Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(16):4041-4056. doi:10.7150/thno.19989 This issue Cite
Research Paper
Notch3 Maintains Luminal Phenotype and Suppresses Tumorigenesis and Metastasis of Breast Cancer via Trans-Activating Estrogen Receptor-α
Xiao-Wei Dou1, 2*, Yuan-Ke Liang1, 2, 3*, Hao-Yu Lin1, 2, 4, Xiao-Long Wei1, 2, 5, Yong-Qu Zhang1, 2, Jing-Wen Bai1, 2, Chun-Fa Chen1, 2, Min Chen2, Cai-Wen Du6, Yao-Chen Li1, 2, Jie Tian7, Kwan Man8, Guo-Jun Zhang1, 2 ![]()
1. The Breast Center, the Cancer Hospital of Shantou University Medical College (SUMC), China;
2. ChangJiang Scholar's Laboratory, the Cancer Hospital of Shantou University Medical College (SUMC), China;
3. Department of Medical Oncology, University of Groningen, University Medical Center Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands;
4. Department of Breast and Thyroid Surgery, the First Affiliated Hospital of SUMC;
5. Department of Pathology, the Cancer Hospital of Shantou University Medical College (SUMC), China;
6. Department of Breast Medical Oncology, the Cancer Hospital of Shantou University Medical College (SUMC), China;
7. Institute of Automation, Chinese Academy of Science, China;
8. Department of Surgery, Hong Kong University Li Ka-Tsing faculty of Medicine, Hong Kong, China.
* These authors contributed this work equally.
Received 2017-3-8; Accepted 2017-8-11; Published 2017-9-20
Abstract

The luminal A phenotype is the most common breast cancer subtype and is characterized by estrogen receptor α expression (ERα). Identification of the key regulator that governs the luminal phenotype of breast cancer will clarify the pathogenic mechanism and provide novel therapeutic strategies for this subtype of cancer. ERα signaling pathway sustains the epithelial phenotype and inhibits the epithelial-mesenchymal transition (EMT) of breast cancer. In this study, we demonstrate that Notch3 positively associates with ERα in both breast cancer cell lines and human breast cancer tissues. We found that overexpression of Notch3 intra-cellular domain, a Notch3 active form (N3ICD), in ERα negative breast cancer cells re-activated ERα, while knock-down of Notch3 reduced ERα transcript and proteins, with alteration of down-stream genes, suggesting its ability to regulate ERα. Mechanistically, our results show that Notch3 specifically binds to the CSL binding element of the ERα promoter and activates ERα expression. Moreover, Notch3 suppressed EMT, while suppression of Notch3 promoted EMT in cellular assay. Overexpressing N3ICD in triple-negative breast cancer suppressed tumorigenesis and metastasis in vivo. Conversely, depletion of Notch3 in luminal breast cancer promoted metastasis in vivo. Furthermore, Notch3 transcripts were significantly associated with prolonged relapse-free survival in breast cancer, in particular in ERα positive breast cancer patients. Our observations demonstrate that Notch3 governs the luminal phenotype via trans-activating ERα expression in breast cancer. These findings delineate the role of a Notch3/ERα axis in maintaining the luminal phenotype and inhibiting tumorigenesis and metastasis in breast cancer, providing a novel strategy to re-sensitize ERα negative or low-expressing breast cancers to hormone therapy.
Keywords: Notch3, estrogen receptor α, epithelial-mesenchymal transition, breast cancer, luminal phenotype.
Introduction
The majority of breast cancer deaths result from metastasis at distant sites [1]. One of the pivotal processes that induces tumor metastasis is conversion of epithelial cells to a mesenchymal phenotype, or the epithelial-mesenchymal transition (EMT) [2]. ERα, a typical marker of the luminal epithelial phenotype in breast cancer cells, is a good indicator of breast cancers that respond to endocrine therapy. E2/ERα signaling promotes the differentiation of mammary epithelia along a luminal/epithelial lineage, in part through transcriptional activation of luminal/epithelial-related transcription factors [3]. For instance, in ERα positive breast cancer cells, knockdown of ERα decreased expression of the luminal epithelial marker E-cadherin and promoted invasion and motility [4-7]. ERα is thought to inhibit invasion by repressing Slug transcription and by phosphorylating and inactivating GSK-3β [4]. Thus, loss of ERα could promote metastasis of breast cancer cells [8, 9]. However, the mechanisms that regulate ERα in breast cancer cells are still poorly understood. Thus, elucidating the regulatory mechanisms of ERα may aid in understanding breast cancer metastasis and facilitate development of novel targets for breast cancer therapy.
Notch family members play a critical role in the tumorigenesis and metastasis of breast cancers, and development of therapeutic agents that target the key steps in the Notch signaling pathway may provide strategies to inhibit tumor growth [10]. Increasing evidence demonstrates that Notch family proteins have distinct activities and biological functions [11]. Hao et al. demonstrated that Notch1 promotes growth of ERα-positive breast cancer [12]. Harrison et al. showed that Notch4 and Notch1 enhance breast cancer stem cell activity, and inhibition of Notch4 or Notch1 reduces tumor formation in vivo [13]. Yen et al. confirmed that suppression of Notch2/Notch3 inhibits tumor growth and reduces the number of stem cells in breast, lung, ovarian, and pancreatic cancers [14-16].
Little is known about Notch3's potential roles in cancer development. Bouras et al. found that Notch3 levels are specifically elevated in mouse mammary luminal progenitor and epithelial cells, but not in mammary stem cells [17], and similar results were also reported by transcriptome analysis [18]. In transformed breast cells, Notch3 is required for breast luminal filling by inhibiting apoptosis [19]. In transgenic mice, Notch3 promotes lobular-alveolar epithelial cell expansion and leads to tumor formation [20]. As ERα promotes mammary luminal/epithelial cell differentiation [3], these results suggest a close correlation between Notch3 and ERα expression in the luminal epithelial differentiation during breast development and breast cancer. However, Notch3 has been shown to play different or even opposing roles in modulating EMT in various cancers. For example, Notch3 induces EMT and promotes metastasis and invasion in glioma, hepatocellular carcinoma and ovarian cancer cells [21-23]. In contrast, Notch3 was shown to promote glandular differentiation and squamous differentiation in gastric and esophageal cancers, respectively, and reverses EMT or promotes mesenchymal epithelial transition (MET) [24, 25]. Recently, our group showed that Notch3 inhibits cell cycle progression by inducing Cdh1 and suppresses EMT through activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells [26,27], suggesting Notch3's role in tumorigenesis and metastasis in breast cancers.
These collective findings prompted us to investigate the role of Notch3 in tumorigenesis/metastasis and ERα regulation of breast cancer, particularly its relationship to molecular subtypes of breast cancer. In this study, we investigated the regulatory mechanisms of EMT via Notch3 and ERα expressions in breast cancer.
Materials and Methods
Cell culture and transfection
MCF-7, T47D, SKBR3, BT-549 and MDA-MB-231 cell lines were obtained from the American Type Tissue Collection and cultured in DMEM medium supplemented with 10% FCS and penicillin/streptomycin. For Notch3 knockdown in MCF-7 cells, siRNAs targeting Notch3 and control siRNAs were synthesized by GenePharma (Suzhou, China). Oligonucleotide siRNAs are listed in Supplementary Table S2. For Notch3 overexpression in MDA-MB-231 cells, pCLE-N3ICD (Plasmid 26894) and control vector pCLE (Plasmid 17703) were obtained from Addgene (MA, USA). A stable Notch3-silenced cell line, MCF-7shN3, was generated by transfection of pGPU6/GFP/Neo-shNotch3#1 containing oligonucleotides specifically targeting Notch3, while its control cell line was also established with vector containing scramble sequence (see Supplementary Table S2) (GenePharma, Suzhou, China). A stable Notch3 ICD over-expressing cell line, MDA-MB-231-N3ICD, was generated by transfection of the pCLE-N3ICD vector. Lipofectamine 2000 was used for transfection according to the manufacturer's instructions. To establish stable MCF-7-shN3 and MDA-MB-231-N3ICD cell lines, 1 μg/mL of G418 was added to the medium, for selection, 2 days following transfection.
RNA purification and real-time PCR analysis
RNA purification and cDNA synthesis were performed using Takara kits according to the manufacturer's instructions. To detect mRNA expression, quantitative real-time PCR was performed with SYBR Select Master Mix (Thermo Fisher, MA, USA) and the CFX96 Real-time PCR Detection System (Bio-Rad, CA, USA). Primer sequences used in real-time PCR are listed in Supplementary Table S2.
