX
Cells were seeded at a density of 75,000 cells per cm2 in an 8-chamber slide (Lab-Tek II, Nunc) and incubated in blocking buffer (PBS supplemented with 5 mM MgCl2, 0.1 mg/mL tRNA, 0.1 mg⁄mL salmon sperm DNA, and 5% FBS) at room temperature for 20 min. After two washes with PBS, cells were cultured in full DMEM medium (phenol red free) for another 2 h with 50 nM LysoTracker (Life Technologies) in the first 90 min, followed by the incubation with 100 nM folded EpCAM aptamer or 100 nM FITC-EpCAM antibody for 30 min. Cells were then counterstained with 3 ng/mLHoechst 33342 (Sigma). Images were analysed using Image-Pro software (Media Cybernetics).
To determine the internalization of Apt-DOX, cells were prepared and blocked as described above prior to incubation with free DOX and Apt-DOX at an equivalent DOX concentration of 1.5 µM for 30 min and 2 h at 37˚C, (time course)followed by counterstaining with Hoechst 33342. At each time point, solution of DOX or aptamer-DOX conjugates was removed and the cells washed three times prior to visualization via confocal microscopy. As for evaluating the retention of Apt-DOX within tumour cells, HT-29 tumour cells were incubated with phenol red-free DMEM for a further 2 h and 24 h before being imaged. The co-localization of DOX in nucleus was quantified using Image-Pro software.
To quantitatively determine the accumulation of Apt-DOX inside cancer cells, cellular uptake and retention of Apt-DOX compared to that of free DOX was determined. After 10 and 30 min incubation of HT29 sphere cells with free DOX, Apt-DOX and Ctrl-Apt-DOX (at an equivalent concentration of 1.5 µM DOX), cells were washed with PBS and incubated with phenol red-free media red for a further 2 and 24 h, respectively.The dose-dependent analysis was performed by incubating cells with DOX and aptamer-DOX at concentrations equivalent to 0.5, 1 and 2 µM DOX. Untreated cells were used as a control.
Analysis of tumoursphere penetration of Apt-DOX
Two thousand HT29 and HEK293T cells were plated out in ultralow attachment wells (Corning, Germany) and allowed to form tumourspheres for 3-5 days in stem cell media (DMEM/F12 media supplemented with B27 (100 units/mL), Insulin (10 µg/mL), EGF (20 ng/mL) and bFGF (20 ng/mL). After being washed three times in PBS containing 5 mM MgCl2 and blocked for 20 min using blocking buffer, the tumourspheres were then incubated with 100 nM of Apt-DOX for 30 min followed by three washes with PBS prior to visualization using confocal microscopy.
Tumour implantation and evaluation
To establish xenograft tumours, single HT29 cell suspension was harvested after trypsinization. The cells (1 × 105) resuspended in DMEM and Matrigel (50:50 = V:V) were injected into the flank of NOD/SCID mouse. Once tumours arose, mice were randomized into treatment groups of 4 mice per group. Treatment was initiated when the tumour volume reached 150 mm3. Tumour fragments were archived in 10% neutral buffered formalin and subjected to immunohistochemistry analysis (double staining of anti-CD31 and aptamer or antibody).
Chromatographic instrumentation and system
A high performance liquid chromatography (HPLC) system consisting of a Waters e2695 Separation Module and a Waters 2475 Multi λ Fluorescence Detector was used with the excitation and emission wavelengths being 470 nm and 585 nm, respectively. Chromatographic separation was performed using a Nova-Pak® C18 column (3.9 × 150 mm i.d., 4 μm, Waters, USA) using the chromatographic process described previously 76.
