Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Evolution of models of cancer...
Aptamer-mediated drug transport...
Aptamer-guided drug delivery to...
Aptamer-guided delivery of...
Aptamer-guided co-delivery of...
Aptamer-guided delivery of...
Conclusions and challenges
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(16):3948-3961. doi:10.7150/thno.20725 This issue Cite
Review
Aptamer-Based Therapeutic Approaches to Target Cancer Stem Cells
Gang Zhou1, Olivier Latchoumanin1, Mary Bagdesar1, Lionel Hebbard1,2, Wei Duan3, Christopher Liddle1, Jacob George1, Liang Qiao1 ![]()
1. Storr Liver Centre, Westmead Institute for Medical Research, University of Sydney and Westmead Hospital, Westmead, NSW 2145, Australia;
2. Department of Molecular and Cell Biology, Centre for Comparative Genomics, The Centre for Biodiscovery and Molecular Development of Therapeutics, James Cook University, Australian Institute of Tropical Health and Medicine, Townsville, QLD 4811, Australia;
3. School of Medicine, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3217, Australia.
Received 2017-4-25; Accepted 2017-7-31; Published 2017-9-13
Abstract

Cancer stem cells (CSCs) are believed to be a principal cellular source for tumour progression and therapeutic drug resistance as they are capable of self-renewal and can differentiate into cancer cells. Importantly, CSCs acquire the ability to evade the killing effects of cytotoxic agents through changes at the genetic, epigenetic and micro-environment levels. Therefore, therapeutic strategies targeting CSCs hold great potential as an avenue for cancer treatment. Aptamers or “chemical antibodies” are a group of single-stranded nucleic acid (DNA or RNA) oligonucleotides with distinctive properties such as smaller size, lower toxicity and less immunogenicity compared to conventional antibodies. They have been frequently used to deliver therapeutic payloads to cancer cells and have achieved encouraging anti-tumour effects. This review discusses progress in CSC evolution theory and the role of aptamers to target CSCs for cancer treatment. Challenges of aptamer-mediated CSC targeting approaches are also discussed.
Keywords: aptamers, CSCs, cancer, drug resistance, antibodies, nanoparticles.
Introduction
Despite significant progresses in therapy, effective cancer treatment still remains an unmet clinical need. This is mainly due to the failure of present treatments to eradicate a subset of rare but important cells known as cancer stem cells (CSCs) [1]. CSCs have similar properties to adult stem cells such as the ability for unlimited self-renewal and differentiation and are believed to be a major source of cancer initiation, progression and drug resistance [2]. It is now understood that standard cancer treatments only eliminate rapidly proliferating cancer cells (non-CSCs), whereas the relatively quiescent CSCs can escape cell death and even be enriched in the remaining or recurrent tumour. These CSCs are thought to be the “root” of cancer relapse and metastasis. Thus, developing therapeutic strategies that not only kill non-CSC, but also eliminate CSCs has potential for achieving better treatment outcomes.
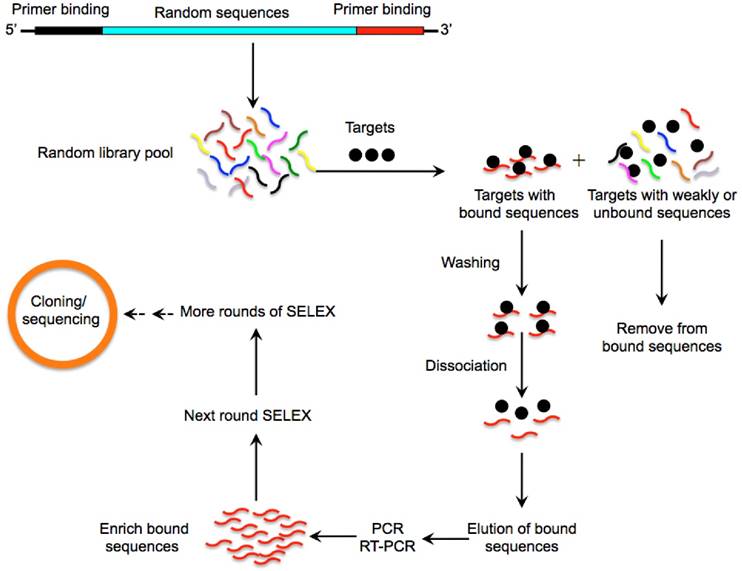
Over the past few years, many agents and strategies targeting CSCs have been developed [3-5]. However, successful penetration and accumulation of therapeutic agents into the tumour core particularly to CSCs is extremely difficult due to complicated drug resistance mechanisms. In addition, current anti-CSC reagents face numerous obstacles such as poor retention and bioavailability and strong immunogenicity and high toxicity which greatly hinders their clinical application [6, 7]. Aptamers are short single-stranded oligonucleotides that are selected from pools of random nucleic acid oligonucleotides by a systematic evolution of ligands by exponential enrichment (SELEX) (Fig. 1) [8, 9]. Aptamers can fold into unique tertiary structures and bind to their cognate targets with high affinity and specificity, hence the term “chemical antibodies” [10]. Importantly, aptamers exhibit multiple characteristics that are superior to conventional antibodies such as low toxicity and non-immunogenicity (Table 1) [10]. Furthermore, the smaller size of aptamers greatly facilitates their diffusion and penetration into tumour cores favouring the transport of therapeutic agents and nanoparticles (NPs) to their targets [11]. These properties make aptamers a prominent vehicle to accomplish specific drug delivery to cancer cells. Currently, a variety of aptamers targeting specific CSC surface markers have been developed and aptamer-based nano-medicine systems have achieved excellent anti-CSCs effects in preclinical studies [12-16]. In this review we provide readers with an understanding of recent developments on the subject of CSC evolution and discuss aptamers that target CSCs and their potential use for cancer treatment.
A schematic illustration of the SELEX method for aptamer selection. A random oligonucleotide library consisting of a large array of different sequence motifs (in the range of 1014-15) is designed. Two primer-binding sequences on both ends (5' and 3') of the randomized sequences allow for PCR amplification. The oligonucleotide library is then incubated with a pool of targets (such as proteins, nucleic acids or whole cells) under appropriate buffer and temperature conditions. Following stringent washing steps, a very small fraction of sequences with high binding affinities and specificities towards the target protein is separated from unbound or weakly bound sequences via various partitioning strategies. The tight binders are then eluted by dissociating the DNA or RNA-target complex and amplified by PCR to enrich into a new oligonucleotide pool which is then subjected to the next round of SELEX. This process is typically repeated for 10-15 rounds to enrich the sequences binding to the corresponding targets with highest affinity and specificity. To enhance the enrichment efficiency and specificity of aptamers, the stringency of the selection conditions is progressively increased in the later rounds through using effective binding competitors or decreasing the amount of proteins. The most promising sequences are randomly cloned and sequenced to obtain individual aptamers that are specifically involved in target recognition.
Comparison of aptamers and conventional antibodies
| Aptamers | Antibodies | |
|---|---|---|
| Targets | Wider range (nucleic acids, metal ions, organic molecules, amino acids, antibiotics, proteins, virus, whole cells, organisms) | Mostly larger molecules |
| Size | 10~25 kD | 50~100 kD |
| Production process | In vitro selection | In vivo selection |
| Toxicity Immunogenicity | Very low No or minimal | Relatively higher Relatively stronger |
| Cost of production | Cost-effective | Expensive |
| Modification | Easy to modify with imaging agents or therapeutic components | Difficult to modify |
| Stability | Stable in a harsh tumour micro-environment | Lower stability in physiological environments |
| Inter-batch variability | Low | Significant |
Evolution of models of cancer stem cell behaviour
The discovery of phenotypically distinct CSC pools with an extraordinary capacity to sustain long-term clonal growth and re-populate competitive sub-clones to survive therapies represents a rational explanation for the functional diversity of tumour cells and treatment resistance [17]. It is now believed that CSC evolution involves a dynamic equilibrium that requires both CSCs and the conventional clonal model acting in concert (Fig. 2A-B) [17]. Although CSCs can occasionally be replaced by normal stem cells [18], oncogenic mutations and epigenetic changes in CSCs at the early phase of tumour growth render these special clones less likely to be substituted by wild-type stem cells. Interestingly, the competition between normal stem cells and primary CSCs is closely dependent on the tumour microenvironment. For example, P53 mutated CSCs had similar proliferative capacities to wild-type normal stem cells in physiological conditions while they exhibited stronger progression than wild-type stem cells in inflamed intestine [18]. The combinatory effects of diverse advantageous mutations, epigenetic reprograming and micro- environmental factors results in multiple pools of rare primary CSCs with various self-renewal capabilities and variable aggressive properties. Within these pools there may be initiating, quiescent or slow cycling CSCs [19]. Indeed, distinct CSCs with diverse phenotypes and functions have been identified in leukemia and other solid organ tumours [7, 20-22].
