Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Amyloid β (Aβ)...
Tau hypothesis
Other hypothesis of AD
Current therapeutic molecules...
Nucleic-acid based molecules for...
Recent progress in modified...
Conclusions and Future...
Abbreviations
Acknowledgements
References
Author Biography
Introduction
Amyloid β (Aβ)...
Tau hypothesis
Other hypothesis of AD
Current therapeutic molecules...
Nucleic-acid based molecules for...
Recent progress in modified...
Conclusions and Future...
Abbreviations
Acknowledgements
References
Author Biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(16):3933-3947. doi:10.7150/thno.21529 This issue Cite
Review
Nucleic Acid-Based Theranostics for Tackling Alzheimer's Disease
Madhuri Chakravarthy1, 2, Suxiang Chen1, 2, Peter R. Dodd3, Rakesh N. Veedu1, 2, 3 ![]()
1. Centre for Comparative Genomics, Murdoch University, Murdoch, Perth, Australia 6150;
2. Perron Institute for Neurological and Translational Science, QEII Medical Centre, Nedlands, Perth, Australia 6005;
3. School of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, Brisbane, Australia 4072.
Received 2017-6-20; Accepted 2017-7-28; Published 2017-9-5
Citation:
Chakravarthy M, Chen S, Dodd PR, Veedu RN. Nucleic Acid-Based Theranostics for Tackling Alzheimer's Disease. Theranostics 2017; 7(16):3933-3947. doi:10.7150/thno.21529. https://www.thno.org/v07p3933.htm
Other stylesAbstract

Nucleic acid-based technologies have received significant interest in recent years as novel theranostic strategies for various diseases. The approval by the United States Food and Drug Administration (FDA) of Nusinersen, an antisense oligonucleotide drug, for the treatment of spinal muscular dystrophy highlights the potential of nucleic acids to treat neurological diseases, including Alzheimer's disease (AD). AD is a devastating neurodegenerative disease characterized by progressive impairment of cognitive function and behavior. It is the most common form of dementia; it affects more than 20% of people over 65 years of age and leads to death 7-15 years after diagnosis. Intervention with novel agents addressing the underlying molecular causes is critical. Here we provide a comprehensive review on recent developments in nucleic acid-based theranostic strategies to diagnose and treat AD.
Keywords: nucleic acids, Alzheimer's disease, amyloid beta peptides, tau peptide, chemically modified oligonucleotides, nucleic acid therapeutics.
Introduction
Nucleic acid-based technologies typically use synthetic oligonucleotides ̴8-50 nucleotides in length, most of which bind to RNA through Watson-Crick base pairing to alter the expression of the targeted RNA and protein. Novel chemical modifications and conjugation strategies have been developed to improve pharmacokinetics and tissue-specific delivery. Vitravene, Kynamro, Nusinersen and Eteplirsen are antisense oligonucleotides (AOs) approved by the FDA to treat cytomegalovirus retinitis, familial hypercholesterolemia, spinal muscular atrophy, and Duchenne muscular dystrophy respectively [1-3]. The nucleic acid aptamer drug Macugen was approved for age-related macular degeneration [4]. These successful clinical translations demonstrate the potential of nucleic acid-based technologies and provide scope for developing novel therapeutics for AD. AD is the most common form of dementia; it accounts for 70% of cases with that diagnosis. Globally there are ~47 million current cases; 7.7 million new cases are added each year [5]. AD is characterized by a progressive loss of memory and cognitive function [6]. Patients eventually need 24-hour care that places emotional and economic burdens on the community. There is no cure for AD, nor any treatment that addresses its underlying molecular cause [5]. Current treatments use cholinesterase inhibitors [7] and N-methyl-D-aspartate receptor (NMDA) antagonists [8] that improve cognitive function and reduce symptoms temporarily but do not stop the progression of the disease. The current approach to diagnosis relies on a combination of cognitive and clinical assessment, genetic profiling, and magnetic resonance imaging to measure anatomical changes in the brain [9], but confirmation relies on post-mortem neuropathological assessment and misdiagnosis is common [6]. Two hallmarks of the disease are extracellular amyloid-β (Aβ) plaques (mainly an agglomeration of Aβ peptides) and intracellular neurofibrillary tangles (hyperphosphorylated tau peptides). In this review we focus on the potential of nucleic acid therapeutic, diagnostic, and research strategies that target both Aβ and tau pathologies to help diagnose and treat AD.
Amyloid β (Aβ) hypothesis
The Aβ hypothesis states that there is an imbalance of toxic Aβ peptide production and clearance [10-12]. The main Aβ species, Aβ1-40 and Aβ1-42, can aggregate to form fibrils and plaques [10-12]. Aβ1-40 and Aβ1-42 are produced by the aberrant splicing of amyloid precursor protein (APP) by β-site APP cleaving enzyme 1 (BACE1) and γ-secretase (Figure 1) [11-15]. Mutations in the APP and Presenilin genes (PSEN1 codes for the catalytic subunits of γ-secretase) increase Aβ1-42 levels [10-12, 14, 16-18] and lead to early-onset familial AD. Down syndrome cases have an extra copy of chromosome 23, and hence of the APP gene, and develop Aβ plaques early in adulthood [19]. Oligomers of Aβ promote synaptic loss, neuronal dysfunction, and cell death [20, 21]. Aβ1-42 inhibits the maintenance of hippocampal long-term potentiation, resulting in altered memory function [10, 22] and reduced synaptic neurotransmission through NMDA receptor-mediated signaling [10, 22, 23]. Aβ toxicity has also been implicated in inflammation [11], oxidative stress [11, 24], cholinergic transmission [23], glucose metabolism [25, 26], and cholesterol metabolism [27].
Tau hypothesis
Microtubule-associated protein tau (tau), predominantly expressed in neuronal axons, is involved in microtubule assembly and stability. Tau is regulated by phosphorylation [28, 29]. Hyperphosphorylation decreases the ability of tau to bind to microtubules, leading to reduced trafficking, destabilization of microtubules, and synaptic loss [29, 30] (Figure 2). Abnormal tau can aggregate into paired helical filaments to form neurofibrillary tangles [31] in the cytosol and sequester normal tau to inhibit microtubule assembly [29]. Alternatively, tau aggregation may be a protective mechanism to stop hyperphosphorylated tau sequestering normal tau and inhibit microtubule assembly [29]. Tau hyperphosphorylation is detrimental in various neurodegenerative diseases termed “tauopathies” [28, 32]. Hyperphosphorylation of tau correlates with neurodegeneration and cognitive decline [29, 32]. Other post-translational modifications of tau, including abnormal glycosylation and reduced β-linked acylation of N-acetylglucosamine, increase hyperphosphorylation [29, 33]. Inhibition of the ubiquitin-proteasome system may also increase the aggregation of hyperphosphorylated tau [31].
Other hypothesis of AD
Drugs currently approved by the FDA for the treatment of AD are Donepezil, Rivastigmine, Galantamine and Memantine (Table 1) [34-37]. These agents enhance cholinergic and glutamatergic neurotransmission and improve cognitive function temporarily. However, they do not slow the progression of the disease. Oxidative stress [38], inflammation [39], insulin impairment [40, 41] and abnormal cholesterol metabolism [27] may also play roles (Table 1), but will not be considered in depth here.
Figure 1
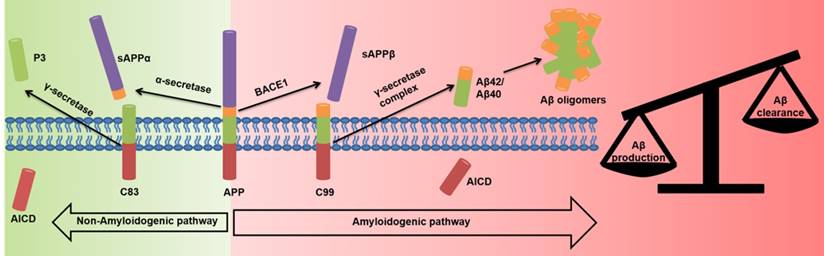
Non-amyloidogenic and amyloidogenic pathways in AD neurons. In the amyloidogenic pathway the APP is aberrantly spliced by BACE1 and γ-secretase to overproduce toxic Aβ species.
Figure 2
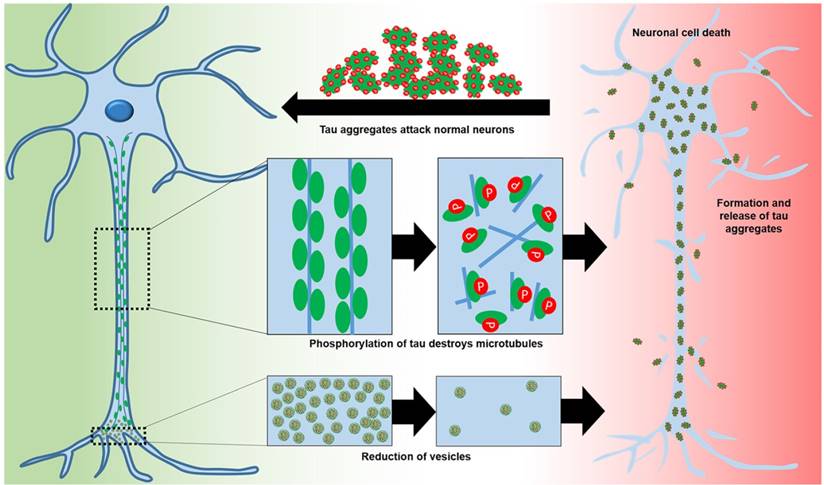
The roles of tau in normal neurons and of hyperphosphorylation in AD neurons that lead to neuronal toxicity.