Western blot analysis
Protein extraction and western blot were performed as described previously [28]. Characteristics of the antibodies used are listed in Supplementary Table S3.
Immunofluorescence
After 48 h of transfection, cells were fixed with 4% paraformaldehyde for 10 min, then washed in cold PBS 3 times. Then, cells were permeabilized with 0.5% Triton-X-100 in PBS for 20 min. Cells were washed with cold PBS 3 times and blocked in 3% BSA in PBS for 1 h at room temperature and then incubated overnight at 4°C with primary antibody diluted in blocking buffer. After that, cells were washed with cold PBS 3 times and incubated with appropriate secondary antibodies (Alexa Fluor 594 Donkey Anti-Rabbit IgG, Alexa Fluor 488 Donkey Anti-Mouse IgG). Cells were washed again in PBS 3 times, and counterstained for 10 min with DAPI (Life, USA). Images were visualized with an inverted microscope.
IHC and scoring
Tissue sample processing and immunohistochemistry were performed as previously described [29]. Briefly, the sample was processed by deparaffinization, rehydration, antigen retrieval, IHC staining and pathology scoring. Scores include the proportion of positive-staining tumor cells and staining intensities. The proportion of positive tumor cells were given scores of 0 (no tumor cells stained), 1 (<10%), 2 (10%-50%), or 3 (>50%). The intensity of staining was scored as 0 (no staining), 1 (light yellow), 2 (yellow), or 3 (brown). The score of intensity × proportion was regarded as low expression (<3) and high expression (≥3). Interpretation of IHC results were evaluated by two independent observers. ERα was scored as negative (-) and positive (+) staining according to expression. The antibodies used in immunohistochemistry staining are listed in Supplementary Table S3.
Wound healing assay
Cells were pretreated with mitomycin C (25 mg/mL) 30 min before a scratch wound was applied. The wound was made with a 2 mm-wide tip on cells plated in culture dishes at 90% confluency. After rinsing with PBS, cells were allowed to migrate in complete medium, and photographs were taken (×40) after 24 h (MDA-MB-231) or 48 h (MCF-7). An average of five random widths along the injury line were measured for quantitation.
Transwell migration and invasion assay
Cell culture inserts (8 μM pore size; BD) and Matrigel invasion chambers (BD) were used to perform migration and invasion assays, respectively. Transfected cells were serum-starved for 24 h and 2×104 MDA-MB-231 cells or 5×104 MCF-7 cells in serum-free medium were inoculated in the upper chamber. Complete medium was added to the bottom chamber. Cells were stained with 0.1% crystal violet for migration assays at 48 h (MDA-MB-231) or 48 h (MCF-7) and 48 h (MDA-MB-231and MCF-7) for invasion assays. Each assay was performed in triplicate. The number of cells from 5 fields in each well was counted by two independent investigators.
Transient transfection and dual-luciferase reporter assays
The ERα promoter (-928 upstream of exon1 and extending to +72 bp) was cloned into the NheI/BglII sites of a luciferase reporter vector (Panomics, USA). The ERα promoter deletion constructs Δ2CSL (-928 to -389 bp upstream of exon1) and ΔCSL (-928 to -169 bp upstream of exon1) were engineered into the same reporter vector. Transfection was performed using Lipofectamine 2000 according to the manufacturer's instructions. pRL-SV40 (Promega, WI, USA) was used as control vector to normalize transfection efficiency. To detect the influence of silenced or overexpressed Notch3 on ERα promoter activity, the ERα reporter construct or its deletion constructs were co-transfected into MCF-7 cells with siRNA or expression vectors. A dual luciferase reporter assay kit (Promega, WI, USA) was employed to detect luciferase activity. Results were obtained by dual luciferase reporter assay or BLI using a Xenogeny IVIS Kinetic system.
Chromatin immunoprecipitation (ChIP) assay
MCF-7 cells were cultured in a 100 mm dish to 80% confluence. CHIP assays were performed as previously described [21]. Cell lysates were incubated with IgG (Santa Cruz Biotechnology) as an IP control or an antibody specific to Notch3 (CST). Cell lysates (10%) were used for input PCR amplification. The PCR reaction amplified the 239 bp (located -527∽-298 bp, ERαP2) and 230 bp products (located -328∽-90bp, ERαP1) from the ERα promoter containing CSL binding elements (located -380∽-384 bp and -159∽-155 bp upstream of exon 1). A 243 bp PCR reaction product (located -628∽-386 bp), which was near the CSL binding elements, but did not contain them, was used as a negative control. Primers and antibodies used in ChIP assays are listed in Supplementary Tables S2 and S3, respectively.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were obtained from MCF-7 cells. Oligonucleotides containing CSL binding elements were used in EMSAs. EMSA was performed according to manufacturer's instructions (Viagene, FL, USA). In competition assays, excessive amounts of unlabeled competitor were added 20 min before addition of the labeled probes. For the supershift assay, anti-Notch3 monoclonal antibody (2 μg, CST) was added and incubated at 4°C for 60 min. Probe sequences and mutated competitors used in EMSA are shown in Supplementary Tables S2.
Tumor xenograft models
All animal protocols were approved by the Animal Care and Use Committee of Shantou University Medical College (SUMC). We used 6-week-old Nu/Nu female mice (purchased from Vital River, Beijing, China) for all the experiments. Briefly, MDA-MB-231NC-Fluc or MDA-MB-231N3ICD-Fluc cells were injected via the lateral tail vein (1×106 cells) or unilaterally injected into the fourth mammary fat pad (2×106 cells) of each mouse. Tumor growth was monitored by measuring the length and width of tumors twice a week. Tumor volumes were estimated using the formula: length×width2×0.5. The development of whole-body metastasis was monitored by an IVIS Kinetic imaging system (PerkinElmer, MA, USA) once every 3 days. 15 min before imaging, mice were injected i.p. with 150 mg/kg D-luciferin (PerkinElmer, MA, USA). Mice were euthanized after 31 days following vein tail injection, or 42 days following mammary fat pad injection. At the time of killing, organs were harvested and visualized by the IVIS Kinetic imaging system. Tumor xenografts, lungs and livers were excised, fixed in 4% paraformaldehyde and embedded in paraffin. HE staining and immunohistochemistry were performed for tumor phenotype or detection of Notch3, ERα, E-cadherin and vimentin expression. For MCF-7 cells, Nu/Nu female mice were anesthetized and 2×106 MCF-7shNC or MCF-7shN3 cells were injected via the tail vein. Mice were euthanized after 8 weeks. Lungs and livers were excised and fixed in 4% paraformaldehyde and embedded in paraffin. H&E staining was performed on the harvested organs. Tumor formation was identified by experts of the Department of Pathology in the Cancer Hospital of SUMC.
Kaplan Meier analysis of Notch3 expression in breast cancer patients
A publicly accessible online clinical database (http://kmplot.com) was used to assess the association between Notch3 expression and recurrence-free survival information in 4142 breast cancer patients [30, 31]. Kaplan-Meier survival curves, Hazard ratio (HR), 95 % confidence intervals and log-rank P were calculated and displayed on the webpage according to Notch3 expression status.
Statistical analysis
All experiments were performed in triplicate. Data are shown as the mean ± SEM, unless otherwise stated, and were statistically analyzed using a two-sided Student's t-test. P<0.05 was considered statistically significant. Kaplan-Meier survival curve, HR with 95 % confidence intervals and log-rank P value were calculated and plotted in R using Bio-conductor packages.
Study approval
The study was approved by the ethics committee of the Cancer Hospital of Shantou University Medical College, and a written informed consent was obtained for all patients enrolled in this study. The protocol for animal study was approved by the Institutional Animal Care and Use Committee of SUMC.
Results
Notch3 expression is correlated with the luminal epithelial phenotype in breast cancer
To investigate the potential role of Notch signaling in breast cancer, we examined Notch expression in breast cancer cell lines. Western blotting showed that both full-length (FL) Notch3 and Notch3 intracellular domain (ICD) were mainly expressed in breast cancer cells with a luminal epithelial phenotype, as shown by increased ERα and E-cadherin expression in MCF-7 and T-47D cell lines (Figure 1A). However, Notch3 expression was not detected in either basal-like MDA-MB-231 and BT-549 breast cancer cell lines, or the HER2-positive SK-BR-3 breast cancer cell line, which expresses the stromal cell marker vimentin (Figure 1A). We further detected Notch3 and ERα mRNA expression by real-time PCR in several breast cancer cell lines. As expected, high levels of Notch3 and ERα mRNA were expressed in the ERα-positive MCF-7 and T-47D breast cancer cell lines, but not in the ERα-negative MDA-MB-231, BT-549 and SK-BR-3 breast cancer cell lines (Figure 1B, C). In contrast, Notch1 and Notch2 were relatively highly expressed both in ERα-positive and -negative cells, and Notch4 was primarily expressed in ERα-negative cells at protein (Figure 1D) and mRNA levels (Figure 1E-G). Immunofluorescence further revealed that Notch3 was expressed in the ERα-positive MCF-7 and T47D cell lines, but not in ERα-negative MDA-MB-231 and BT-549 cells (Figure 1H, Supplementary Figure 1A). Our results, in general, suggest a possible link between Notch3 and the luminal epithelial phenotype in breast cancer.
