Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3595-3607. doi:10.7150/thno.18974 This issue Cite
Research Paper
Inhibition of Bone Marrow-Derived Mesenchymal Stem Cells Homing Towards Triple-Negative Breast Cancer Microenvironment Using an Anti-PDGFRβ Aptamer
Simona Camorani1, Billy Samuel Hill2, 3, Raffaela Fontanella2, 3, Adelaide Greco4, 2, 3, Matteo Gramanzini2, 3, Luigi Auletta5, Sara Gargiulo2, 3, Sandra Albanese2, 3, Enrico Lucarelli5, Laura Cerchia1* ![]() , Antonella Zannetti2, 3*
, Antonella Zannetti2, 3* ![]()
1. Istituto per l'Endocrinologia e l'Oncologia Sperimentale "G. Salvatore", CNR, Naples, Italy;
2. Istituto di Biostrutture e Bioimmagini-CNR, Naples, Italy;
3. Ceinge, Advanced Biotechnology, Scarl, Naples, Italy;
4. Dipartimento di Scienze Biomediche Avanzate, Università degli Studi di Napoli "Federico II", Naples, Italy;
5. IRCCS SDN, Naples, Italy;
6. Istituto Ortopedico Rizzoli, Bologna, Italy.
* L. Cerchia and A. Zannetti contributed equally to this work.
Received 2016-12-29; Accepted 2017-6-20; Published 2017-8-22
Abstract

Bone marrow-derived mesenchymal stem cells (BM-MSCs) are shown to participate in tumor progression by establishing a favorable tumor microenvironment (TME) that promote metastasis through a cytokine networks. However, the mechanism of homing and recruitment of BM-MSCs into tumors and their potential role in malignant tissue progression is poorly understood and controversial. Here we show that BM-MSCs increase aggressiveness of triple-negative breast cancer (TNBC) cell lines evaluated as capability to migrate, invade and acquire stemness markers. Importantly, we demonstrate that the treatment of BM-MSCs with a nuclease-resistant RNA aptamer against platelet-derived growth factor receptor β (PDGFRβ) causes the inhibition of receptor-dependent signaling pathways thus drastically hampering BM-MSC recruitment towards TNBC cell lines and BM-MSCs trans-differentiation into carcinoma-associated fibroblast (CAF)-like cells. Moreover, in vivo molecular imaging analysis demonstrated the aptamer ability to prevent BM-MSCs homing to TNBC xenografts. Collectively, our results indicate the anti-PDGFRβ aptamer as a novel therapeutic tool to interfere with BM-MSCs attraction to TNBC providing the rationale to further explore the aptamer in more complex pre-clinical settings.
Keywords: Bone marrow-derived mesenchymal stem cells, aptamer, platelet-derived growth factor receptor β, triple-negative breast cancer.
Introduction
Mesenchymal stem cells (MSCs) are multi-potent cells that in response to a large variety of bioactive molecules, growth factors, cytokines and chemokines, are recruited to injured or inflamed tissue to support their repair contributing to stem cells homeostasis and immunomodulation [1]. Similar to site of injury, tumors through production of paracrine and endocrine signals enrolled MSCs in their microenvironment (TME) that is seen as a "wound that never heals" [2]. The ability of MSCs to migrate to tumors makes them potential cargo for anticancer agents [3]. Once reached the tumors, MSCs may exert opposite roles depending on tumor types, thus supporting cancer progression, as reported in breast [2], melanoma [4], prostate [5] and colon [6] carcinomas or suppressing it, as in malignant glioma [7]. The main source of MSCs in adult is bone marrow, even if additional tissues have been identified as font [8]. Bone marrow-derived mesenchymal stem cells (BM-MSCs) along with fibroblasts, pericytes, adipocytes, macrophages and immune cells play a crucial role into microenvironment of breast cancers, promoting extracellular matrix remodeling, cell migration and invasion, neo-angiogenesis, drug resistance and evasion of immunosurveillance [9, 10]. Once in TME, BM-MSCs can trans-differentiate into cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs) or vascular and perivascular cells switching from neutral role toward pro-tumorigenic behavior [11]. Recently, we reported the ability of BM-MSCs to promote migration, invasion and epithelial-mesenchymal transition (EMT) of osteosarcoma and hepatocellular carcinoma cells through chemokine receptor type 4 (CXCR4) [12]. In the last decade, different biomolecules have been found involved in BM-MSCs recruitment and activity into breast cancer including chemokine C-C motif ligand 5 (CCL5) [2]; transforming growth factor β (TGF-β) and interleukin 17B [13]; CXCL10 and placental growth factor [14]. Recently, it has been reported that platelet-derived growth factor receptor β (PDGFRβ) signaling plays an important role in recruitment of MSCs towards tumor sites [15]. In addition, Dhawan et al. [16] showed that platelet-derived growth factor BB (PDGF-BB) along with basic fibroblast growth factor (bFGF) are involved in bone marrow support in the dissemination of breast cancer cells.
Triple-negative breast cancers (TNBCs), representing ~15% of all breast cancers, are characterized by the absence of estrogen and progesterone receptors and ErbB2, excluding the possibility of specific intervention for these tumors. They are biologically more aggressive and have a very poor outcome compared to other breast tumors [17].
In this study, we investigated the role of PDGFRβ in BM-MSC homing and activity into the highly malignant and invasive mesenchymal-like (ML) TNBC subtype [17]. We have recently demonstrated that cells of this subtype have a strong tendency to vasculogenic mimicry (VM) both in vitro and in vivo, and that targeting epidermal growth factor receptor (EGFR)-integrin αvβ3 interaction by an anti-EGFR aptamer, results in inhibition of VM and tumor growth in ML TNBC [18].
To interfere with the PDGFRβ-mediated cross-talk between BM-MSCs and tumor cells, we used a nuclease-resistant RNA aptamer, named Gint4.T, that we have previously characterized as high specific inhibitor of the receptor in human glioblastoma [19, 20]. The aptamer binds at high affinity (Kd: 9.6 nmol/l) to the extracellular domain of the PDGFRβ and impedes the ligand-dependent activation of the receptor both in vitro and in vivo. Further, it has been validated in vivo for its tumor targeting efficacy, resulting neither toxic nor immunogenic [19, 21].
Here we show that Gint4.T aptamer inhibits the migration of BM-MSCs towards TNBC cells and their trans-differentiation into CAFs. In addition, we used in vivo cell-tracking method to monitor instantly, noninvasively and in real time the inhibitory effect of the aptamer on BM-MSC homing to breast cancer xenografts, which ultimately results in interfering with BM-MSC-dependent pro-invasive activity. Our findings suggest a novel therapeutic agent that blocks either the recruitment of BM-MSCs into TME and the tumor cell-stroma interactions thus hampering TNBC aggressiveness.
Materials and Methods
Cell lines and culture condition
All human breast cancer cell lines came from American Type Culture Collection and were grown in appropriate medium supplemented with 10% fetal bovine serum (FBS), 100 U/ ml penicillin and 100 μg/ml streptomycin. The cells were maintained in a humidified incubator in 5% CO2 at 37 °C. MCF-7 and U87MG cells were grown in Dulbecco's modified Eagle's medium (DMEM); MDA-MB-231 and BT-549 cells were grown in RPMI 1640.