Table 1
Therapeutic molecules in clinical trials, their targets, and trial outcomes.
| Drug molecule | Role/ Target | Trial stage | Results | References |
|---|---|---|---|---|
| Donepezil (Pfizer) | Cholinesterase inhibitor | FDA approved- Although they improve the symptoms temporarily these drugs do not stop the progression of the disease. | [34-37] | |
| Rivastigmine (Novartis) | Cholinesterase inhibitor | |||
| Galantamine (Jansen-Cilag) | Cholinesterase inhibitor | |||
| Memantine (Lundbeck) | NMDA receptor antagonist | |||
| Tramiprosate | Aβ aggregation inhibitor | Phase III | No significant benefit. May promote abnormal tau aggregation | [47-49] |
| Colostrinin | Aβ aggregation inhibitor | Phase III | Modest improvements not sustained | [50-52] |
| Scyllo-inositol | Stabilizes Aβ aggregates and inhibits toxicity | Phase II | No statistically significant effect. Reduced Aβ in cerebrospinal fluid | [53] |
| Aβ vaccination | Aβ aggregation inhibitor | Phase II | Halted because patients developed meningo-encephalitis | [54] |
| Bapineuzumab | Aβ aggregation inhibitor | Phase III | End points not significantly different | [55] |
| Solanezumab | Aβ aggregation inhibitor | Phase III | End points not significantly improved | [56] |
| Anti-amyloid Ab | Aβ aggregation inhibitor | Phase III | No positive primary outcome | [57] |
| Other mAbs | Aβ aggregation inhibitor | Various | No positive outcome | [42, 58, 59] |
| Tarenflurbil | γ-secretase inhibitor | Phase III | No significant improvement | [60-62] |
| LY450139 (Eli Lilly) | γ-secretase inhibitor | Phase III | Discontinued: no Aβ40/42 reduction | [63] |
| BMS-708163 (B-M Squibb) | γ-secretase inhibitor | Phase II | Terminated due to lack of favorable pharmacodynamics | [42, 64] |
| Verubecestat | BACE1 inhibitor | Phase III | Currently running | [65] |
| Rogiglitazone | BACE1 inhibitor and Type 2 diabetes drug | Phase III | No positive outcome | [66] |
| Pioglitaozone | BACE1 inhibitor and Type 2 diabetes drug | Phase III | No positive outcome | [66] |
| Methyl thionium chloride | Tau aggregation inhibitor | Phase II | Significantly improved cognitive function | [67, 68] |
| Tideglusib | GSK3β | Phase IIb | No positive outcome | [68, 69] |
| Davunetide | Microtubule stabilizer | Phase III | No significant improvement | [30] |
| Antioxidants | ROS | Phase III | No positive outcome | [42, 70] |
| Anti-inflammatories | Inflammation | Phase III | No significant improvement | [25, 39, 59, 71-73] |
| Intranasal insulin | Insulin impairment | Pilot | Improvement in patients without APOE-ε4 allele | [40, 74] |
| Other anti-diabetics | Insulin impairment | Phase III | Currently running | |
| Statins | Cholesterol metabolism | Phase III | Preliminary results positive; mechanism unknown. | [27, 75] |
Current therapeutic molecules and clinical trials for the treatment of AD
Many disease-modifying therapeutics show positive outcomes in animal models but disappointing results in clinical trials (for drug candidates and ongoing trials see Table 1). Current strategies have been comprehensively reviewed [42]. Poor outcomes might have arisen because each agent is targeting a single pathway, whereas AD is a complex disease and it may be important to aim at multiple targets [43, 44]. Success in developing a suitable therapeutic approach is challenging because the pathogenesis of AD is unknown [45]. Trials might be affected by factors such as genetics, metabolism, and diet [46], but there is clearly a need to develop novel therapeutics for this disease.
Nucleic-acid based molecules for tackling AD
Unlike conventional small-molecule drugs, nucleic acid-based therapeutic agents such as AOs, small-interfering RNAs (siRNAs), microRNA moieties that target oligonucleotides (antimiRs and miRNA mimics), and DNAzymes/ribozymes can regulate the expression of key proteins by selectively targeting their mRNAs. The outcome is mRNA cleavage, repair, or steric blockade (Figure 3). The class of modified nucleic acids called aptamers can target proteins and inhibit their function (Figure 3). Nucleic acid-based strategies could be an effective alternative to drug development for AD because they can target a range of pathological features.
Figure 3
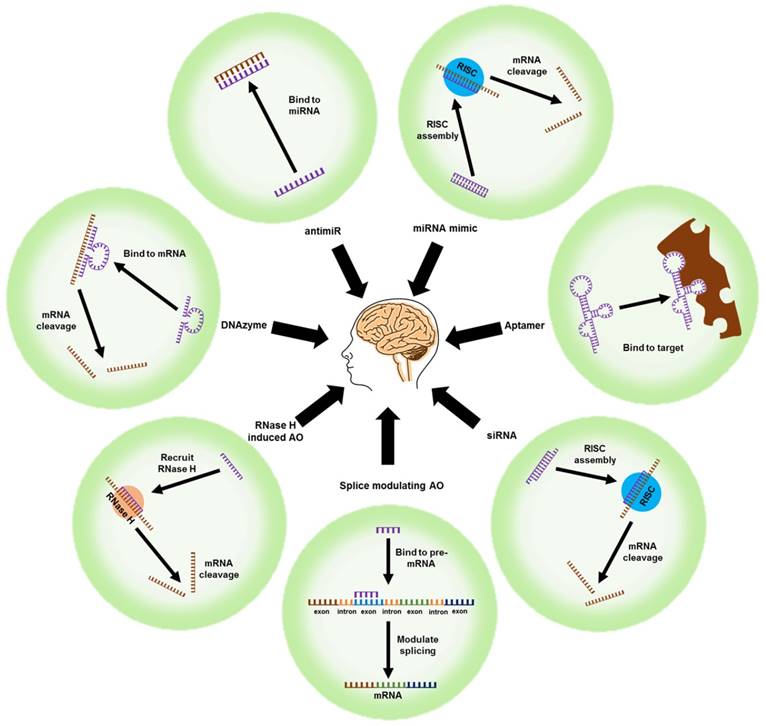
Nucleic acid-based therapeutic strategies. mRNA: messenger RNA; RNase H: ribonuclease H; siRNA: small interfering RNA; RISC: RNA inducing silencing complex; AO: antisense oligonucleotide; antimiR: anti-microRNA; miRNA mimic: microRNA mimic
Figure 4
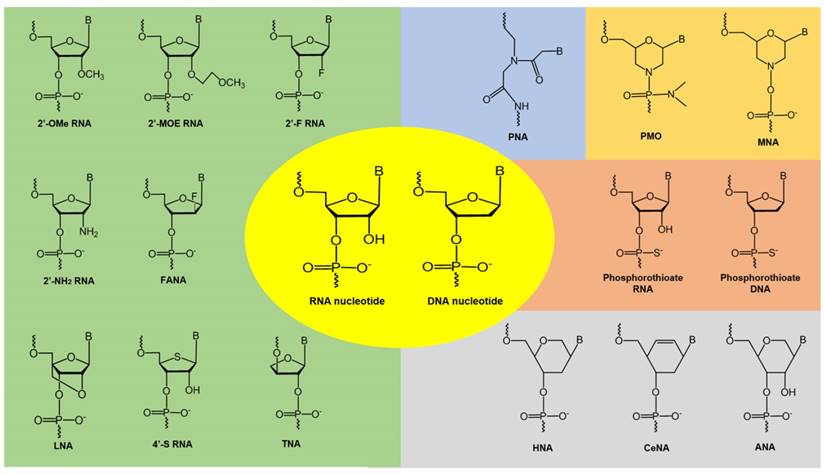
Examples of chemically-modified nucleotide analogues. 2'-OMe: 2'-O-methyl; 2'-MOE:2'-O-methoxyethyl; 2'-F: 2'-fluoro; 2'-NH2: 2'-amino; FANA: fluoroarabinonucleotide; LNA: locked nucleic acid; TNA: threose nucleic acid; PNA: peptide nucleic acid; PMO: phosphorodiamidate morpholino oligomer; MNA: morpholino nucleic acid; HNA: hexitol nucleic acid; CeNA: cyclohexenyl nucleic acid; ANA: anhydrohexitol nucleic acid
Improving the stability and efficacy of nucleic acid-based therapeutics
Therapeutic oligonucleotides composed of naturally occurring nucleotides are rapidly degraded in vivo, which makes them unsuitable for drug development. To improve their pharmacokinetic properties, chemically modified nucleotide analogues with high resistance to nucleases are normally used. A number of analogues have been developed by modifying the base or sugar moieties, or the inter-nucleotide linkages (see Figure 4) [76-78]. Phosphorothioate DNA [79], 2'-O-methyl (2'-OMe) RNA [80], 2'-fluoro (2'-F) RNA [81], 2'-O-methoxyethyl (2'-MOE) RNA [82], and phosphorodiamidate morpholino (PMO) [83] analogues have been successfully utilized in FDA-approved oligonucleotide drugs. Analogues such as locked nucleic acids (LNA) [84, 85], peptide nucleic acids (PNA) [86], tricyclo-DNA (tcDNA) [87], and cyclohexenyl nucleic acids (CeNA) [88] also show excellent biophysical properties and offer further scope for novel oligonucleotide development. These chemistries can be used to construct fully modified or mixmer oligonucleotides. Aptamers can be modified during the selection or post-selection stages to improve their affinity and bioavailability [77]. Another challenge in the clinical utilization of unmodified oligonucleotides is rapid renal clearance from the blood due to their small size that falls under the renal filtration threshold. To increase their bioavailability, oligonucleotides can be conjugated with polyethylene glycol (PEG) to increase their size, which could also improve their resistance to nucleases [89]. Several PEGylated drugs have been approved by the FDA for clinical use [89, 90]. Other strategies include conjugating the oligonucleotides to albumin, which has a size of around 7 nm and shows reduced renal clearance and can therefore increase the circulation half-life of the oligonucleotides. Phosphorothioate modified oligonucleotides also showed reduced renal clearance by binding to plasma proteins like albumin to avoid glomerular filtration [91]. Another strategy is the synthesis of neutral siRNA (masking the negative charge on the phosphate backbone). Neutral siRNA showed reduced renal clearance [92].
