Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Principles and instrumentation
Application of MS in cancer...
Challenges in biomarker...
Conclusion
Acknowledgements
References
Introduction
Principles and instrumentation
Application of MS in cancer...
Challenges in biomarker...
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3559-3572. doi:10.7150/thno.20797 This issue Cite
Review
Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: approaches and application
Rongrong Huang1, Zhongsi Chen1, Lei He1, Nongyue He1,4 ![]() , Zhijiang Xi2, Zhiyang Li1,3, Yan Deng1,4
, Zhijiang Xi2, Zhiyang Li1,3, Yan Deng1,4 ![]() , Xin Zeng5
, Xin Zeng5 ![]()
1. State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, China;
2. School of Medicine, Yangtze University, Jingzhou 434023, China;
3. Department of Clinical Laboratory, the Affiliated Drum Tower Hospital of Nanjing University Medical School, Nanjing 210008, China;
4. Economical Forest Cultivation and Utilization of 2011 Collaborative Innovation Center in Hunan Province, Hunan Key Laboratory of Green Chemistry and Application of Biological Nanotechnology; Hunan University of Technology, Zhuzhou 412007, China;
5. Nanjing Maternity and Child Health Medical Institute, Obstetrics and Gynecology Hospital Affiliated to Nanjing Medical University, Nanjing 210004, China
Received 2017-4-29; Accepted 2017-7-12; Published 2017-8-18
Citation:
Huang R, Chen Z, He L, He N, Xi Z, Li Z, Deng Y, Zeng X. Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: approaches and application. Theranostics 2017; 7(14):3559-3572. doi:10.7150/thno.20797. https://www.thno.org/v07p3559.htm
Other stylesAbstract

There is a critical need for the discovery of novel biomarkers for early detection and targeted therapy of cancer, a major cause of deaths worldwide. In this respect, proteomic technologies, such as mass spectrometry (MS), enable the identification of pathologically significant proteins in various types of samples. MS is capable of high-throughput profiling of complex biological samples including blood, tissues, urine, milk, and cells. MS-assisted proteomics has contributed to the development of cancer biomarkers that may form the foundation for new clinical tests. It can also aid in elucidating the molecular mechanisms underlying cancer. In this review, we discuss MS principles and instrumentation as well as approaches in MS-based proteomics, which have been employed in the development of potential biomarkers. Furthermore, the challenges in validation of MS biomarkers for their use in clinical practice are also reviewed.
Keywords: mass spectrometry, proteomics, cancer biomarkers
Introduction
Cancer remains a major life-threatening disease with about 14.1 million new cases and 8.2 million cancer-associated mortalities reported in 2012 [1]. The global demographic and epidemiologic transitions signal an ever-increasing cancer burden over the next decades [2]. Cancer is a multigene disease and each tumor is composed of a variety of cell populations with distinct morphologies and behaviors [3]. Biomarkers such as proteins or biomolecular chemical modifications are quantifiable indicators of a specific biological state. In this respect, cancer-associated biomarkers are useful for studying disease, identifying patients at different clinical stages, and developing adaptive therapies [4]. For example, recent studies have demonstrated that long noncoding RNAs, circular RNAs [5], circulating tumor DNAs [6], and non-essential amino acids that support numerous metabolic processes crucial for the growth and survival of proliferating cells [7] can serve as biomarkers for cancers. Also, epidermal growth factor receptor, which is associated with the development of certain types of cancers [8], is regarded as a useful tool for cancer detection (Figure 1).
Cancer biomarkers can be classified into two categories including disease-related biomarkers and drug related biomarkers [9]. A biomarker should be (i) a mediator of the disease pathology, (ii) present at low and stable expression levels in healthy individuals and higher expression levels in patients, and (iii) simple and quick to evaluate [10]. Such a biomarker can be assayed and linked to cancer using a defined mechanism [11].
Figure 1
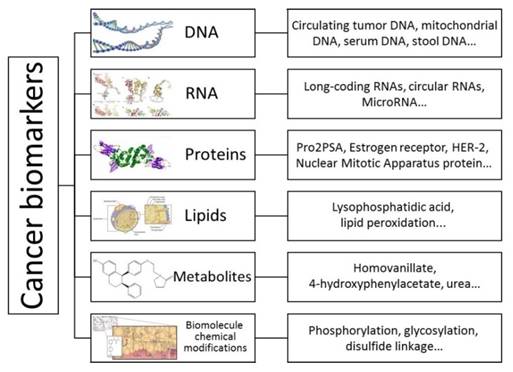
Schematic illustration of biomarkers for various types of cancers. Biomarkers are quantitative indicators of a specific biological state; therefore, cancer-associated biomarkers are useful for understanding the molecular basis of disease, early detection, identifying patients at different clinical stages, and developing a personal therapy.
Figure 2
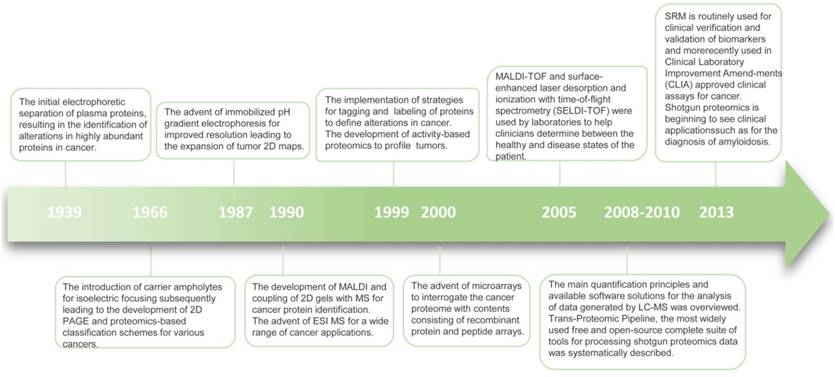
Timeline of progress in proteomics.
Recently, advanced molecular methods have been used in clinical diagnostic laboratories. Most novel techniques are based on transcriptional profiling and DNA methylation. However, compared with the genome and transcriptome, the proteome is more complex and dynamic [12]. The term “proteome” was first used in 1994 to indicate all time- and condition-specific proteins that are simultaneously produced by a cell or a tissue [12]. Proteins are often subject to proteolytic cleavage or post-translational modifications. Although genomics and transcriptomics can provide valuable information, they do not always reflect the variation of encoded proteins. Also, the association between mRNAs and protein expression levels is low compared with that of cell surface proteins [13]. Since proteins are the functional molecules in an organism and may be most ubiquitously affected in disease, therapy response, and recovery, proteomics holds special promise in detecting pathological conditions, predicting the efficacy of treatment, and tailoring personalized medicine (Figure 2) [14].
In a typical clinical proteomic study for diagnostic biomarker discovery, measurement of a large number of proteins in various samples is the first step. The initial protein candidates are proteins that are differentially expressed in patient and control samples [15]. By confirmation of differential protein abundance in clinically useful samples, candidates can be progressively credentialed to yield a few specific proteins [15]. Candidate biomarker verification should be included in the biomarker development pipeline (Figure 3) to provide reproducible and sensitive quantitative assays [16].