For production of MDA-MB-231 expressing green fluorescent protein (GFP), lentiviral constructs and infection of cells were conducted as previously described [22]. Briefly, MDA-MB-231 cells were plated on 12-well plates and infected with lentiviruses in the presence of 10% FBS and 8 μg/ml Polybrene (hexadimethrine bromide; Sigma-Aldrich, St. Louis, MO). After 48 hours of incubation, infection efficiency was determined by analyzing GFP expression by flow cytometry.
To obtain conditioned medium (CM) cells were grown in medium with 1% FBS for 48 hours. The medium was then collected, centrifuged at 1000 × g for 10 minutes, and filtered through 0.22-μm filters (Millipore,Billerica, MA) before being used.
The concentration of PDGF-BB in CM of MDA-MB-231 and BT-549 cells was measured using the ELISA method as reported [23].
Human BM-MSC isolation and culture
Human BM-MSCs were provided by Dr. Lucarelli. Samples were taken from patients after obtaining informed consent according to a protocol approved by the Ethics Committee of Rizzoli Orthopaedic Institute. Isolation and culture condition of human BM-MSC was performed as previously described [12]. BM-MSCs immunophenotypic characterization was performed by flow cytometry using antibodies against CD44, CD73, CD90, CD105, CD146 (staminal positive markers) and CD34 and CD45 (staminal negative markers). The BM-MSCs were used between passages 3-5.
BM-MSC differentiation into osteoblasts and adipocytes has been evaluated as reported [24].
BM-MSCs grown in the presence of MDA-MB-231 CM for 48 hours are referred as "BM-MSCMDA-MB 231".
RNA isolation and Real time-PCR
RNA isolation and Real time-PCR was performed as previously described [12]. Briefly, total RNA from BM-MSCs was extracted using TRIzol reagent (Invitrogen/Life Technologies, Carlsbad, CA, USA) and equal amount of total RNA (200 ng) was reverse-transcribed to cDNA using Superscript II RNase H-reverse transcriptase according to the manufacturer's instructions (Invitrogen/Life Technologies, Carlsbad, CA, USA). Quantitative PCR analysis was performed using SYBR Green reaction mixture (Bio-Rad, Hercules, CA), and an ABI Prism 7000 (robocycler was used for amplification Applied Biosystems, Carlsbad, CA, USA). The gene-specific primers used for the amplifications were as follows:
PDGFRβ
5'-AGGACACGCAGGAGGTCAT-3' (forward)
5'-TTCTGCCAAAGCATGATGAC-3' (reverse)
α-smooth muscle actin (α-SMA)
5'-ACCCAGCACCATGAAGATCA-3' (forward)
5'-AGAGACAGAGAGGAGCAGGA-3' (reverse)
Fibroblast-specific Protein 1 (FSP-1)
5'-CTCTCCTCAGCGCTTCTTCT-3' (forward)
5'-GGGTCAGCAGCTCCTTTAGT-3' (reverse)
Fibroblast Activation Protein (FAP)
5'-TACGTTTCATCACTGGCCCT-3' (forward)
5'-CATCTGCTGTTCCGTGGATG-3' (reverse)
β-actin
5'-CAAGAGATGGCCACGGCTGCT-3' (forward)
5'-TCCTTCTGCATCCTGTCGGCA-3' (reverse)
Sox2
5'-AGAAGGATAAGTACACGCTGC -3' (forward)
5'-TCCAGCCGTTCATGTGC -3' (reverse)
Nanog
5'-GAAATACCTCAGCCTCCAGC -3' (forward)
5'-GCGTCACACCATTGCTATTC -3' (reverse)
Aptamers and treatments
2′F-Py RNA aptamers (Gint4.T and Scrambled), developed by Camorani et al. [19], were synthesized by TriLink Biotechnologies and purchased from Tebu-bio srl (Magenta, Milan, Italy). Before each treatment, the aptamers were subjected to a short denaturation-renaturation step as reported [20].
Gint4.T aptamer, PDGFRβ inhibitor: 5'-UGUCGUGGGGCAUCGAGUAAAUGCAAUUCGACA-3'. Aptamer used as a negative control and herein indicated as Scr: 5'-UUCGUACCGGGUAGGUUGGCUUGCACAUAGAACGUGUCA-3'.
Cell lysate preparation and western blot analyses
Whole-cell lysates and Western blots analyses were performed as previously reported [25]. The following primary antibodies were used: rabbit polyclonal anti-phospho-PDGFRβ (CST-3173, indicated as pPDGFRβ), rabbit monoclonal anti-PDGFRβ (CST-3169), rabbit polyclonal anti-phospho-AKT (CST-9271, indicated as pAKT), rabbit polyclonal anti-AKT (CST-9272), rabbit polyclonal anti anti-phospho44/42 MAPK (CST-9101, indicated as pERK) (Cell Signaling Technology Inc., Danvers, MA); rabbit polyclonal anti-α-SMA (ab5694, Abcam, Cambridge, MA); mouse monoclonal anti-β-actin (A4700, Sigma-Aldrich, St. Louis, MO); rabbit polyclonal anti-Erk1 (sc-93) and goat polyclonal anti-vinculin (sc-7649) (Santa Cruz Biotechnology, Santa Cruz, CA). Detection of the targeted antibody was revealed by ECL reagent according to the manufacturer's recommendations. Densitometric analyses were performed on at least two different expositions to assure the linearity of each acquisition using ImageJ software (v1.46r). Blots shown are representative of at least three independent experiments.
Cell migration and invasion assays
Cell migration and cell invasion were performed using a 24-well Boyden chambers (Corning, NY) containing inserts of polycarbonate membranes with 8 μm pores [26]. For migration assays, all cells tested (0.3 x 105 in 100 μl serum-free medium per well) were plated into each upper chamber in the presence of different chemoattractants (lower chamber). After incubation for 24 hours at 37°C in a humidified incubator in 5% CO2, the non-migrating cells were removed from the top chamber using a cotton swab, and the cells that had migrated to the lower surface of the membrane insert were visualized by staining with 0.1% crystal violet in 25% methanol and counted in 10 random fields/filter at 200x magnification. Results are expressed as the percentage of migrating cells considering the untreated control sample as 100%.
To perform invasion assays, the top compartment of Boyden chambers was coated with 50 μl of diluted (1:3 in PBS) Matrigel (BD Biosciences, San Jose, CA). After coating, chambers were incubated at 37°C for 1 hour to allow Matrigel to solidify. MDA-MB-231 and BT-549 cells (0.5 × 105 in 100 μl serum-free medium per well) were then added to each top chamber. After incubation for 72 hours at 37°C in a humidified incubator in 5% CO2, invaded cells were visualized and analyzed as described above.
Analysis of BM-MSC trans-differentiation into CAF
BM-MSCs and TNBC cells were co-cultured in trans-well chambers with microporous-3µm membrane (Corning, NY). To this aim, BM-MSCs (1 x 106 cells) treated or no with Gint4.T and Scr were seeded into the lower chamber in a 6-multiwell. After incubation for 24 hours at 37°C in a humidified incubator in 5% CO2, MDA-MB-231 and BT-549 cells (50 x 105 cells) were added to each top chamber. After 5 days at 37°C in a humidified incubator in 5% CO2, total RNA was extracted from BM-MSCs and analyzed for CAF markers: α-SMA, FAP and FSP-1.