Pharmacokinetics (PK) study
Healthy male Sprague-Dawley rats (200 to 250 g, n=3) were injected i.v. with agents at an equivalent dose of DOX 5 mg/kg. After injection, blood was serially collected from animals tail vein into heparinised tubes from the tail vein at the time points of 10 min, 0.5 h, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 10 h, 12 h, 14 h and 24 h. Blood samples (200 μL) were centrifuged at 3,000 g at 4 °C for 10 min to separate the plasma. The supernatant was stored at -20 °C until determination of DOX by HPLC. The pharmacokinetic parameters were analysed using the DAS 2.0 software (Mathematical Pharmacology Professional Committee of China).
Bio-distribution assay
HT29 tumour (an average volume of 150 mm3)-bearing NOD/SCID mice were randomly divided into three treatment groups as above with 3 mice per group. Agents were delivered via tail vein injection with an equivalent dose of DOX 5 mg/kg. Tissues and tumours were collected 3 h, 6h and 24 h post treatments. The concentration of DOX was determined using HPLC77 while the aptamer concentration was quantified by aptamer-ELISA78.
Tumour dissociation
HT29 xenograft tumour tissues were washed thoroughly with Hank's buffer (containing 1% penicillin/strep), and minced into approximately 2 - 4 mm3 followed by additional three washes in Hank's buffer. The minced tissues were digested with 50-100 U/mL collagenase in stem cell media, with a medium to tissue ratio of 6 mL/gram at 37 °C for 3 h for cell sorting or overnight for replating. The samples were then pipetted 10 times and filtered through 40 µm cell strainers. Cells were collected through centrifugation at 1000 g for 5 min, resuspended and washed in PBS twice followed by cell viability assay using Trypan blue staining.
Fluorescence-activated cell sorting
Cells were analysed using FACS-Canto II or sorted using the FACSAria flow cytometer as described in our previous publication78. The antibodies used were: human-specific antibodies to PE-CD24 (1:25 dilution, BD Biosciences, Cat. No: 555428), V450-CD44 (1:100 dilution, BD Biosciences, Cat. No: 561292) and FITC-EpCAM (1:10 dilution) (BD Biosciences, Cat No: 347197). Cells were routinely sorted twice, and an aliquot of sorted the cells (1 × 105) was reanalyzed for purity, which typically was > 95%. For surface marker analysis, a minimum of 10,000 events were analysed for each sample from three independent experiments.
Tumoursphere formation and ex vivo tumorigenicity assays
The colonosphere assay was conducted according to previously reported protocols 79. Briefly, cells were harvested at 80% confluence with trypsin digestion and resuspended as single cells in stem cell medium. Cells were plated into round-bottom 96-well ultralow attachment plates (Corning) at a density of 1, 5, 10, 100, and 200 cells per well, or at 8000 cells /well in 6-well flat-bottom ultralow attachment plates. Six experiment groups were used: saline, free aptamer, free DOX treatment (1 µM), Ctrl-Apt-DOX and Apt-DOX treatment (equivalent to 1 µM free DOX) and salinomycin (1 µM). The frequency of CSCs was calculated using the ELDA website (http://bioinf.wehi.edu.au/software/elda/index.html). The tumoursphere formation frequency in 6-well plates was calculated according to the formula F= Numbers of forming tumourspheres / Number of single cells plated. For secondary and tertiary tumoursphere formation, single cell suspension prepared from the previous generation of tumourspheres were re-plated under the same conditions as the first generation.
For LDA, HT29 cells were cultured in ultra-low attachment plate and maintained in stem cell medium. Following 5 - 7 days of treatment with free Aptamer, free DOX and Apt-DOX (2 µM), single cell preparation of HT29 sphere-forming cells were injected subcutaneously (s.c.) into NOD/SCID mice at a dose of 1x 104 and 1 x 105 in serum-free DMEM/Matrigel (V:V = 1:1). Tumour volume was monitored and calculated as described77.