Schematic illustration of the clonal evolution and dynamic plastic CSC model. A. Clonal evolution is a non-hierarchical model that acquires the successive accumulation of mutations to confer special clones with a competitive growth advantage under the pressures of cancer therapy, resulting in the expansion of surviving homogeneous sub-clones. B. The integration of clonal and the dynamic plastic CSC model. Primary mutations in progenitor cells endow them with stem cell properties to become initiating CSCs. Some of the mutated cells can remain quiescent and become metastatic CSCs, while other initiating CSCs transform to aggressive CSCs that eventually give rise to competitive metastatic CSCs upon serial mutations. The CSC populations gradually expand and dominate tumour sites although they eventually crash down upon cancer remission. Bio-directional plastic inter-conversion can occur between different CSCs or between the CSCs and non-CSCs, leading to complicated tumour heterogeneity. C. Therapeutic strategies targeting CSCs. Current chemotherapy can only kill rapidly dividing differentiated cancer cells in the tumour bulk but resistant CSCs are largely spared. The combination of traditional chemotherapy with CSC-targeted therapy is more likely to destroy non-CSCs and CSCs with better therapeutic outcomes. As residual non-CSCs can still convert to CSCs resulting in tumour relapse, and since tumour micro- environmental or epigenetic factors also contribute to the conversion of non-CSCs to CSCs, effective cancer therapy may require multiple approaches targeting CSCs, non-CSCs, and their dynamic conversion.
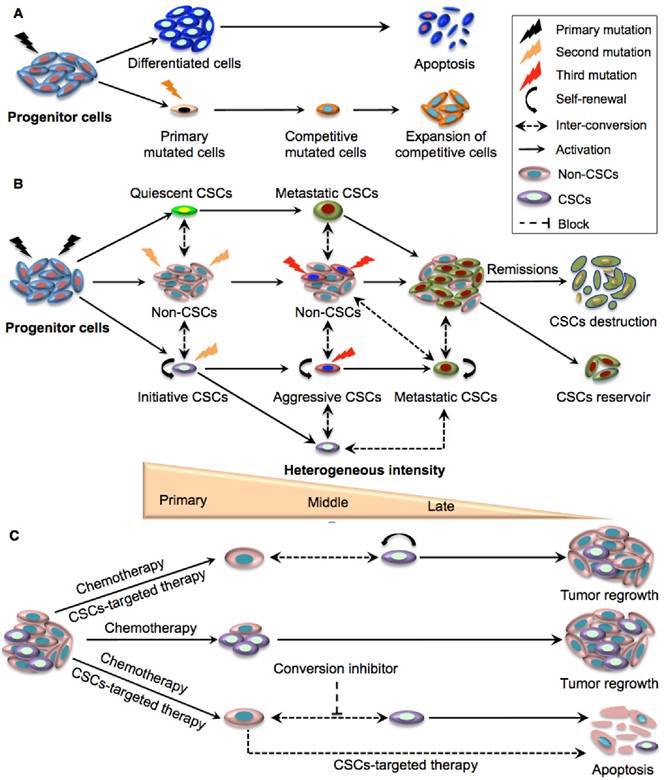
As tumours evolve and progress, additional advantageous mutations and micro-environmental pressures (e.g., as a result of cancer therapies) may impair the maturation process and cause competitions among diverse CSCs, leading to the expansion of increasingly competitive CSCs with even higher self-renewal capacity and more aggressive phenotypes [23]. When tumours progress to an advanced stage, the frequency of competitive CSCs with superior fitness will increase to become functionally and phenotypically homogeneous [23]. In this process, metastatic CSCs may emerge at the apex of tumour evolution upon overcoming other competing CSCs with less pioneering capacity. Interestingly, the last step of tumour evolution is always accompanied by a reduction of CSC expansion when the patient is in remission or ultimately succumbs to the tumour. This suggests that uncontrolled CSC expansion eventually leads to their destruction and is referred to as “CSC overshot”, a phenomenon similar to what occurs in ecosystems when the number of a species exceeds the maximum bearing capacity of their surroundings [24] (Fig. 2B). It is now understood that the number of CSCs within the same tumour dynamically changes during the entire malignant process, developing along a steep hierarchy in the primary stage consisting of a small number of heterogeneous CSCs and a larger number of differentiated non-CSCs, towards a hierarchy containing continually increasing proportions of CSCs, and constant progress to a very shallow hierarchy in the metastatic stage where the proportion of CSCs reach a plateau [23, 24] (Fig. 2B). Thus, CSC heterogeneity may apply more correctly to the early- rather than the more advanced stage of tumour development in which dominant clones probably drive tumour growth.
The complexity of tumour evolution is further reflected by the plastic inter-conversion between CSCs and non-CSCs. This is thought to result from a combination of micro-environmental pressure, genetic mutations and epigenetic reprogramming [25, 26-28] (Fig. 2B). Interestingly, the relationship between CSCs and the microenvironment is bidirectional in that the niche can determine the fate of CSCs and conversely, CSCs can alter the local environment. Indeed, glioblastoma (GBM) CSCs have been found to secret VEGF to stimulate the expansion of vasculature, whereas tumour endothelial cells may secret nitric oxide that can activate Notch signalling in glioma cells, a key regulatory mechanism for CSCs [29]. Surprisingly, some studies even demonstrate functional and phenotypic inter-conversions between different subsets of CSCs [29].
Taken together, the selective survival advantage of quiescent CSCs and expansion of aggressive CSCs upon several cycles of chemotherapy results in a relative increase in the CSC population in the residual tumour. Besides, conversion of non-CSCs to CSCs following targeted CSC therapies may be partially responsible for the resurgence of CSCs. Therefore, a combination of CSC-targeting approaches and therapeutic strategies that can ablate epigenetic or micro-environmental factors responsible for the transformation of progenitor cells into CSCs may open new avenues for inhibiting cancer recurrence (Fig. 2C).
Aptamer-mediated drug transport to cancer stem cells (CSCs)
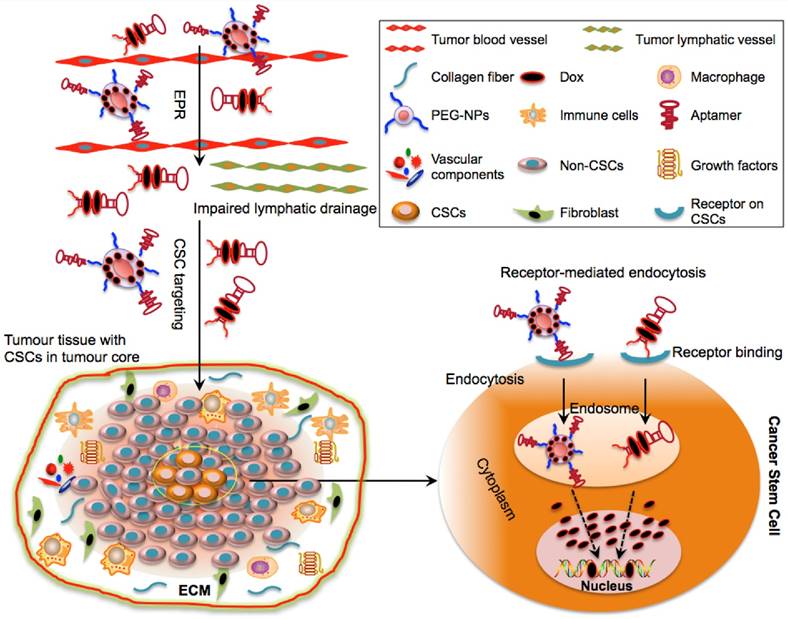
Most CSCs have been found to reside in the tumour center far away from tumour blood vessels. This means that therapeutic agents have to cross various biological obstacles in order to reach their targets. These obstacles may include leaky blood/lymphatic vessels, extracellular matrix (ECM) in which therapeutic agents may accumulate and tumour cell membranes that the agents have to penetrate through before reaching the peri-nuclear regions of CSCs [30]. These barriers may lead to impaired drug uptake, excessive drug degradation and short drug circulation within the tumour [24, 31]. In addition, agents without cancer targeting moieties may indiscriminately diffuse into both normal and tumour blood vessels leading to unwanted side effects [31]. Therefore, effective delivery approaches are needed to not only specifically transport therapeutic agents to CSCs but also to maintain their bioavailability and pharmacokinetics in blood. In this perspective, active targeting strategies which depend both on the enhanced permeability and retention (EPR) effect and aptamer-guided specific targeting have been explored.