NIR fluorescent BM-MSC-labeling
VivoTrack 680 Fluorescent Cell Labeling Agent (MW: 1173 gmol-1; A: 676 nm; Em: 696 nm) was commercially obtained from PerkinElmer (Waltham, MA), dissolved in 1.3 mL of warm sterile PBS (final volume of 2.0 mL). BM-MSCs (1 x 106) were re-suspended in 200 µL of sterile PBS and incubated with VivoTrack 680 (ratio 1:1, v/v) for 30 minutes at 37°C under agitation in the dark. Afterwards, the suspension was washed three times with PBS containing 1% FBS to remove excess cell labeling agent. At that time, VivoTrack 680-labeled cell viability was assessed microscopically by Tripan-blue exclusion (GibcoTM). To verify that the cells were successfully labelled, they were examined by flow cytometry (BD Accuri™ C6), using appropriate lasers and filters for detection of 680 nm wavelength.
Animal Tumor Models and treatment
All experimental procedures complied with the European Communities Council directives (2010/63/EU) and national regulations (D.L. 116/92) and were performed in accordance with National Institutes of Health (NIH) recommendations. The present study was approved by the Italian Ministry of Health (authorization number 38/2015-01-28). MDA-MB-231 cells (10 × 106) were re-suspended in 0.1 ml of 1:1 mix of physiological saline and Matrigel and subcutaneously injected into the flank of five-week-old Female Balb/c nude mice which weighed around 18-20 g (Charles River, Milan, Italy). Once tumors became palpable (established), approximately 50 mm3 [volume = 0.5 × long diameter × (short diameter)2], nude mice were randomized into 2 groups (3 mice for each group) for imaging studies [27].
In order to develop a system that models the entire metastatic process, we injected MDA-MB-231/GFP cells (1×106) in presence or absence of BM-MSCs (1×106) in 150 µl of PBS Matrigel mixture (1:1, v:v) orthotopically into the mammary fat pads of severely immunocompromised NOD scid gamma (NSG) mice. BM-MSCs were treated with Gint4.T (1600 pmoles) for 3 hours prior injection. Mice (3 mice for each group) were monitored for development of primary xenograft tumor with high-frequency ultrasound system (Vevo 2100 equipment; FUJIFILM VisualSonics, Inc., Toronto, Ontario, Canada). Sixty days post injection, mice were euthanized and tumors and organs were removed. They were either stored in 10% neutral buffered formalin for paraffin embedding/sectioning and H&E staining, or they were embedded in O.C.T. (Optimal Cutting Temperature) compound and 10 µm frozen sections were incubated with DAPI for 5 minutes to stain nuclei blue and subsequently prepared for fluorescent microscopy of GFP.
In vivo tracking of BM-MSC to TNBC xenografts by Fluorescence Molecular Tomography (FMT)
For FMT studies, tumor-bearing mice were maintained on a diet with a purified, alfalfa-free rodent chow for 15 days before fluorescent imaging to minimize fluorescence in the gut. VivoTrack 680 alone or BM-MSCs (1 × 106) labeled with VivoTrack 680 and treated with Gint4.T or the Scr aptamer (1600 pmoles) for 3 hours were administered to MDA-MB-231 xenografts by caudal vein injection. Mice under isoflurane anesthesia were analyzed at different time points by FMT 4000 imaging system (PerkinElmer, Waltham, MA). Epifluorescence (2D) and tomography (3D) datasets were both acquired and analyzed by FMT system software (TrueQuantTM v4.0) from PerkinElmer (Waltham, MA). 3D regions of interest (ROIs) were drawn around tumor regions, and a threshold was applied equal to 30% of the maximum value of fluorescence in the adjoining non-tumor area. The total amount (pmoles) of fluorochrome was automatically calculated relative to internal standards generated with known concentrations of the appropriate dye. After imaging studies, mice were euthanized and tumors were excised and then harvested for FACS analysis of labeled BM-MSCs.
Statistical analysis
Results were obtained from at least three independent experiments and are expressed as the mean ±SD. Statistical values were defined using GraphPad Prism version 6.00 for Windows by t-test. P value < 0.05 was considered significant for all analyses.
Results
BM-MSCs promote TNBC cells migration and invasion
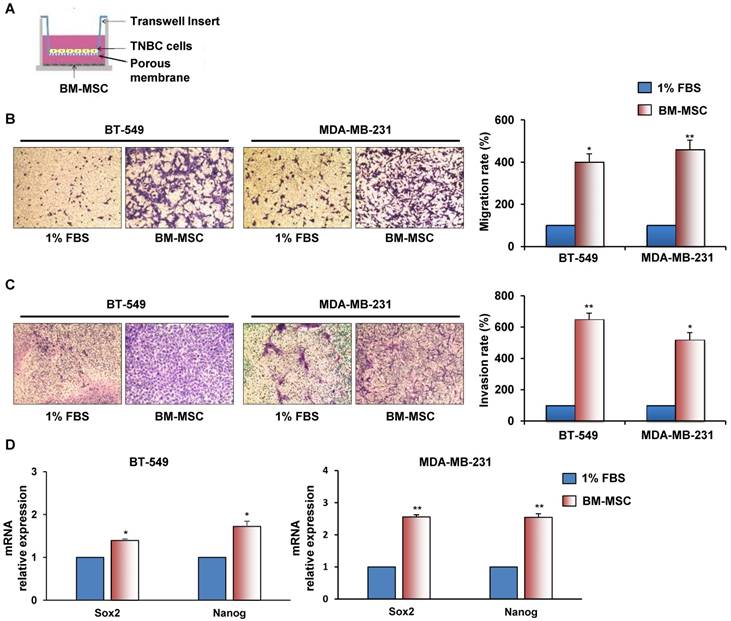
To investigate whether BM-MSCs have the ability to promote migration and invasion of ML MDA-MB-231 and BT-549 cells a trans-well assay was performed, in which each cancer cell line was exposed to BM-MSCs, added in the lower chamber (Figure 1A). We found that TNBC cells migrated much avidly when co-cultured with BM-MSCs for 24 hours (Figure 1B). Similarly, when TNBC cells were seeded on top compartment of trans-well coated with Matrigel a strong stimulation of their invasion was observed in the presence of BM-MSCs (Figure 1C). We also found increased expression of stem cell markers, such as Sox2 and Nanog in TNBC cells upon BM-MSCs co-culture for 5 days (Figure 1D).
BM-MSCs augment migration, invasion and stemness of TNBC cells. (A) Representation of BT-549 or MDA-MB-231 cells co-cultured with BM-MSCs using Boyden chambers. (B) Migration of BT-549 and MDA-MB-231 cells was analyzed by Boyden chamber containing inserts of polycarbonate membranes with 8 μm pores. TNBC cells were seeded into upper chamber and exposed to BM-MSCs grown in medium supplemented with 1% FBS or medium containing 1% FBS as a chemoattractant (lower chamber) for 24 hours. (C) Invasion of BT-549 and MDA-MB-231 cells was performed at 72 hours as described above with membranes of trans-well coated with Matrigel. Representative photographs of at least three different experiments are shown. The results are expressed as percent of migrated/invaded cells and cells vs 1% FBS are reported as 100%. (D) For stemness markers analysis BT-549 and MDA-MB-231 cells were co-cultured with BM-MSCs for 5 days using a Boyden chamber containing inserts of polycarbonate membranes with 3 μm pores. Nanog and Sox2 mRNA levels were analyzed by Real Time-PCR. All experiments were performed at least three times. *P < 0.01 **P < 0.001 compared to control (medium with 1% FBS).
The anti-PDGFRβ aptamer inhibits PDGFRβ-mediated signaling pathway in BM-MSCs
In agreement with previous reports [28], we found that BM-MSCs express high levels of PDGFRβ protein and mRNA (Figure 2A). Human PDGFRβ-positive glioblastoma U87MG and PDGFRβ-negative breast cancer MCF-7 cells [19] were used as positive and negative control, respectively, in western blotting (left panel) and Real Time-PCR (right panel) analyses.