Because of the limited availability and accessibility of suitable reagents, most proteins in a species cannot be detected and quantified by affinity-based assays [17]. Therefore, almost all currently available proteomic procedures and strategies use mass spectrometry (MS) techniques, which are capable of high-throughput profiling of complex samples. Nowadays, non-targeted MS methods have emerged as suitable tools to perform relative quantitation of a large number of proteins to discover novel protein biomarker candidates while targeted MS mode are applied to identify peptides of interest [18, 19]. A variety of MS-based proteomic methods have been developed to identify and quantify proteins in biological and clinical samples [20-23] to obtain biomarker candidates. The present study describes various currently used MS-based proteomic approaches and their applications. Also, the challenges of biomarker validation for their use in clinical practice are discussed.
Principles and instrumentation
MS analysis utilizes electromagnetic fields in a vacuum, where the molecular mass of the charged particle is determined [3]. MS is used to evaluate the molecular mass of a polypeptide or to determine additional structural features [17]. Tandem MS/MS is performed in the latter case to determine detailed structural features of peptides. Moreover, MS-based proteomic methods can also be applied to characterize protein complexes [22]. For example, protein conformation in solution and structural characterization of therapeutic proteins can be studied by hydrogen/deuterium exchange mass spectrometry (HDX-MS) [23].
MS instrumentation
In general, during MS analysis, the analyte is ionized in the gas phase, and the ions are subsequently separated according to their mass-to-charge ratio (m/z). Electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) are two methods widely used to perform the protein ionization. Both techniques hold great potential for the characterization of biomolecules.
A mass analyzer is an instrument that determines the m/z of ions and the number of ions corresponding to a particular m/z is recorded by a detector. Quadrupole (QD), ion trap (IT), time-of-flight (TOF), orbitrap, and Fourier transform ion cyclotron resonance (FTICR) are common types of mass analyzers. Numerous mass analyzers are often combined to achieve maximum performance [24]. For example, Muntel et al. used a quadrupole orbitrap instrument for urine protein biomarker discovery [25]. Moreover, the workflow of a MALDI imaging mass spectrometer (MALDI IMS) enables the histology-directed analysis of the mass spectra using tissues [26, 27]. In addition, optical density mass analyzers, known for their tolerance of high pressure, are particularly suited to the pulsed nature of ESI.
Figure 3
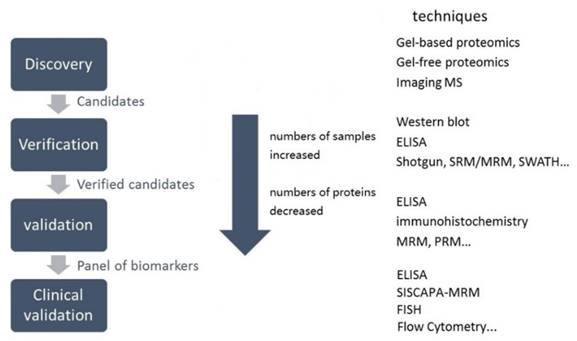
Schematic representation of the various stages in the biomarker pipeline. SISCAPA is the acronym for Stable Isotope Standards and Capture by Antipeptide Antibodies. FISH is short for fluorescent in situ hybridization.
Figure 4
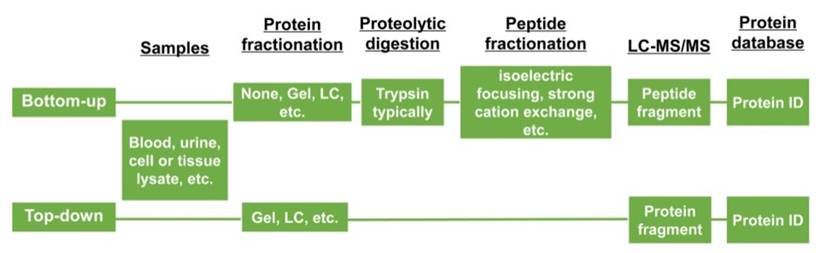
Two categories of proteomic experiments.
MS methodologies
Two-dimensional electrophoresis (2-DE) and chromatography-based proteomics
There are two main approaches to identify proteins applying gel-based proteomics, including bottom-up and top-down proteomics. In the former approach, proteins separated by two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) or in some instances, such as shot-gun proteomics wherein the fractionation step is left out, are digested in gel and then analyzed by MS [28, 29]. Which means the proteins are digested using chemicals or enzymes before introducing them into MS. Needless to say, this strategy may have several problems including the occurrence of modifications on disparate peptides. while the top-down approach, on the other hand, both the intact proteins and fragment ions masses can be measured [30] (Figure 4).
2-DE has been applied in proteomic research since its introduction in 1975. For example, Klein et al. used the 2DE-MS approach to analyze the nuclear proteome of human gastric cancer cell lines with and without inactivation of hypoxia-inducible factor 1 [31, 32]. The shortcomings of this strategy include a limited dynamic range and low-throughput analysis [3]. Although 2-D gel is still a powerful technique in proteomic analyses [33, 34], such as alternative detection for modification of specific proteins [35], attempts have been made to alleviate these drawbacks by using other techniques such as three-dimensional gel electrophoresis [36].
Shotgun based proteomics
Shotgun proteomics, also referred to as discovery proteomics, is a successfully used method [37]. It is based on employing a liquid chromatography-tandem MS (LC-MS/MS) for data-dependent acquisition (DDA) or in some certain occasions data-independent acquisition (DIA) mode. In DDA mode, peptide fragmentation is guided by the abundance of peptide ions detected in a survey scan. The recorded information of specific ions is searched against a protein database to determine the peptide sequence and protein identity [38]. In addition to its exquisite specificity, DDA-based proteomics has numerous other advantages, including unbiased and free-from hypotheses [39]. DIA offers advantages over conventional DDA methods as it overcomes the stochastic, intensity-based selection of peptide precursors [40].
One of the applications of the shotgun approach is to generate spectral libraries for mass spectrometric reference maps [41, 42]. It has also been used for the analysis of unique types of samples with biological and clinical importance including serum [43] and plasma [44, 45]. In a previous study, shotgun proteomics was applied to detect changes in protein profiles related to lung cancer [46].
Although many MS-based proteomic studies were performed using shotgun proteomics, the stochastic sampling of this technique markedly affects reproducible detection [47]. Furthermore, in traditional shotgun proteomics experiments, a large number of MS/MS spectra are collected. Peptide sequences are assigned using database searching algorithms, such as Sequest and PepExplorer, which use rigorous pattern recognition to assemble a list of homologous proteins [48]. However, not all spectra acquired are matched to peptides. To investigate this problem, Chick et al. identified unassigned peptides and demonstrated that at least one-third of unmatched spectra arise from peptides with substoichiometric modifications [49].