In vivo tumorigenicity assay
HT29 tumour-bearing NOD/SCID mice (60 mm3) were randomly assigned to six groups (n=4) and received i.v. injection of agents indicated. One day after the last treatment, viable single cell suspensions dissociated from each tumour were inoculated into four sub-groups of mice using cell doses of 1 × 105, 1 × 104, 1 × 103 and 1 × 102/mouse, 4 mice per sub-group. The growth of tumour was evaluated daily over a 3-month period. The animal ethics endpoint was tumour reaching a size of 17 mm or the loss of 10% weight. The CSC frequency was derived as described above.
Immunohistochemistry
The tumour tissues dissected from tumour-bearing mice were subjected to immunohistochemistry analysis using methods described in our previous publication 80. The antibodies used include mouse anti-human Ki-67 antibody (Abcam, Cat No: ab15580, 1:100 dilution) and goat anti-mouse HRP-labelled secondary antibody (Thermo Fisher, Cat No: 31430) as well as DAB peroxidase substrate solution (Vector Laboratories, Burlingame, CA). For performing the TUNEL assay, the indicated slides were performed using the ApopTag Red In Situ Apoptosis Detection Kit (Millipore) according to the manufactural instruction.
Evaluation of antitumour efficacy and survival rate
HT29 tumour (50 mm3) bearing mice were grouped and treated using the same method as that for in vivo tumorigenicity assay. The mice were evaluated daily for disease-free survival and disease-related events. The Kaplan-Meier Survival Curves were derived using a Log-Rank test with a 95% confidence interval.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 6.0 (San Diego, CA, USA). An unpaired t test was used for comparisons between two experimental groups, and ANOVA was used for comparisons of more than two groups. Unless otherwise indicated, all results were averaged from biological triplicates and values are reported as means ± SEM. P < 0.05 was considered statistically significant.
Acknowledgements
D. Xiang was supported by a Deakin University Postgraduate Research Scholarship. This work was supported by grants from Indo-Australia Science and Technology Fund. (Grant No. ST040007), Victorian Cancer Agency Platform Technology Capacity Building Grant (Grant No. PTCP-02) and CASS Foundation (Australia), and from Natural Science Foundation of Shanghai, China (No. 16ZR1425900).
Author Contributions
W.D. conceived and designed the research. S.S. contributed to the design of the experiments. D. X. A.B, M.B., H.A.S, T.W, M.M and W.Y. performed experiments. D.X., L.K., A.H. and Y.L. collected and analyzed the data. W.D., A.H., K.L., S.Z. and Y.H., wrote the manuscript. A.H. Y.L., R.B, P.Z., P.T., A.B., provided materials and/or revised the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cardinale D, Colombo A, Lamantia G. et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol. 2010;55:213-220
2. MacKay JA, Chen M, McDaniel JR. et al. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater. 2009;8:993-999
3. Paoletti X, Le Tourneau C, Verweij J. et al. Defining dose-limiting toxicity for phase 1 trials of molecularly targeted agents: results of a DLT-TARGETT international survey. Eur J Cancer (Oxford, England: 1990). 2014;50:2050-2056
4. Le Tourneau C, Razak ARA, Gan HK. et al. Heterogeneity in the definition of dose-limiting toxicity in phase I cancer clinical trials of molecularly targeted agents: a review of the literature. Eur. J. Cancer (Oxford, England: 1990). 2011;47:1468-1475
5. Shackleton M, Quintana E, Fearon ER & Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822-829
6. O'Connor ML, Xiang D, Shigdar S. et al. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014;344:180-187
7. Visvader JE & Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755-768