Notch3 expression is associated with the luminal epithelial phenotype in breast cancer in cell lines and clinical cases. (A) Expression of Notch3, ERα, and EMT-related markers, such as E-cadherin and vimentin, was detected by western blot in breast cancer cell lines MCF-7, T47D, MDA-MB231, SKBR3 and BT549. (B and C) Relative ERα and Notch3 mRNA levels were quantified in breast cancer cell lines by real-time PCR. (D) N1ICD, N2ICD and N4ICD protein levels were detected by western blot in breast cancer cell lines. (E, F and G) Notch1, Notch2 and Notch4 mRNA levels were determined by real-time PCR in breast cancer cell lines. (H) Immunofluorescence staining for Notch3 and ERα was performed in MCF-7 (upper panel) and T47D (bottom panel) cells, and nuclei were stained with DAPI (4', 6-diamidino-2-phenylindole). A merged image was obtained by combining Notch3, ERα and DAPI fluorescent staining images. The scale bar represents 50 μm. (I) ERα and Notch3 protein expression were immunohistochemically stained in ERα-positive (a, b) and ERα-negative (c, d) human breast cancer tissues.
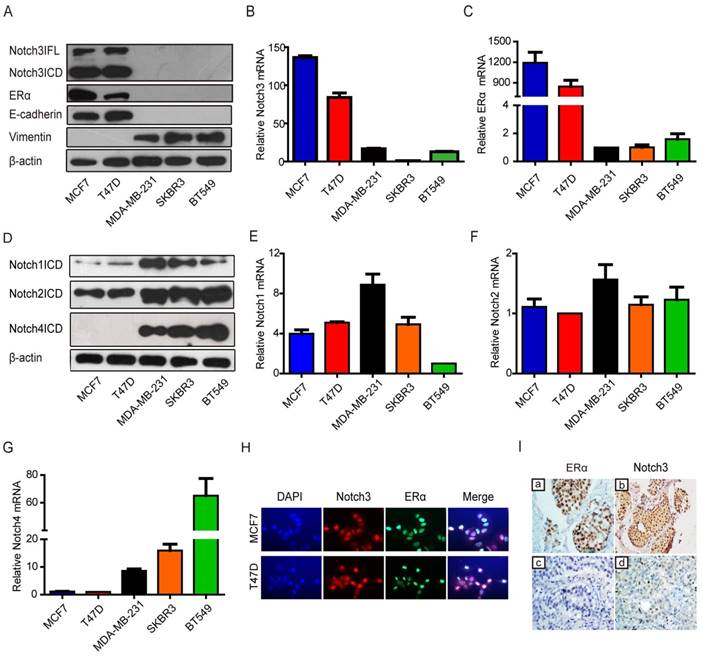
To determine the relationship between Notch3 and the luminal phenotype in clinically diagnosed breast cancer, we examined Notch3 expression patterns in human tumor samples from luminal A and triple-negative (TNBC) breast cancers. Using immunohistochemistry, we observed that Notch3 was highly expressed in luminal A compared with TNBC tissue (Figure 1I and Supplementary Table S1).
Notch3 up-regulates ERα expression at the protein and mRNA levels
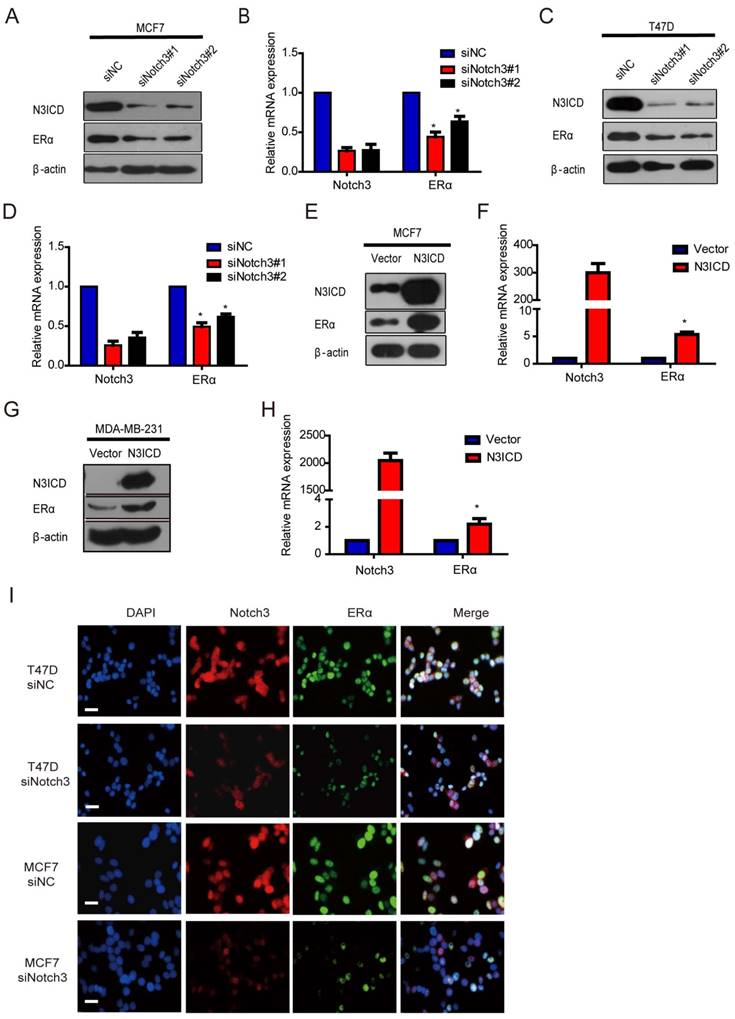
Because Notch3 was highly expressed in ERα-positive breast cancer cells and Notch3-expressing cells acquired the characteristics of mammary luminal progenitors that give rise to the ERα-positive luminal lineage [17, 32, 33], we speculated that Notch3 regulates ERα expression in breast cancer. To examine this hypothesis, we silenced and overexpressed Notch3 in breast cancer cells. As expected, downregulation of Notch3 by siRNA decreased ERα expression at both the mRNA and protein levels in ERα-positive MCF-7 and T47D cells (Figure 2A-D). Conversely, overexpression of the active form of Notch3, N3ICD, resulted in upregulation of ERα at both the mRNA and protein levels in ERα-positive MCF-7 and ERα-negative MDA-MB-231 cells (Figure 2E-H). Immunofluorescence confirmed that Notch3 and ERα co-localized in the nuclei of ERα-positive MCF-7 and T47D cells, whereas Notch3 silencing also resulted in decreased ERα expression (Figure 2I). These results suggest that Notch3 up-regulates ERα expression in breast cancer cells.
Knockdown or over-expression of Notch3 down- or up-regulates ERα gene transcription in breast cancer cells. (A and B) ERα-positive MCF-7 cells were transiently transfected with 2 different siRNAs against Notch3 (#1 and #2) for 48 h, and then Notch3 and ERα protein and mRNA expression levels were detected and quantified by western blot (a) and quantitative real-time PCR (B). (C and D) ERα-positive T47D cells were transiently transfected with 2 different siRNAs against Notch3 (#1 and #2) for 48 h, and Notch3 and ERα protein and mRNA expression levels were detected and quantified by western blot (C) and quantitative real-time PCR (D). (E and F) Protein (E) or mRNA (F) levels for Notch3 and ERα were determined by western blot or quantitative real-time PCR in MCF-7 cells transfected with an N3ICD-expressing or control vector as indicated. (G and D) Protein (G) or mRNA (H) levels for Notch3 and ERα were determined by western blot or quantitative real-time PCR in MDA-MB-231 cells transfected with the N3ICD-expressing or control vector as indicated. (I) Confocal fluorescence microscopy of DAPI/Notch3/ERα staining in T47D (upper) and MCF-7 (bottom) cells treated with control siRNA or siNotch3. The scale bar represents 50 μm.