SRM-based proteomics
The adaptation of targeted data acquisition in the form of selected reaction monitoring (SRM), approximately a decade ago, was initially motivated by the requirement for robust and sensitive quantification of proteins [50]. Numerous LC-MS workflows employ shotgun LC-MS; however, many others require a significantly higher reproducibility, sensitivity, accuracy, and precision of SRM [51]. SRM, also known as multiple reaction monitoring, uses triple Quadrupole (QD) (Figure 5), where molecular ions are selected in Q1, collision-activated dissociation fragmentation is performed in Q2, and unique fragments ions are evaluated in Q3 [52]. SRM is an attractive choice for sample analysis due to its sensitivity [53].
Advances in SRM have led to the discovery of numerous allergens in food complexes and cancer-related proteins [54, 55, 56]. Recently, by adding an isotopically labeled protein (15N-α-S1-casein), accuracy of SRM analysis was increased [57]. In addition, absolute quantitation (AQUA), which has benefits of linearity over four orders of magnitude [58] and inter-laboratory comparability, also demands its use in allergen quantitation [59].
SRM has also been applied in biological fields [60], metabolic processes [61], signaling pathways [62], and validation of potentially interesting proteins [63]. As protein-protein interaction networks are significantly important in biological processes, it is essential to develop a computational method to predict protein-protein interactions. For example, Huang et al. proposed an efficient strategy that used a weighted sparse representation-based classifier model and novel feature extraction to sequence proteins for construction of protein-protein networks. [64]. Since investigation of phosphorylation events may serve an important role in biological research, Angeleri et al. developed an efficient strategy to obtain information regarding the phosphorylated sites [65].
Targeted data acquisition by SRM has been successful; however, the technique has intrinsic limitations. For example, the sensitivity of SRM currently cannot achieve the entire space of all organisms. Furthermore, the isolation width of Q1 can lead to false positive identifications [66]. Recent improvements, including time-scheduled SRM or intelligent SRM, have increased the scale and improved the quality of SRM evaluations [67]. In addition, parallel reaction monitoring has been developed markedly in instrumentation and software [68, 69].
Sequential window acquisition of all theoretical mass spectra (SWATH)-based proteomics
SWATH, a recently developed methodology [70, 71, 72] that relies on peptide spectral libraries, can be established by shotgun or obtained from community data repositories. Therefore, in contrast to SRM, SWATH-MS can quantify unlimited number of peptides that are included in spectral libraries.
SWATH-MS can be used in quantitative interaction proteomics [73, 74, 75]. For example, Ortea et al. provided evidence that LC-MS/MS combined pre-treatment and SWATH-MS was effective to identify lung cancer biomarker candidates [76]. SWATH-MS is also useful for the identification of candidate biomarkers, which will be further discussed in the following section [77, 78].
Additionally, there have been attempts to optimize the SWATH-MS workflow. The generation of a reference assay library is one of the key challenges and limitations of this approach [79]. It has been demonstrated that combined assay libraries can be used for SWATH data extraction [78], and certain software tools have been proposed for creating combined assay libraries [80, 81]. The parameters of MS detection were also optimized to increase the size of the library and decrease systematic errors [82]. These developments have broadened the application of SWATH.
Multiplexed MS/MS
In SWATH and other DIA approaches, peptides and their modified forms are difficult to distinguish because of the width of the window used for the isolated precursor. Egertson et al. introduced and improved the DIA framework, multiplexed MS/MS, to overcome the constraint on the scanning speed of the instrument [83]. The authors also suggested that this method may exploit other strengths of DIA [84].
Figure 5
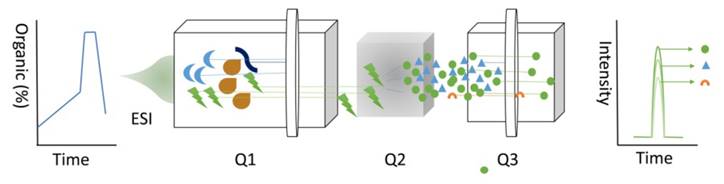
SRM technique.
Multiplexed MS/MS has certain disadvantages. It is more suitable for complex samples rather than simple mixtures due to its likely effect on the detection of low abundance peptides. Furthermore, the de-multiplexing and reconstruction of multiplexed MS/MS data may be a time-consuming process [85].
Application of MS in cancer biomarker discovery
Gastric, pancreatic, and liver cancers
Gastric cancer has one of the highest mortality rates worldwide [86, 87] urgently requiring its early detection [88, 89]. Studies of gastric cancer biomarkers mainly focus on tissues [90], blood [91], and biological fluids to identify protein, RNA [92], and DNA [93]. MS-based proteomics can aid in the identification of protein biomarkers and help study the mechanisms underlying gastric cancer [94]. Using MALDI-TOF-MS, Yang et al. analyzed serum samples obtained from 70 patients with gastric cancer and 72 healthy volunteers and identified two peptides (P < 0.001) related to gastric cancer [95]. Quantitative MS-based proteomic approaches include SCI techniques or label-free strategies in gastric cancer research. A variety of sources have been used to identify gastric cancer biomarkers, such as serum, gastric fluid [96, 97], cells obtained from tumor sections [98], cancer stem cells, circulating tumor cells [99], plasma membrane [100], saliva, plasma [101] and cancer tissues [102].
Cancer stem cells (CSCs) have been suggested to be extremely resistant to chemotherapy [103, 104]. Therefore, the identification of CSC markers has become a novel therapeutic perspective. Yashiro et al. used CSC-like side population cells to identify novel biomarkers of gastric CSCs [105].
Pancreatic cancer has been described as one of the most lethal tumors [106] with 45220 new cases and 38460 mortalities reported in the US in 2013 [107, 108]. There is a critical need for developing clinically useful biomarkers for pancreatic cancer detection. Carcinoma antigen 19-9 (CA19-9) is a biomarker which has been shown to be significant in the diagnosis, prognosis, and management of pancreatic ductal adenocarcinoma [109]. However, CA19-9 reacts with the sialylated Lewisa blood group antigen present in the glycoprotein serum fraction [110]. 5-10% of the general population has the Lewisa-b- phenotype; therefore, CA19-9 is not an appropriate biomarker for these individuals [111]. To overcome this problem, Yoneyama et al. identified insulin-like growth factor-binding protein 2 (IGFBP2) (AUC value of 0.706) and IGFBP3 (AUC value of 0.766) as plasma biomarkers for early detection of invasive ductal adenocarcinoma of pancreas [112]. In another biomarker study, Zhong et al. described a 2D-MALDI-TOF-TOF-MS/MS combined strategy for isolating and identifying membrane proteins. Immunohistochemical staining experiment demonstrated that the biomarker candidate they discovered was downregulated in pancreatic cancer tissue (P < 0.05) [113]. In another example, Tatsuyuki et al. identified novel prognostic markers by applying MS-based proteomic analysis [114].
HCC is the most common primary liver malignant disease [115]. HCC-associated mortality is high due to numerous contributing factors [116]. Therefore, there is an urgent need to develop clinical biomarkers that enable early detection for HCC [117]. Megger et al. performed 2-DE and label-free ion intensity-based quantification by applying MS and LC to identify differential protein abundance in HCC and control tissues [118]. Later, the same group combined previous results with label-free analysis [119]. In another study, Wang et al. analyzed five HCC subline variants using 2-DE coupled with MALDI-TOF MS [120].