8. Bao S, Wu Q, McLendon RE. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-760
9. Diehn M, Cho RW, Lobo NA. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780-783
10. Eyler CE & Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839-2845
11. Davis ME, Chen ZG & Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-782
12. Cabral H, Nishiyama N & Kataoka K. Supramolecular nanodevices: from design validation to theranostic nanomedicine. Acc Chem Res. 2011;44:999-1008
13. Bertrand N, Wu J, Xu X, Kamaly N & Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2-25
14. Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750-763
15. Modery-Pawlowski CL & Gupta AS. Heteromultivalent ligand-decoration for actively targeted nanomedicine. Biomaterials. 2014;35:2568-2579
16. Harding FA, Stickler MM, Razo J & DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256-265
17. Schrama D, Reisfeld RA & Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147-159
18. Radom F, Jurek PM, Mazurek MP, Otlewski J & Jelen F. Aptamers: molecules of great potential. Biotechnol Adv. 2013;31:1260-1274
19. Panowksi S, Bhakta S, Raab H, Polakis P & Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6:34-45
20. Keefe AD, Pai S & Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537-550
21. Lyu Y, Chen G, Shangguan D. et al. Generating Cell Targeting Aptamers for Nanotheranostics Using Cell-SELEX. Theranostics. 2016;6:1440-1452
22. Wu X, Zhao Z, Bai H. et al. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics. 2015;5:985-994
23. Huang YF, Shangguan D, Liu H. et al. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem. 2009;10:862-868
24. Xiong X, Liu H, Zhao Z. et al. DNA aptamer-mediated cell targeting. Angew Chem Int Ed Engl. 2013;52:1472-1476
25. Shigdar S, Lin J, Yu Y. et al. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102:991-998
26. Shigdar S, Qiao L, Zhou SF. et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330:84-95
27. Yang F, Teves SS, Kemp CJ & Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 2014;1845:84-89
28. Macdonald J, Henri J, Goodman L, Xiang D, Duan W & Shigdar S. Development of a bi-functional aptamer targeting the transferrin receptor and EpCAM for the treatment of brain cancer metastases. ACS Chem Neurosci. 2017;8:777-784
29. Xu W, Siddiqui IA, Nihal M. et al. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted therapy of prostate cancer. Biomaterials. 2013;34:5244-5253
30. Suva ML, Rheinbay E, Gillespie SM. et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580-594
31. Gupta PB, Chaffer CL & Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010-1012
32. Choi HS, Liu W, Misra P. et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165-1170
33. Han Y, Liu Y, Nie L, Gui Y & Cai Z. Inducing cell proliferation inhibition, apoptosis, and motility reduction by silencing long noncoding ribonucleic acid metastasis-associated lung adenocarcinoma transcript 1 in urothelial carcinoma of the bladder. Urology. 2013;81:209 e201-207
34. Mo J, Sun B, Zhao X. et al. The in-vitro spheroid culture induces a more highly differentiated but tumorigenic population from melanoma cell lines. Melanoma Res. 2013;23:254-263
35. Illa-Bochaca I, Fernandez-Gonzalez R, Shelton DN. et al. Limiting-dilution transplantation assays in mammary stem cell studies. Methods Mol Biol. 2010;621:29-47
36. Hu Y & Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70-78
37. Sahlberg SH, Spiegelberg D, Glimelius B, Stenerlow B & Nestor M. Evaluation of cancer stem cell markers CD133, CD44, CD24: association with AKT isoforms and radiation resistance in colon cancer cells. PLoS One. 2014;9:e94621
38. Abdul Khalek FJ, Gallicano GI & Mishra L. Colon cancer stem cells. Gastrointest Cancer Res. 2010:S16-23