Recent progress in modified nucleic acids for AD
Antisense oligonucleotides
A classical nucleic acid approach to controlling the expression of proteins is to use AOs, short single-stranded synthetic oligonucleotides, which can precisely target an mRNA transcript to regulate the expression of the protein it codes for. Antisense mechanisms include RNase H recruitment and cleavage of mRNA, modulation of splicing in pre-mRNA, and steric blockade of either mature or pre-mRNAs (Figure 3). RNase H-mediated cleavage involves designing a short DNA oligonucleotide that binds to the target mRNA to form a RNA-DNA duplex [93]. The duplex is recognized and cleaved by endogenous RNase H. AOs that modulate pre-mRNA splicing can be used to repair defective RNA and eliminate disease-associated splice variants [94]. Many pre-mRNA transcripts are alternatively spliced to produce different mRNA, and hence protein, variants [94].
APP
Many groups have designed AOs that target APP to reduce APP expression. An early study by Allinquant et al. [95] developed AOs that successfully blocked rat APP synthesis. Administration of the AOs showed that APP played a role in axonal and dendritic growth, and thus in neuronal differentiation [95]. ISIS Pharmaceuticals (now Ionis Pharma) have patented (US 2003/0232435 A1) 78 gapmer AOs with 2'-MOE wings and central DNA region. The AOs target various regions of APP mRNA and inhibit 39-82% of APP protein expression [96].
Kumar and colleagues [97] developed phosphorothioated DNA AOs against sequences that correspond to the Aβ region of APP (17-42 amino acids). Administration of the AOs led to improved cognitive function in senescence-accelerated mouse-prone 8 (SAMP8) mice. SAMP8 mice have a natural mutation that leads to APP over-expression, impaired Aβ removal, and loss of memory with increasing age. The AOs that target the mid-Aβ region reduced APP levels by 43-68% in the amygdala, septum and hippocampus [97]. The mice showed improvement in acquisition and retention in the footshock avoidance paradigm, which reversed their deficits in learning and memory [97]. AOs that target the sequences that correspond to the region of APP coding for the first 17-30 amino acids of Aβ were the subject of intellectual property protection [98]. Banks and colleagues [99] showed that a radioactively tagged phosphorothioate DNA AO targeting the Aβ region of APP could transit the blood-brain barrier (BBB) of mice to enter the cerebrospinal fluid. When a 100-fold higher dose of the AO was injected into the brain by intracerebroventricular injection it reversed the learning and memory deficits in SAMP8 mice, possibly through reduced oxidative stress. Poon et al. [100] used proteomics to show that lower Aβ levels result in reduced oxidative stress in brain.
Opazo et al. [101] transfected the AOs described by Kumar and colleagues [97] into the CTb cell line, a neuronal line from mice that overexpresses APP, and the CNh cell line from normal mice. The AOs resulted in APP knockdown in CTb cells by 36%, 40% and 50% compared with normal CNh cells after 24 h, 48 h and 72 h respectively [101]. By 72 h after AO transfection, choline uptake was similar to that in CNh cells and there was increased choline release in response to glutamate, nicotine and KCl depolarization, which reached similar levels to those observed in CNh cells. The CTb cells come from a Down syndrome mouse model, which show some learning deficits and cholinergic dysfunction that are similar to those found in AD [102]. Similarly, Rojas et al. [103] showed that APP overexpression reduced the expression and retrograde transport of nerve growth factor. This reduced nicotine-induced stimulation of α3β2 nicotinic acetylcholine receptor and in consequence lowered intracellular Ca2+ responses in CTb cells. The effects of APP overexpression were restored close to normal by treatment with AOs targeting APP expression.
Chauhan and colleagues [104] designed gapmer AOs with 2'-OMe and DNA nucleotides on a phosphorothioate backbone that target the β-secretase cleavage site of APP and found that they reduced brain Aβ40 and Aβ42 levels in a mouse model of AD. The AOs were delivered intracerebroventricularly and showed rapid uptake and retention for 30 minutes. They efficiently crossed cell membranes into the nuclear and cytoplasmic compartments of neuronal and non-neuronal cells. Chauhan and Siegel [105] designed two additional AOs targeting the β- and γ-secretase site of APP in the Tg2576 mouse model that expresses APP. The AO targeting the mutated β-secretase site increased soluble APPα by 43% and decreased soluble Aβ40 and Aβ42 levels by 39%, whereas the AO targeting the γ-secretase site had no effect. The AO targeting β-secretase also inhibited acetylcholinesterase activity, increasing acetylcholine by five-fold in cortex compared with controls.
Erickson et al. [106] peripherally administered an APP AO to SAMP8 mice. This resulted in a 30% increase in APP levels but no change in soluble Aβ levels. The treated mice showed improved memory. They also showed [107] that AO-mediated APP knockdown in Tg2576 mouse brains reduced cytokine expression and improved learning and memory. Attenuating APP overexpression may improve learning and memory by reducing inflammation (also implicated in AD pathology).
BACE1
Yan et al. [108] developed two AOs that target β-secretase aspartyl protease and found that they reduced the release of Aβ40 and Aβ42 by 50-80%. Vassar and colleagues [109] also used AOs that target β-secretase to reduce Aβ40 and Aβ42 production by around 30%. These studies showed that β-secretase is important for the production of Aβ40 and Aβ42 and highlighted BACE1 as an important target for AD. Wolfe et al. [110] designed splice-modulating AOs to target BACE1, since alternatively spliced transcript variants at exons 2 and 3 do not show β-secretase activity. The AOs reduced Aβ production significantly in cells without altering total BACE1 mRNA.
Presenilin 1 (PSEN1)
Refolo et al. [111] found that AOs targeting PSEN1 in a human cell line reduced PSEN1 holoprotein by 80% 12 days after treatment and by 90% after 14 days. This was correlated with a two-fold increase in Aβ42 levels. Grilli et al. [112] found that hippocampal primary neurons overexpressing mutant PSEN1 were vulnerable to excitotoxic and hypoxia-hypoglycemic damage and increased cell death. They designed two phosphorothioate AOs targeting PSEN1 in wild type mice. In contrast to Refolo et al. [111] they found that lower PSEN1 expression reduced cell death and provided neuroprotection [112]. Fiorini et al. [113] administered AOs targeting PSEN1 to aged SAMP8 mice and found they reduced brain oxidative stress biomarkers. In the T-maze foot shock avoidance and novel object recognition tests the mice showed a reversal of learning and memory deficits.
Tau
Caceres et al. [114] showed that an AO targeting the 5' end of the tau gene, in the region before the start codon, showed strong inhibition of neurite elongation in primary rat neurons. Immunoblotting revealed that the tau protein level was reduced in AO-treated mice but not in control mice. The effect of AO treatment on cognition needs to be assessed. DeVos et al. [115] screened 80 AOs targeting tau and selected the three that showed the best knockdown of tau to test in vivo. The latter reduced tau mRNA levels by more than 75%. The best AO was selected for further testing in mice; it lowered brain tau mRNA and protein significantly in a dose-dependent manner. Behavioral impacts and neurotoxicity were not measured. Kalbfuss et al. [116] developed splice-modulating AOs modified with 2'-OMe nucleotides to target the tau exon 10 splice junctions to reduce exon 10 inclusion. Exclusion of exon 10 increases the ratio of tau proteins lacking the microtubule-binding domain. In consequence, the microtubule cytoskeleton becomes destabilized as observed in frontotemporal dementia and parkinsonism.
Peacey et al. [117] designed bipartite AOs that bound to the hairpin structure at the boundary between exon 10 and intron 10 of tau to inhibit exon 10 splicing, reversing the effect of disease-causing mutations in cells. Liu et al. [118] developed a small-molecule (mitoxantrone) conjugated to a bipartite AO that binds to the tau RNA hairpin structure. The conjugate also inhibited exon 10 splicing in cell-free conditions more effectively than mitoxantrone or the bipartite AO alone, but induced cytotoxicity. The same group used a PNA-modified bipartite AO conjugated to mitoxantrone that inhibited tau splicing but was also cytotoxic [110].
Sud et al. [119] developed PMO AOs to modulate the splicing of tau and tau expression. The AOs were designed to target sequences at the donor and acceptor splice sites, the splicing branch points, and splicing enhancers and inhibitors to induce exon skipping. Exons 0, 1, 4, 5, 7, 9, and 10 were targeted. Exons 1, 4, 5, 7, and 9 are found in all 6 isoforms of tau while exon 10 is present in only three of the six isoforms. Of the 31 AOs tested, AO E1.4 targeting the splice donor site at the exon 1 - intron 1 junction reduced tau mRNA expression by 50%. The other AOs effective in this region were a combination of AOs that targeted the splice donor and acceptor sites and the start codon. AO E5.3 targeted the splice donor site at the exon 5 -intron 5 junction and reduced total tau mRNA expression by 29-46%. It also reduced tau protein level by 58-62%. The resulting transcript was missing exons 4 to exon 7 using the normal splice sites. AO E7.7 targeted the splice donor site on exon 7; it reduced tau mRNA expression by 30% and tau protein levels by 67%. E5.3 injected into mice in vivo produced lower tau mRNA levels than in non-injected regions.
GSK-β
Farr et al. [120] showed that a phosphorothioated AO that targets GSK-3β decreased GSK-3β protein levels in the cortex of SAMP8 mice. There were improvements in learning and memory, reduced oxidative stress, increased levels of the antioxidant transcription factor nuclear factor erythroid-2 related factor 2, and decreased tau phosphorylation.
Acetylcholinesterase (AChE)
Fu et al. [121] found that AOs against human AChE mRNA reduced AChE activity in an AD mouse model after 8 h; the effect lasted till 42 h. Lower enzymic activity was accompanied by improvement on behavioral tasks, which showed increased memory retention and improved water maze performance (shorter swimming time).