Colorectal cancer (CRC)
CRC is the third most common cancer diagnosed and one of leading causes of cancer-related deaths in the US and [121]. The survival of patients with CRC is primarily associated with the stage of cancer [122]. However, limited number of CRC biomarkers have been developed [123]. Prognostic biomarkers could help the management of CRC [124]. Tomonaga et al. used the isobaric tags for relative and absolute quantitation (iTRAQ) shotgun method to discover biomarker candidates, which were subsequently validated by SRM [125]. Bosch et al. identified potential cancer markers to improve the diagnostic accuracy of the fecal immunochemical test to detect small traces of the blood protein, hemoglobin [126]. In another study investigating CRC, Peltier et al. combined iTRAQ technology [128, 129, 130] with reversed-phase liquid chromatography and MALDI-TOF/TOF to perform quantitative proteomic analysis of adenoma, CRC, and healthy control serum samples [127].
Glycosylation is important in many biological processes, such as immune surveillance for tumors [131, 132, 133]. Protein glycosylation commonly occur with the addition of specific glycan residues to asparagine (N-linked glycosylation) [134]. Sethi et al. utilized LC-MS/MS-based N-glycoproteomics to map the N-glycome landscape associated with a panel of colorectal cell lines and described a novel method to identify disease-associated markers [135]. In another study investigating CRC, a fluorogenic derivatization-LC-MS/MS approach was utilized to perform a differential proteomic analysis of normal and cancer cells [136].
Lung cancer
Lung cancer can be classified into small cell (SCLC) and non-small cell lung cancer (NSCLC) [137, 138]. Numerous previous studies have demonstrated that pleural effusions contain proteins of potential diagnostic value [139, 140]. Recently, the proteome of pleural effusion in patients with NSCLC was investigated using pleural fluid from 20 patients with NSCLC and 10 patients with tuberculosis (Figure 6a) [141].The homodimeric glycoprotein stanniocalcin 2 was reported to serve numerous roles in a variety of cancer subtypes. By applying MS/MS analysis on tissue samples from 53 cancer patients, Na et al. revealed that stanniocalcin 2 was upregulated in lung cancer cells [142].
MALDI-TOF-MS has been used in numerous cancer studies [143]. It has been shown that endothelial cells (ECs) play an important role in the tumor microenvironment [144, 145] and the properties of tumor-derived ECs are different from normal ECs [146]. Zhuo et al. isolated ECs from lung squamous cell carcinoma using magnetic beads (Figure 6b) [147]. Using the same method, Jin et al. discovered a protein candidate which was related to the histological presence of lymph node metastasis and neural invasion (p < 0.01) [148].
Toxicity and drug resistance remain major challenges facing cancer therapy. Efforts have been made to discover ideal biomarkers to improve the treatment efficiency. Rovithi et al. developed a serum peptide algorithm to classify cancer patients with regard to their clinical outcome [149]. To guide the radiotherapeutic method and avoid severe toxicity, Walker et al. investigated the alterations in blood during therapy [150].
The collection of saliva is less invasive compared with collection of the blood [151] or tissue making it an attractive biological fluid for diagnosis. Xiao et al. used 2D-MS to analyze two pooled samples. The results indicated that saliva analyses might be established for lung cancer detection [152].
Advances in MS help in mapping a large number of mass spectrophotometric peaks to reference libraries [153]. Using LC-SRM, 17 circulating proteins could be identified as potential cancer biomarkers in plasma samples collected from 72 patients [154]. However, despite extensive efforts in lung cancer diagnosis, it remains challenging to move protein candidates in the clinic [155-158].
Melanoma
Melanoma is a skin cancer with a high mortality rate [159]. Besides serum, urine, and cell lines, proteomics can also be used for quantitative analysis on formalin-fixed paraffin-embedded (FFPE) tissues. For example, Byrum et al. used label-free quantitative MS to analyze FFPE to identify potential targets for the therapy of melanoma [160]. Qendro et al. performed LC-MS/MS to profile five melanoma cell lines, a tissue sample of metastatic melanoma, and a benign melanocyte cell strain [161]. Bioinformatics analysis was performed with each group of proteins to assign over-represented Gene Ontology terms.
Extracellular vesicles including exosomes are one of the mechanisms used for cell-cell communication. Exosomes are initially defined as reticulocyte-secreted vesicles secreted by many cell types [161, 162]. Exosomes play an important role in cancer progression [163]. Previous studies demonstrated that melanoma exosomes may influence disease progression by enhancing immunosuppression [164], angiogenesis, and tumor metastasis [165, 166]. Lazar et al. performed proteomic analysis of seven melanoma cell lines and demonstrated that exosomes may be a potential biomarker for melanoma classification [167].
Uveal melanoma (UM) is a primary malignancy of eye the etiology of UM remains poorly understood. According to clinical, histopathological, and genetic features of these tumors, patients with UM can be classified into low-risk and high-risk metastatic groups [168]. Crabb et al. performed global quantitative proteomic analysis of UM to increase our understanding of UM metastasis processes and to identify biomarkers of UM metastasis [169]. MS-based proteomics using the untargeted MS method to discover novel protein biomarker candidates and the targeted MS mode to identify peptides of interest, has been a useful tool in melanoma research [170].
Breast cancer
Breast cancer contributes to approximately 14% of the cancer-associated mortality [171]. Although 5-year survival rates have improved, ≥20% of all patients continue to develop metastatic disease with an associated poor outlook [172]. Hormone receptor positive, erb-b2 receptor tyrosine kinase 2 (ErbB2) positive, and hormone (estrogen or progesterone) receptor and ErbB2 negative breast cancers are the four main types of this aggressive disease [173].
Breast milk is an appropriate cancer microenvironment for identifying breast cancer biomarkers. Aslebagh et al. used a nanoLC-MS/MS to analyze breast milk samples collected from patients with cancer and controls. The results demonstrated that sample-specific bands were present between the two groups [174]. Besides milk, serum is also used for identifying breast cancer-specific markers [175-182]. Dowling et al. combined metabolomics and proteomics platforms to analyze cancer and non-cancer serum samples [175]. High mobility group protein HMG-I/HMG-Y (HMGA1) abundance level was found to be associated with breast cancer clinicopathological features. Maurizio et al. utilized label-free shotgun MS to analyze the proteins extracted from HMGA1-silenced cells and control breast cancer cell line MDA-MB-231 [176]. Ning Qing Liu et al. evaluated numerous approaches for global proteome quantification and proteins involved in a signaling pathway in breast cancer tissues were identified (Figure 6c) [177]. Yang et al. collected serum samples from 183 breast cancer patients and 64 healthy controls to extract peptides using magnetic beads and analyzed by them MALDI-TOF-MS [178]. Besides serum, urine was also used in proteomic studies to analyze its feasibility as a potential source for breast cancer biomarkers [183].