39. Xiang D, Shigdar S, Qiao G. et al. Nucleic Acid Aptamer-Guided Cancer Therapeutics and Diagnostics: the Next Generation of Cancer Medicine. Theranostics. 2015;5:23-42
40. Shi Y, Moon M, Dawood S, McManus B & Liu PP. Mechanisms and management of doxorubicin cardiotoxicity. Herz. 2011;36:296-305
41. Terris B, Cavard C & Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol. 2010;52:280-281
42. Yamashita T, Forgues M, Wang W. et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451-1461
43. Calcagno AM, Fostel JM, To KK. et al. Single-step doxorubicin-selected cancer cells overexpress the ABCG2 drug transporter through epigenetic changes. Br J Cancer. 2008;98:1515-1524
44. Zhuang X, Zhang W, Chen Y. et al. Doxorubicin-enriched, ALDHbr mouse breast cancer stem cells are treatable to oncolytic herpes simplex virus type 1. BMC Cancer. 2012;12:1-16
45. Liu H, Lv L & Yang K. Chemotherapy targeting cancer stem cells. Am J Cancer Res. 2015;5:880-893
46. Zheng XQ, Cui D, Xu SH, Brabant G & Derwahl M. Doxorubicin fails to eradicate cancer stem cells derived from anaplastic thyroid carcinoma cells: Characterization of resistant cells. Int J Oncol. 2010;37:307-315
47. Maugeri-Sacca M, Di Martino S & De Maria R. Biological and clinical implications of cancer stem cells in primary brain tumors. Front Oncol. 2013;3:6
48. Maugeri-Sacca M, Vigneri P & De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17:4942-4947
49. Opyrchal M, Salisbury JL, Iankov I. et al. Inhibition of Cdk2 kinase activity selectively targets the CD44(+)/CD24(-)/Low stem-like subpopulation and restores chemosensitivity of SUM149PT triple-negative breast cancer cells. Int J Oncol. 2014;45:1193-1199
50. Mercado-Lubo R, Zhang Y, Zhao L. et al. A Salmonella nanoparticle mimic overcomes multidrug resistance in tumours. Nat Commun. 2016;7:12225
51. Yalcintepe L, Halis E & Ulku S. Effect of CD38 on the multidrug resistance of human chronic myelogenous leukemia K562 cells to doxorubicin. Oncol Lett. 2016;11:2290-2296
52. Todaro M, Alea MP, Di Stefano AB. et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389-402
53. Shmelkov SV, Butler JM, Hooper AT. et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118:2111-2120
54. Damia G & Garattini S. The pharmacological point of view of resistance to therapy in tumors. Cancer Treat Rev. 2014;40:909-916
55. Chan KS. Molecular Pathways: Targeting Cancer Stem Cells Awakened by Chemotherapy to Abrogate Tumor Repopulation. Clin Cancer Res. 2016;22:802-806
56. Zhao J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol Ther. 2016;160:145-158
57. Prasad V. Perspective: The precision-oncology illusion. Nature. 2016;537:S63
58. Patil RR, Guhagarkar SA & Devarajan PV. Engineered nanocarriers of doxorubicin: a current update. Crit Rev Ther Drug Carrier Syst. 2008;25:1-61
59. Barenholz Y. Doxil(R)-the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117-134
60. Das D, Rameshbabu AP, Ghosh P. et al. Biocompatible nanogel derived from functionalized dextrin for targeted delivery of doxorubicin hydrochloride to MG 63 cancer cells. Carbohydr Polym. 2017;171:27-38
61. Ma Z, He H, Sun F. et al. Selective targeted delivery of doxorubicin via conjugating to anti-CD24 antibody results in enhanced antitumor potency for hepatocellular carcinoma both in vitro and in vivo. J Cancer Res Clin Oncol. 2017
62. Zhang H, Jiang W, Liu R. et al. Rational Design of Metal Organic Framework Nanocarrier-Based Codelivery System of Doxorubicin Hydrochloride/Verapamil Hydrochloride for Overcoming Multidrug Resistance with Efficient Targeted Cancer Therapy. ACS Appl Mater Interfaces. 2017;9:19687-19697