Aptamers are 15-20 times smaller than antibodies and possess a range of advantages over conventional antibodies. Hence they have now emerged as a class of ideal drug delivery agents. Studies have shown that aptamer-based therapeutic drug delivery offers excellent tissue-penetrating properties and improved access of cytotoxic agents to CSCs within hypoxic regions [10]. Incorporation of aptamers on the surface of drug-carrying NPs can greatly enhance the efficiency of receptor-mediated endocytosis since the non-covalent binding of aptamers to their cognate receptors allows rapid dissociation of the aptamer-receptor complexes and recycling of the receptors to the cell surface in an intact configuration upon aptamer internalization [12, 14]. In addition, the conjugation of NPs with aptamers can augment the entrance and localization of NPs into the acidic environment of endosomes or lysosomes [32]. Since drugs release more readily from NP carriers in the acidic microenvironment than in the neutral condition, endosomal localization of aptamer-tailed NPs allows more effective release of functional free drug to the cytoplasm [33]. For example, a CD30 aptamer previously loaded with Doxorubicin (Dox) and surface engineered with a gold nanosphere (HAuNS) was used to generate a NPs-based chemotherapeutic system termed “apt-HAuNS-Dox”; this complex was able to rapidly release 80% of coupled Dox within 2 h at pH 5.0 while only 55% of Dox could be released from HAuNS-Dox without aptamer conjugation [34, 35] Furthermore, pH-triggered drug delivery systems such as aptamer-single-walled carbon nanotubes [35] and endosomal escape units like PEG-charge-conversion polymers allow endosomal membrane destabilization [33] and greatly minimizes the adhesion of NPs to endosomes leading to more effective endosomal escape of the therapeutic cargo.
Indeed, aptamer-tagged macromolecules were recently found to effectively enter and retained in tumours [36]. For example, an aptamer against EpCAM (a commonly used CSC marker) demonstrated 8-fold higher concentration in endosomes than EpCAM antibody [35]. Additional studies showed that EpCAM aptamers penetrated into the center of tumour spheres within 30-240 min and remained in the tumour cores for at least 26 h whereas the signal of EpCAM antibody was barely detectable after 4 h of incubation [36]. Besides, PEGylated aptamers could retain in animal tumours for more than 26 h, 4.3-fold longer than what could be achieved by an antibody. PEGylated aptamers also showed stronger tissue penetration capacity than antibody in that PEGylated aptamers were detectable 150-200 μm away from the blood vessels of tumour tissues whereas the antibody was mainly distributed near blood vessels [36]. In a very recent study, aptamer-anchored polymersome vesicles exhibited favourable tumour penetration ability in both 3D tumour spheroids and tumour-bearing mice; these vesicles were shown to selectively accumulate Dox to tumour sites leading to decreased systemic toxicity and significant suppression of breast cancer in vivo [37]. These encouraging results suggest that aptamers can be used as promising escorting tools to target CSCs.
Aptamer-guided drug delivery to CSCs
Selectively delivering cytotoxic agents using aptamers to CSCs has been explored in cancer therapy and various aptamers that can specifically and effectively target CSC surface markers have been developed (Table. 2) (12-16; 38-46). These aptamer moieties are able to deliver free drugs or drug-loaded NPs to CSCs with negligible harmful effects to normal cells (Table. 3). Upon entering into tumour tissues by the EPR effect, the aptamer complex may cross the barrier of ECM interstitia without undergoing degradation and are selectively internalized into CSCs via receptor-mediated endocytosis, an important mechanism capable of bypassing ATP-binding cassette transporters (Fig. 3).
Aptamers targeting surface markers of CSCs
| Aptamers (apt) | Targets | Binding affinity (Kd) | Cell lines tested | Refs |
|---|---|---|---|---|
| EpDT3/EP166/SYL3C (RNA) | EpCAM | 6 nM-67 nM | HT29/Hep3B/MCF7 /HepG2/KatoIII/MDA-MB-231 | [12, 16, 38, 39] |
| A15/B19 (RNA) | CD133 | 34-145 nM | HT29/Hep3B | [15] |
| TA1/TA6 (DNA) /Apt1/MS03(RNA) | CD44 | 21.5-285 nM | SKOV3/IGROV/A2780/A549/MCF7/MCF10A | [39-42] |
| Kit-129/Apt1-4 (DNA) | CD117 | 7.1-36.2 nM | BJAB/HEL | [43, 44] |
| A12/35 (RNA) | ABCG-2 | 16.7 nM | MCF-7/MDA-MB23 | [45] |
| A1/A4 (DNA) | GBM CSCs | 0.27-3.8 nM | CD133+ GBM cells | [13] |
| CSC13/17/22 (RNA) | Prostate CSCs | 2.2-31.3 nM | DU145/PC3/LNCaP | [14] |
A549: lung cancer cell line; BJAB: Burkitt lymphoma B cell line; DU145, PC3, LNCaP: prostate cancer cell lines; HEL: human erythroleukemia cell lines; Hep3B, HepG2: liver cancer cell lines; HT29: colon cancer cell lines; KatoIII: gastric cancer cell line; MCF7, MDA-MB-231: breast cancer cell lines; SKOV3, IGROV, A2780: ovarian cancer cell lines;
Binding Kd is defined as the concentration of aptamer at which 50% of the cell receptor sites are occupied.
Aptamer-mediated delivery of therapeutic agents to CSCs
| Aptamers (DNA or RNA) | Targets | Cytotoxic agents | Delivery vehicle | Anti-CSCs effect | Refs |
|---|---|---|---|---|---|
| Apt4 (DNA) | CD117 | MTX | Apt4 | Induce apoptosis in AML CSCs in vitro and in vivo | 44 |
| EpDT3 (RNA) | EpCAM | Dox | EpDT3 | Inhibit proliferation of retinoblastoma CSCs in vitro | 46 |
| Anti-EpCAM (RNA) | EpCAM | Dox | PLGA-PEG NPs | Induce significant cytotoxicity to breast CSCs in vitro | 48 |
| Anti-EpCAM (RNA) | EpCAM | Dox | PLGA-PEG NPs | Induce cytotoxicity to lung cancer CSCs in vitro, inhibit tumour growth in vivo | 49 |
| Anti-EpCAM (RNA) | EpCAM | Curcumin | PLGA-lecithin-PEG | Induce substantial cytotoxicity to colon CSCs in vitro | 37 |
| Anti-EpCAM (RNA) | EpCAM | Nutlin-3a | PLGA-QD | Induce cytotoxicity to breast and ovarian CSCs in vitro and in vivo | 50 |
| Anti-EpCAM (RNA) | EpCAM | Dox | MSNs | Inhibit proliferation and induce apoptosis in colon CSCs in vitro | 54 |
| Anti-EpCAM (RNA) | EpCAM | Fe-blf | AEC-CP | Inhibit tumour growth of colon CSCs in vivo | 56 |
| CSC13 (DNA) | Prostate CSCs | AuNRs | CSC13 | Induce apoptosis of prostate CSCs in vitro and in vivo | 57 |
| EpDT3 (RNA) | EpCAM | Ad5-PTEN | EpDT3 | Inhibit tumour growth of liver CSCs in vitro and in vivo | 63 |
| EpDT3 (RNA) | EpCAM | Survivin siRNA | EpDT3 | Reverse chemoresistance of breast CSCs, and inhibit tumour growth in vitro and in vivo | 66 |
| A19 (RNA) | EpCAM | PLK1 siRNA | A19 | Inhibit tumourigenicity of TNBC CSCs and tumour growth in vitro and in vivo | 67 |
| Ep (RNA) | EpCAM | EpCAM siRNA | EpApt | Inhibit proliferation of RB and breast CSCs in vitro, regresses growth of breast cancer in vivo | 68 |
| Ep (RNA) | EpCAM | EpCAM siRNA | PEI NPs | Inhibit proliferation of RB and breast cancer CSCs in vitro | 69 |
| EpDT3 (RNA) | EpCAM | BCL9l | SWNT-PEI NPs | Induce apoptosis of breast CSCs in vitro | 70 |
| HER-2 apt (DNA) | HER-2 | MED1 siRNA | pRNA-HER2apt | Reduce breast CSCs population and inhibit tumour growth and metastasis in vivo | 72 |
| MRP1 apt (RNA) | MRP1 | CD28 apt | MRP1 apt | Enhance anti-tumour immunity to MRP1 melanoma CSCs | 87 |
Schematic illustration of drug delivery to CSCs via aptamer-based targeting. The optimal molecular size of aptamer-Dox-PEG conjugate or aptamer-guided NPs can facilitate their passive diffusion into the leaky vasculature of tumour tissues based on their enhanced permeability and retention (EPR) effect. Due to the impairment of tumour lymphatic drainage, these aptamer-based conjugates can accumulate in the vicinity of the tumour bulk and may selectively penetrate into the tumour core (where CSCs reside) via active targeting of aptamers. Aptamer-based conjugates can specifically bind to the surface receptors of CSCs and can be taken up by CSCs via receptor-mediated endocytosis. Following internalization and escape from endosomes, the released drugs engage their cytoplasmic or nucleic targets to destroy CSCs.