Ovarian and uterine cervical cancers
Ovarian cancer consists of numerous distinct subtypes [184, 185]. However, the gold-standard biomarker, CA125, only performs well in one of these. A number of novel protein biomarkers relevant to ovarian cancer have been identified using MS-based proteomics [186]. Nepomuceno et al. applied LC-MS/MS on tissues obtained from chickens that developed ovarian tumors spontaneously as an emerging experimental model to investigate the ovarian cancer proteome and reported the upregulation of an inhibitor in tumors (p = 0.0005) [187]. Also, Poersch et al. performed LC-MS/MS on tumor fluids to identify ovarian cancer-associated protein biomarkers [188].
Drug resistance is a major challenge for ovarian cancer chemotherapeutic treatments. Therefore, it is essential to discover biomarkers that can distinguish chemosensitive and chemoresistant ovarian cancer patients [189]. Based on the LC-MS/MS results acquired from epithelial ovarian cancer, Chappell et al. hypothesized that mitochondrial proteome changes were required to develop chemotherapy drug cisplatin resistance [190]. In another study, Zhang et al. analyzed the protein abundance level in chemotherapy drug paclitaxel-resistant ovarian cancer cells and tissues [191].
Figure 6
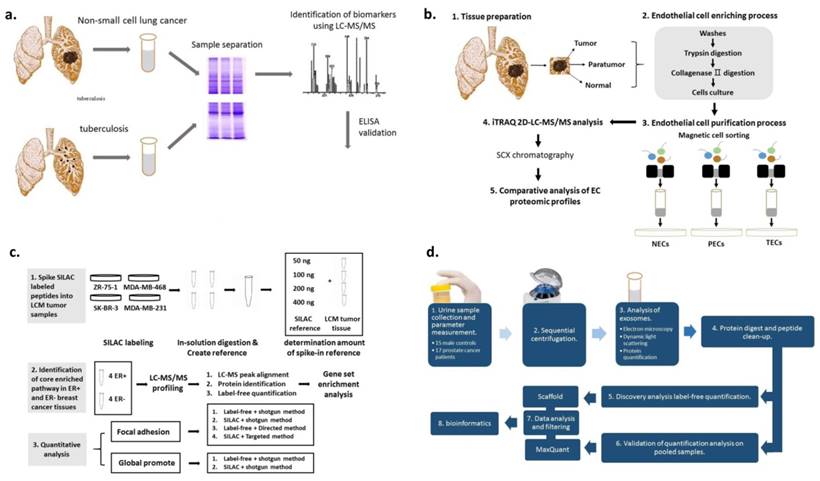
(A) Schematic illustration of proteome screening of pleural effusions to identify biomarkers for NSCLC. 1D SDS-PAGE was performed to separate proteins in pleural fluids. ELISA was used for the validation of protein candidates. (B) Schematic diagram of the experimental design. Normal, para-tumor-, and tumor-derived cluster of differentiation (CD) 105+ endothelial cells (ECs) were isolated, followed by iTRAQ-2DLC-MS/MS-based protein abundance profiling and comparative analysis of profiles. (C) Schematic diagram of the experimental design of systematic comparison between various quantitative methods for quantification of proteins within one pathway. (D) Schematic diagram of the experimental design for the isolation and characterization of urinary exosomes.
Using iTRAQ and LC-MS platform, Shetty et al. revealed that major histocompatibility complex class 1 (p < 0.01) may be related to ovarian cancer drug resistance [192]. In addition, the mechanism underlying somatic genome effects on the cancer proteome and associations between post-translational modification levels of proteins and clinical outcomes in high-grade serous carcinomas have been investigated [193].
Since Papanicolaou (Pap) test was approved by the US Food and Drug Administration (FDA) in 1996, a vast majority of cervical cancer screening has used liquid-based Pap test [194, 195]. Boylan et al. examined the proteins present in residual Pap test fixative samples from females with normal cervical cytology by 2-D-MS/MS and created a “Normal Pap test Core Proteome” [196]. More recently, the same group used iTRAQ to quantify the proteins in Pap test samples from patients with ovarian cancer compared with healthy controls or patients with benign gynecological disease [197]. The labeled samples were analyzed by 2D-LC-MS/MS. The results demonstrated that Pap test samples may be a valuable source for the identification of ovarian cancer biomarkers [197].
Urinary cancers
Urinary cancers include kidney, bladder, prostate, and testicular cancers [198]. Sensitive and accurate MS quantitative analyses have been introduced for biomarker discovery in these cancers [199]. Zhao et al. performed quantitative proteomic analysis on clear cell renal cell carcinoma (RCC) and adjacent kidney tissues using LC-MS/MS [200]. Urine is a rich resource for the investigation of kidney physiology as well as diagnosis of glomerulonephritis, hypertensive nephropathy, and renal cancer [201]. Sandim et al. investigated the proteins in urine samples collected from 64 patients with clear cell RCC and compared them with the healthy controls [202], whereas Neely et al. combined proteo-transcriptomic analysis and investigated alterations in protein abundance [203].
Prostate cancer is among the most common types of adult malignancies with an estimated 220,000 American males diagnosed with the disease annually [204]. Sensitive biomarkers would improve the efficiency of diagnosis, prognosis, and personalized therapy of prostate cancer. Øverbye et al. identified proteins with differential abundance in 16 prostate cancer patients compared to 15 healthy controls by MS-based proteomics (Figure 6d) [205]. Kim et al. developed SRM-MS assays in post-digital rectal examination urine samples. The results demonstrated that this strategy may accurately identify non-invasive biomarkers [206].
Urine is also considered to be an attractive source for bladder cancer biomarkers identification [204, 208]. Guo et al. proposed a strategy to identify urine proteins associated with bladder cancer [209]. In Europe and North America, a majority of bladder cancers are urothelial carcinomas [210]. Lin et al. used MALDI-TOF spectrometry on urinary exosomes for the determination of urothelial biomarkers [211].
MS-based proteomics has also been used to identify testicular cancer biomarkers. Liu et al. used the proteomics platform to identify proteins that participate in spermatogenesis and can, therefore, serve as novel targets for the treatment of male infertility and cancer [212]. The proteins they identified may also be used for personalized therapy for patients with testicular cancer.
Challenges in biomarker implementation and future prospects
Cancer progression is a comprehensive event that makes biomarker development a challenging task. Despite rapid advances in academia and industry, not many biomarkers move on to clinical practice [213]. Failure of cancer biomarkers appears to be due to several distinct challenges depicted in Figure 7 [214].
Figure 7
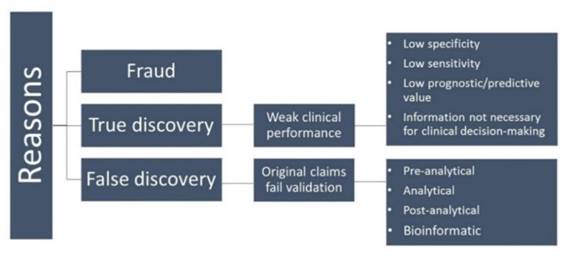
Schematic illustration of potential reasons for failure of biomarkers in clinical practice.