Gint4.T aptamer inhibits PDGFRβ-mediated downstream signaling pathway in BM-MSCs. (A) Lysates from U87MG, MCF-7 and BM-MSCs were analyzed by immunoblot with anti-PDGFRβ antibody, and equal loading was confirmed by immunoblot with anti-actin antibody (left). PDGFRβ mRNA levels were analyzed by Real Time-PCR (right). (B) BM-MSCs cells were serum-starved for 18 hours and then left untreated or stimulated for 15 minutes with 100 ng/ml PDGF-BB in the absence or in the presence of 200 nmol/l Gint4.T or Scr, as indicated. Cell lysates were immunoblotted with anti-pPDGFRβ, anti-pERK, anti-pAkt antibodies. Filters were stripped and reprobed with anti-PDGFRβ, anti-Erk and anti-Akt antibodies, as indicated. Equal loading was confirmed by immunoblot with anti-vinculin antibody. The histogram indicates pPDGFRβ/PDGFRβ, pERK/ERK and pAKT/AKT ratio of densitometric signals, reported as relative to Scr-treated cells, arbitrarily set to 1. Depicted results represent one of three typical experiments performed. Molecular weights of indicated proteins are reported (A and B).
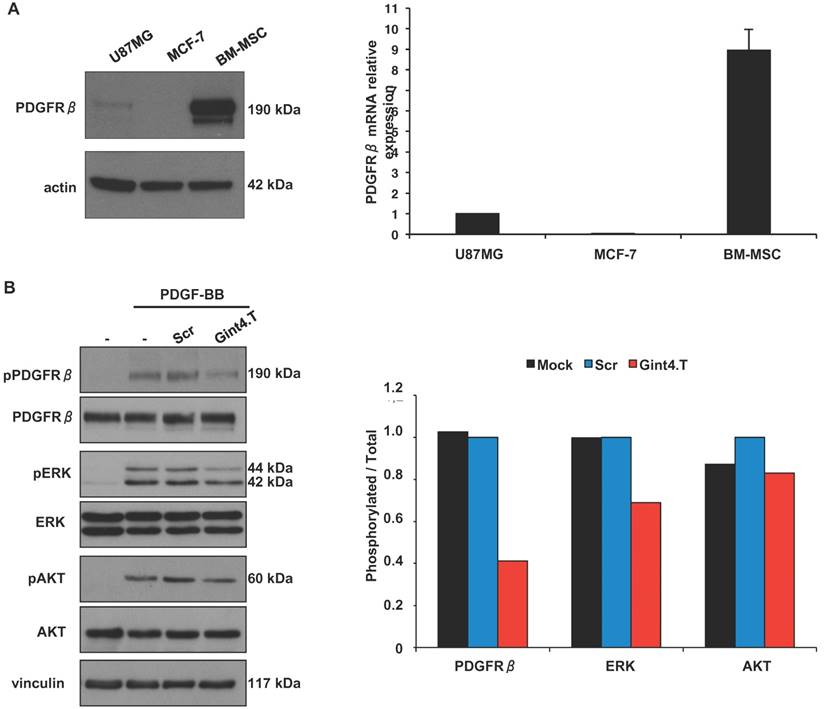
Since several evidences show a critical role for PDGFRβ in BM-MSCs homing to tumor sites including malignant glioma [29], chronic lymphocytic leukemia (CLL) [30] and colon carcinomas [6] we wondered whether the Gint4.T RNA aptamer, that we previously validated as a specific inhibitor of PDGFRβ [19, 20], is able to interfere with this phenomenon. The aptamer binds to the extracellular domain of human PDGFRβ thus interfering with PDGF-BB ligand stimulation of the receptor [19, 20]. Firstly, we verified the aptamer ability to inhibit ligand-dependent PDGFRβ activation and downstream phosphatidyl-3-kinase (PI3K)/AKT and extracellular signal-regulated kinase 1/2 (ERK1/2) pathways in BM-MSCs upon PDGF-BB stimulation (Figure 2B).
It has been reported that inhibition of PDGFRβ and downstream ERK and AKT signaling inhibited human MSC osteogenesis in vitro [31]. Accordingly, we found that the aptamer hampered BM-MSC differentiation into osteoblasts but not into adipocytes (Figure S1).
The anti-PDGFRβ aptamer inhibits BM-MSCs migration towards TNBC cells
Next, we determined whether the aptamer-mediated inhibition of intracellular signaling initiated by PDGFRβ results in impairment of BM-MSCs migration. As shown in Figure 3A, treating BM-MSCs with the anti-PDGFRβ aptamer strongly reduced cell migration stimulated by serum, causing 44% inhibition as compared with the Scr aptamer. A similar effect was elicited by imatinib mesylate, a tyrosine kinase inhibitor acting on different tyrosine protein kinases including PDGFRβ [32].
Gint4.T aptamer inhibits BM-MSC migration towards TNBC cells. Migration of BM-MSCs was analyzed by Boyden chamber. (A) BM-MSCs in the absence or in the presence of 400 nmol/l Gint4.T, 400 nmol/l Scr or 5µmol/l Imatinib mesylate were placed into upper chamber whereas 10% FBS was used as inducer of migration (lower chamber) for 24 hours. (B, C) BM-MSCs in the absence or in the presence of 400 nmol/l Gint4.T or Scr were placed into upper chamber whereas 100 ng/ml PDGF-BB (B), MDA-MB-231 (C, upper panels) and BT-549 cells (C, lower panels) were added to the lower chambers. Representative photographs of at least three different experiments are shown. The results are expressed as percent of migrated cells and reported as relative to mock-treated cells. *P < 0.01,**P < 0.001 compared to mock-treated cells; #P < 0.01, ##P < 0.001 compared to Scr aptamer.
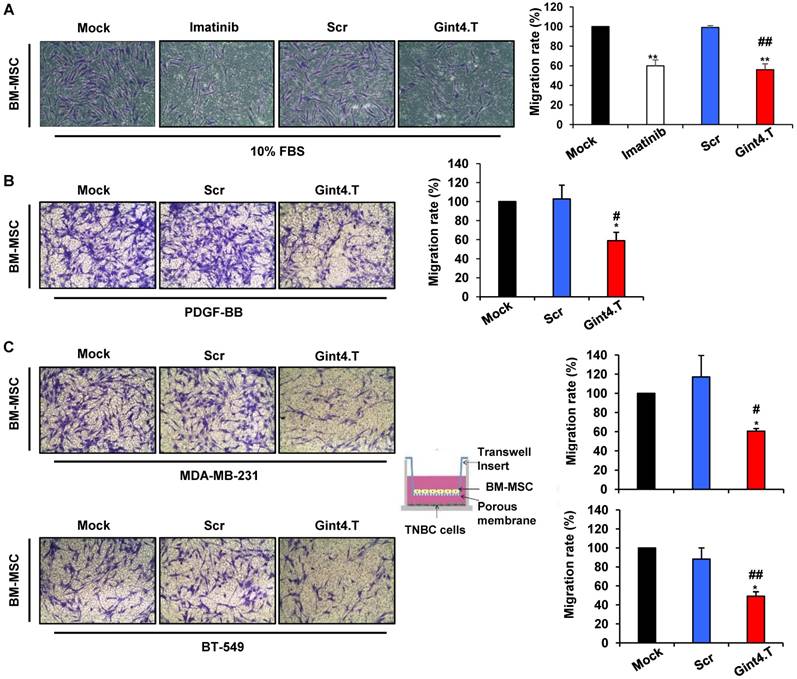
MSCs have been recently described to home into breast carcinoma in response to numerous endocrine and paracrine signals [2, 13, 14]. Importantly, the treatment of BM-MSCs with Gint4.T aptamer caused a dramatic reduction of their migration toward PDGF-BB ligand as compared with controls treatment (Mock and Scr) (Figure 3B). Next, we analyzed the secretion of PDGF-BB in conditioned medium of MDA-MB-231 and BT-549 by ELISA. The data showed that PDGF-BB detected in BT-549 was 12,84 ±1.52 pg/ml whereas in MDA-MB-231 was 6.46 ±1,46 pg/ml. Accordingly, Gint4.T strongly inhibited BM-MSCs migration to both TNBC cell lines (Figure 3C).