Notch3 activates ERα expression by binding to CSL binding elements in the ERα promoter
To explore the mechanism underlying transcriptional regulation of ERα by Notch3, the ERα promoter (~1 kb) was cloned into the upstream of a luciferase reporter gene, and luciferase activity was determined in the absence or presence of the active Notch3 (N3ICD). Luciferase activity of the ERα promoter was decreased after silencing Notch3 in MCF-7 cells (Figure 3A), and was activated in a dose-dependent manner by N3ICD overexpression in MDA-MB-231 cells (Figure 3B). We identified 2 CSL-binding motifs located at the upstream (-159∽-155bp and -380∽-384bp) of exon 1 in the ERα promoter, to which Notch3 can potentially bind (Figure 3C). We compared full length ERα promoter activity with mutant ERα promoter vectors, containing deletions in one (ΔCSL2 construct) or both (ΔCSL1&2 construct) CSL binding elements. We found that the mutant reporters with either one CSL or two CSL deletions showed significantly decreased luciferase activities by 50% or 75%, respectively, following co-transfection with N3ICD into MDA-MB-231 cells (Figure 3C and D). These results show that Notch3 up-regulates ERα promoter activity through binding to CSL binding elements.
Notch3 regulates ERα expression by binding to CSL binding elements in the ERα promoter. (A) MCF-7 cells, silenced by control siRNA or siNotch3, were co-transfected with an ERα promoter-driven firefly luciferase (ERα-Luc) and an internal control plasmid pRL-SV40 expressing Renilla luciferase. Promoter activity was expressed as the ratio of firefly luciferase/Renilla luciferase. (B) MDA-MB-231 cells were transiently co-transfected with ERα-Luc and N3ICD at incremental doses as indicated, and Renilla luciferase was transfected as an internal control. (C) Schematic representation of wild-type ERα-Luc and mutant constructs ΔCSL (deletion of one CSL binding element) or Δ2CSL (deletion of both CSL binding elements) used in reporter assays. (D) Luciferase reporter assay with ERα-Luc or mutant ERα-Luc were performed in the absence or presence of N3ICD in MDA-MB-231 cells. Promoter activity was expressed as the ratio of firefly luciferase/Renilla luciferase. (E and F) ChIP assays. Schematic representation of the 2 CSL binding element-containing regions (Region 2 and Region 3) and negative control region (Region 1) used in the ChIP assay. PCR products were detected in the presence of anti-Notch3 antibody. (G) Sequences of probes 1 and 2 containing the GGGAA core element for EMSA. (H) EMSA using probes 1 and 2. Competition assays were performed by adding either 100-fold (lane 3) excess of unlabeled oligonucleotides containing the core CSL binding element, or 100-fold excess of unlabeled mutant oligonucleotides in the core CSL binding element (lane 4). Each reporter assay was performed in triplicate.
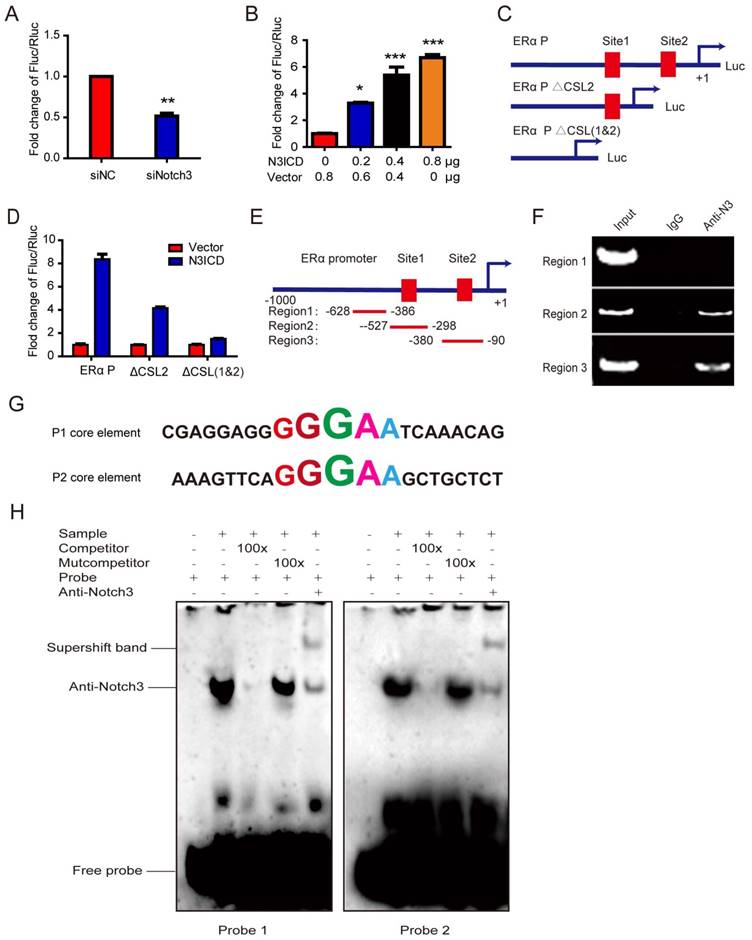
To examine whether Notch3 binds directly to the ERα promoter, we performed chromatin immunoprecipitation assays (ChIPs), using antibody against Notch3, on MCF-7 cells with IgG as a negative control. Two pairs of primers were designed to span the CSL binding elements located at the upstream (-328 - 90 bp, ERα P1 and -527 - 298 bp, ERα P2) of exon 1 in the ERα promoter (Figure 3E). Significant binding was observed in the two regions containing CSL binding elements, but not in the control region (located at -628-386bp), which does not contain CSL binding elements (Figure 3F).
To further investigate the mechanism by which Notch3 regulates ERα expression, electrophoretic mobility shift assays (EMSAs) were employed to analyze core elements in the ERα promoter for Notch3 binding (Figure 3G), showing that Notch3 binds both core elements. Competition assays for Notch3 binding, using an excess of unlabeled oligonucleotides (100×), abolished the Notch3-shift band. An excess of unlabeled mutation oligonucleotide (TGTCT, 100×) did not compete nor abolish the Notch3-shifted band (Figure 3H). The supershifted band was noted after an anti-Notch3 specific antibody and labeled probe were added to MCF-7 nuclear extracts, confirming that Notch3 can bind to the core element (GGGAA) of the ERα promoter. Based on previous reports [34, 35], our findings demonstrate that Notch3 binds as part of a complex to the ERα promoter.
Overexpressed N3ICD in MDA-MB-231 cells reverses EMT
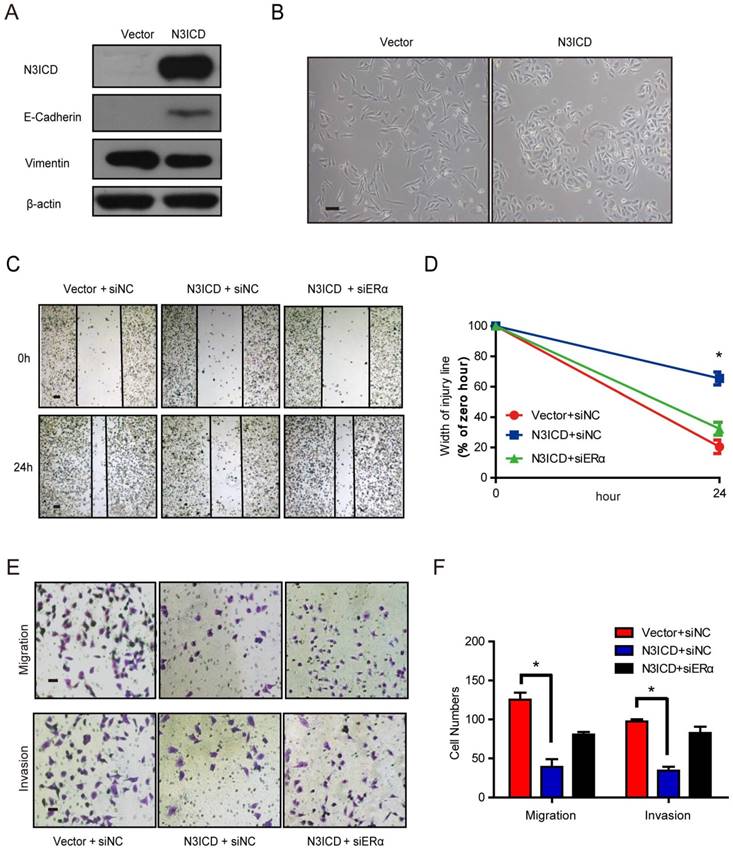
Because ERα is a marker of the luminal epithelial phenotype in breast cancer cells, we speculated that Notch3 regulates the shift of EMT markers. To investigate Notch3's role in EMT regulation, we examined the shift in EMT markers after Notch3 overexpression in MDA-MB-231 cells. As expected, overexpressed N3ICD in MDA-MB-231 cells downregulated the mesenchymal marker vimentin, and upregulated the epithelial cell marker E-cadherin (Figure 4A). In two-dimensional cell culture, MDA-MB-231-vector-transfected cells had a more spindle-shaped, mesenchymal morphology, whereas MDA-MB-231 cells stably expressing N3ICD exhibited a more epithelial phenotype (Figure 4B). In a wound healing assay, MDA-MB-231-control cells migrated as single cells to close the wound, whereas MDA-MB-231-N3ICD cells migrated as a collective sheet. After 24 h, the gap in MDA-MB-231-N3ICD cells was 65.5% of the 0 h injury width, which is much higher than the 20.5% displayed by MDA-MB-231-control cells. This shows that Notch3 overexpression inhibits cellular motility in MDA-MB-231 cells. The inhibitory effect disappeared (percentage was reduced to 32.3%) when ERα was knocked down by siRNA (Figure 4C-D, Supplementary Figure 1B). Cell migration assays (transwell) showed fewer MDA-MB-231-N3ICD cells migrated at ~30.4% of vector-transfected control cells after 48 h. However, when ERα was knocked down by siRNA, the number of migrated cells increased more than 2-fold compared with MDA-MB-231-N3ICD cells. Similar results were observed in a cell invasion assay (transwell) (Figure 4E and F). These data suggest that Notch3 inhibits the motility and invasiveness of MDA-MB-231 cells via upregulation of ERα.