The first category of fraud is quite rare [215]. False discovery is a major reason for failure of biomarkers to reach the clinic. These biomarkers fail at independent reproduction in the validation phase [216, 217]. Small sample size as well as control samples used in the experiments that are not matched for age, sex, and race can lead to deceptive results [218]. Other important issues to be considered include, criteria for selection and inclusion of samples, strict standards for collecting and handling samples, suitability of the methodology, for the analysis of the data obtained, and, most importantly, independent validation of the identified biomarkers [219, 220].
Although few cancer biomarkers have entered clinical use, there are numerous ways to improve the situation. For biomarkers with low specificity and sensitivity that are not suitable for clinical use, it is possible to combine a panel of different biomarkers to identify clinical scenarios [214]. For example, a novel ovarian cancer biomarker, human epididymis protein 4, is not superior to CA125, which is an FDA-approved marker for ovarian cancer [221]. However, by combining human epididymis protein 4 and CA125, diagnosis of malignant versus benign pelvic masses can be improved [222]. For false discovery or artefactual biomarkers, understanding of the biological and molecular heterogeneity of disease states is required to guide the experimental design [223]. In addition, efforts should be taken made for improving the MS technologies to explore proteins with lower abundance [224].
Conclusion
Because of recent advances in MS-based proteomics together with streamlined sample preparation, improved instrumentation, and combination of various analytical platforms, numerous cancer biomarkers have been identified with diagnostic and prognostic values. The challenge is to realize the diagnostic and prognostic potential of these biomarkers in the clinical practice.
Acknowledgements
This research was financially supported by NSFC (61527806, 61401217 and 61471168), Chinese 863 Project (2015AA020502), the National Key Research and Development Program of China (2017YFA0205300), Open Funding of State Key Laboratory of Oral Diseases (SKLOD2017OF04) China Postdoctoral Science Foundation (2016T90403), and the Economical Forest Cultivation and Utilization of 2011 Collaborative Innovation Center in Hunan Province [(2013) 448].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-E86
2. Stewart B, Wild CP. World cancer report 2014. World. 2016
3. Mäbert K, Cojoc M, Peitzsch C, Kurth I, Souchelnytskyi S, Dubrovska A. Cancer biomarker discovery: current status and future perspectives. Int J Radiat Biol. 2014;90:659-77
4. Jin C, Qiu L, Li J, Fu T, Zhang X, Tan W. Cancer biomarker discovery using DNA aptamers. Analyst. 2016;141:461-6
5. Li P, Chen S, Chen H, Mo X, Li T, Shao Y. et al. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta. 2015;444:132-6
6. Tabernero J, Lenz H-J, Siena S, Sobrero A, Falcone A, Ychou M. et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015;16:937-48
7. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650-62
8. Ilkhani H, Sarparast M, Noori A, Zahra Bathaie S, Mousavi MF. Electrochemical aptamer/antibody based sandwich immunosensor for the detection of EGFR, a cancer biomarker, using gold nanoparticles as a signaling probe. Biosens Bioelectron. 2015;74:491-7
9. Goossens N, Nakagawa S, Sun X, Hoshida Y. Cancer biomarker discovery and validation. Transl Cancer Res. 2015;4:256
10. Batrla R, Jordan BW. Personalized health care beyond oncology: new indications for immunoassay-based companion diagnostics. Ann N Y Acad Sci. 2015;1346:71-80
11. Shin J KG, Lee J W. et al. Identification of ganglioside GM2 activator playing a role in cancer cell migration through proteomic analysis of breast cancer secretomes. Cancer Sci. 2016;107:828-35
12. Hudler P, Kocevar N, Komel R. Proteomic approaches in biomarker discovery: new perspectives in cancer diagnostics. The Scientific World J. 2014. 2014
13. Bausch-Fluck D, Hofmann A, Bock T, Frei AP, Cerciello F, Jacobs A. et al. A mass spectrometric-derived cell surface protein atlas. PLoS One. 2015;10:e0121314
14. Hanash S, Taguchi A. The grand challenge to decipher the cancer proteome. Nat Rev Cancer. 2010;10:652-60
15. Skates SJ, Gillette MA, LaBaer J, Carr SA, Anderson L, Liebler DC. et al. Statistical Design for Biospecimen Cohort Size in Proteomics-based Biomarker Discovery and Verification Studies. J Proteome Res. 2013;12:5383-94
16. Boja E, Rivers R, Kinsinger C, Mesri M, Hiltke T, Rahbar A. et al. Restructuring proteomics through verification. Biomark Med. 2010;4:799-803
17. Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat Biotechnol. 2010;28:710-21
18. Andrew G Chambers AJP, Romain Simon & Christoph H Borchers. MRM for the verification of cancer biomarker proteins: recent applications to human plasma and serum. Expert Rev Proteomic. 2014;11:137-48
19. Jedrychowski Mark P, Wrann Christiane D, Paulo Joao A, Gerber Kaitlyn K, Szpyt J, Robinson Matthew M. et al. Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab. 2015;22:734-40
20. Parker CE, Borchers CH. Mass spectrometry based biomarker discovery, verification, and validation — Quality assurance and control of protein biomarker assays. Mol Oncol. 2014;8:840-58
21. Popescu ID, Codrici E, Albulescu L, Mihai S, Enciu A-M, Albulescu R. et al. Potential serum biomarkers for glioblastoma diagnostic assessed by proteomic approaches. Proteome Sci. 2014;12:47
22. Tran BQ, Goodlett DR, Goo YA. Advances in protein complex analysis by chemical cross-linking coupled with mass spectrometry (CXMS) and bioinformatics. Biochim Biophys Acta. 2016;1864:123-9
23. Wei H, Mo J, Tao L, Russell RJ, Tymiak AA, Chen G. et al. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov Today. 2014;19:95-102
24. Jaiswal M, Washburn MP, Zybailov BL. Mass Spectrometry-Based Methods of Proteome Analysis. Reviews in Cell Biology and Molecular Medicine. 2015
25. Muntel J, Xuan Y, Berger ST, Reiter L, Bachur R, Kentsis A. et al. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. J Proteome Res. 2015;14:4752-62
26. Schwamborn K, Caprioli RM. Molecular imaging by mass spectrometry — looking beyond classical histology. Nat Rev Cancer. 2010;10:639-46
27. Aichler M, Walch A. MALDI Imaging mass spectrometry: current frontiers and perspectives in pathology research and practice. Lab Invest. 2015;95:422-31