Apolipoprotein E receptor 2 (ApoER2)
ApoER2 may be a primary risk factor for late-onset AD [122, 123]. Dysregulation of ApoER2 splicing may result in impaired synaptic homeostasis. Cerebral injection of mice with AOs targeting the adjacent introns enhanced exon 19 inclusion, an effect that persisted for up to 6 months [123]. The mice showed improvement in Aβ-induced cognitive defects. It was postulated that the AOs bind to the splicing factor SRF1 to reduce its expression and increase the inclusion of exon 19, thereby increasing the level of the active form of ApoER2 to enhance NMDA receptor phosphorylation.
siRNA
These are short synthetic double stranded RNA oligonucleotides that target complementary mRNA and silence gene expression through the assembly of the RNA-induced silencing complex (RISC) (Figure 3) [93, 124]. Chemical modifications can be introduced into the siRNA to increase its stability against nucleases and increase its selectivity for the target (Figure 4).
APP
Miller and colleagues [125] found that siRNAs against the Swedish mutant in APP that causes a familial form of AD silenced the expression of mutant alleles. The siRNAs were designed to ensure that they bound specifically to the mutant alleles and not the wild-type. The mutation was placed in the central region of the siRNA duplex to achieve high silencing efficiency.
BACE1
McSwiggen and colleagues [126] patented 325 siRNAs that target BACE (NCBI ID: NM_012104). The patent covers sequences with various chemically modified siRNAs that include 2'-deoxy, 2'-F and 2'-OMe pyrimidine and purine nucleotides, phosphorothioate internucleotide linkages and inverted deoxyabasic caps. Four of the siRNAs reduced BACE expression by 40-90% at 25 nM concentration, but there was no data on whether this altered Aβ40 and Aβ42 expression. Basi et al. [127] made a siRNA that reduced the BACE1 mRNA level by 50% and BACE protein level by more than 90%. It decreased the secretion of Aβ peptide without affecting BACE2 expression, indicating specificity for BACE1. Kao et al. [128] also designed siRNAs, where two of the siRNAs reduced BACE1 mRNA by more than 90% and Aβ production by 36-41%. Pretreatment of neurons with the siRNA increased neuroprotection against hydrogen peroxide-induced oxidative stress. Modarresi et al. [129] injected LNA-modified siRNAs targeting BACE1 antisense transcripts into the third ventricle of Tg-19959 mice to downregulate BACE1 and BACE1 antisense transcripts, which led to lower BACE1 protein levels and less Aβ production and aggregation in the brain. Notably, Cai et al. [130] showed that siRNAs targeting BACE1 inhibited it in mice and increased choroidal neovascularization: BACE1 is also expressed in the neural retina and in in vitro and in vivo angiogenesis. Although BACE1 inhibition may be therapeutically beneficial in AD, it may contribute to retinal pathologies and exacerbate conditions such as age-related macular degeneration.
Heterogeneous nuclear ribonucleoprotein H
A G-rich region in exon 3 of BACE1 may form a G-quadruplex structure and recruit a splicing regulator, heterogeneous nuclear ribonucleoprotein H, that regulates splicing to increase generation of the BACE1 501 isoform. Fisette et al. [131] reported that siRNA and short hairpin RNA candidates that target heterogeneous nuclear ribonucleoprotein H reduced its expression and thereby decreased BACE1 501 isoform levels and Aβ production.
AntimiRs and miRNA mimics
miRNAs are short non-coding RNAs that regulate protein expression post-transcriptionally. miRNA mimics can modulate RNA and protein expression by acting like their endogenous miRNA counterparts. AntimiRs can modulate RNA and protein expression by inhibiting endogenous miRNA similar to AOs (Figure 3). miRNAs silence gene expression by translational repression and/or mRNA degradation [132, 133]. miRNAs are first transcribed by RNA polymerases II or III to form long primary miRNA with a 5' CAP and a poly(A) tail [133-135]. These are then processed in the nucleus into short 70-nucleotide hairpin structures called precursor miRNAs (pre-miRNA) by the microprocessor complex [133-135]. The pre-miRNAs are exported to the cytoplasm by Exportin 5 and processed by Dicer into double-stranded miRNA duplexes, which are approximately 22 nucleotides long [133-135].
BACE1
An endogenous non-coding BACE1 antisense transcript stabilizes the BACE1 transcript and may upregulate BACE1 in AD cases. BACE1 antisense binds to BACE1 at the miR-485-5p binding site and suppresses BACE1 expression. Faghihi et al. [136] found that LNA-antimiRs that target miR-485-5p decreased miRNA-induced suppression of BACE1 and increased BACE1 antisense expression. Hebért et al. [137] showed that miR-29a/b-1 cluster was significantly reduced in sporadic AD patients and correlated with increased BACE1 expression and Aβ generation, and therefore may be potential targets for miRNA mimics as a therapeutic strategy for AD.
Tau
Mi34a reduces endogenous tau expression at both the mRNA and protein level in M17D cells by binding to the 3' UTR region of tau [110], whereas miR-34c levels are elevated in the hippocampus of AD patients and mouse AD models [138]. Wolfe et al. [110] used LNA antimiRs to inhibit miR-34a,‑34b and -34c and found increased tau expression. Zovolis et al. [138] found that an antimiR that targets miR-34c rescued learning in mouse models.
Acetyl-CoA acyl transferase
Acetyl-CoA acyl transferase has a role in lipid metabolism that has been implicated in the pathogenesis of AD. Murphy et al. [139] inhibited Acetyl-CoA acyl transferase using an artificial miRNA to reduce Aβ plaque burden and improve cognition in a mouse model of AD. The miRNA also reduced full-length human APP levels.
Brain-derived neurotropic factor
Brain-derived neurotropic factor regulates synaptic plasticity and memory and is decreased in AD brains [140-142], while miR-206 suppresses brain-derived neurotropic factor levels and memory function in AD mice [143]. Lee et al. [143] injected an anti-miR candidate AM-206 that targets miR-206 into the third ventricle of Tg2576 mice. It increased brain levels of brain-derived neurotropic factor, enhanced hippocampal synaptic density, neurogenesis, and memory. Intranasally administered AM-206 also reached the brain and had similar effects to the injected AM-206.
DNAzymes/Ribozymes as therapeutic candidates for AD
A target RNA can be cleaved to reduce its expression using catalytic oligonucleotides such as DNAzymes and ribozymes [144]. The arms of these enzymes hybridise with the target RNA and cleave their targets through the catalytic loop in the middle. DNAzymes and ribozymes cleave the phosphodiester bond at the purine-pyrimidine or purine-purine junction (Figure 3).
BACE1
Nawrot et al. [145] designed RNA-cleaving hammerhead ribozymes that downregulated BACE1 mRNA expression by more than 90% in HEK293 and SH-SY5Y cells and reduced Aβ40 and Aβ42 production by more than 80%. They also showed that a DNAzyme with the 10-23 catalytic loop reduced BACE mRNA expression by 70%. However, whether the reduced BACE mRNA expression leads to reduced Aβ production is unknown and requires validation.
Nucleic acid aptamers
Aptamers are short single stranded RNA or DNA oligonucleotides with unique three-dimensional structure that bind to targets with high affinity and specificity. Aptamers can be developed against a variety of targets ranging from small molecules to complex proteins over whole cells. Aptamers can be used for therapeutic, diagnostic (biosensors and molecular imaging), and targeted drug delivery applications. They are typically selected from large DNA and RNA oligonucleotide libraries through a process called Systematic Evolution of Ligands by EXponential enrichment (SELEX) [146, 147].
Aβ
Ylera et al. [148] were the first to report novel RNA aptamers that bound to Aβ1-40 fibrils with high affinity (29-48 nM). Bunka et al. [149] made aptamers against amyloid-like fibrils from β2-microglobulin. They bound to the target with high affinity, but also bound to other amyloid fibrils including, but not confined to, those found in dialysis-related amyloidosis patients. Rahimi et al. [150] also developed RNA aptamers against Aβ fibrils, but these also interacted with other amyloidogenic proteins by binding to a common β-sheet motif. They bound to fibrils with ≥15-fold higher sensitivity than thioflavin-T, suggesting that aptamers might be diagnostic tools for AD. Takahashi et al. [151] isolated two RNA aptamers, N2 and E2, that bound to monomeric Aβ40 with dissociation constants of 21.6 and 10.9 µM respectively. Though the affinities were quite low for clinical use, enzyme-linked immunosorbent assay (ELISA) showed that they could inhibit Aβ aggregation efficiently. When conjugated to AuNP gold nanoparticles, N2 and E2 bound to both Aβ monomers and oligomers. Mathew et al. [152] showed that the N2 aptamer conjugated to curcumin-polymer nanoparticles enhanced binding to, and disaggregated, amyloid plaques, which were then cleared by phagocytosis. The study targeted peripheral amyloid as peripheral organs may also generate amyloid proteins, which have also been implicated in AD. Targeting peripheral amyloid is easier due to the challenges in the brain delivery of aptamers.
Farrar et al. [153] developed a fluorescently tagged aptamer that bound to Aβ oligomers in both AD and transgenic mouse brain tissue. The aptamer may be useful for Aβ imaging, which has diagnostic implications. Similarly, Babu and colleagues [154] developed an aptamer complexed with ruthenium that binds to, and inhibits the formation of, Aβ oligomers. The aptamer-ruthenium interaction increases luminescence intensity, which is reduced when the aptamer binds to Aβ monomer or oligomers.
BACE1
Rentmeister et al. [155] made an RNA aptamer that binds to the short cytoplasmic tail of BACE1. It is a good research tool to investigate the biological function of the cytoplasmic tail without interfering with BACE1 transport and localization. Liang et al. [156] developed two DNA aptamers, A1 and A4, that bind to the extracellular domain of BACE1 with high affinity (Kd 15-69 nM) and specificity. They have similar affinities to the anti-BACE1 antibody. In vitro, APP Swedish mutant cells treated with A1 showed lower Aβ40 and Aβ42 levels than control cells. sAPPβ expression decreased with A1 treatment compared with untreated controls.