N3ICD overexpression in MDA-MB-231 cells reverses EMT. (A) Western blot showing the protein levels of E-cadherin and Vimentin after overexpressing N3ICD in MDA-MB-231 cells. (B) Morphology of MDA-MB-231 cells transfected with N3ICD or a control vector, Scale bars represent 50 μm. (C and D) Representative images (C) and quantitative (D) wound recovery data after 24 h in MDA-MB-231 cells transfected with N3ICD and ERα siRNA. Scale bars represent 50 μm. (E and F) Representative micrographs and quantitative data of Matrigel-coated or non-coated transwell assays. Stably transfected N3ICD MDA-MB-231 cells were co-transfected with siERα or siNC. Invading or migrating cells were counted in 5 random fields. Scale bars represent 50 μm. All experiments were performed at least three times and data were statistically analyzed by two-sided t-test. *P < 0.05 versus control. Error bars indicate SEM.
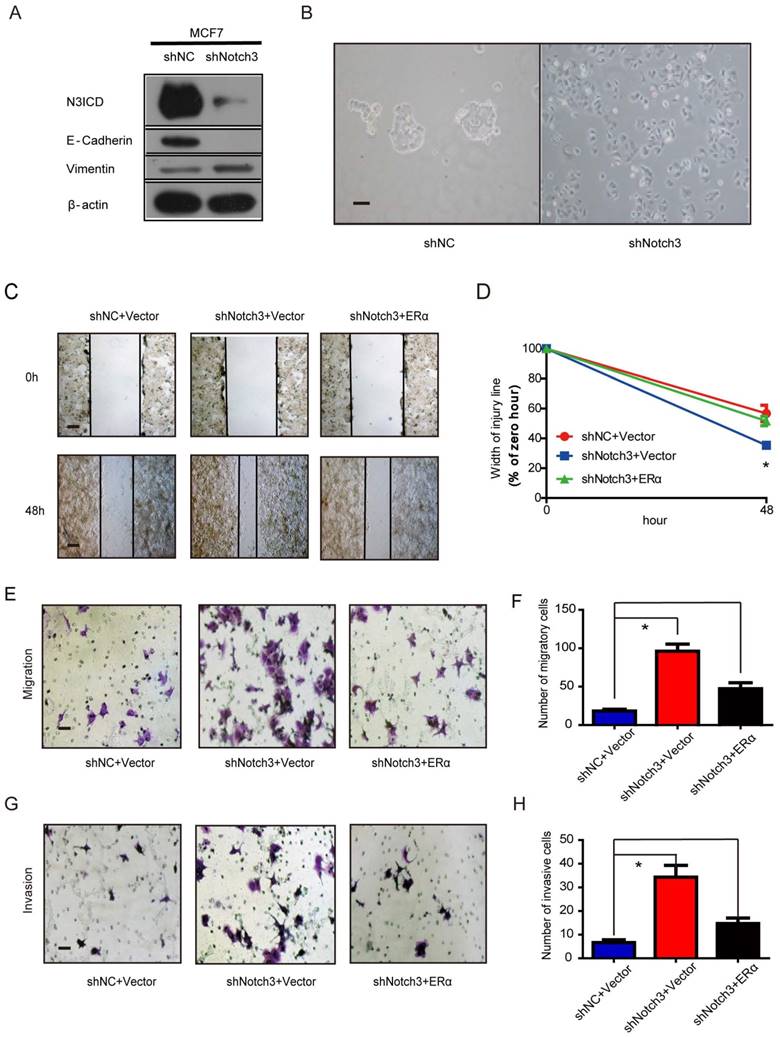
Knock-down of Notch3 in MCF-7 cells promotes EMT
In addition, Notch3 knockdown in MCF-7 cells decreased expression of the epithelial cell marker E-cadherin compared to control cells. In contrast, expression of the mesenchymal marker vimentin was increased in MCF-7 cells by silencing Notch3 (Figure 5A). We also observed morphological changes associated with EMT in MCF-7 cells following stable Notch3 knockdown. Control cells maintained their cobblestone-like phenotype with strong cell-cell adhesion, while some cells with Notch3 shRNA had a fibroblast-like morphology and showed cellular scattering (Figure 5B). We examined the effect of Notch3 silencing on cell motility using a wound healing assay. After 48 h in cell culture, the gap in shNotch3 MCF-7 cells was 35.4% of that at 0 hour compared with control transfected shNC MCF-7 cells (56.7%), indicating that Notch3 knockdown enhanced cellular motility. The residual open gap was back to 56.1% by transfecting an expression plasmid encoding ERα, showing that this change could be rescued by ERα overexpression (Figure 5C and D, Supplementary Figure 1C). The motility potential of Notch3-silenced MCF-7 cells was determined using a transwell migration assay. Notch3 shRNA led to a 5-fold increase in cell migration, which was partially rescued by ERα overexpression (Figure 5E and F). Similar results also occurred in the cell invasion assay (Figure 5G and H). These data suggest that Notch3 knockdown in MCF-7 cells promotes cellular motility and invasiveness via downregulation of ERα.
Notch3 knockdown in MCF-7 cells promotes EMT. (A) Notch3 knockdown by shRNA in MCF-7 cells downregulated the epithelial cell marker E-cadherin and upregulated the mesenchymal cell marker vimentin. (B) Morphology of MCF-7 cells stably transfected with control shRNA and shNotch3. (C and D) Wound healing assay showed that Notch3 knockdown enhanced cellular motility in MCF-7 cells; these changes were rescued by transfection with an expression plasmid encoding ERα. Representative images (C) and quantitative (D) wound recovery data after 48 h in cell culture. (E-H) Notch3 knockdown increased cellular migration and invasion of MCF-7 cells, and these changes were rescued by transfection with an ERα plasmid. Representative images (E and G) and quantitative data (F and H) from migration or invasion assays. All experiments were performed at least three times and data were statistically analyzed by a two-sided t-test. *P<0.05 versus control. Error bars indicate SEM.
Notch3 inhibits breast cancer tumorigenesis and metastasis in vivo
To explore the role of Notch3 in tumorigenesis and metastasis in breast cancers, we employed MDA-MB-231 cells overexpressing N3ICD or Notch3-knockdown MCF-7 cells in tumor xenograft models. Orthotopic implantation of MDA-MB-231 N3ICD or control cells in mammary fat pads was followed either by caliper measurement or BLI monitoring. After 6 weeks, mice implanted with N3ICD-overexpressing MDA-MB-231 cells developed fewer xenograft tumors (5/11) that were also smaller in size and weighed less than those in the control group (9/11), indicating that Notch3 inhibited breast cancer tumorigenesis and delayed the onset of breast cancer formation (Figure 6A-C). Similarly, BLI monitoring showed that mice bearing N3ICD-overexpressing MDA-MB-231 tumors consistently had an ~100-fold decreased tumor burden at 6 weeks after transplantation compared with the control group (Figure 6D and E). After transplants were harvested, immunohistochemistry showed that tumors derived from control cells expressed higher levels of vimentin, but that tumors derived from Notch3-overexpressing cells expressed higher levels of Notch3, ERα and E-cadherin, suggesting that the Notch3-mediated shift in expression of mesenchymal makers to epithelial markers was retained in vivo (Figure 6F). BLI analysis of various organs ex vivo showed that livers in 4 out of 11 mice derived from control cells developed metastases, but no metastases were found in livers derived from Notch3-overexpressing MDA-MB-231 cells (Figure 6G and H). These results demonstrate that Notch3 inhibits tumorigenesis, shifts mesenchymal markers to epithelial markers and inhibits breast cancer metastasis in vivo.