28. Milman BL. General principles of identification by mass spectrometry. Trends Analyt Chem. 2015;69:24-33
29. Antohe F. Mass Spectrometry based proteomics. Acta Endocrinol (Copenh). 2015;11:139-42
30. Catherman AD, Skinner OS, Kelleher NL. Top Down proteomics: Facts and perspectives. Biochem Biophys Res Commun. 2014;445:683-93
31. Klein O, Rohwer N, de Molina KF, Mergler S, Wessendorf P, Herrmann M. et al. Application of two-dimensional gel-based mass spectrometry to functionally dissect resistance to targeted cancer therapy. Proteomics Clin Appl. 2013;7:813-24
32. Mao L. Seeing through the trick of cancer cells via 2D gels. Proteomics Clin Appl. 2013;7:723-4
33. Rabilloud T, Lelong C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J Proteomics. 2011;74:1829-41
34. Oliveira BM, Coorssen JR, Martins-de-Souza D. 2DE: The Phoenix of Proteomics. J Proteomics. 2014;104:140-50
35. Rogowska-Wrzesinska A, Le Bihan M-C, Thaysen-Andersen M, Roepstorff P. 2D gels still have a niche in proteomics. J Proteomics. 2013;88:4-13
36. Colignon B, Raes M, Dieu M, Delaive E, Mauro S. Evaluation of three-dimensional gel electrophoresis to improve quantitative profiling of complex proteomes. Proteomics. 2013;13:2077-82
37. Liu Y, Hüttenhain R, Collins B, Aebersold R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert Rev Mol Diagn. 2013;13:811-25
38. Frese CK, Altelaar AM, Hennrich ML, Nolting D, Zeller M, Griep-Raming J. et al. Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. J Proteome Res. 2011;10:2377-88
39. Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347-55
40. Wu JX, Song X, Pascovici D, Zaw T, Care N, Krisp C. et al. SWATH Mass Spectrometry Performance Using Extended Peptide MS/MS Assay Libraries. Mol Cell Proteomics. 2016;15:2501-14
41. Picotti P, Clement-Ziza M, Lam H, Campbell DS, Schmidt A, Deutsch EW. et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013;494:266-70
42. Kim M-S, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R. et al. A draft map of the human proteome. Nature. 2014;509:575-81
43. Zhang Q, Fillmore TL, Schepmoes AA, Clauss TR, Gritsenko MA, Mueller PW. et al. Serum proteomics reveals systemic dysregulation of innate immunity in type 1 diabetes. J Exp Med. 2013;210:191-203
44. Overgaard AJ, Thingholm TE, Larsen MR, Tarnow L, Rossing P, McGuire JN. et al. Quantitative iTRAQ-based proteomic identification of candidate biomarkers for diabetic nephropathy in plasma of type 1 diabetic patients. Clin Proteom. 2010;6:105
45. Chan KCA, Jiang P, Zheng YWL, Liao GJW, Sun H, Wong J. et al. Cancer Genome Scanning in Plasma: Detection of Tumor-Associated Copy Number Aberrations, Single-Nucleotide Variants, and Tumoral Heterogeneity by Massively Parallel Sequencing. Clin Chem. 2013;59:211-24
46. Yu L, Shen J, Mannoor K, Guarnera M, Jiang F. Identification of ENO1 As a Potential Sputum Biomarker for Early-Stage Lung Cancer by Shotgun Proteomics. Clin Lung Cancer. 2014;15:372-8.e1
47. Shao S, Guo T, Aebersold R. Mass spectrometry-based proteomic quest for diabetes biomarkers. Biochim Biophys Acta. 2015;1854:519-27
48. Leprevost FV, Valente RH, Lima DB, Perales J, Melani R, Yates JR. et al. PepExplorer: A Similarity-driven Tool for Analyzing de Novo Sequencing Results. Mol Cell Proteomics. 2014;13:2480-9
49. Chick JM, Deepak Kolippakkam, David P. Nusinow, Bo Zhai, Ramin Rad, Edward L. Huttlin, and Steven P. Gygi.. An ultra-tolerant database search reveals that a myriad of modified peptides contributes to unassigned spectra in shotgun proteomics. Nat Biotechnol. 2015;33:743-9
50. Aebersold R, Bensimon A, Collins BC, Ludwig C, Sabido E. Applications and Developments in Targeted Proteomics: From SRM to DIA/SWATH. Proteomics. 2016;16:2065-7
51. Manes NP, Nita-Lazar A. The development of SRM assays is transforming proteomics research. Proteomics. 2016
52. Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9:555-66
53. Gallien S, Bourmaud A, Kim SY, Domon B. Technical considerations for large-scale parallel reaction monitoring analysis. J Proteomics. 2014;100:147-59
54. Ahsan N, Rao RSP, Gruppuso PA, Ramratnam B, Salomon AR. Targeted proteomics: Current status and future perspectives for quantification of food allergens. J Proteomics. 2016;143:15-23
55. Liu NQ, Dekker LJM, Van Duijn MM, Umar A. Use of Universal Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-Based Selected Reaction Monitoring (SRM) Approach for Verification of Breast Cancer-Related Protein Markers. In: Martins-de-Souza D, editor. Shotgun Proteomics: Methods and Protocols. New York, NY: Springer New York. 2014:307-22
56. Liebler DC, Zimmerman LJ. Targeted Quantitation of Proteins by Mass Spectrometry. Biochemistry. 2013;52:3797-806
57. Newsome GA, Scholl PF. Quantification of Allergenic Bovine Milk αS1-Casein in Baked Goods Using an Intact 15N-Labeled Protein Internal Standard. J Agric Food Chem. 2013;61:5659-68
58. Gallien S, Duriez E, Domon B. Selected reaction monitoring applied to proteomics. J Mass Spectrom. 2011;46:298-312
59. Croote D, Quake SR. Food allergen detection by mass spectrometry: the role of systems biology. NPJ Syst Biol Appl. 2016;2:16022
60. Costenoble R, Picotti P, Reiter L, Stallmach R, Heinemann M, Sauer U. et al. Comprehensive quantitative analysis of central carbon and amino-acid metabolism in Saccharomyces cerevisiae under multiple conditions by targeted proteomics. Mol Syst Biol. 2011:7
61. Hu LZ, Zhang WP, Zhou MT, Han QQ, Gao XL, Zeng HL. et al. Analysis of Salmonella PhoP/PhoQ regulation by dimethyl-SRM-based quantitative proteomics. Biochim Biophys Acta. 2016;1864:20-8
62. Gingras AC, Wong CJJ. Proteomics approaches to decipher new signaling pathways. Curr Opin Struct Biol. 2016;41:128-34
63. Wohlgemuth I, Lenz C, Urlaub H. Studying macromolecular complex stoichiometries by peptide-based mass spectrometry. Proteomics. 2015;15:862-79
64. Huang YA, You ZH, Li X, Chen X, Hu P, Li S. et al. Construction of reliable protein-protein interaction networks using weighted sparse representation based classifier with pseudo substitution matrix representation features. Neurocomputing. 2016;218:131-8
65. Angeleri M, Muth-Pawlak D, Aro EM, Battchikova N. Study of O-Phosphorylation Sites in Proteins Involved in Photosynthesis-Related Processes in Synechocystis sp. Strain PCC 6803: Application of the SRM Approach. J Proteome Res. 2016