These results indicate that inhibiting PDGFRβ with Gint4.T hampers the homing of BM-MSCs to TNBC cell lines.
The anti-PDGFRβ aptamer inhibits BM-MSC trans-differentiation into Cancer Associated Fibroblasts
Because CAFs are well known to play a key role in tumor progression and metastatic spread we wondered whether BM-MSCs when co-cultured with TNBC cells acquire the expression of CAF-specific markers. Interestingly, a significant increase of mRNA levels of α-SMA, FAP and FSP-1 was observed when BM-MSCs were exposed up to 5 days to MDA-MB-231 and BT-549 cells, thus indicating that TNBC are able to induce BM-MSCs trans-differentiation into CAF-like cells (Figure 4A). Importantly, Gint4.T treatment strongly reduced CAF markers mRNA expression in BM-MSCs, co-cultured with MDA-MB-231 (Figure 4B, left) and BT-549 (Figure 4B, right) compared to Scr negative control, thus suggesting the aptamer ability to prevent BM-MSC differentiation into CAFs in TNBC microenvironment. Further, in order to assess the effect of the aptamer on the pro-invasive function of CAF-like BM-MSCs, BM-MSCs were grown for 48 hours in the presence of CM from MDA-MB-231 (BM-MSCMDA-MB 231) to induce a CAF-like phenotype (Figure 4C). Next, MDA-MB-231 cells invasiveness was assessed by using CM from naïve BM-MSCs and from BM-MSCMDA-MB-231 as chemoattractant in the transwell invasion assay. As shown (Figure 4D), CM from BM-MSCMDA-MB-231 strongly increased the invasion rate of MDA-MB-231 cells compared to CM from naïve BM-MSCs and, importantly, Gint4.T interfered with this event.
Gint4.T inhibits BM-MSC trans-differentiation into Cancer Associated Fibroblasts. (A) BM-MSCs were co-cultured with MDA-MB 231 or BT-549 cells for 5 days using a Boyden chamber containing inserts of polycarbonate membranes with 3 μm pores. α-SMA, FAP and FSP-1 mRNA levels were detected using Real Time-PCR. *P < 0.01,**P < 0.001 compared to BM-MSCs. (B) BM-MSCs incubated with 400 nmol/l Gint4.T or Scr were co-cultured with MDA-MB 231 or BT-549 cells as described above. α-SMA, FAP and FSP-1 mRNA levels were quantified using Real Time-PCR and reported as relative to Scr-treated cells, used as negative control. #P < 0.01, ##P < 0.001 compared to Scr aptamer. (C) Lysates from BM-MSCs grown for 48 hours in medium supplemented with 1% FBS or in CM from MDA-MB-231 (BM-MSCMDA-MB 231) in the absence or in the presence of 400 nmol/l Gint4.T, were immunoblotted with anti-α-SMA and anti-vinculin antibodies. Values below the blot indicate signal levels relative to BM-MSCs grown in 1% FBS medium, arbitrarily set to 1 (labeled with asterisk). Molecular weights of indicated proteins are reported. (D) MDA-MB-231 cells were placed into upper matrigel-coated compartment of Boyden chamber and exposed to conditioned media from BM-MSCs grown as in (C) as chemoattractants (lower chamber) for 24 hours. Representative photographs of at least three different experiments are shown. The results are expressed as percent of invaded cells and reported as relative to BM-MSC CM.
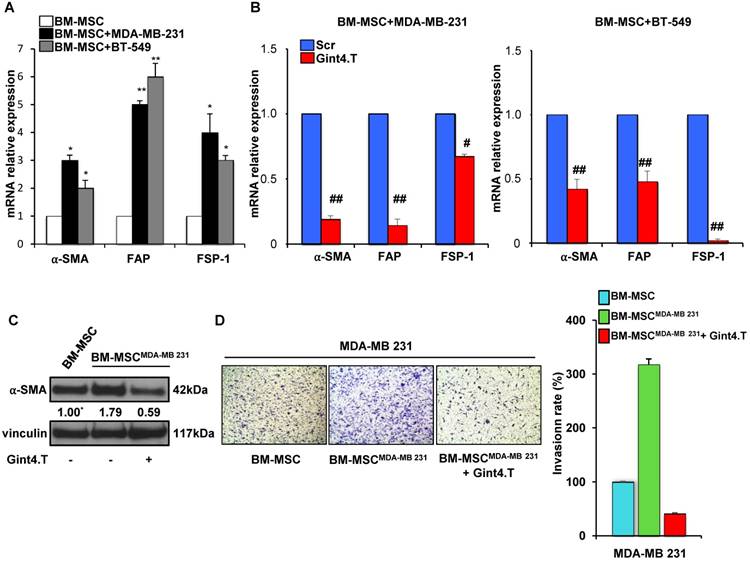
These results indicate that blocking PDGFRβ on BM-MSCs with Gint4.T aptamer not only decreased their ability to migrate towards TNBC cells but also their capability to trans-differentiate in CAFs and to induce a pro-invasive phenotype of TNBC cells.