Notch3 reserves EMT, tumorigenesis and metastasis in a humanized mouse model. (A) Control vector or Notch3-expressing MDA-MB-231 cells (2×106 cells) were injected into the mammary fat pad tissue of immunodeficient NU/NU mice (n=11). Tumor size was measured twice a week. Mice were euthanized and examined for metastasis when the primary tumor grew to 1.5 cm in diameter. Mice implanted with either vector (upper panel) or N3ICD transfected cells (middle panel) at 42 days after implantation. Primary tumors dissected from each group of mice are shown in the lower panel. (B) The time course of tumor growth is shown with error bars representing ±SEM. (C) Weight of primary tumors in the different groups. Error bars show ±SEM. (D) Representative bioluminescence imaging and (E) quantitative analysis of imaging was performed. (F) Immunohistochemical staining for Notch3, ERα, and EMT markers, as well as HE staining, were performed in dissected tumors. (G) Livers from both control and N3ICD expressing groups ex vivo were imaged with BLI (upper panel), and HE staining was also performed (lower panel). (H) Quantitative analysis of liver imaging. (I) 30 days after tail-vein injection with MDA-MB-231 N3ICD or control cells, whole body BLI was performed. (J) Quantitative analysis of lung imaging in vivo. (K-L) Lungs from mice injected either with vector-transfected or MDA-MB-231 cells expressing N3ICD were imaged with BLI after euthanization. (M) Metastatic lesions were detected with HE staining in lungs from mice with vector control cells (left panel) or N3ICD-expressing MDA-MB-231 cells (right panel). (N) HE staining shows blood vessel invasion in lung metastases from vector transfected cells. (O) HE staining was applied to detect lung metastases in either vector control (left panel) or Notch3-silenced MCF-7 cells (right panel).
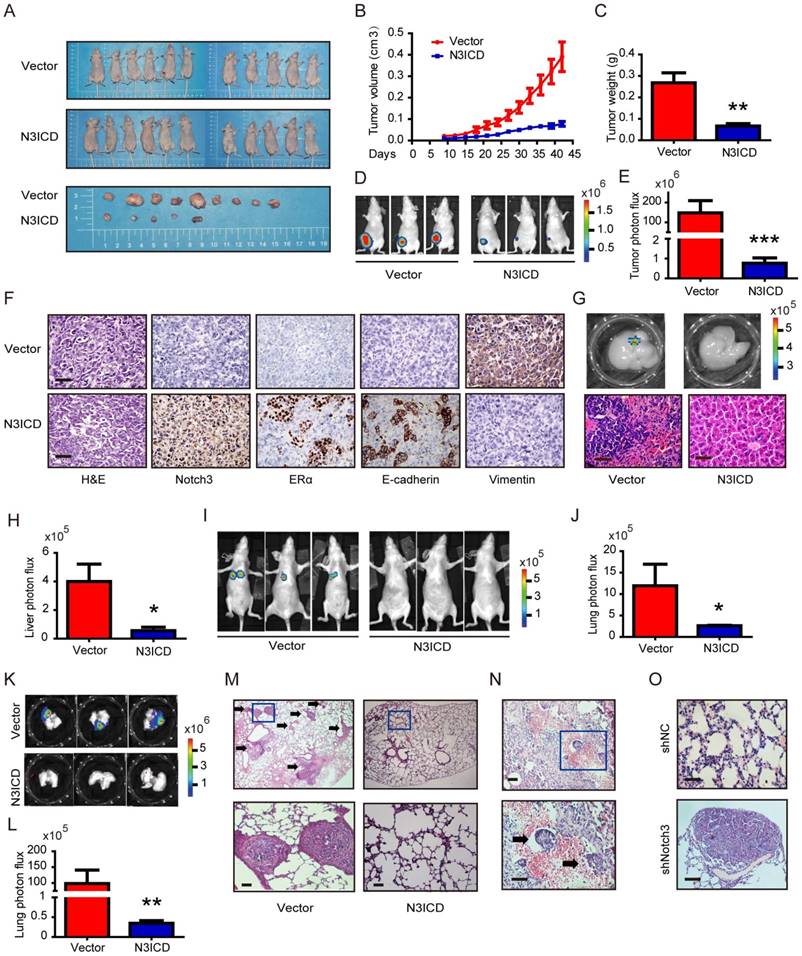
Moreover, we injected MDA-MB-231 cells via tail vein to examine whether Notch3 affects breast cancer metastasis. Thirty-one days after transplantation of N3ICD-overexpressing MDA-MB-231 cells or control cells, mice were euthanized and organs were harvested. Lung metastases were detected in 5/6 mice harboring vector-transfected MDA-MB-231 cells, as determined by BLI analysis, while no metastases were observed in mice implanted with N3ICD-overexpressing cells (n = 6) (Figure 6I and J). With ex vivo BLI imaging and HE staining, we further confirmed that Notch3 overexpression suppressed lung metastasis and vessel invasion in our breast cancer model (Figure 6K-N).
We then investigated whether Notch3 knockdown in MCF-7 cells increased tumor metastasis in vivo. Mock- and Notch3 shRNA-transfected MCF-7 cells were injected into the tail vein of Nu/Nu mice, and formation of tumor metastases was allowed to proceed for 8 weeks. As shown in Figure 6O, lungs from mice (2/7) injected with Notch3-silenced MCF-7 cells displayed breast cancer metastasis, whereas lungs from mice injected with mock-transfected MCF-7 cells did not (0/9). The statistical analysis showed a difference between the 2 groups (Chi-Square Test, p = 0.074). We did not find breast cancer metastasis in the livers from both Notch3-silenced and mock-transfected MCF-7 cells. These results further demonstrate that Notch3 inhibits breast cancer metastasis in vivo.
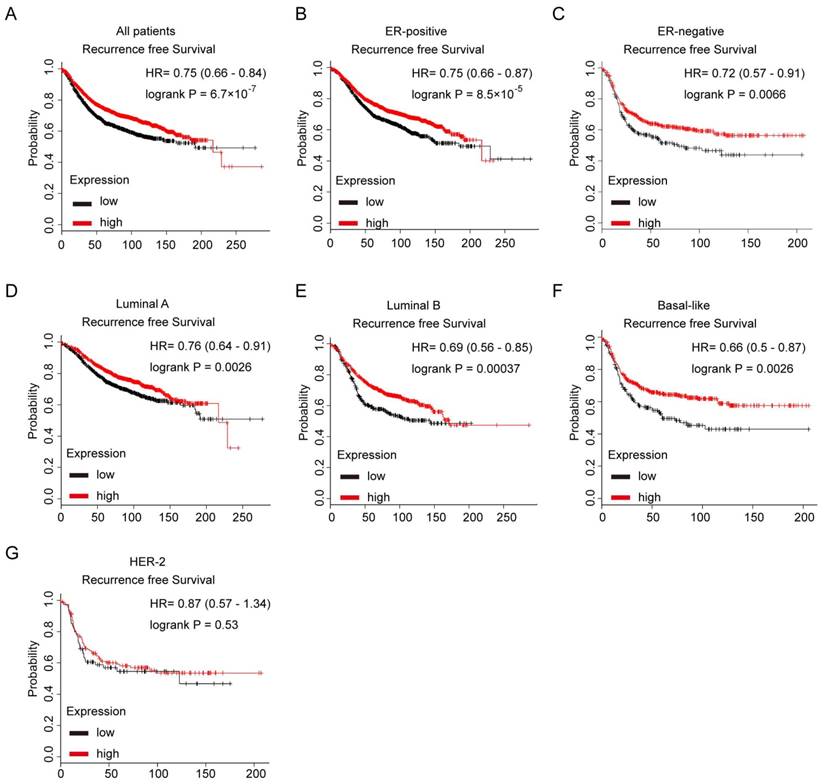
Notch3 mRNA is a favorable prognostic factor in breast cancer patients
We further evaluated the prognostic value of low or high expression of Notch3 mRNA in a large online clinical microarray database of breast tumors from 4142 patients. Elevated expression of Notch3 mRNA in tumor was associated with better recurrence-free survival (RFS) (n = 3554, p = 6.7×10-7, HR = 0.75) in all breast cancer patients (Figure 7A). In ERα-positive subgroup of patients, higher Notch3 expression indicated better prognosis with respect to RFS (n = 2766, p = 8.5×10-5, HR = 0.75) as expected (Figure 7B). In the ERα-negative subgroup of patients, who often develop metastatic disease and have a poor prognosis, elevated expression of Notch3 in tumors was associated with longer RFS (n = 788, p = 0.0066, HR = 0.72) (Figure 7C). When the analysis was restricted to the intrinsic subtype of breast cancer, higher expression of Notch3 mRNA was found to be indicative of favorable prognoses for recurrence-free survival in luminal A (n = 1764, p = 0.0026, HR = 0.7), luminal B (n = 1002, p = 0.00037, HR = 0.69) and basal-like (n = 580, p = 0.0026, HR = 0.66) subtypes (Figure 7D-F), but not in the HER-2 overexpressing subtype (n = 208, p = 0.53, HR = 0.87).