Application of aptamers in the delivery of free drug to CSCs
Both DNA and RNA aptamers are known to form tertiary structures that contain short double stranded regions through intra-molecule base pairing. Chemotherapeutic agents such as Dox can be preferably inserted into the GC pairs of the double strand domain of aptamers through non-covalent intercalation to generate a therapeutic conjugate. Utilizing this physical conjugation method, a chimeric EpCAM aptamer-Dox conjugate was found to specifically deliver Dox to EpCAM+ retinoblastoma CSCs and to selectively inhibit proliferation [46]. Similarly, a DNA aptamer against a putative leukemia CSC marker CD117 was conjugated with methotrexate (MTX). The resultant CD117 apt-MTX complex could be selectively taken up and internalized to CD117+ AML cells, leading to potent inhibition of tumour growth [44]. However, the weak non-covalent connection between drug and aptamer makes drug encapsulation and uptake efficiency unpredictable. Moreover, these conjugates with small physical size are also flawed by rapid renal clearance and inefficient drug retention due to lack of a sustainable drug release system [47].
Application of aptamers in the delivery of nanoparticle (NP)-based drugs to CSCs
Nano-materials that have superior drug encapsulation and controllable drug release capacity have proved to be more effective and safer for anti-CSC therapy. NPs including organic polymers such as poly lactic-co-glycolic acid (PLGA) and inorganic NPs have been loaded with therapeutic agents and linked with long-living aptamers specific to unique surface markers of CSCs. These aptamer-guided NP systems enhance the selective uptake and retention of therapeutic drugs within tumours, achieving promising and specific CSC-killing.
Delivery of organic nanoparticle (NP) drugs to CSCs
Liposomes are one of the most successful NP-based drug delivery tools due to advantages such as high biocompatibility and stability, ease of synthesis and low toxicity. PEGylated liposomal Dox was the first FDA approved NP-based drug. In a recent study, PEGylated liposomal NPs were prepared by modifying them with aptamers against the CSC marker CD44. The resultant aptamer-liposome complex demonstrated high selectivity and affinity to CD44+ lung CSCs [39]. However, such a liposomal formulation could not provide continuous release of loaded drugs, leading to short drug retention and undesirable in vivo therapeutic efficacy [42]. Subsequently, poly (D, L-lactide-co-glycolide) (PLGA)-PEG NP have been investigated as a more promising approach owing to their favourable biocompatibility and sustained drug release properties [48]. This nano-scale system is composed of PLGA which is able to form a hydrophobic core for encapsulating various drugs, a PEG shell to the prolong circulating half-life in vivo, and aptamers attached on the surface to target CSCs. Recently, a RNA aptamer against EpCAM covalently coupled to the surface of a PLGA-PEG nanopolymersome was successfully loaded with Dox with an encapsulation efficiency of more than 90%. In this system, Dox could be constantly released from the aptamer-NP after the initial burst of release. Moreover, the resultant complex exhibited more efficient internalization and penetration into tumour spheroids resulting in potent selective inhibition in the growth of EpCAM+ breast cancer in vitro and lung cancer both in vitro and in vivo [48, 49]. In another study, an anti-EpCAM aptamer conjugated PLGA-PEG NP was used to encapsulate curcumin (CUR), a natural anti-tumour compound with poor solubility and limited bioavailability [36]. In order to improve the stability and drug-loading capacity of PLGA-PEG, lecithin known as a good dispersing agent was coated onto the PEG core. Due to the sustainable drug release from the polymer matrix after drug absorption, the resultant CUR-NPs exhibited an approximately 6-fold increase in half-life and a 3-fold increase in mean retention time in vivo compared to free CUR. This led to significant inhibition of proliferation of EpCAM+ colon CSCs in vitro and colon cancer growth in vivo [36].
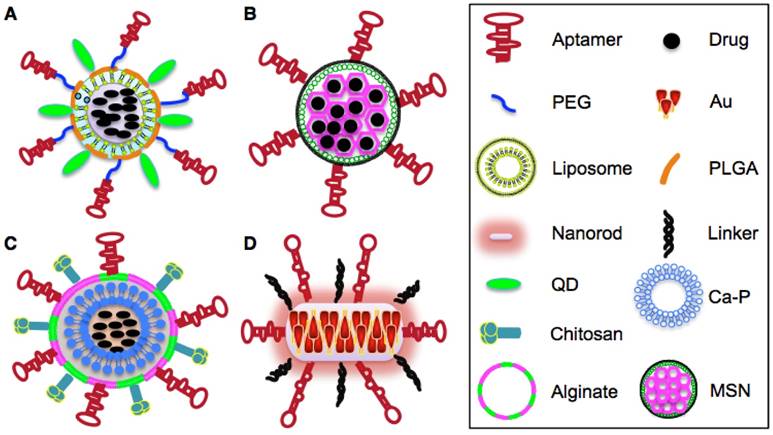
Apart from their role in targeted and sustained drug delivery, aptamer-guided NPs can be used to simultaneously detect cancer cells and assess therapeutic response. In this setting, a multi-functional nanotheranostic system was formulated by conjugating EpCAM aptamers and imaging quantum dots (QD) onto the surface of PLGA NPs that were pre-loaded with a chemotherapeutic agent nutlin-3a (Fig. 4A) [50]. This nutlin-3a-loaded apt-QD-PLGA nano-system demonstrated better accumulation and deeper penetration into QD-visualizing EpCAM+ breast and ovarian cancer spheroids than the untargeted NPs counterparts and showed promising therapeutic effects against EpCAM+ tumours cells [50]. Although PLGA NPs have an encapsulation efficiency of 60~90% (percentage of the amount of loaded drug relative to total amount of drug used for the formulation), the drug incorporation efficiency of PLGA (percentage of the amount of the loaded drug relative to the total amount of NPs) is generally unsatisfactory, being around 10% [51]. This is mainly attributed to the water solubility of hydrophilic drugs which can lead to rapid partitioning into the aqueous phase and hence decrease their entrapment into NPs during polymer deposition [52]. Besides, the large porous area of PLGA geometry also contributes to enhanced drug leakage into the aqueous phase. The low drug incorporation efficiency constitutes a major hurdle for commercialization of PLGA-based NPs since a large quantity of expensive PLGA with well-defined characteristics need to be required to achieve desirable drug concentrations at targeted sites [52].
Schematic illustration of aptamer-based NPs. A. Structure of aptamer-Nut-QD-PLGA- PEG NPs. B. Structure of aptamer-Dox-MSNs NPs. C. Structure of aptamer-alginate-chirosan-Ca- Fe-blf NPs. D. Structure of aptamer-AuNRs NPs.
Delivery of inorganic nanoparticle (NP) drugs to CSCs
In order to overcome the drug leaking issue of organic polymer NPs, some inorganic NPs such as mesoporous silica NPs (MSNs) have recently been developed. MSNs as mesostructures have several advantages over polymer NPs including controllable configuration, larger surface area and smaller but higher organized pore networks. These features greatly reduce the leakage of anti- cancer drugs from the pore tunnels and increases drug loading capacity [53]. Recently, Dox-loaded MSNs were surface conjugated with an anti-EpCAM aptamer to selectively transport Dox to EpCAM-expressing colon cancer cells (Fig. 4B). The resulting apt-MSN-Dox NPs showed high Dox incorporation efficiency of 41.6% and increased uptake by EpCAM+ but not by the EpCAM- colon cancer cells leading to more cytotoxicity and apoptosis on targeted cells than the non-targeted MSN-Dox control [54].
Iron-saturated bovine lactoferrin (Fe-blf) is a multifunctional pleiotropic natural glycoprotein that exhibits potent anti-cancer activity in breast, lung, bladder, colon and liver cancers [55]. In order to overcome the harmful effects of Fe-blf to normal cells and further enhance their bioavailability to tumour sites, an alginate-enclosed, chitosan-coated (AEC), calcium phosphate (Ca-P) inorganic nanocarrier (NCs) was formulated to encapsulate Fe-blf and surface modified with an EpCAM aptamer (Fig. 4C) [56]. The rate of uptake of the modified NCs by the EpCAM+ cells was 2.84-fold higher than that by the EpCAM- cells. Additionally, the resulting AEC-CP-Fe-blf-apt nano-formulation was found to significantly regress the growth of xenograft tumours derived from EpCAM+/CD133+/CD44+ colon CSCs in mice and to decrease CSCs marker expression. Only 10% of mice fed with AEC-CP-Fe-blf-apt showed tumour recurrence compared to 30% tumour recurrence in mice fed on non-targeted NCs [56]. In addition to delivering chemotherapeutic payloads to CSCs, aptamers have been utilized to selectively induce photothermal destruction of CSCs. Previously, an aptamer called CSC13 targeting a subpopulation of prostate CSCs was conjugated with inorganic gold nanorods (AuNRs) and was found to convert near-infrared (NIR) laser to localized thermal energy resulting in photo- thermal destruction of adjacent cells (Fig. 4D) [57]. Upon selective uptake and internalization, AuNRs were released from CSC13-AuNRs and generated sufficient thermal heat enabling a sharp increase of temperature from 25°C to 55°C in the target cells (prostate CSCs) while the temperature of the untargeted cells was barely affected. This specific laser-irradiation effect of CSC13-AuNRs selectively induced apoptosis of prostate CSCs with minimal harmful effect on surrounding normal tissues [57].