The anti-PDGFRβ aptamer inhibits BM-MSC recruitment into TNBC xenografts as assessed by Fluorescence Molecular Tomography
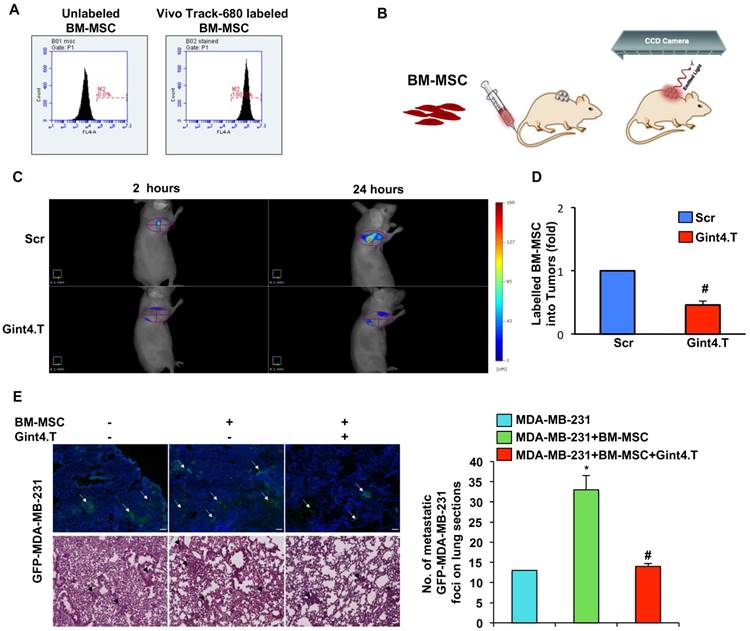
To follow the homing of BM-MSCs toward TNBC in vivo, we labeled the cells with VivoTrack 680 Fluorescent Cell Labeling Agent. As assessed by flow cytometry analysis, a total labeling efficiency was obtained at the experimental condition used (Figure 5A). First, we checked the absence of cell viability loss after labeling (data not shown). Then, nude mice bearing MDA-MB-231 xenografts, were intravenously injected with VivoTrack 680-labeled BM-MSCs, treated with Gint4.T or the Scr aptamer, and analyzed by Fluorescence Molecular Tomography at different time-points (Figure 5B). 3D images were reconstructed and volumes of interest (VOIs) were generated around the tumor. The fluorescence signal of BM-MSCs was visualized in the tumor at 2 hours post-injection and remained visualized up to 24 hours (Figure 5C). Because VivoTrack 680 emits at NIR wavelength where autofluorescence and tissue absorbance are minimal, a low fluorescence was observed in the tissues around the tumor. As shown, the treatment of BM-MSCs with Gint4.T aptamer strongly reduced BM-MSC recruitment into MDA-MB-231 xenografts compared to the negative control (Figure 5C). Moreover, FACS analysis of NIR labeled BM-MSCs in cell suspension from tumors, revealed a significant decrease of BM-MSCs recruitment in tumors after their treatment with Gint4.T aptamer (Figure 5D), thus confirming the results of in vivo imaging. These findings prove the ability of the aptamer to inhibit tropism of BM-MSCs to TNBC targeting PDGFRβ.
Representative images of BM-MSC recruitment into TNBC xenografts by fluorescence tomography and BM-MSCs pro-metastatic activity. (A) BM-MSCs were incubated with VivoTrack 680 for 30 minutes and the dye uptake was analyzed by flow cytometry. (B-C) Nude mice bearing MDA-MB-231 xenografts were i.v. injected with VivoTrack 680-labeled BM-MSCs treated with Gint4.T or Scr aptamer and analyzed with FMT. (D) FACS analysis of NIR labeled BM-MSCs in cell suspension from tumors (#P < 0.01, compared to Scr aptamer). (E) Representative fluorescence (upper panels) and H&E (lower panels) images of lung sections from mice injected with MDA-MB-231/GFP cells alone or mixed with BM-MSCs, left untreated or treated with Gint4.T for 3 hours prior to injection. Multifocal metastatic lesions expressing GFP or H&E-stained are indicated by white and black arrows, respectively. Nuclei were stained with DAPI. All images had a magnification of 5.41x from the initial slide scan. Scale bar = 100 μm. Metastatic foci in lung sections were counted on at least 5 random fields per section (n = 3 mice, *P < 0.01 compared to MDA-MB-231/GFP; #P < 0.01 compared to MDA-MB-231/GFP +BM-MSCs).
The anti-PDGFRβ aptamer inhibits BM-MSC induced TNBC metastasis in vivo
In agreement with previous report [2] we found that the growth of MDA-MB-231 cells is not affected when breast cancer cells were co-injected with BM-MSCs in mammary fat pad of immunocompromised mice (Figure S2). Conversely, BM-MSCs increased the metastatic potential of MDA-MB-231 cells in orthotopic model [2] thus correlating with our in vitro findings. To investigate whether the anti-PDGFRβ aptamer has the ability to inhibit BM-MSC induced TNBC metastasis in vivo, GFP-labelled MDA-MB-231 were mixed with BM-MSCs, left untreated or prior treated with Gint4.T, and injected in mammary fat pad of mice. After 8 weeks, mice were sacrificed and the lungs were extracted for analysis. Although no visible macro-metastatic lesions were observed in each experimental mouse group, GFP fluorescence was evident in the lung sections demonstrating microscopic metastasis. As shown (Figure 5E), BM-MSCs induced a significant enhancement in the number of detectable multifocal metastatic lesions that was counteracted by Gint4.T treatment.
Discussion
Since the tumor microenvironment contributes to many aspects of carcinogenesis and cancer progression, the different stromal cell types, which play key roles in determining or enhancing several hallmark capabilities in different TMEs, should be taken into account to stimulate future anticancer drug development. In particular, the recent discovery that MSCs can be recruited into TME and promote cancer progression has led to consider MSCs as a suitable target for anti-cancer therapy. Importantly, tumor associated-MSCs contribute to tumor cell growth and metastatic behavior in a variety of cancers, including breast [2], prostate [5, 33], osteosarcoma [12, 26, 34] and colon [6] cancers. The high tropism of MSCs for malignant tumors is largely similar to what observed for inflamed and injured tissues and involve the same growth factors, chemokines and cytokines [8]. Tumor cells are able to produce a chemotactic gradient consisting of different chemokines such as CCL2, CCL5, CXCL12 and CXCL16, which is able to recruit MSCs [35].Jung et al. (2013) showed that MSCs were both recruited as well as transformed in CAFs by CXCL16 produced by prostate cancer cells. In turn, MSC-like CAFs secreted CXCL12 that binding to CXCR4 on tumor cells induced an EMT, which ultimately promoted metastasis to secondary tumor sites [5].
Many growth factors such as PDGF, vascular endothelial growth factor (VEGF), insulin-like growth factor 1 (IGF-1), TGF-β and basic fibroblast growth factor (bFGF) have been found to effectively mediate MSC homing to TME [36].Interestingly, several studies reported PDGF-BB as a critical mediator of MSC tropism to gliomas [37-39]. It has been reported that recruitment of MSCs towards pancreatic tumors in response to PDGF, EGF and VEGF, can be inhibited by Glivec, Erbitux and Avastin, respectively [40]. Furthermore, it has been demonstrated that bFGF and downstream ERK/Smad3 signaling pathway are involved in BM-MSC tropism to 4T1 breast cancer cells by using a specific neutralizing antibody [41].