Kaplan-Meier analysis of the probability of correlation between Notch3 expression and patient survival in a cohort of 4142 human breast cancer patients in Kaplan Meier Plotter database. (A) Recurrence-free survival (RFS) in breast cancer patients according to Notch3 expression. (B, C) RFS of ER-positive and ER-negative breast cancer patients. (D - G) RFS in luminal A, luminal B, basal-like or Her-2 subtype breast cancer patients. P values were computed by log-rank test.
Discussion
In this study, we demonstrate for the first time that Notch3 transcriptionally upregulates ERα expression and inhibits EMT in breast cancers. In breast development, Notch3 expression is highly elevated in mammary luminal-restricted progenitor/mature luminal cells compared with bi-potent mammary stem cells, indicating that Notch3 might play a crucial role in the regulation of ERα and luminal epithelial development [18, 20]. In the present study, we found that Notch3 upregulates epithelial molecular markers, such as E-cadherin, and silencing Notch3 increases mesenchymal biomarkers, such as vimentin, in breast cancers.
Using a conditional Notch3-induced transgenic mouse strain, Lafkas et al. demonstrated that Notch3 is expressed in highly clonogenic and transiently quiescent luminal progenitor cells [33]. When the active form of Notch3, N3ICD, is conditionally expressed, p21 and p16INK are induced in luminal mammary cells [33]. Similarly, N3ICD overexpression was found to result in a very significant expansion of a sub-set of cells, the CD24highCD29low cell subpopulations, which are positive for CK6/CK18 and reported to contain luminal progenitors [17]. All of these findings, together with the present study, strongly suggest that Notch3 is most likely a biomarker of luminal epithelial cells in breast cancers.
We show a strong correlation between Notch3 and ERα both in breast cancer cells and human breast cancer tissues. In particular, ectopic overexpression of Notch3 results in activation of ERα in ERα-negative breast cancer cell lines, whereas silencing Notch3 in ERα-positive cells downregulates ERα. In ERα-positive breast cancer cells, estrogen/ERα signaling has been shown to antagonize EMT-promoting transcription factors, such as Slug and Snail, and activates MET-promoting factors, such as GATA3 and FoxA1 [3]. Taking into consideration that loss of ERα might correlate to distant metastasis, exploring the role of Notch3 in regulating ERα in breast cancer cells would be of significance, e.g., for elucidating ERα generation, increasing breast cancer therapeutic efficacy and preventing metastasis [36, 37]. With regard to how ERα is regulated via Notch3, our results demonstrate that N3ICD specifically binds to CSL binding elements in the ERα promoter and transactivates ERα expression. However, the mechanisms through which Notch3 transactivates ERα, either by specificity of the TAD domain or by inducing its ligands, remains unclear.
In addition to the shift from luminal epithelial markers to mesenchymal markers, we observed that silencing Notch3 drastically promoted the EMT phenotype, including migration and invasion in vitro and metastasis in vivo, and increased the proportion of cancer stem cells in breast cancers (unpublished data). In contrast, Notch3 overexpression suppressed EMT as well as migration and invasion in ERα-negative cells. Notably, ectopic ERα expression significantly reversed EMT transition induced by silencing Notch3, and conversely, knock-down of ERα almost completely reversed MET induced by Notch3 overexpression. The above observations strongly suggest that Notch3, at least in part, inhibits EMT through ERα activation/upregulation. Given that ERα is an indicator of effective endocrine therapy in breast cancers, more extensive studies are needed to focus on the relationship between Notch3 status and sensitivity/responses to endocrine therapy in both ERα-positive and -negative breast cancers.
In an ERα-negative cell line, Notch3 overexpression inhibited tumorigenesis and tumor growth in an orthotopic mouse model, as determined by both caliper measurement and non-invasive monitoring with molecular imaging. MDA-MB-231 cells overexpressing Notch3, after injection via tail vein, demonstrated significantly reduced metastasis to lungs detected with bioluminescence imaging, revealing that metastatic potential was suppressed by Notch3. In ex vivo assays, increased luciferase activity was detected in resected lungs or livers, consistent with the results of in vivo imaging showing the lack of metastasis when Notch3-overexpressing cells were implanted. We further histologically confirmed liver or lung metastases in both orthotopic and tail vein models with H&E staining. Moreover, immunohistochemical staining demonstrated upregulation of ERα expression and epithelial biomarkers, suggesting that metastatic inhibition by Notch3 overexpression is likely caused via modulation of ERα. Although recent reports indicate EMT may not be required for invasion and metastasis, as evidenced with genetically engineered mouse models, EMT is still thought to be important for metastasis [38, 39]. In combination with the observation that lung metastasis is promoted by silencing Notch3, Notch3 might play an important role in inhibiting tumorigenesis and metastasis in breast cancer through inducing ERα and suppressing EMT, at least in part.
Notch3 high expression detected with immunohistochemistry has shown an inconsistent prognostic predictive value, e.g., poor prognosis in hepatocellular carcinoma, non-small cell lung cancer, medullary thyroid cancer, and ovarian cancer [40-44]; favorable prognosis in small intestine and gastric cancers [24, 45]. In a large clinical microarray database, Notch3 transcripts were significantly associated with better RFS in clinically diagnosed breast cancer patients, especially for those with ERα-positive tumors. In terms of sub-type of breast cancers, Notch3 did show a favorable RFS in luminal type breast cancers, but no association with the Her2 overexpressed type. However, interestingly, in ER-negative or triple-negative subtypes, higher expression of Notch3 also predicted more favorable prognosis than in those subtypes expressing lower Notch3. It seems that Notch3 would be a better prognostic value independent of ERα in those ER-negative breast cancer patients. Mechanistic analysis suggested that Notch3 could function as a tumor suppressor by regulating complex signaling pathways. For example, our previous studies and a study by Cui et al demonstrated that Notch3 functions as a tumor suppressor by controlling cellular senescence and inhibits breast cancer cell proliferation by up-regulating p21[46], by inducing Cdh1 expression [27], and by activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells [26]. Taken together, clinical data suggest Notch3's role in predicting recurrence for breast cancer patients, in particular, for those with the ERα-positive luminal phenotype, supporting the experimentally described role for Notch3 as a potential tumor suppressor by suppressing EMT through ERα activation. Moreover, future determination of Notch3 at the protein level and its sub-cellular localization would provide more insights on its prognostic significance. Jaskula-Sztul et al. demonstrated that genetic and/or pharmacological induction of Notch3 had a tumor-suppressor role in medullary thyroid carcinoma. AB3, an HDAC inhibitor, induced both full length Notch3 and its activated form NICD3, and inhibited cellular proliferation by inducing apoptosis in MZ-CRC-1 and TT cell lines [47]. Thus, it may be useful to explore AB3's potential as a combined therapy option.
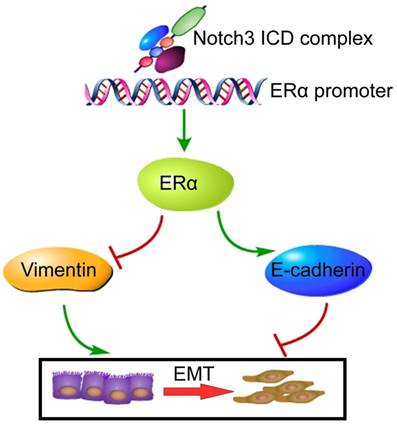
In summary, the present study demonstrates that Notch3 suppresses EMT through transcriptionally activating ERα expression (Figure 8). These findings delineate the role of a Notch3/ERα axis in maintaining the luminal phenotype and inhibiting tumorigenesis and metastasis in breast cancer, providing a novel strategy to re-sensitize ERα negative or low expressing breast cancers to hormone therapy.