Delivery of nanoparticles (NP) drugs to both CSCs and non-CSCs
Due to the plasticity of CSCs in that non-CSCs may dedifferentiate into CSCs in response to therapeutic pressure, selectively depleting CSCs would not be sufficient to prevent tumour recurrence if the residual differentiated non-CSCs are still present. Hence, more effective therapeutic approaches targeting both CSCs and non-CSCs is essential. One of the plausible approaches would be to utilize aptamers targeting common receptors that are overexpressed in both tumour bulk cells and CSCs. For example, epidermal growth factor receptor (EGFR) is known to be over-expressed in the majority of tumour cells and CSCs of breast, brain and pancreatic cancers [58]. In previous studies, cetuximab (an EGFR-specific antibody) was used as a targeting agent to specifically deliver gold NPs loaded with gemcitabine (GEM) to pancreatic cancer cells and achieve significant inhibitory effects on tumour growth in vivo [59]. In order to attenuate the severe toxicities of cetuximab, another group subsequently utilized a RNA aptamer against EGFR (E07) to escort GEM to pancreatic cancer cells. GEM as a nucleoside analog was incorporated into the selection oligonucleotide pool and annealed to the EGFR aptamer to create a nucleic acid compound with two functional domains composed of EGFR aptamer and GEM-containing polymer [60]. The E07-GEM polymer exhibited selective internalization to EGFR+ cells and stronger inhibitory effects on EGFR+ pancreatic cells in vitro than the non-escorted GEM polymer [60]. In addition, a new DNA aptamer (HB5) against HER2 (over-expressed in both differentiated breast cancer cells and breast CSCs) was shown to specifically carry Dox into HER2+ breast cancer cells and selectively regress tumour growth in vitro [61, 62].
Aptamer-guided delivery of therapeutic nucleic acids to CSCs
Apart from delivering poorly soluble drugs, aptamers can be used to selectively transport tumour suppressor gene to CSCs. For example, a recombinant adenovirus coupling the tumour suppressor gene PTEN (Ad5-PTEN) was shown to be an effective anti-tumour agent for liver cancer [63]. Through conjugating with EpCAM aptamer (EpDT3) and PEG, the serum retention of Ad5-PTEN could be increased 16-fold resulting in a significant inhibition of tumour growth in vitro and in vivo of EpCAM-positive liver cancer [63]. SiRNA and miRNA which function as crucial post-transcriptional suppressors of target genes via RNA interference (RNAi) can knockdown vital oncogenic and anti-apoptotic genes that are involved in drug resistance of CSCs [64]. However, clinical application of therapeutic siRNA and miRNA is limited by several shortcomings such as low cellular uptake, poor pharmacokinetic profiles and systemic toxicity due to their nuclease-labile and hydrophilic characteristics [65]. Thus, more efficient aptamer-based delivery systems that can protect siRNAs and miRNAs from nuclease degradation and facilitate their selective intracellular transport and accumulation in tumour cores to target CSCs are needed (Table 3). Currently several aptamers against CSC surface markers have been developed to achieve specific siRNA and miRNA delivery to CSCs of various tumours (Fig. 5).
Schematic illustration of aptamer-mediated nucleic acid delivery to CSCs. Exogenous therapeutic siRNAs or miRNAs can be directly linked with aptamers or encapsulated within NPs that is surface functionalized with aptamers. Aptamer-siRNA/miRNA or aptamer-NPs can bind to and internalize into CSCs via receptor-mediated endocytosis, followed by entry into the endosome complex. Under the acidic environment, siRNAs/miRNAs are released and escape from endosomes and are then incorporated into the RNA-induced silencing complex. The mature siRNAs and most miRNAs interact with their cytoplasmic target mRNA while a few miRNAs such as miR29b are predominantly localized to nuclei, leading to mRNA degradation, translational and transcriptional regulation.
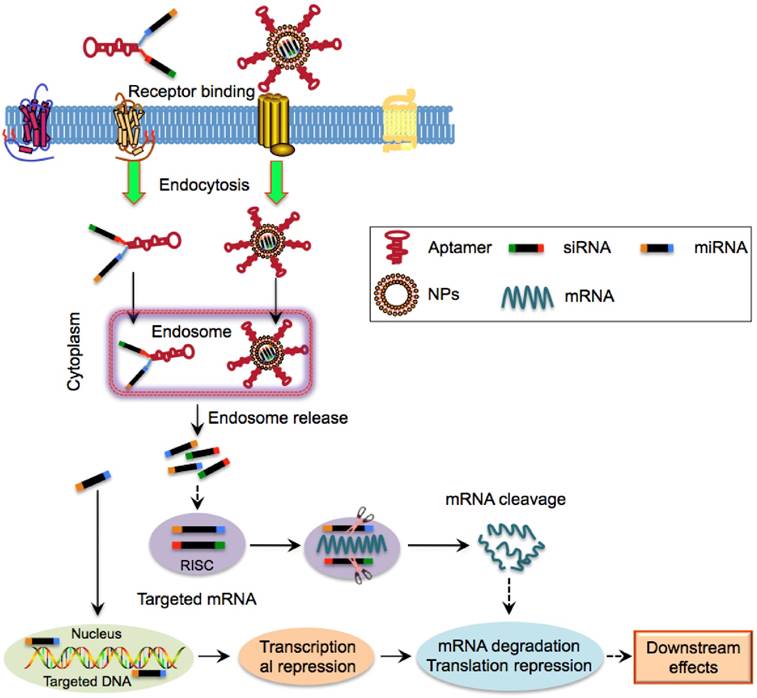
Aptamer-guided delivery of siRNAs to CSCs
Survivin is an important pro-survival protein involved in the promotion of tumour angiogenesis and chemo-resistance. An EpCAM-specific aptamer has been utilized to specifically deliver survivin siRNAs to breast CSCs leading to a decrease of endogenous survivin by more than 80% in EpCAM+ breast cancer cells [66]. Moreover, this aptamer-siRNA chimera-mediated survivin silencing reversed chemo-resistance such that combined treatment with Dox significantly inhibited tumour growth and prolonged survival of mice bearing chemo-resistance tumours accompanied by the reduction of CSC populations and impairment of self-renewal capacity [66]. In another interesting example, an EpCAM aptamer-siRNA chimera known as AsiC was used to specifically deliver polo like kinase 1 (PLK1) siRNA to triple-negative breast cancer (TNBC, which are poorly differentiated breast cancers lacking the expression of estrogen, progesterone and HER2 receptors). In the AsiC chimera, the EpCAM aptamer was connected to the PLK1 siRNA sense strand via a U-U-U linker and then annealed to the anti-sense strand of siRNA. This PLK1 EpCAM-AsiC could efficiently knockdown PLK1 expression and significantly attenuated the tumour initiating and self-renewal ability of EpCAM+ TNBC CSCs in vitro, and regressed the growth of xenograft tumours derived from EpCAM+ but not EpCAM- TNBCs [67].
EpCAM is not only a well-known CSC marker but also an important transcriptional target of oncogenic Wnt/β-catenin signalling. EpCAM can be cleaved by a tumour-necrosis-factor alpha converting enzyme (TACE/ADAM17) leading to the release of an intracellular domain (EpICD) of EpCAM into the cytoplasm. EpICD can migrate into the nucleus where it forms a complex with four and a half LIM domain 2 (FHL2), β-catenin and lymphoid enhancer binding factor 1 (Lef1). This complex in turn induces the activation of target genes (such as c-myc and cyclins) of Wnt/β-catenin signalling [68]. In a recent study, an EpCAM aptamer was coupled with siRNA against EpCAM and the resulting chimera (EpApt-siEp) successfully knocked down EpICD expression in EpCAM+ cancer cells [68]. Moreover, this chimera was shown to significantly and specifically inhibit the proliferation of EpCAM+ retinoblastoma (RB) cells and epithelial breast cancer cells in vitro, and completely regress the growth of epithelial breast cancer in vivo [68]. In order to improve the stability and tumour retention of the therapeutic EpCAM siRNA, the same group further incorporated siRNA into a cationic PEI polymeric carrier that was surface coupled with the EpCAM aptamer. The resulting PEI-EpApt-SiEp nano-complex selectively silenced EpCAM expression and exerted stronger anti-proliferative effects than EpApt-siEp to EpCAM+ RB cells and breast cancer cells in vitro [69].
Another recently synthesized single-walled carbon nanotube (SWNT) with a unique nano-needle structure was shown to efficiently cross the plasma membrane by an endocytosis-dependent pathway [70]. To overcome the poor solubility and low biocompatibility of SWNT, SWNT was conjugated to a hydrophobic piperazine-PEI derivative that was surface linked with an anti-EpCAM aptamer (EpDT3) for specific delivery of siRNA against B cell lymphoma 9-like (BCL9L), an oncogene required for Wnt activation. This SWNT-PEI-EpDT3 nano- complex exhibited 10-fold higher siRNA delivery efficiency than PEI alone in EpCAM+ breast cancer cells. Consequently, efficient silencing of BCL9L specifically induced significant apoptosis in EpCAM+ breast cancer cells [70].