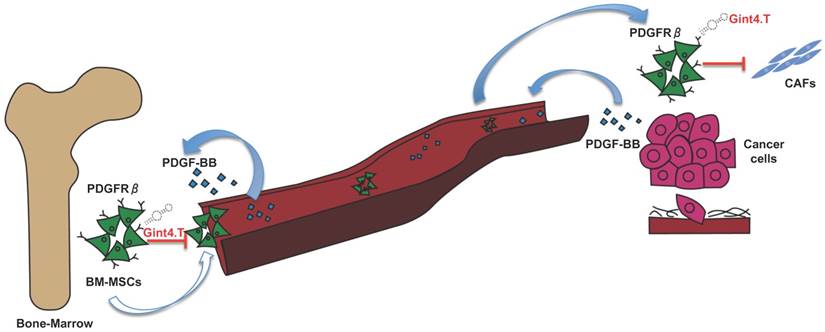
An increasing number of evidence has shown that MSCs in primary tumors can differentiate in active components of the tumor microenvironment such as CAFs, which are known to play a key role in tumor progression [42], neo-angiogenesis [43], EMT [44] and metastatic spread [45]. Therefore, great interest has been shown in developing therapeutic targeting strategies aimed to inhibit MCS support thus improving therapeutic efficacy in treating cancer [46, 47]. Thus, in this scenario, the starting point for MSC targeted therapy is the identification of specific molecular pathways involved in pro-tumorigenic cross-talk between MSCs and cancer cells. The delineation of key molecular pathways that underlie this important interplay has improved the knowledge of the biology of TME, tumor spreading, and carcinogenesis. Over past years, several studies have shown that MSCs express high levels of PDGFRβ [28] suggesting that it could play a crucial role in processes in which MSCs are involved. In fact, it is widely reported that PDGFRβ is involved in specifying MSC commitment to mesenchymal lineages as well as in MSC proliferation and self-renewal [48]. Furthermore, PDGFRβ signaling has emerged as a predominant pathway in recruitment of MSCs towards tumor sites [15]. Hata et al. [29], for example, have demonstrated that PDGFRβ is involved in MSC tropism for malignant gliomas, providing direct evidence that tumor is capable of attracting MSCs in an intracranial glioma model through PDGFRβ pathway. Furthermore, it has been reported that PDGFRβ-mediated MSC recruitment into TME, contributed to enhance the aggressive and invasive properties of many other tumors, including chronic lymphocytic leukemia [30] and colon carcinomas [6]. However, although PDGFRβ inhibitors have already been tested for their capability to target and interfere with receptor activity in MSCs, no studies have still demonstrated the possibility to target PDGFRβ to prevent pro-tumorigenic effect of MSCs into tumor microenvironment. Moreover, the current class of PDGFRβ drugs consists of small molecule TKIs that show limited specificity and modest efficacy whereas no antibodies have entered the clinic. This study reports, for the first time, an innovative therapeutic tool, an anti-PDGFRβ aptamer [19, 20] able to prevent BM-MSC recruitment and activity into TNBC microenvironment. Recent evidences have widely shown that BM-MSCs can integrate into the tumor-associated stroma and promote the progression of TNBC through the activation of different mechanisms involving HIFs [14], TGF-β [13] and CCL5 [2]. Up to now, no work has demonstrated the involvement of PDGFRβ pathway in MSC homing toward TNBC microenvironment, as well as MSC-mediated TNBC progression. Interestingly, in this study we show that the anti-PDGFRβ aptamer strongly reduces both in vitro BM-MSC migration stimulated by two different TNBC cell lines and in vivo BM-MSC recruitment into TNBC xenografts, as assessed by FMT. These results underline the importance of PDGFRβ in mediating MSC migration towards TNBC site and highlights Gint4.T aptamer to specifically prevent PDGFRβ-mediated MSC tropism for TME. Finally, recent evidence has demonstrated that MSCs selectively proliferate in TME and trans-differentiate in pro-tumor supporting CAFs [10]. Although the effect of MSCs on growth of breast cancer is controversial and probably depends on the experimental setting of the animal model [49], conversely, it is well established that MSCs have a significant impact on promoting breast cancer metastasis [50]. Our data demonstrated that Gint4.T aptamer not only hampers PDGFRβ downstream signaling in BM-MSCs, but it also interferes with their recruitment into TNBC and with their differentiation into CAF thus preventing metastatic lung lesions (Figure 6).
Schematic illustration of Gint4.T effect on recruitment and activity of BM-MSCs into triple negative breast cancer microenvironment. Gint4.T targeting PDGFRβ inhibits BM-MSCs homing to cancer cells and their trans-differentiation into cancer associated fibroblasts (CAFs) as well as BM-MSC pro-metastatic function.
Conclusions
Tumor microenvironment recruits MSCs from the circulation, adjacent tissues and bone-marrow thus educating these cells to adopt pro-tumorigenic phenotypes. MSCs in tumor cause a formation of an immunosuppressive microenvironment and can also enhance the proportions of cancer stem cells, promote EMT, and stimulate tumor angiogenesis. These effects contribute to tumor growth, progression, and metastasis. Our aptamer-based approach to interrupt the cross-talk between cancer cells, MSCs and the inflammatory tumor microenvironment will likely provide new anti-cancer opportunities.
Abbreviations
α-SMA: α-smooth muscle actin
bFGF: basic fibroblast growth factor
BM-MSCs: bone marrow-derived mesenchymal stem cells
CAF: carcinoma-associated fibroblast
CCL5: chemokine C-C motif ligand 5
CLL: chronic lymphocytic leukemia
CXCL10: C-X-C motif chemokine 10
CXCR4: chemokine receptor type 4
DMEM: Dulbecco's modified Eagle's medium
EMT: epithelial-mesenchymal transition
ERK 1/2: extracellular signal-regulated kinase 1/2
FAP: fibroblast activation protein
FBS: fetal bovine serum
FMT: Fluorescence Molecular Tomography
FSP-1: Fibroblast-specific Protein 1
ML: mesenchymal-like
MSCs: mesenchymal stem cells
PDGF-BB: platelet-derived growth factor BB
PDGFRβ: platelet-derived growth factor receptor β
PI3K: phosphatidyl-3-kinase
ROIs: regions of interest
TAMs: tumor-associated macrophages
TME: tumor microenvironment
TNBC: triple-negative breast cancer
TGF-β: transforming growth factor β
VOIs: volumes of interest
Acknowledgements
We are grateful to Maria Napolitano for FACS analysis; Pietro Formisano and Vittoria Esposito for ELISA analysis; Mr Massimiliano Sorbillo and Mrs Sara Ugramin for their technical support. We thank Monica Fedele and Elvira Crescenzi for helpful discussions. This work was supported by grant from Associazione Italiana per la Ricerca sul Cancro (AIRC) IG 18753 (L.C.).