Proposed model of how Notch3 and ERα mediate EMT in breast cancer, where the Notch3 ICD complex directly binds to the ERα promoter, and ultimately transactivates ERα and its downstream signaling, permitting the repression of E-cadherin and up-regulation of vimentin to induce EMT in breast cancer.
Abbreviations
N3ICD: Notch3 intracellular domain; ERα: Estrogen receptor alpha; EMT: epithelial-mesenchymal transition; CSL: CBF1/RBP-Jκ in humans, Su(H) in Drosophila, and Lag-1 in Caenorhabditis elegans; IHC: immunohistochemistry; ChIP: Chromatin immunoprecipitation assay; EMSA: Electrophoretic mobility shift assay.
Acknowledgements
We are grateful to Dr. William Jr. Kaelin for critically reading the manuscript. We also thank Drs. De He and Haolong Ding for advice or technical assistance in the tail vein injection procedure.
Financial support
This work was supported in part by grants from the Major State Basic Research Development Program of China (No. 2011CB707705), Major International Collaborative Research Project of NSFC (81320108015), Team project of Guangdong Natural Science Foundation Committee (2016A030312008), and Guangdong Provincial Key Laboratory on Breast Cancer Diagnosis and Treatment Research.
Author contributions
XWD, and GJZ designed the study and conceived the project. XWD, YKL, XLW, YQZ, JWB, CFC, and HYL performed the experiments and acquired the data. CWD, YCL, MC and JT analyzed the data. XWD, YKL, and HYL drafted the manuscript. GJZ wrote and finalized the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nature reviews Cancer. 2005;5:591-602
2. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559-64
3. Guttilla IK, Adams BD, White BA. ERalpha, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends in endocrinology and metabolism: TEM. 2012;23:73-82
4. Ye Y, Xiao Y, Wang W, Yearsley K, Gao J, Shetuni B. et al. ERα signaling through slug regulates E-cadherin and EMT. Oncogene. 2010;29:1451-62
5. Rochefort H, Platet N, Hayashido Y, Derocq D, Lucas A, Cunat S. et al. Estrogen receptor mediated inhibition of cancer cell invasion and motility: an overview. The Journal of steroid biochemistry and molecular biology. 1998;65:163-8
6. Al Saleh S, Al Mulla F, Luqmani YA. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS One. 2011;6:e20610
7. Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F. et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nature cell biology. 2007;9:470-8
8. Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast cancer research and treatment. 2004;83:249-89
9. Tyson JJ, Baumann WT, Chen C, Verdugo A, Tavassoly I, Wang Y. et al. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nature reviews Cancer. 2011;11:523-32
10. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature reviews Clinical oncology. 2011;8:97-106
11. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nature reviews Cancer. 2011;11:338-51
12. Hao L, Rizzo P, Osipo C, Pannuti A, Wyatt D, Cheung LW. et al. Notch-1 activates estrogen receptor-alpha-dependent transcription via IKKalpha in breast cancer cells. Oncogene. 2010;29:201-13
13. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR. et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer research. 2010;70:709-18
14. Yen WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B. et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2084-95
15. Graziani I, Eliasz S, De Marco MA, Chen Y, Pass HI, De May RM. et al. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer research. 2008;68:9678-85
16. Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D. et al. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer research. 2004;64:7787-93
17. Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR. et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell stem cell. 2008;3:429-41
18. Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M. et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell stem cell. 2008;3:109-18
19. Pradeep CR, Kostler WJ, Lauriola M, Granit RZ, Zhang F, Jacob-Hirsch J. et al. Modeling ductal carcinoma in situ: a HER2-Notch3 collaboration enables luminal filling. Oncogene. 2012;31:907-17
20. Hu C, Dievart A, Lupien M, Calvo E, Tremblay G, Jolicoeur P. Overexpression of activated murine Notch1 and Notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. The American journal of pathology. 2006;168:973-90
21. Gupta N, Xu Z, El-Sehemy A, Steed H, Fu Y. Notch3 induces epithelial-mesenchymal transition and attenuates carboplatin-induced apoptosis in ovarian cancer cells. Gynecologic oncology. 2013;130:200-6
22. Zhou L, Zhang N, Song W, You N, Li Q, Sun W. et al. The significance of Notch1 compared with Notch3 in high metastasis and poor overall survival in hepatocellular carcinoma. PLoS One. 2013;8:e57382
23. Pierfelice TJ, Schreck KC, Dang L, Asnaghi L, Gaiano N, Eberhart CG. Notch3 activation promotes invasive glioma formation in a tissue site-specific manner. Cancer research. 2011;71:1115-25
24. Kang H, An HJ, Song JY, Kim TH, Heo JH, Ahn DH. et al. Notch3 and Jagged2 contribute to gastric cancer development and to glandular differentiation associated with MUC2 and MUC5AC expression. Histopathology. 2012;61:576-86
25. Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H. et al. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer research. 2011;71:6836-47
26. Zhang X, Liu X, Luo J, Xiao W, Ye X, Chen M. et al. Notch3 inhibits epithelial-mesenchymal transition by activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells. Oncogenesis. 2016;5:e269
27. Chen CF, Dou XW, Liang YK, Lin HY, Bai JW, Zhang XX. et al. Notch3 overexpression causes arrest of cell cycle progression by inducing Cdh1 expression in human breast cancer cells. Cell Cycle. 2016;15:432-40
28. Liang YK, Zeng D. et al. MCAM/CD146 promotes tamoxifen resistance in breast cancer cells through induction of epithelial-mesenchymal transition, decreased ERα expression and AKT activation. Cancer Letters. 2017;386:65-76
29. Liu J, Wei XL. et al. Cytoplasmic Skp2 Expression Is Associated with p-Akt1 and Predicts Poor Prognosis in Human Breast Carcinomas. PloS one. 2012
30. Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Szasz MA. et al. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast cancer research and treatment. 2013;140:219-32
31. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment. 2010;123:725-31
32. Buono KD, Robinson GW, Martin C, Shi S, Stanley P, Tanigaki K. et al. The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Developmental biology. 2006;293:565-80
33. Lafkas D, Rodilla V, Huyghe M, Mourao L, Kiaris H, Fre S. Notch3 marks clonogenic mammary luminal progenitor cells in vivo. The Journal of cell biology. 2013;203:47-56
34. Wilson JJ, Kovall RA. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell. 2006;124:985-96
35. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216-33
36. Shang Y. Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nature reviews Cancer. 2006;6:360-8
37. Brabletz T. To differentiate or not-routes towards metastasis. Nature reviews Cancer. 2012;12:425-36
38. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
39. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
40. Hu W, Liu T, Ivan C, Sun Y, Huang J, Mangala LS. et al. Notch3 pathway alterations in ovarian cancer. Cancer research. 2014;74:3282-93
41. Ozawa T, Kazama S, Akiyoshi T, Murono K, Yoneyama S, Tanaka T. et al. Nuclear Notch3 Expression is Associated with Tumor Recurrence in Patients with Stage II and III Colorectal Cancer. Annals of Surgical Oncology. 2014;21:2650-8
42. Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T. et al. Notch3 Overexpression as Potential Therapeutic Target in Advanced Stage Chemoresistant Ovarian Cancer. American Journal of Clinical Pathology. 2012;138:535-44
43. Shen ZP, Hou XW, Chen B, Chen P, Zhang Q. NOTCH3 Gene Polymorphism is Associated With the Prognosis of Gliomas in Chinese Patients. Medicine. 2015:94
44. Ye YZ, Zhang ZH, Fan XY, Xu XL, Chen ML, Chang BW. et al. Notch3 overexpression associates with poor prognosis in human non-small-cell lung cancer. Medical Oncology. 2013:30
45. Eom DW, Hong SM, Kim J, Kim G, Bae YK, Jang KT. et al. Notch3 signaling is associated with MUC5AC expression and favorable prognosis in patients with small intestinal adenocarcinomas. Pathology, research and practice. 2014;210:501-7
46. Cui H, Kong Y, Xu M, Zhang H. Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer research. 2013;73:3451-9
47. Jaskula-Sztul R, Eide J, Tesfazghi S, Dammalapati A, Harrison AD, Yu XM. et al. Tumor-suppressor role of Notch3 in medullary thyroid carcinoma revealed by genetic and pharmacological induction. Mol Cancer Ther. 2015;14:499-512
Author contact
![]() Corresponding author: Guo-Jun Zhang, MD, PhD. Tel.: +86(754)88556826; E-mail: guoj_zhangcom.
Corresponding author: Guo-Jun Zhang, MD, PhD. Tel.: +86(754)88556826; E-mail: guoj_zhangcom.