63. Gao N, Bozeman EN, Qian W. et al. Tumor Penetrating Theranostic Nanoparticles for Enhancement of Targeted and Image-guided Drug Delivery into Peritoneal Tumors following Intraperitoneal Delivery. Theranostics. 2017;7:1689-1704
64. Qiao S, Zhao Y, Geng S. et al. A novel double-targeted nondrug delivery system for targeting cancer stem cells. Int J Nanomedicine. 2016;11:6667-6678
65. Fonseca NA, Rodrigues AS, Rodrigues-Santos P. et al. Nucleolin overexpression in breast cancer cell sub-populations with different stem-like phenotype enables targeted intracellular delivery of synergistic drug combination. Biomaterials. 2015;69:76-88
66. Chen J, Liu Q, Xiao J & Du J. EpCAM-Antibody-Labeled Noncytotoxic Polymer Vesicles for Cancer Stem Cells-Targeted Delivery of Anticancer Drug and siRNA. Biomacromolecules. 2015;16:1695-1705
67. Hu K, Zhou H, Liu Y. et al. Hyaluronic acid functional amphipathic and redox-responsive polymer particles for the co-delivery of doxorubicin and cyclopamine to eradicate breast cancer cells and cancer stem cells. Nanoscale. 2015;7:8607-8618
68. Alibolandi M, Ramezani M, Sadeghi F, Abnous K & Hadizadeh F. Epithelial cell adhesion molecule aptamer conjugated PEG-PLGA nanopolymersomes for targeted delivery of doxorubicin to human breast adenocarcinoma cell line in vitro. Int J Pharm. 2015;479:241-251
69. Subramanian N, Raghunathan V, Kanwar JR. et al. Target-specific delivery of doxorubicin to retinoblastoma using epithelial cell adhesion molecule aptamer. Mol Vis. 2012;18:2783-2795
70. Heuser M & Humphries RK. Biologic and experimental variation of measured cancer stem cells. Cell Cycle. 2010;9:909-912
71. O'Brien CA, Kreso A & Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16:3113-3120
72. Ghajar CM & Bissell MJ. Metastasis: Pathways of parallel progression. Nature. 2016
73. Lennox KA, Owczarzy R, Thomas DM, Walder JA & Behlke MA. Improved Performance of Anti-miRNA Oligonucleotides Using a Novel Non-Nucleotide Modifier. Mol Ther Nucleic Acids. 2013;2:e117
74. Wan WB & Seth PP. The Medicinal Chemistry of Therapeutic Oligonucleotides. J Med Chem. 2016;59:9645-9667
75. Xiang D, Zheng C, Zhou SF. et al. Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors. Theranostics. 2015;5:1083-1097
76. Alvarez-Cedron L, Sayalero ML & Lanao JM. High-performance liquid chromatographic validated assay of doxorubicin in rat plasma and tissues. J Chromatogr B Biomed Sci Appl. 1999;721:271-278
77. Li L, Xiang D, Shigdar S. et al. Epithelial cell adhesion molecule aptamer functionalized PLGA-lecithin-curcumin-PEG nanoparticles for targeted drug delivery to human colorectal adenocarcinoma cells. Int J Nanomedicine. 2014;9:1083-1096
78. Wang T, Gantier MP, Xiang D. et al. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics. 2015;5:1456-1472
79. Shaw FL, Harrison H, Spence K. et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia. 2012;17:111-117
80. Shigdar S, Lv L, Wang L & Duan W. Application of Aptamers in Histopathology. Methods Mol Biol. 2016;1380:191-196
Author contact
![]() Corresponding author: Dr. Aina He, Department of Oncology, Affiliated Sixth People's Hospital, Shanghai Jiaotong University, No. 600, Yishan Road, Shanghai 200233, P. R. China. Tel: +86 21 2405 8430; E-mail: ai_na_hecom (A. He). Professor Wei Duan, School of Medicine, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria, 3217, Australia. Tel: +61 3 52271149; Fax: +61 3 52272945; E-mail: wduanedu.au (W. Duan)
Corresponding author: Dr. Aina He, Department of Oncology, Affiliated Sixth People's Hospital, Shanghai Jiaotong University, No. 600, Yishan Road, Shanghai 200233, P. R. China. Tel: +86 21 2405 8430; E-mail: ai_na_hecom (A. He). Professor Wei Duan, School of Medicine, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria, 3217, Australia. Tel: +61 3 52271149; Fax: +61 3 52272945; E-mail: wduanedu.au (W. Duan)