Due to the high plasticity of CSCs, delivering therapeutic nucleic acids to both differentiated and undifferentiated tumour cells (CSCs) would be a desirable approach for cancer therapy. In this regard, aptamers targeting the HER-2 gene have been employed as the delivery vehicle of siRNA against an anti-apoptotic gene, B cell lymphoma 2 (BCL-2). This aptamer-conjugated BCL-2 siRNA effectively increased the chemo-sensitivity of HER-2+ breast cancer cells to cisplatin-induced apoptosis [71]. Another recent study generated a novel multifunctional RNA NP using the three-way junction (3WJ) motif derived from bacteriophage phi29 packaging RNA (pRNA) as the scaffold core [72]. The pRNA NPs were further coupled with a HER2 aptamer and siRNA against estrogen receptor (ER) coactivator mediator subunit 1 (MED1) to form a pRNA-HER2apt-siMED1 complex. In addition to their capacity to significantly reduce tumour growth and metastasis, the resulting complex greatly depleted CSCs of HER2-expressing breast tumour when combined with tamoxifen treatment in vivo [72]. Although a direct effect on CSC fate has not been claimed, the anti-tumour effects from these proof-of-concept studies indicates a novel direction in aptamer-based delivery of chemo-sensitizing siRNAs to cancer cells.
Aptamer-guided delivery of miRNAs to CSCs
miRNAs have been demonstrated to play both oncogenic and tumour suppressive roles and are a key regulatory factor in the treatment resistance of CSCs [73]. To this end, specific delivery of tumour-suppressive miRNA such as miRNA126 and miRNA34 to CSCs through aptamer-based approaches may represent a promising therapeutic strategy [10].
Transferrin receptor (TfR) is overexpressed on both CSCs and non-CSCs of various cancers including those from lung, breast, brain and pancreas [74]. Recently, a TfR-specific aptamer which elongated at the 3' end with the passenger strand of the siRNA sequence via a GC-rich “stick” complementary linker and further annealed to a guide strand of siRNA was developed. Such sticky-end annealing linkage not only ensures correct folding of the aptamers, but also reinforces the non-covalent binding between the miRNA and the aptamer [75]. Based on this approach, the TfR aptamer was shown to specifically deliver tumour suppressor miR126 into breast cancer cells leading to a significant inhibition of cell proliferation in vitro [76]. In a similar report, tumour suppressor miR222 was delivered into PDGF+ glioblastoma (GBM) cells by Gint 4.T, an aptamer against platelet derived growth factor receptor (PDGFR) which is extensively expressed in both differentiated tumour bulk cells and CSCs, resulting in marked reduction of tumour growth in vitro and in vivo [77].
In order to increase the retention and stability of therapeutic miRNAs in the circulation, aptamer-conjugated nano-carriers that can sustainably release encapsulated miRNAs into desired tumour sites have been exploited. For instance, polyamidoamine (PAMAM) that is cationic with dendritic structures are widely employed as non-viral gene vectors to transfect exogenous DNA or RNA into cells. In a recent study, PAMAM-PEG NPs were utilized to efficiently encapsulate miR34a (a potent endogenous tumour suppressor), and the miR34a-encapsulated NPs were conjugated with S6, an aptamer against A549 lung cancer cells. The resulting chimera (PAM-Ap/pMiR-34a NPs) was shown to inhibit the growth and migration of lung cancer cells [78]. In another report, a novel multifunctional RNA NP was generated using the three-way junction (3WJ) motif [74]. This pRNA-3WJ core was further modified to form trifunctional NPs harboring three functional modules including EGFR aptamers as the targeting ligand to TNBC, therapeutic anti-miR-21, and Alexa-647 as an imaging module [79]. The resultant chimera 3WJ-EGFRapt/anti-miR-21 was shown to successfully navigate across the tumour micro-environment barriers, significantly down-regulating oncogenic miRNA-21 and up-regulating its down-stream targets such as tumour suppressor phosphatase and tensin homolog (PTEN) and programmed cell death 4 (PDCD4), resulting in efficient eradication of TNBC CSCs and inhibition of tumour growth both in vitro and in vivo [79].
Aptamer-guided co-delivery of therapeutics to CSCs
As multiple mechanisms are involved in the development of drug resistance of CSCs, blocking one mechanism with a single anti-cancer agent frequently activates and strengthens alternative pathways leading to ongoing drug resistance and tumour relapse [57]. Therefore, approaches that target multiple mechanisms such as co-administration of drug cocktails or combined drug and nucleic acid therapy may exert synergistic inhibitory effects on CSCs. Previously, multi-functional NP systems such as PLGA-PEG, liposomes and micelles that can simultaneously incorporate multiple therapeutic agents into a single NP have been investigated. These therapeutic complexes are specifically delivered into tumour cells under the guidance of targeting aptamer moieties with promising therapeutic efficacy [80-82]. However, most of these studies were only tested in vitro and whether these multifunctional aptamer-NPs can improve specific cytotoxicity to CSCs and regress tumour re-growth is still unknown. In order to achieve the best possible therapeutic effect, development of smart aptamer-coupled nano-carriers that can selectively deliver drug combinations to CSCs and comprehensively evaluating their CSC-targeting efficacy is necessary.
Aptamer-guided delivery of immunotherapeutic drugs to CSCs
The interaction of co-stimulatory molecules (such as 4-1BB, CD28 and OX40) with their cognate ligands is essential for the activation of T lymphocytes [83]. The reduction of co-stimulatory ligands within the tumour microenvironment greatly compromise the ability of T cells to exert anti-tumour immunity [83]. Some agonistic aptamers against CD28 and 4-1 BB were found to co-stimulate T cells and promote tumour immunity [84, 85]. To minimize the non-specific targeting of these agonists, a bi-specific aptamer that can simultaneously target prostate specific membrane antigen (PSMA) and the 4-1 BB receptor was developed and shown to potentiate T-cell dependent tumour immunity at prostate tumour sites [86]. However, the effects of these co-stimulatory aptamers on CSCs are poorly understood. In this regard, a new multidrug resistance-associated protein 1 (MRP1) aptamer targeting chemotherapy-resistant melanoma CSCs was first developed and utilised to form a new bi-specific MRP1-CD28 aptamer [87]. The resulting bivalent aptamer was able to specifically deliver the protective CD28 co-stimulatory signals to MRP1-expressing melanoma CSCs eliciting a potent anti-tumour immune response and reduced tumour growth in vivo than conventional CD28 agonist [87]. Moreover, a whole-cell vaccine (aptvax) consisting of irritated melanoma tumour cells coated with MRP1-CD28 aptamer was shown to boost anti-tumour immunity and significantly inhibit tumour growth in vivo [87].
Conclusions and challenges
It is now well-accepted that the evolution of cancer follows Darwin's principle of “survival of the fittest”, uncovering the unique competitiveness and aggressive nature of CSCs in adapting to the pressures of cancer treatment. Moreover, CSCs within a heterogeneous tumour displays complex plasticity by reversibly transitioning between CSCs and differentiated cancer cells. The tumour- initiating and chemo-resistant capabilities of CSCs neccesitate the development of effective anti-CSC therapeutic strategies that can selectively destroy resistant CSCs. Through the combination of anti-CSCs and traditional therapeutic approaches, both residual tumour cells and CSCs are more likely to be depleted, avoiding the frequent switching of non-CSCs to CSCs and tumour relapse. To achieve these goals, a number of NP systems have been incorporated with specific aptamers targeting CSC surface markers and has demonstrated promising therapeutic efficacy for CSCs in pre-clinical studies.
However, several key problems need to be resolved before aptamer-NP conjugates can be widely tested in clinical settings. First, there is a lack of a specific marker for CSCs and the currently available markers for CSCs are also present on normal stem cells and even normal cells. Therefore, aptamer-nano-carriers aimed at targeting CSCs may exert non-specific toxicity to normal stem cells. One potential and feasible approach is to develop aptamers that specifically target stemness-enriched cancer cells (such as tumour sphere cells generated from patients-derived tumour cells) rather than CSC markers using the whole-cell SELEX method. To select unique CSC-specific aptamers, the living tumour sphere cells are utilised as targets and negative selection is performed using normal cancer cells to eliminate non-CSC associated aptamers. This was exemplified by a very recent study that developed two pancreatic CSC-associated DNA aptamers using tumour sphere cells as targets in the Cell-SELEX selection [88]. Second, the tumour penetration potential of aptamer-guided conjugates has mostly been tested in in vitro studies but rarely in relevant animal models. Whether aptamer-based drug delivery actually facilitates drug accumulation and release into the deep layers of tumours is still unclear. Therefore, comprehensive evaluation of tumour-penetrating and cargo-delivering characteristics of aptamer-targeted conjugates in vivo using living animal imaging systems is necessary in future studies. Third, in most studies, the approach for evaluating the therapeutic efficacy of anti-CSC therapies in preclinical models is based on tumour volume reduction. However, tumour eradication and prevention of tumour relapse that genuinely reflects an effect on CSC elimination has rarely been assessed. In this aspect, developing clinically-relevant mouse models (such as patient-derived orthotopic models) that can not only closely recapitulate the pathologic tumour microenvironment but can also mimic tumour metastasis and regrowth may be a viable approach to precisely assess the therapeutic efficacy of anti-CSC agents. In conclusion, although aptamer-mediated anti-CSC therapy provides a promising strategy to eliminate the “culprit” of cancers, more robust pre-clinical and clinical studies are needed to explore their full therapeutic potential.