Tau
Kim et al. [157] used recombinant his-tagged tau40 to select aptamers from an RNA library through SELEX. 12 rounds of selection produced a tau-1 aptamer, which represented ~76% of identified aptamers, that reduced the levels of oligomeric tau (by ~94%) in vitro in a dose-dependent manner. However, it could not de-oligomerize pre-existing tau oligomers and had no effect on tau degradation. The aptamer bound to tau protein and inhibited its oligomerization, unlike control aptamers. Primary neurons treated with tau-1 aptamer showed less cytotoxicity than controls but no difference in membrane integrity or viability; there was little effect on normal tau function. Primary rat cortical neurons administered tau oligomers and treated with tau-1 aptamers showed significantly less oligomeric tau phosphorylation at Ser199/202 but there was no effect on monomeric tau. Extracellular tau oligomers also stress neighboring neurons. Administration of tau oligomers leads to severe neurotoxicity, which was reduced by tau-1 aptamer treatment. Tau-1 aptamers can prevent or reverse cytotoxicity mediated by tau oligomerization both in a non-neuronal cell line and in primary rat cortical neurons. Unfortunately, the tau-1 aptamers isolated by Kim et al. bound only to one of the six isoforms of tau. Therefore, the effects of tau-1 aptamers observed in mice may not translate clinically, because six isoforms are prone to aggregation and implicated in neurodegeneration. To be successful clinically, the aptamers must be able to cross the BBB and the neuronal cell membrane, and disaggregate the neurofibrillary tangles after binding [158]. Kim et al. [159] reported a DNA aptamer-antibody sandwiched to the tau-381 isoform that detected tau in human plasma at femtomolar concentrations by surface plasmon resonance.
The ubiquitin-proteasome system
Lee et al. [160] developed an aptamer against USP14, an enzyme that delays protein degradation by the ubiquitin-proteasome system. Recombinant USP14 was incubated with a random RNA library for SELEX. Three aptamers, USP14-1, USP14-2 and USP-14-3, were identified, all of which bound to USP14 with high affinity. USP14-3 showed the strongest inhibition of deubiquitination, which may be due to its ability to bind both USP14 and UCH37. UCH37 is a protein that also slows protein degradation in the proteasome. The aptamers have yet to be tested in mice for their effect on tau oligomerization and degeneration.
Prion protein
Mashima et al. [161] isolated aptamers against bovine prion protein by SELEX that may have therapeutic potential in prion diseases and AD. Aβ oligomers bind to the prion protein to block long-term potentiation. Thus, prion protein may mediate Aβ oligomer-induced synaptic dysfunction.
Brain delivery of nucleic acid molecules
Receptor-mediated endocytosis or nanoparticle conjugation strategies
The barrier for any successful drug to treat neurological diseases is its failure to cross the BBB. Various approaches have been made to overcome this issue, and in many instances the drug can be conjugated with other molecules to improve brain delivery. Lipophilic molecules under 500 Da can cross the BBB by simple diffusion. Therapeutic nucleic acids are typically too large to cross the BBB, although nanotechnology is now starting to overcome this problem. This was the subject of a comprehensive review by Kanwar et al. [162]. Nucleic acids can be transported through the BBB by receptor-mediate endocytosis when conjugated to molecules such as transferrin, insulin, leptin, and insulin-like growth factor 1: these bind to their receptors on the BBB, which allows them to cross the BBB. Many studies have used nucleic acids conjugated to molecules that target the transferrin receptor. Transferrin-conjugated nanoparticles, or nanoparticles conjugated to transferrin receptor antibodies, can transport drugs across the BBB [163, 164]. An aptamer that targeted the mouse transferrin receptor allowed a lysosomal enzyme to enter cells via endocytosis; this can be applied to drug transport [165]. Two aptamers that bound to epithelial cell adhesion molecule and transferrin receptor were fused together; the product could bind to cancer cells expressing the epithelial cell adhesion molecule after crossing the BBB by transferrin-receptor targeting [166].
Cell-penetrating peptide-based delivery systems
The use of cell-penetrating peptide-based delivery systems is another approach for transporting nucleic acids across the BBB. These systems generally contain between 8 and 30 amino acids. Fluorescein isothiocyanate-labeled cell-penetrating peptides were conjugated to a morpholino AO targeting the mutant ataxia telangiectasia gene and were able to cross the BBB [167]. Heitz et al. [168] reviewed the development of cell-penetrating peptides.
Intracerebroventricular infusion
Nucleic acids can also be introduced directly into the cerebrospinal fluid by intracerebroventricular infusion. The advantage of this over the use of targeting molecules is that the drugs are delivered at therapeutic concentrations quickly. However, intracerebroventricular infusions are highly invasive and rely on diffusion of the drugs throughout the ventricular system. The drugs can then enter the blood stream, because the cerebrospinal fluid turns over every 4-5 hours [169]. Pardridge et al. [169] described the advantages and disadvantages of different drug delivery methods to the brain. Intranasal methods are non-invasive and can deliver nucleic acids directly to the brain. They have been used successfully to deliver insulin to AD patients [170]. Various molecules delivered intranasally have improved cognitive function in a mouse model of AD and in clinical trials, as summarized in the review by Hanson et al. [171]. Many reviews describe how nose-to-brain delivery occurs, the different drugs that have been successfully delivered this way, and its potential for treating neurodegenerative diseases such as AD [172, 173].
Conclusions and Future Perspectives
Nucleic acid-based approaches offer great promise for developing novel therapeutics for AD, a complex neurodegenerative disease with several pathological features. Confounds include genetic factors, metabolic disorders including high cholesterol levels, insulin resistance due to impaired glucose metabolism, and dysfunction in various molecular pathways. Existing therapies only treat AD symptoms, not the underlying molecular causes. Although many drug molecules have shown success in cell and animal models, this effect often cannot be replicated in human trials. There is an unmet need for better theranostic strategies. The drug Nusinersen, recently approved by the FDA for spinal muscular atrophy, shows that nucleic acids have potential for the treatment of neurological diseases, including AD. Their efficacy in targeting several pathways that underlie AD highlights their potential to be developed as novel therapeutics for AD.
Abbreviations
FDA: Food and Drug Administration; AD: Alzheimer's disease; AO: antisense oligonucleotides; NMDA: N-methyl-D- aspartate receptor; Aβ: amyloid β; APP: amyloid precursor protein; BACE1: β-site amyloid precursor protein cleaving enzyme 1; PSEN: Presenilin; tau: microtubule associated protein; siRNA: small interfering RNA; antimiR: anti-microRNA; 2'-OMe: 2'-O-methyl; 2'-MOE: 2'-O-methoxyethyl; 2'-F: 2'-fluoro; LNA: locked nucleic acids; PNA: peptide nucleic acids; PMO: phosphorodiamidate morpholino; tcDNA: tricyclo-DNA; CeNA: cyclohexenyl nucleic acid; PEG: polyethylene glycol; mRNA: messenger RNA; SAMP8: senescence- accelerated mouse-prone 8; BBB: blood brain barrier; AChE: acetylcholinesterase; ApoER2: Apolipoprotein E receptor 2; RISC: RNA induced silencing complex; SELEX: systemic evolution of ligands by exponential enrichment.