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Melzer C, Yang Y, Hass R. Interaction of MSC with tumor cells. Cell Commun Signal. 2016;14:20
2. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557-63
3. Bergfeld SA, DeClerck YA. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010;29:249-61
4. Suzuki K, Sun R, Origuchi M, Kanehira M, Takahata T, Itoh J. et al. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol. Med. 2011;17:579-87
5. Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J. et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013;4:1795
6. Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Kodama M, Higashi Y. et al. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int. J. Cancer. 2010;127:2323-33
7. Dasari VR, Velpula KK, Kaur K, Fassett D, Klopfenstein JD, Dinh DH. et al. Cord blood stem cell-mediated induction of apoptosis in glioma downregulates X-linked inhibitor of apoptosis protein (XIAP). PLoS One. 2010;5:e11813
8. Yagi H, Kitagawa Y. The role of mesenchymal stem cells in cancer development. Front Genet. 2013;4:26
9. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309-22
10. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18:84
11. Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73:4965-77
12. Fontanella R, Pelagalli A, Nardelli A, D'Alterio C, Ieranò C, Cerchia L. et al. A novel antagonist of CXCR4 prevents bone marrow-derived mesenchymal stem cell-mediated osteosarcoma and hepatocellular carcinoma cell migration and invasion. Cancer Lett. 2016;370:100-7
13. Goldstein RH, Reagan MR, Anderson K, Kaplan DL, Rosenblatt M. Human bone marrow-derived MSCs can home to orthotopic breast cancer tumors and promote bone metastasis. Cancer Res. 2010;70:10044-50
14. Chaturvedi P, Gilkes DM, Wong CC, Kshitiz W, Luo W, Zhang H. et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J. Clin. Invest. 2013;123:189-205
15. Veevers-Lowe J, Ball SG, Shuttleworth A, Kielty CM. Mesenchymal stem cell migration is regulated by fibronectin through α5β1-integrin-mediated activation of PDGFR-β and potentiation of growth factor signals. J Cell Sci. 2011;124:1288-300
16. Dhawan A, von Bonin M, Bray LJ, Freudenberg U, Pishali Bejestani E, Werner C. et al. Functional Interference in the Bone Marrow Microenvironment by Disseminated Breast Cancer Cells. Stem Cells. 2016;34:2224-35
17. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;12:2750-67
18. Camorani S, Crescenzi E, Gramanzini M, Fedele M, Zannetti A, Cerchia L. Aptamer-mediated impairment of EGFR-integrin αvβ3 complex inhibits vasculogenic mimicry and growth of triple-negative breast cancers. Sci Rep. 2017;7:46659
19. Camorani S, Esposito CL, Rienzo A, Catuogno S, Iaboni M, Condorelli G. et al. Inhibition of receptor signaling and of glioblastoma-derived tumor growth by a novel PDGFRβ aptamer. Mol Ther. 2014;22:828-41
20. Camorani S, Crescenzi E, Colecchia D, Carpentieri A, Amoresano A, Fedele M. et al. Aptamer targeting EGFRvIII mutant hampers its constitutive autophosphorylation and affects migration, invasion and proliferation of glioblastoma cells. Oncotarget. 2015;6:37570-8
21. Monaco I, Camorani S, Colecchia D, Locatelli E, Calandro P, Oudin A. et al. Aptamer Functionalization of Nanosystems for Glioblastoma Targeting through the Blood-Brain Barrier. J Med Chem. 2017;60:4510-16
22. Crescenzi E, Raia Z, Pacifico F, Mellone S, Moscato F, Palumbo G. et al. Down-regulation of wild-type p53-induced phosphatase 1 (Wip1) plays a critical role in regulating several p53-dependent functions in premature senescent tumor cells. J Biol Chem. 2013;288:16212-24
23. Esposito V, Perna A, Lucariello A, Carleo MA, Viglietti R, Sangiovanni V. et al. Different Impact of Antiretroviral Drugs On Bone Differentiation In An In Vitro Model. J Cell Biochem. 2015;116:2188-94
24. Pierini M, Lucarelli E, Duchi S, Prosperi S, Preve E, Piccinini M. et al. Characterization and cytocompatibility of a new injectable multiphasic bone substitute based on a combination of polysaccharaide gel-coated OSPROLIFE(®) HA/TTCP granules and bone marrow concentrate. Biomed Mater Res B Appl Biomater. 2016;104:894-902
25. Zannetti A, Iommelli F, Fonti R, Papaccioli A, Sommella J, Lettieri A. et al. Gefitinib induction of in vivo detectable signals by Bcl-2/Bcl-xL modulation of inositol trisphosphate receptor type 3. Clin Cancer Res. 2008;14:5209-19
26. Pelagalli A, Nardelli A, Fontanella R, Zannetti A. Inhibition of AQP1 Hampers Osteosarcoma and Hepatocellular Carcinoma Progression Mediated by Bone Marrow-Derived Mesenchymal Stem Cells. Int J Mol Sci. 2016:17 pii: E110
27. Zannetti A, Iommelli F, Speranza A, Salvatore M, Del Vecchio S. 3′-deoxy-3′-18F-fluorothymidine PET/CT to guide therapy with epidermal growth factor receptor antagonists and Bcl-xL inhibitors in non-small cell lung cancer. J Nucl Med. 2012;53:443-50
28. Spitzer TL, Rojas A, Zelenko Z, Aghajanova L, Erikson DW, Barragan F. et al. Perivascular human endometrial mesenchymal stem cells express pathways relevant to self-renewal, lineage specification, and functional phenotype. Biol Reprod. 2012;86:58
29. Hata N, Shinojima N, Gumin J, Yong R, Marini F, Andreeff M. et al. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery. 2010;66:144-56
30. Ding W, Knox TR, Tschumper RC, Wu W, Schwager SM, Boysen JC. et al. Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood. 2010;116:2984-93
31. Fierro F, Illmer T, Jing D, Schleyer E, Ehninger G, Boxberger S. et al. Inhibition of platelet-derived growth factor receptorbeta by imatinib mesylate suppresses proliferation and alters differentiation of human mesenchymal stem cells in vitro. Cell Prolif. 2007;40:355-66
32. Roberts WG, Whalen PM, Soderstrom E, Moraski G, Lyssikatos JP, Wang HF. et al. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res. 2005;65:957-66
33. Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V. et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-β1. Stem Cells. 2016;34:2536-47
34. Tu B, Du L, Fan QM, Tang Z, Tang TT. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer Lett. 2012;325:80-8
35. Shi Y, Du L, Lin L, Wang Y. Tumor-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017;16:35-52
36. Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730-8
37. Cheng P, Gao ZQ, Liu YH, Xue YX. Platelet-derived growth factor BB promotes the migration of bone marrow-derived mesenchymal stem cells towards C6 glioma and up-regulates the expression of intracellular adhesion molecule-1. Neurosci Lett. 2009;451:52-6
38. Hata N, Shinojima N, Gumin J, Yong R, Marini F, Andreeff M. et al. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery. 2010;66:144-56
39. Doucette T, Rao G, Yang Y, Gumin J, Shinojima N, Bekele BN. et al. Mesenchymal stem cells display tumor-specific tropism in an RCAS/Ntv-a glioma model. Neoplasia. 2011;13:716-25
40. Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J. et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. 2008;99:622-31
41. Yang X, Hao J, Mao Y, Cao R, Liu XH, Ding XL. et al. BFGF promotes migration and induces cancer-associated fibroblast differentiation of mouse bone mesenchymal stem cells to promote tumor growth. Stem Cells Dev. 2016;25:1629-1629
42. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392-401
43. Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012;31:195-208
44. Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L. et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945-56
45. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G. et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33:2423-31
46. Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z. et al. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells. 2012;30:2810-9
47. Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Onoyama M, Ohnishi M. et al. Stroma-directed imatinib therapy impairs the tumor-promoting effect of bone marrow-derived mesenchymal stem cells in an orthotopic transplantation model of colon cancer. Int J Cancer. 2013;132:813-23
48. Gharibi B, Ghuman MS, Hughes FJ. Akt- and Erk-mediated regulation of proliferation and differentiation during PDGFRβ-induced MSC self-renewal. J Cell Mol Med. 2012;16:2789-801
49. Wang H, Cao F, De A, Cao Y, Contag C, Gambhir SS. et al. Trafficking mesenchymal stem cell engraftment and differentiation in tumor-bearing mice by bioluminescence imaging. Stem Cells. 2009;27:1548-58
50. Rowan BG, Gimble JM, Sheng M, Anbalagan M, Jones RK, Frazier TP. et al. Human adipose tissue-derived stromal/stem cells promote migration and early metastasis of triple negative breast cancer xenografts. PLoS One. 2014;9:e89595
Author contact
![]() Corresponding author: Zannetti Antonella, Istituto di Biostrutture e Bioimmagini-CNR, Via T. De Amicis, 95 80145 Naples, Italy; Tel.:+390812203431; E-mail: antonella.zannettiit or Laura Cerchia, Istituto di Endocrinologia e Oncologia Sperimentale "G.Salvatore", CNR, Via S. Pansini 5, 80131 Naples, Italy; Tel. +390815455751; E-mail: cerchiait
Corresponding author: Zannetti Antonella, Istituto di Biostrutture e Bioimmagini-CNR, Via T. De Amicis, 95 80145 Naples, Italy; Tel.:+390812203431; E-mail: antonella.zannettiit or Laura Cerchia, Istituto di Endocrinologia e Oncologia Sperimentale "G.Salvatore", CNR, Via S. Pansini 5, 80131 Naples, Italy; Tel. +390815455751; E-mail: cerchiait