Acknowledgements
This work was partially supported by the Robert W. Storr Bequest to the Sydney Medical Foundation, University of Sydney; a Cancer Council NSW grant to CL and LQ (ID: APP1070076).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2014;20:648-59
2. Siegel RD, Naishadham D. Cancer statistics. CA Cancer J Clin. 2013;63:11-30
3. Zhao Y, Alakhova DY, Kabanov AV. Can nanomedicines kill cancer stem cells. Adv Drug Delivery Rev. 2013;65:1763-83
4. O'Connor ML, Xiang D, Shigdar S, Macdonald J, Li Y, Wang T. et al. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014;344:180-7
5. Malhi S, Gu X. Nanocarrier-mediated drugs targeting cancer stem cells: an emerging delivery approach. Expert Opin Drug Deliv. 2014;12:1177-1201
6. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond). 2012;7:597-615
7. Kuhlmann JD, Hein L, Kurth L, Wimberger P, Dubrovska A. Targeting cancer stem cells: promises and challenges. Anti-cancer Agent Me. 2016;16:38-58
8. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505-10
9. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;30:818-22
10. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;30:182-9
11. Dua P, Kim S, Lee DK. Nucleic acid aptamers targeting cell-surface proteins. Methods. 2011;54:215-25
12. Shigdar S, Lin J, Yu Y, Pastuovic M, Wei M, Duan W. et al. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102:991-8
13. Kim YM, Wu Q, Hamerlik P, Hitomi M, Sloan AE, Barnett GH. et al. Aptamer identification of brain tumor-initiating cells. Cancer Res. 2013;73:4923-36
14. Sefah K, Bae KM, Phillips JA, Siemann DW, Su Z, McClellan S. et al. Cell-based selection provides novel molecular probes for cancer stem cells. Int J Cancer. 2013;132:2578-88
15. Shigdar S, Qiao L, Zhou SF, Xiang DX, Wang T, Li Y. et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330:84-95
16. Kim JW, Kim EY, Kim SY, Byun SK, Lee D, Oh KJ. et al. Identification of DNA aptamers toward epithelial cell adhesion molecule via cell- SELEX. Mol Cells. 2014;37:742-56
17. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;28:755-68
18. Vermeulen L, Morrissey E, Heijden M, Nicholson AM, Sottoriva A, Buczacki S. et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995-1008
19. Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Natl Acad Sci USA. 2011;108:6468-73
20. Dieter SM, Ball CR, Hoffmann CM, Nowrouzi A, Herbst F, Zavidij O. et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell. 2011;9:357-65
21. Longo DL. Tumor heterogeneity and personalized medicine. N Engl J Med. 2012;366:956-67
22. Leon G, MacDonagh L, Finn SP, Cuffe S, Barr MP. Cancer stem cells in drug resistant lung cancer: targeting cell surface markers and signaling pathways. Pharmacol Ther. 2016;158:71-90
23. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK. et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem cell-like properties. Cell. 2013;152:25-38
24. Di Francesco AM, Toesca A, Cenciarelli C, Giordano A, Gasbarrini A, Puglisi MA. Metabolic modification in gastrointestinal cancer stem cells: characteristics and therapeutic approaches. J Cell Physiol. 2016;231:2081-7
25. Guo H, Nagy T, Pierce M. Post-translational glycoprotein modifications regulate colon cancer stem cells and colon adenoma progression in Apc(min/+) mice through altered Wnt receptor signalling. J Biol Chem. 2014;289:31534-49
26. Yu G, Jing Y, Kou X, Ye F, Gao L, Fan Q. et al. Hepatic stellate cells secreted hepatocyte growth factor contributes to the chemoresistance of hepatocellular carcinoma. PLoS One. 2013;28:e73312
27. Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM. et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;189:510-23
28. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
29. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO. et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950-5
30. Li H, Xu F, Li S, Zhong A, Meng X, Lai M. The Tumor microenvironment: an irreplaceable element of tumor budding and epithelial-mesenchymal transition-mediated cancer metastasis. Cell Adh Migr. 2016;3:434-46
31. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011;108:1397-402
32. Sun H, Zu Y. Aptamers and their applications in nanomedicine. Small. 2015;11:2352-64
33. Xu Y, Wang J, Li X, Liu Y, Dai L, Wu X. et al. Selective inhibition of breast cancer stem cells by gold nanorods mediated plasmonic hyperthermia. Biomaterials. 2014;35:4667-77
34. Zhao NX, You J, Zeng ZH, Li C, Zu YL. An ultra pH-sensitive and aptamer-equipped nanoscale drug system for selective killing of tumor cells. Small. 2013;20:3477-84
35. Wei X, Senanayake TH, Warren G, Vinogradov SV. Hyaluronic acid-based nanogel-drug conjugates with enhanced anticancer activity designed for the targeting of CD44-positive and drug-resistant tumors. Bioconjugate Chem. 2013;24:658-68
36. Xiang DX, Zheng CL, Zhou SF, Qiao SX, Tran PH, Pu CW. et al. Superior performance of aptamer in tumor penetration over antibody: implication of aptamer-based theranostics in solid tumors. Theranostics. 2015;5:1083-97
37. Li L, Xiang DX, Shigdar S, Yang WY, Li Q, Lin J. et al. Epithelial cell adhesion molecule aptamer functionalized PLGA-lecithin-curcumin-PEG nanoparticles for targeted drug delivery to human colorectal adenocarcinoma cells. Int J Nanomed. 2014;9:1083-96
38. Song YL, Zhu Z, An YT, Zhang WT, Zhang HM, Liu D. et al. Selection of DNA aptamers against epithelial cell adhesion molecule for cancer cell imaging and circulating tumor cell capture. Anal Chem. 2013;84:4141-9
39. Somasunderam A, Thiviyanathan V, Tanaka T, Li X, Neerathilingam M, Lokesh GL. et al. Combinatorial selection of DNA thioaptamers targeted towards the HA binding domain of human CD44. Biochemistry. 2010;49:9106-12
40. Ababneh N, Alshaer W, Allozi O, Mahafzah A, El-Khateeb M, Hillaireau H. et al. In vitro selection of modified RNA aptamers against CD44 cancer stem cell marker. Nuclei Acid Ther. 2013;23:401-7
41. Lu M, Zhou L, Zheng XH, Quan Y, Wang XL, Zhou XN. et al. A novel molecular marker of breast cancer stem cells identified by cell-SELEX method. Cancer Biomark. 2015;15:163-70
42. Alshaer W, Hillaireau H, Vergnaud J, Ismail S, Fattal E. Functionalizing liposomes with anti-CD44 aptamer for selective targeting of cancer cells. Bioconjugate Chem. 2015;26:1307-13
43. Meyer S, Maufort JP, Nie J, Stewart R, Mclntosh BE, Conti LR. et al. Development of an efficient targeted cell-SELEX procedure for DNA aptamer reagents. PLoS One. 2013;8:e71798
44. Zhao NX, Pei SN, Qi JJ, Zeng ZH, Lyer SP, Lin P. et al. Oligonucleotide aptamer-drug conjugates for targeted therapy of acute myeloid leukemia. Biomaterials. 2015;67:42-51
45. Palaniyandi K, Pockaj BA, Gendler SJ, Chang XB. Human breast cancer stem cells have significantly higher rate of clathrin-independent and caveolin-independent endocytosis than the differentiated breast cancer cells. J Cancer Sci Ther. 2011;4:214-22
46. Subramanian N, Raghunathan V, Kanwar JR, Kanwar RK, Elchuri SV, Khetan V. et al. Target-specific delivery of doxorubicin to retinoblastoma using epithelial cell adhesion molecule aptamer. Mol Vis. 2012;18:2783-95
47. Bagalkot V, Farokhzad OC, Langer R, Jon S. An aptamer-Doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew Chem Int Ed Engl. 2006;11:8149-52
48. Alibolandi M, Ramezani M, Sadeghi F, Abnous K, Hadizadeh F. Epithelial cell adhesion molecule aptamer conjugated PEG-PLGA nanopolymersomes for targeted delivery of doxorubicin to human breast adenocarcinoma cell line in vitro. Int J Pharm. 2015;479:241-51
49. Alibolandi M, Ramezani M, Abnous K, Sadeghi F, Atyabi F, Asouri M. et al. In vitro and in vivo evaluation of therapy targeting epithelial-cell adhesion-molecule aptamers for non-small cell lung cancer. J Control Release. 2015;209:88-100