Acknowledgements
RNV thanks the funding from the McCusker Charitable Foundation and Perron Institute for Neurological Diseases and Translational Science. RNV and MC thank the funding support from Greg and Dale Higham.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J. et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. 2016;79:257-271
2. Crooke ST, Geary RS. Clinical pharmacological properties of mipomersen (Kynamro), a second generation antisense inhibitor of apolipoprotein B. Br J Clin Pharmacol. 2013;76:269-276
3. Vitravene Study Group. A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol. 2002;133:467-474
4. Macugen Diabetic Retinopathy Study Group. A phase II randomized double-masked trial of pegaptanib, an anti-vascular endothelial growth factor aptamer, for diabetic macular edema. Ophthalmology. 2005;112:1747-1757
5. Prince M, Comas-Herrera A, Knapp M, Guerchet M, Karagiannidou M. World Alzheimer Report 2016: Improving healthcare for people living with dementia. In: (ed.) Rees G. World Alzheimer Report. London, UK: London School of Economics and Political Sciences. 2016:140
6. Alzheimers Association Update. Alzheimers Dement (Amst). 2015;11:104-105
7. Birks JS. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev. 2006
8. Olivares D, Deshpande VK, Shi Y, Lahiri DK, Greig NH, Rogers JT. et al. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease. Curr Alzheimer Res. 2012;9:746-758
9. Robinson SW, Fernandes M, Husi H. Current advances in systems and integrative biology. Comput Struct Biotechnol J. 2014;11:35-46
10. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8:101-112
11. Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741-766
12. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. The Lancet. 2006;368:387-403
13. Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid β-peptide. Cell. 1993;75:1039-1042
14. Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23-A31
15. Wilquet V, De Strooper B. Amyloid-β precursor protein processing in neurodegeneration. Curr Opin Neurol. 2004;14:582-588
16. Selkoe DJ. Normal and abnormal biology of the β-amyloid precursor protein. Annu Rev Neurosci. 1994;17:489-517
17. Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545-555
18. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184-185
19. Lahiri DK, Farlow MR, Greig NH, Sambamurti K. Current drug targets for Alzheimer's disease treatment. Drug Dev Res. 2002;56:267-281
20. Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812-818
21. Sindi IA, Tannenberg RK, Dodd PR. Role for the neurexin-neuroligin complex in Alzheimer's disease. Neurobiol Aging. 2014;35:746-756
22. Ondrejcak T, Klyubin I, Hu N-W, Barry AE, Cullen WK, Rowan MJ. Alzheimer's disease amyloid β-protein and synaptic function. Neuromolecular Med. 2010;12:13-26
23. Danysz W, Parsons CG. Alzheimer's disease, β-amyloid, glutamate, NMDA receptors and memantine-searching for the connections. Br J Pharmacol. 2012;167:324-352
24. Schelterns P, Feldman H. Treatment of Alzheimer's disease; current status and new perspectives. Lancet Neurol. 2003;2:539-547
25. Ferreira ST, Clarke JR, Bomfim TR, De Felice FG. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer's disease. Alzheimers Dement (Amst). 2014;10:S76-S83
26. Hoyer S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004;490:115-125
27. Maulik M, Westaway D, Jhamandas J, Kar S. Role of cholesterol in APP metabolism and its significance in Alzheimer's disease pathogenesis. Mol Neurobiol. 2013;47:37-63
28. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121-1159
29. Iqbal K, Alonso AdC, Chen S, Chohan MO, El-Akkad E, Gong C-X. et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta Mol Bas Dis. 2005;1739:198-210
30. Medina M, Avila J. New perspectives on the role of tau in Alzheimer's disease. Implications for therapy. Biochem Pharmacol. 2014;88:540-547
31. Hernandez F, Avila J. Tauopathies. Cell Mol Life Sci. 2007;64:2219-2233
32. Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta Mol Bas Dis. 2005;1739:331-354
33. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825-858
34. Feldman H, Gauthier S, Hecker J, Vellas B, Emir B, Mastey V. et al. Efficacy of donepezil on maintenance of activities of daily living in patients with moderate to severe Alzheimer's disease and the effect on caregiver burden. J Am Geriatr Soc. 2003;51:737-744
35. McKeith I, Del Ser T, Spano P, Emre M, Wesnes K, Anand R. et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. The Lancet. 2000;356:2031-2036
36. Raskind M, Peskind E, Wessel T, Yuan W, Galantamine USA Study Group. Galantamine in AD A 6-month randomized, placebo-controlled trial with a 6-month extension. Neurology. 2000;54:2261-2268
37. Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317-324
38. Wang X, Wang W, Li L, Perry G, Lee H-g, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta Mol Bas Dis. 2014;1842:1240-1247
39. Pimplikar SW. Neuroinflammation in Alzheimer's disease: from pathogenesis to a therapeutic target. J Clin Immunol. 2014;34:64-69
40. Freiherr J, Hallschmid M, Frey II WH, Brünner YF, Chapman CD, Hölscher C. et al. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505-514
41. Ribarič S. The rationale for insulin therapy in Alzheimer's disease. Molecules. 2016;21:E689
42. Galimberti D, Scarpini E. Progress in Alzheimer's disease. J Neurol. 2012;259:201-211
43. Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702-716
44. Karran E. Current status of vaccination therapies in Alzheimer's disease. J Neurochem. 2012;123:647-651
45. St George-Hyslop PH, Morris JC. Will anti-amyloid therapies work for Alzheimer's disease? The Lancet. 2008;372:180-182
46. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. The Lancet. 2011;377:1019-1031
47. Santa-Maria I, Hernández F, Del Rio J, Moreno FJ, Avila J. Tramiprosate, a drug of potential interest for the treatment of Alzheimer's disease, promotes an abnormal aggregation of tau. Mol Neurodegener. 2007;2:17
48. Aisen PS, Saumier D, Briand R, Laurin J, Gervais F, Tremblay P. et al. A Phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology. 2006;67:1757-1763
49. Gervais F, Chalifour R, Garceau D, Kong X, Laurin J, McLaughlin R. et al. Glycosaminoglycan mimetics: a therapeutic approach to cerebral amyloid angiopathy. Amyloid. 2001;8(Suppl 1):28-35
50. Bilikiewicz A, Gaus W. Colostrinin (a naturally occurring, proline-rich, polypeptide mixture) in the treatment of Alzheimer's disease. J Alzheimers Dis. 2004;6:17-26
51. Leszek J, Inglot AD, Janusz M, Lisowski J, Krukowska K, Georgiades JA. Colostrinin: a proline-rich polypeptide (PRP) complex isolated from ovine colostrum for treatment of Alzheimer's disease. A double-blind, placebo-controlled study. Archivum Immunologiæ et Therapiæ Experimentalis. 1999;47:377-385
52. Popik P, Bobula B, Janusz M, Lisowski J, Vetulani J. Colostrinin, a polypeptide isolated from early milk, facilitates learning and memory in rats. Pharmacol Biochem Behav. 1999;64:183-189
53. Townsend KP, Praticò D. Novel therapeutic opportunities for Alzheimer's disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19:1592-1601
54. Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC. et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553-1562
55. Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M. et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:322-333
56. Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S. et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:311-321
57. Relkin N. Clinical trials of intravenous immunoglobulin for Alzheimer's disease. J Clin Immunol. 2014;34:74-79
58. Moreth J, Mavoungou C, Schindowski K. Passive anti-amyloid immunotherapy in Alzheimer's disease: What are the most promising targets? Immunity and Ageing. 2013;10:18
59. McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126:479-497
60. Kukar T, Prescott S, Eriksen JL, Holloway V, Murphy MP, Koo EH. et al. Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 2007;8:54
61. Galasko DR, Graff-Radford N, May S, Hendrix S, Cottrell BA, Sagi SA. et al. Safety, tolerability, pharmacokinetics, and Aβ levels after short-term administration of R-flurbiprofen in healthy elderly individuals. Alzheimer Disease and Associated Disorders. 2007;21:292-299
62. Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. et al. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomised phase II trial. The Lancet Neurology. 2008;7:483-493
63. Siemers ER, Quinn JF, Kaye J, Farlow MR, Porsteinsson A, Tariot P. et al. Effects of a γ-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology. 2006;66:602-604
64. Coric V, Salloway S, van Dyck C, Kerselaers W, Kaplita S, Curtis C. et al. A phase II study of the γ-secretase inhibitor avagacestat (BMS-708163) in predementia Alzheimer's disease. Alzheimers Dement (Amst). 2013;9:P283
65. Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF. et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer's disease patients. Science Translational Medicine. 2016;8:363ra150
66. Miller BW, Willett KC, Desilets AR. Rosiglitazone and pioglitazone for the treatment of Alzheimer's disease. Ann Pharmacother. 2011;45:1416-1424
67. Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213-11218
68. Martinez A, Gil C, Perez DI. Glycogen synthase kinase 3 inhibitors in the next horizon for Alzheimer's disease treatment. Int J Alzheimers Dis. 2011;2011:280502
69. Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz H-J. et al. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimers Dis. 2015;45:75-88
70. Mecocci P, Polidori MC. Antioxidant clinical trials in mild cognitive impairment and Alzheimer's disease. Biochim Biophys Acta Mol Bas Dis. 2012;1822:631-638
71. Ray B, Lahiri DK. Neuroinflammation in Alzheimer's disease: different molecular targets and potential therapeutic agents including curcumin. Curr Opin Pharmacol. 2009;9:434-444
72. Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005-1015
73. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL. et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388-405
74. Yarchoan M, Arnold SE. Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes. 2014;63:2253-2261
75. Casserly I, Topol EJ. Convergence of atherosclerosis and Alzheimer's disease: inflammation, cholesterol, and misfolded proteins. The Lancet. 2004;363:1139-1146
76. De Mesmaeker A, Haener R, Martin P, Moser HE. Antisense oligonucleotides. Acc Chem Res. 1995;28:366-374
77. Lipi F, Chen S, Chakravarthy M, Rakesh S, Veedu RN. In vitro evolution of chemically-modified nucleic acid aptamers: pros and cons, and comprehensive selection strategies. RNA Biol. 2016;13:1232-1245
78. Chan JH, Lim S, Wong W. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33:533-540
79. Vu H, Hirschbein BL. Internucleotide phosphite sulfurization with tetraethylthiuram disulfide. Phosphorothioate oligonucleotide synthesis via phosphoramidite chemistry. Tetrahedron Lett. 1991;32:3005-3008
80. Majlessi M, Nelson NC, Becker MM. Advantages of 2′-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998;26:2224-2229
81. Kawasaki AM, Casper MD, Freier SM, Lesnik EA, Zounes MC, Cummins LL. et al. Uniformly modified 2'-deoxy-2'-fluoro-phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J Med Chem. 1993;36:831-841
82. Geary RS, Watanabe TA, Truong L, Freier S, Lesnik EA, Sioufi NB. et al. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J Pharmacol Exp Ther. 2001;296:890-897
83. Summerton J, WELLER D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187-195
84. Veedu RN, Wengel J. Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem Biodivers. 2010;7:536-542
85. Veedu RN, Wengel J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009;6:321-323
86. Hyrup B, Nielsen PE. Peptide nucleic acids (PNA): synthesis, properties and potential applications. Bioorg Med Chem. 1996;4:5-23
87. Renneberg D, Leumann CJ. Watson- Crick base-pairing properties of tricyclo-DNA. J Am Chem Soc. 2002;124:5993-6002
88. Wang J, Verbeure B, Luyten I, Lescrinier E, Froeyen M, Hendrix C. et al. Cyclohexene nucleic acids (CeNA): serum stable oligonucleotides that activate RNase H and increase duplex stability with complementary RNA. J Am Chem Soc. 2000;122:8595-8602
89. Ikeda Y, Nagasaki Y. Impacts of PEGylation on the gene and oligonucleotide delivery system. J Appl Polym Sci. 2014:131
90. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly (ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed. 2010;49:6288-6308
91. White PJ, Anastasopoulos F, Pouton CW, Boyd BJ. Overcoming biological barriers to in vivo efficacy of antisense oligonucleotides. Expert Rev Mol Med. 2009:11
92. Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518-6548
93. Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259-293
94. Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125-140
95. Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C, Prochiantz A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol. 1995;128:919-927
96. Dobie K. Antisense modulation of amyloid β protein precursor expression. US: Scios Inc, Fremont, CA (US). 2007
97. Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA. et al. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides. 2000;21:1769-1775
98. Kumar VB. Antisense modulation of amyloid β protein expression. US: St. Louis University, St. Louis, MO (US). 2001
99. Banks WA, Farr SA, Butt W, Kumar VB, Franko MW, Morley JE. Delivery across the blood-brain barrier of antisense directed against amyloid β: reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J Pharmacol Exp Ther. 2001;297:1113-1121
100. Poon HF, Farr SA, Banks WA, Pierce WM, Klein JB, Morley JE. et al. Proteomic identification of less oxidized brain proteins in aged senescence-accelerated mice following administration of antisense oligonucleotide directed at the Aβ region of amyloid precursor protein. Brain Res Mol Brain Res. 2005;138:8-16
101. Opazo P, Saud K, de Saint Pierre M, Cárdenas AM, Allen DD, Segura-Aguilar J. et al. Knockdown of amyloid precursor protein normalizes cholinergic function in a cell line derived from the cerebral cortex of a trisomy 16 mouse: an animal model of Down syndrome. J Neurosci Res. 2006;84:1303-1310
102. Saud K, Arriagada C, Cárdenas AM, Shimahara T, Allen DD, Caviedes R. et al. Neuronal dysfunction in Down syndrome: contribution of neuronal models in cell culture. Journal of Physiology, Paris. 2006;99:201-210
103. Rojas G, Cárdenas AM, Fernández-Olivares P, Shimahara T, Segura-Aguilar J, Caviedes R. et al. Effect of the knockdown of amyloid precursor protein on intracellular calcium increases in a neuronal cell line derived from the cerebral cortex of a trisomy 16 mouse. Exp Neurol. 2008;209:234-242
104. Chauhan NB. Trafficking of intracerebroventricularly injected antisense oligonucleotides in the mouse brain. Antisense Nucleic Acid Drug Dev. 2002;12:353-357
105. Chauhan NB, Siegel GJ. Antisense inhibition at the β-secretase-site of β-amyloid precursor protein reduces cerebral amyloid and acetyl cholinesterase activity in Tg2576. Neuroscience. 2007;146:143-151
106. Erickson MA, Niehoff ML, Farr SA, Morley JE, Dillman LA, Lynch KM. et al. Peripheral administration of antisense oligonucleotides targeting the amyloid-β protein precursor reverses AβPP and LRP-1 overexpression in the aged SAMP8 mouse brain. J Alzheimers Dis. 2012;28:951-960
107. Erickson M, Farr S, Niehoff M, Morley J, Banks W. Antisense directed against the amyloid precursor protein reduces cytokine expression in the brain and improves learning and memory in the Tg2576 mouse model of Alzheimer's disease. Brain Behav Immun. 2012;26:S27
108. Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM. et al. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature. 1999;402:533-537
109. Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P. et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735-741
110. Wolfe MS. Targeting mRNA for Alzheimer's and related dementias. Scientifica. 2014;2014:1-13
111. Refolo LM, Eckman C, Prada CM, Yager D, Sambamurti K, Mehta N. et al. Antisense-induced reduction of Presenilin 1 expression selectively increases the production of amyloid β42 in transfected cells. J Neurochem. 1999;73:2383-2388
112. Grilli M, Diodato E, Lozza G, Brusa R, Casarini M, Uberti D. et al. Presenilin-1 regulates the neuronal threshold to excitotoxicity both physiologically and pathologically. Proc Natl Acad Sci USA. 2000;97:12822-12827
113. Fiorini A, Sultana R, Förster S, Perluigi M, Cenini G, Cini C. et al. Antisense directed against PS-1 gene decreases brain oxidative markers in aged senescence accelerated mice (SAMP8) and reverses learning and memory impairment: a proteomics study. Free Radic Biol Med. 2013;65:1-14
114. Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461-463
115. DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR. et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33:12887-12897
116. Kalbfuss B, Mabon SA, Misteli T. Correction of alternative splicing of tau in frontotemporal dementia and parkinsonism linked to chromosome 17. J Biol Chem. 2001;276:42986-42993
117. Peacey E, Rodriguez L, Liu Y, Wolfe MS. Targeting a pre-mRNA structure with bipartite antisense molecules modulates tau alternative splicing. Nucleic Acids Res. 2012;40:9836-9849
118. Liu Y, Rodriguez L, Wolfe MS. Template-directed synthesis of a small molecule-antisense conjugate targeting an mRNA structure. Bioorg Chem. 2014;54:7-11
119. Sud R, Geller ET, Schellenberg GD. Antisense-mediated exon skipping decreases tau protein expression: a potential therapy for tauopathies. Mol Ther Nucleic Acids. 2014;3:e180
120. Farr SA, Ripley JL, Sultana R, Zhang Z, Niehoff ML, Platt TL. et al. Antisense oligonucleotide against GSK-3β in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic Biol Med. 2014;67:387-395
121. Fu A-L, Zhang X-M, Sun M-J. Antisense inhibition of acetylcholinesterase gene expression for treating cognition deficit in Alzheimer's disease model mice. Brain Res. 2005;1066:10-15
122. Wasser CR, Herz J. Splicing therapeutics for Alzheimer's disease. EMBO Mol Med. 2016;8:308-310
123. Hinrich AJ, Jodelka FM, Chang JL, Brutman D, Bruno AM, Briggs CA. et al. Therapeutic correction of ApoER2 splicing in Alzheimer's disease mice using antisense oligonucleotides. EMBO Mol Med. 2016;8:328-345
124. Hannon GJ. RNA interference. Nature. 2002;418:244-251
125. Miller VM, Gouvion CM, Davidson BL, Paulson HL. Targeting Alzheimer's disease genes with RNA interference: an efficient strategy for silencing mutant alleles. Nucleic Acids Res. 2004;32:661-668
126. McSwiggen J. RNA interference mediated treatment of Alzheimer's disease using short interfering RNA. Terpstra, Anita, J.; McDonnell Boehnen Hulbert & Berghoff, Suite 3200, 300 South Wacker Drive, Chicago, IL 60606 (US). 2002
127. Basi G, Frigon N, Barbour R, Doan T, Gordon G, McConlogue L. et al. Antagonistic effects of β-site amyloid precursor protein-cleaving enzymes 1 and 2 on β-amyloid peptide production in cells. J Biol Chem. 2003;278:31512-31520
128. Kao S-C, Krichevsky AM, Kosik KS, Tsai L-H. BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem. 2004;279:1942-1949
129. Modarresi F, Faghihi MA, Patel NS, Sahagan BG, Wahlestedt C, Lopez-Toledano MA. Knockdown of BACE1-AS nonprotein-coding transcript modulates β-amyloid-related hippocampal neurogenesis. Int J Alzheimers Dis. 2011;2011:929042
130. Cai J, Qi X, Kociok N, Skosyrski S, Emilio A, Ruan Q. et al. β-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol Med. 2012;4:980-991
131. Fisette JF, Montagna DR, Mihailescu MR, Wolfe MS. AG-Rich element forms a G-quadruplex and regulates BACE1 mRNA alternative splicing. J Neurochem. 2012;121:763-773
132. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421-433
133. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509-524
134. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522-531
135. Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1
136. Faghihi MA, Zhang M, Huang J, Modarresi F, Van der Brug MP, Nalls MA. et al. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010;11:R56
137. Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN. et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/β-secretase expression. Proc Natl Acad Sci. 2008;105:6415-6420
138. Zovoilis A, Agbemenyah HY, Agis-Balboa RC, Stilling RM, Edbauer D, Rao P. et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011;30:4299-4308
139. Murphy SR, Chang CC, Dogbevia G, Bryleva EY, Bowen Z, Hasan MT. et al. ACAT1 knockdown gene therapy decreases amyloid-β in a mouse model of Alzheimer's disease. Mol Ther. 2013;21:1497-1506
140. Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311-322
141. Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93:1412-1421
142. Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron. 1991;7:695-702
143. Lee ST, Chu K, Jung KH, Kim JH, Huh JY, Yoon H. et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann Neurol. 2012;72:269-277
144. Achenbach J, Chiuman W, Cruz R, Li Y. DNAzymes: from creation in vitro to application in vivo. Curr Pharm Biotechnol. 2004;5:321-336
145. Nawrot B. Targeting BACE with small inhibitory nucleic acids — a future for Alzheimer's disease therapy? Acta Biochim Pol. 2004;51:431-444
146. Gopinath SCB. Methods developed for SELEX. Anal Bioanal Chem. 2007;387:171-182
147. Stoltenburg R, Reinemann C, Strehlitz B. SELEX — a (r) evolutionary method to generate high-affinity nucleic acid ligands. Biomol Eng. 2007;24:381-403
148. Ylera F, Lurz R, Erdmann VA, Fürste JP. Selection of RNA aptamers to the Alzheimer's disease amyloid peptide. Biochem Biophys Res Commun. 2002;290:1583-1588
149. Bunka DH, Mantle BJ, Morten IJ, Tennent GA, Radford SE, Stockley PG. Production and characterization of RNA aptamers specific for amyloid fibril epitopes. J Biol Chem. 2007;282:34500-34509
150. Rahimi F, Murakami K, Summers JL, Chen C-HB, Bitan G. RNA aptamers generated against oligomeric Aβ40 recognize common amyloid aptatopes with low specificity but high sensitivity. PLoS One. 2009;4:e7694
151. Takahashi T, Tada K, Mihara H. RNA aptamers selected against amyloid β-peptide (Aβ) inhibit the aggregation of Aβ. Mol Biosyst. 2009;5:986-991