50. Das M, Duan W, Sahoo SK. Multifunctional nanoparticle-EpCAM aptamer bioconjugates: A paradigm for targeted drug delivery and imaging in cancer therapy. Nanomed-Nanotechnol. 2015;11:379-89
51. Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3:1377-97
52. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. et al. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2009;12:505-22
53. Yang KN, Zhang CQ, Wang W, Wang PC, Zhou JP, Liang XJ. pH-responsive mesoporous silica nanoparticles employed in controlled drug delivery systems for cancer treatment. Cancer Biol Med. 2014;11:34-43
54. Xie XD, Li FQ, Zhang HJ, Lu YS, Lian S, Lin H. et al. EpCAM aptamer-functionalized mesoporous silica nanoparticles for efficient colon cancer cell-targeted drug delivery. Eur J of Pharm Sci. 2016;83:28-35
55. Roy K, Kanwar RK, Kanwar JR. LNA aptamer based multi-modal, Fe3O4-saturated lactoferrin (Fe3O4-bLf) nanocarriers for triple positive (EpCAM, CD133, CD44) colon tumor targeting and NIR, MRI and CT imaging. Biomaterials. 2015;71:84-99
56. Kanwar JR, Mahidhara G, Roy K, Sasidharan S, Krishnakumar S, Prasad N. et al. Fe-bLf nanoformulation targets survivin to kill colon cancer stem cells and maintains absorption of iron, calcium and zinc. Nanomedicine (Lond). 2015;10:35-55
57. Wang J, Sefah K, Altman MB, Chen T, You MG, Zhao ZL. et al. Aptamer- conjugated nanorods for targeted photothermal therapy of prostate cancer stem cells. Chem Asian J. 2013;8:2417-22
58. Klonisch T, Wiechec E, Hombach-Klonisch S, Ande SR, Wesselborg S, Schulze-Osthoff K. et al. Cancer stem cell markers in common cancers- therapeutic implications. Trends Mol Med. 2008;14:450-60
59. Patra CR, Bhattacharya RW, Wang E, Katarta A, Lau JS, Dutta S. et al. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer Res. 2008;68:1970-8
60. Ray P, Cheek MA, Sharaf ML, Li N, Ellington AD, Sullenger BA. et al. Aptamer-mediated delivery of chemotherapy to pancreatic cancer cells. Nucleic Acid Ther. 2012;22:295-305
61. Garrett JT, Arteaga CL. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications. Cancer Biol Ther. 2011;11:793-800
62. Liu Z, Duan JH, Song YM, Ma J, Wang FD, Lu X. et al. Novel HER2 aptamer selectively delivers cytotoxic drug to HER2-positive breast cancer cells in vitro. J Transl Med. 2012;10:148
63. Xiao S, Liu Z, Deng R, Li C, Fu S, Chen G. et al. Aptamer-mediated gene therapy enhanced antitumor activity against human hepatocellular carcinoma in vitro and in vivo. J Control Release. 2017;28:130-45
64. Esposito CL, Catuogno S, de Franciscis V. Aptamer-mediated selective delivery of short RNA therapeutics in cancer cells. J RNAi Gene Silencing. 2014;13:500-6
65. Ma C, Nong K, Wu B, Dong B, Bai Y, Zhu H. et al. miR-212 promotes pancreatic cancer cell growth and invasion by targeting the hedgehog signalling pathway receptor patched-1. J Exp Clin Cancer Res. 2014;7:33-54
66. Wang T, Michael PG, Xiang DX, Andrew GB, Matthew B, Zhou SF. et al. EpCAM aptamer-mediated survivin silencing sensitized cancer stem cells to doxorubicin in a breast cancer model. Theranostics. 2015;5:1457-72
67. Gilboa-Geffen A, Hamar P, Le MT, Wheeler LA, Trifonova R, Petrocca F. et al. Gene knockdown by EpCAM aptamer-siRNA chimeras suppresses epithelial breast cancers and their tumor-initiating cells. Mol Cancer Ther. 2015;14:2279-91
68. Subramanian N, Kanwar JR, Athalya PK, Janakiraman N, Khetan V, Kanwar RK. et al. EpCAM aptamer mediated cancer cell specific delivery of EpCAM siRNA using polymeric nanocomplex. J Biomed Sci. 2015;22:157-69
69. Subramanian N, Kanwar JR, Kanwar RK, Sreemanthula J, Biswas J, Khetan V. et al. EpCAM aptamer-siRNA chimera targets and regress epithelial cancer. PLoS One. 2015;7:e0132407
70. Mohammadi M, Salmasi Z, Hashemi M, Mosaffa F, Abnous K, Ramezani M. Single-walled carbon nanotubes functionalized with aptamer and piperazine-polyethylenimine derivative for targeted siRNA delivery into breast cancer cells. Int J Pharm. 2015;485:50-60
71. Thiel KW, Hernandez LI, Dassie JP, Thiel WH, Liu X, Stockdale KR. et al. Delivery of chemo-sensitizing siRNAs to HER2- breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012;40:6319-37
72. Zhang Y, Leonard M, Shu Y, Yang Y, Shu D, Guo P. et al. Overcoming tamoxifen resistance of human breast cancer by targeted gene silencing using multifunctional pRNA nanoparticles. ACS Nano. 2017;11:335-46
73. Hendricks BK, Cohen-Gadol AA, Miller JC. Novel delivery methods bypassing the blood-brain and blood-tumor barriers. Neurosurg Focus. 2015;38:E10
74. Miyamoto T, Tanaka N, Eishi Y, Amagasa T. Transferrin receptor in oral tumors. Int J Oral Maxillofac Surg. 1994;23:430-3
75. Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16:440
76. Rohde JH, Weigand JE, Suess B, Dimmeler S. A universal aptamer chimera for the delivery of functional microRNA-126. Nucleic Acid Ther. 2015;25:141-51
77. Catuogno S, Rienzo A, Di Vito A, Esposito CL, de Franciscis V. Selective delivery of therapeutic single strand antimiRs by aptamer-based conjugates. J Control Release. 2015;210:147-59
78. Wang HM, Zhao X, Guo CH, Ren DQ, Zhao YD, Xiao W. et al. Aptamer-dendrimer bioconjugates for targeted delivery of miR-34a expressing plasmid and antitumor effects in non-small cell lung cancer cells. PLoS One. 2015;9:e0139136
79. Shu D, Li H, Shu Y, Xiong GF, William EC, Farzin H. et al. Systemic delivery of anti-miRNA for suppression of triple negative breast cancer utilizing RNA nanotechnology. ACS Nano. 2015;10:9731-40
80. Wu C, Han D, Chen T, Peng L, Zhu G, You M. et al. Building a multifunctional aptamer-based DNA nanoassembly for targeted cancer therapy. J Am Chem Soc. 2013;135:18644-50
81. Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW. et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA. 2006;18:6315-20
82. Porciani D, Tedeschi L, Marchetti L, Citti L, Piazza V, Beltram F. et al. Aptamer-mediated codelivery of doxorubicin and NF-κB decoy enhances chemo-sensitivity of pancreatic tumor cells. Mol Ther Nucleic Acids. 2015;4:1-15
83. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev. 2003;3:939-51
84. McNamara JO, Kolonias D, Pastor F, Mittler RS, Chen L, Giangrande PH. et al. Multivalent 4-1BB binding aptamers costimulate CD8 T cells and inhibit tumor growth in mice. J Clin Invest. 2008;118:376-86
85. Pastor F, Soldevilla MM, Villanueva H, Kolonias D, Inoges S, de Cerio AL. et al. CD28 aptamers as powerful immune response modulators. Mol Ther Nucl Acids. 2013;2:e98
86. Pastor F, Kolonias D, McNamara JO, Gilboa E. Targeting 4-1 BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol Ther. 2011;19:1878-86
87. Soldevilla MM, Villanueva H, Casares N, Lasarte JJ, Bendandi M, Inoges S. et al. MRP1-CD28 bi-specific oligonucleotide aptamers: target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget. 2016;7:23182-96
88. Kim YJ, Lee HS, Jung DE, Kim JM, Song SY. The DNA aptamer binds stemness-enriched cancer cells in pancreatic cancer. J Mol Recognit. 2017;30:34-45
Author contact
![]() Corresponding author: Liang Qiao, Storr Liver Centre, Westmead Institute for Medical Research (WIMR) The University of Sydney and Westmead Hospital, 176 Hawkesbury Road, Westmead, NSW 2145, Australia Tel: +612 8627 3534; Fax: +612 8627 3099; Email: liang.qiaoedu.au
Corresponding author: Liang Qiao, Storr Liver Centre, Westmead Institute for Medical Research (WIMR) The University of Sydney and Westmead Hospital, 176 Hawkesbury Road, Westmead, NSW 2145, Australia Tel: +612 8627 3534; Fax: +612 8627 3099; Email: liang.qiaoedu.au