152. Mathew A, Aravind A, Brahatheeswaran D, Fukuda T, Nagaoka Y, Hasumura T. et al. Amyloid-binding aptamer conjugated curcumin-PLGA nanoparticle for potential use in Alzheimer's disease. Bionanoscience. 2012;2:83-93
153. Farrar CT, William CM, Hudry E, Hashimoto T, Hyman BT. RNA aptamer probes as optical imaging agents for the detection of amyloid plaques. PLoS One. 2014;9:e89901
154. Babu E, Mareeswaran PM, Sathish V, Singaravadivel S, Rajagopal S. Sensing and inhibition of amyloid-β based on the simple luminescent aptamer-ruthenium complex system. Talanta. 2015;134:348-353
155. Rentmeister A, Bill A, Wahle T, Walter J, Famulok M. RNA aptamers selectively modulate protein recruitment to the cytoplasmic domain of β-secretase BACE1 in vitro. RNA. 2006;12:1650-1660
156. Liang H, Shi Y, Kou Z, Peng Y, Chen W, Li X. et al. Inhibition of BACE1 Activity by a DNA Aptamer in an Alzheimer's Disease Cell Model. PLoS One. 2015;10:e0140733
157. Kim JH, Kim E, Choi WH, Lee J, Lee JH, Lee H. et al. Inhibitory RNA aptamers of tau oligomerization and their neuroprotective roles against proteotoxic stress. Mol Pharm. 2016;13:2039-2048
158. Tannenberg RK, Lauridsen LH, Kanwar JR, Dodd PR, Veedu RN. Nucleic acid aptamers as novel class of therapeutics to mitigate Alzheimer's disease pathology. Curr Alzheimer Res. 2013;10:442-448
159. Kim S, Wark AW, Lee HJ. Femtomolar detection of tau proteins in undiluted plasma using surface plasmon resonance. Anal Chem. 2016;88:7793-7799
160. Lee JH, Shin SK, Jiang Y, Choi WH, Hong C, Kim D-E. et al. Facilitated Tau degradation by USP14 aptamers via enhanced proteasome activity. Sci Rep. 2015;5:10757
161. Mashima T, Matsugami A, Nishikawa F, Nishikawa S, Katahira M. Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 2009;37:6249-6258
162. Kanwar JR, Sun X, Punj V, Sriramoju B, Mohan RR, Zhou SF. et al. Nanoparticles in the treatment and diagnosis of neurological disorders: untamed dragon with fire power to heal. Nanomedicine: Nanotechnology, Biology, and Medicine. 2012;8:399-414
163. Ulbrich K, Hekmatara T, Herbert E, Kreuter J. Transferrin- and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood-brain barrier (BBB). Eur J Pharm Biopharm. 2009;71:251-256
164. Gan CW, Feng SS. Transferrin-conjugated nanoparticles of poly(lactide)-D-alpha-tocopheryl polyethylene glycol succinate diblock copolymer for targeted drug delivery across the blood-brain barrier. Biomaterials. 2010;31:7748-7757
165. Chen CH, Dellamaggiore KR, Ouellette CP, Sedano CD, Lizadjohry M, Chernis GA. et al. Aptamer-based endocytosis of a lysosomal enzyme. Proc Natl Acad Sci USA. 2008;105:15908-15913
166. Macdonald J, Henri J, Goodman L, Xiang D, Duan W, Shigdar S. Development of a bifunctional aptamer targeting the transferrin receptor and epithelial cell adhesion molecule (EpCAM) for the treatment of brain cancer metastases. ACS Chem Neurosci. 2017;8:777-784
167. Du L, Kayali R, Bertoni C, Fike F, Hu H, Iversen PL. et al. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum Mol Genet. 2011;20:3151-3160
168. Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195-206
169. Pardridge WM. Drug targeting to the brain. Pharm Res. 2007;24:1733-1744
170. Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS. et al. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer's disease. J Alzheimers Dis. 2013;35:789-797
171. Hanson LR, Frey WH 2nd. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9(Suppl 3):S5
172. Dhuria SV, Hanson LR, Frey WH 2nd. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99:1654-1673
173. Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64:614-628
Author Biography
 Madhuri Chakravarthy obtained her Bachelor's (2011) in Biomedical Science and Master's (2014) in Biomedical Science from the University of Auckland, New Zealand. She is currently a PhD student under the supervision of Dr. Rakesh N. Veedu at the Murdoch University, Western Australia. Her research is centered on developing novel nucleic acid technologies to tackle neurological diseases.
Madhuri Chakravarthy obtained her Bachelor's (2011) in Biomedical Science and Master's (2014) in Biomedical Science from the University of Auckland, New Zealand. She is currently a PhD student under the supervision of Dr. Rakesh N. Veedu at the Murdoch University, Western Australia. Her research is centered on developing novel nucleic acid technologies to tackle neurological diseases.
 Suxiang Chen received his Bachelor degree of Agriculture in 2010 in South China Agricultural University. He then obtained his Master degree in Biotechnology (Advanced) and a second Master degree in Technology and Innovation Management from the University of Queensland in 2013 and 2014 respectively. Later, he worked in Darui Biotechonology Co. Ltd and Gene Denovo Biotechnology Co. Ltd in Guangzhou, China as a core technician and technical support team member respectively. Since 2016, he has been pursuing a PhD degree at Murdoch University under the supervision of Dr. Rakesh N. Veedu. His current research focuses include the development of novel RNA targeting therapies for tackling Duchenne muscular dystrophy and Type-2 Diabetes.
Suxiang Chen received his Bachelor degree of Agriculture in 2010 in South China Agricultural University. He then obtained his Master degree in Biotechnology (Advanced) and a second Master degree in Technology and Innovation Management from the University of Queensland in 2013 and 2014 respectively. Later, he worked in Darui Biotechonology Co. Ltd and Gene Denovo Biotechnology Co. Ltd in Guangzhou, China as a core technician and technical support team member respectively. Since 2016, he has been pursuing a PhD degree at Murdoch University under the supervision of Dr. Rakesh N. Veedu. His current research focuses include the development of novel RNA targeting therapies for tackling Duchenne muscular dystrophy and Type-2 Diabetes.
 A/Prof. Peter R. Dodd obtained his PhD from Imperial College, University of London, and is currently the Director of Queensland Brain Bank at the School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Australia. His lab uses human autopsy tissues to study amino acid neurotransmission, and has developed protocols to prepare good-quality mRNA, miRNA, and proteins for mapping and quantification. His lab studied phenotype-genotype interactions in alcoholics and dementia cases, partitioned by sex and comorbid disease, with key findings including altered expression of GABA receptors in alcoholics without comorbid disease and of glutamate receptors in cirrhotic alcoholics, and altered expression of glutamate transporters and receptors in Alzheimer disease. Prof. Dodd's group was the first to use microarrays to study the human brain transcriptome. Overall, his lab researches the pathogenesis of human neurological diseases.
A/Prof. Peter R. Dodd obtained his PhD from Imperial College, University of London, and is currently the Director of Queensland Brain Bank at the School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Australia. His lab uses human autopsy tissues to study amino acid neurotransmission, and has developed protocols to prepare good-quality mRNA, miRNA, and proteins for mapping and quantification. His lab studied phenotype-genotype interactions in alcoholics and dementia cases, partitioned by sex and comorbid disease, with key findings including altered expression of GABA receptors in alcoholics without comorbid disease and of glutamate receptors in cirrhotic alcoholics, and altered expression of glutamate transporters and receptors in Alzheimer disease. Prof. Dodd's group was the first to use microarrays to study the human brain transcriptome. Overall, his lab researches the pathogenesis of human neurological diseases.
 Rakesh N. Veedu is currently leading the precision nucleic acid theranostics group at Murdoch University and Perron Institute for Neurological and Translational Science. He obtained his PhD in synthetic organic chemistry in 2006 from The University of Queensland, Australia under the supervision of Prof. Curt Wentrup after completing his MSc from Griffith University, Australia. He then continued his postdoctoral career under the supervision of Prof. Jesper Wengel at the Nucleic Acid Center, University of Southern Denmark in the field of nucleic acid chemical biology. Later in 2009, he was appointed as a Research Associate Professor within the Nucleic Acid Center. He then returned to The University of Queensland in mid-2010 and established his independent research career in Functional Nucleic Acid Theranostics Development. His current research is focused on developing novel precision nucleic acid theranostics for tackling solid cancers, neurological diseases including Alzheimer's disease and infectious diseases using nucleic acid aptamers, antisense oligonucleotides, siRNA, antimiRs, molecular beacons and DNAzymes.
Rakesh N. Veedu is currently leading the precision nucleic acid theranostics group at Murdoch University and Perron Institute for Neurological and Translational Science. He obtained his PhD in synthetic organic chemistry in 2006 from The University of Queensland, Australia under the supervision of Prof. Curt Wentrup after completing his MSc from Griffith University, Australia. He then continued his postdoctoral career under the supervision of Prof. Jesper Wengel at the Nucleic Acid Center, University of Southern Denmark in the field of nucleic acid chemical biology. Later in 2009, he was appointed as a Research Associate Professor within the Nucleic Acid Center. He then returned to The University of Queensland in mid-2010 and established his independent research career in Functional Nucleic Acid Theranostics Development. His current research is focused on developing novel precision nucleic acid theranostics for tackling solid cancers, neurological diseases including Alzheimer's disease and infectious diseases using nucleic acid aptamers, antisense oligonucleotides, siRNA, antimiRs, molecular beacons and DNAzymes.
![]() Corresponding author: Rakesh N. Veedu, PhD. Centre for Comparative Genomics, Murdoch University, Building 390 Discovery Drive, Perth, Western Australia, Australia 6150. Email: R.Veeduedu.au
Corresponding author: Rakesh N. Veedu, PhD. Centre for Comparative Genomics, Murdoch University, Building 390 Discovery Drive, Perth, Western Australia, Australia 6150. Email: R.Veeduedu.au
Citation styles
APA
Chakravarthy, M., Chen, S., Dodd, P.R., Veedu, R.N. (2017). Nucleic Acid-Based Theranostics for Tackling Alzheimer's Disease. Theranostics, 7(16), 3933-3947. https://doi.org/10.7150/thno.21529.
ACS
Chakravarthy, M.; Chen, S.; Dodd, P.R.; Veedu, R.N. Nucleic Acid-Based Theranostics for Tackling Alzheimer's Disease. Theranostics 2017, 7 (16), 3933-3947. DOI: 10.7150/thno.21529.
NLM
Chakravarthy M, Chen S, Dodd PR, Veedu RN. Nucleic Acid-Based Theranostics for Tackling Alzheimer's Disease. Theranostics 2017; 7(16):3933-3947. doi:10.7150/thno.21529. https://www.thno.org/v07p3933.htm
CSE
Chakravarthy M, Chen S, Dodd PR, Veedu RN. 2017. Nucleic Acid-Based Theranostics for Tackling Alzheimer's Disease. Theranostics. 7(16):3933-3947.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.