66. Gallien S, Duriez E, Demeure K, Domon B. Selectivity of LC-MS/MS analysis: Implication for proteomics experiments. J Proteomics. 2013;81:148-58
67. Gallien S, Peterman S, Kiyonami R, Souady J, Duriez E, Schoen A. et al. Highly multiplexed targeted proteomics using precise control of peptide retention time. Proteomics. 2012;12:1122-33
68. Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol Cell Proteomics. 2012;11:1475-88
69. Law KP, Lim YP. Recent advances in mass spectrometry: data independent analysis and hyper reaction monitoring. Expert Rev Proteomics. 2013;10:551-66
70. Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L. et al. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol Cell Proteomics. 2012;11:O111.016717
71. Selevsek N, Chang CY, Gillet LC, Navarro P, Bernhardt OM, Reiter L. et al. Reproducible and Consistent Quantification of the Saccharomyces cerevisiae Proteome by SWATH-mass spectrometry. Mol Cell Proteomics. 2015;14:739-49
72. Huang Q, Yang L, Luo J, Guo L, Wang Z, Yang X. et al. SWATH enables precise label-free quantification on proteome scale. Proteomics. 2015;15:1215-23
73. Liu Y, Chen J, Sethi A, Li QK, Chen L, Collins B. et al. Glycoproteomic Analysis of Prostate Cancer Tissues by SWATH Mass Spectrometry Discovers N-acylethanolamine Acid Amidase and Protein Tyrosine Kinase 7 as Signatures for Tumor Aggressiveness. Mol Cell Proteomics. 2014;13:1753-68
74. Liu Y, Hüttenhain R, Surinova S, Gillet LCJ, Mouritsen J, Brunner R. et al. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics. 2013;13:1247-56
75. Gao Y, Wang X, Sang Z, Li Z, Liu F, Mao J. et al. Quantitative proteomics by SWATH-MS reveals sophisticated metabolic reprogramming in hepatocellular carcinoma tissues. Sci Rep. 2017;7:45913
76. Ortea I, Rodríguez-Ariza A, Chicano-Gálvez E, Arenas Vacas MS, Jurado Gámez B. Discovery of potential protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage fluid by SWATH MS data-independent acquisition and targeted data extraction. J Proteomics. 2016;138:106-14
77. Selevsek N, Chang CY, Gillet LC, Navarro P, Bernhardt OM, Reiter L. et al. Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Mol Cell Proteomics. 2015;14:739-49
78. Colgrave M L BK, Blundell M. et al. Comparing Multiple Reaction Monitoring and Sequential Window Acquisition of All Theoretical Mass Spectra for the Relative Quantification of Barley Gluten in Selectively Bred Barley Lines. Anal Chem. 2016;88:9127-35
79. Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC. et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12:258-64
80. Zi J, Zhang S, Zhou R, Zhou B, Xu S, Hou G. et al. Expansion of the ion library for mining SWATH-MS data through fractionation proteomics. Anal Chem. 2014;86:7242-6
81. Röst HL, Rosenberger G, Navarro P, Gillet L, Miladinović SM, Schubert OT. et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32:219-23
82. Schubert OT, Gillet LC, Collins BC, Navarro P, Rosenberger G, Wolski WE. et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc. 2015;10:426-41
83. Egertson JD, Kuehn A, Merrihew GE, Bateman NW, MacLean BX, Ting YS. et al. Multiplexed MS/MS for improved data-independent acquisition. Nat Methods. 2013;10:744-6
84. Weisbrod CR, Eng JK, Hoopmann MR, Baker T, Bruce JE. Accurate Peptide Fragment Mass Analysis: Multiplexed Peptide Identification and Quantification. J Proteome Res. 2012;11:1621-32
85. Law KP, Lim YP. Recent advances in mass spectrometry: data independent analysis and hyper reaction monitoring. Expert Rev Proteomics. 2013;10:551-66
86. Kang C, Lee Y, Lee JE. Recent advances in mass spectrometry-based proteomics of gastric cancer. World J Gastroenterol. 2016;22:8283-93
87. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
88. Cui J, Chen Y, Chou W-C, Sun L, Chen L, Suo J. et al. An integrated transcriptomic and computational analysis for biomarker identification in gastric cancer. Nucleic Acids Res. 2011;39:1197-207
89. Xiao H, Zhang Y, Kim Y, Kim S, Kim JJ, Kim KM. et al. Differential Proteomic Analysis of Human Saliva using Tandem Mass Tags Quantification for Gastric Cancer Detection. Sci Rep. 2016;6:22165
90. Pan S, Brentnall TA, Kelly K, Chen R. Tissue proteomics in pancreatic cancer study: Discovery, emerging technologies, and challenges. Proteomics. 2013;13:710-21
91. Werner S, Chen H, Tao S, Brenner H. Systematic review: Serum autoantibodies in the early detection of gastric cancer. Int J Cancer. 2015;136:2243-52
92. Liu R, Zhang C, Hu Z, Li G, Wang C, Yang C. et al. A five-microRNA signature identified from genome-wide serum microRNA expression profiling serves as a fingerprint for gastric cancer diagnosis. Eur J Cancer. 2011;47:784-91
93. Toiyama Y, Okugawa Y, Goel A. DNA methylation and microRNA biomarkers for noninvasive detection of gastric and colorectal cancer. Biochem Biophys Res Commun. 2014;455:43-57
94. Hu W, Wang J, Luo G, Luo B, Wu C, Wang W. et al. Proteomics-based analysis of differentially expressed proteins in the CXCR1-knockdown gastric carcinoma MKN45 cell line and its parental cell. Acta Biochim Biophys Sin (Shanghai). 2013
95. Yang J, Xiong X, Wang X, Guo B, He K, Huang C. Identification of peptide regions of SERPINA1 and ENOSF1 and their protein expression as potential serum biomarkers for gastric cancer. Tumor Biol. 2015;36:5109-18
96. Wu W, Chung MCM. The gastric fluid proteome as a potential source of gastric cancer biomarkers. J Proteomics. 2013;90:3-13
97. Wu W, Yong WW, Chung MCM. A simple biomarker scoring matrix for early gastric cancer detection. Proteomics. 2016;16:2921-30
98. Catenacci DVT, Liao WL, Zhao L, Whitcomb E, Henderson L, O'Day E. et al. Mass-spectrometry-based quantitation of Her2 in gastroesophageal tumor tissue: comparison to IHC and FISH. Gastric Cancer. 2016;19:1066-79
99. Chen JF, Zhu Y, Lu YT, Hodara E, Shuang H, Agopian VG. et al. Clinical Applications of NanoVelcro Rare-Cell Assays for Detection and Characterization of Circulating Tumor Cells. Theranostics. 2016;6:1425
100. Gao W, Xu J, Wang F, Zhang L, Peng R, Shu Y. et al. Plasma membrane proteomic analysis of human Gastric Cancer tissues: revealing flotillin 1 as a marker for Gastric Cancer. BMC Cancer. 2015;15:367
101. Zhang K, Shi H, Xi H, Wu X, Cui J, Gao Y. et al. Genome-Wide lncRNA Microarray Profiling Identifies Novel Circulating lncRNAs for Detection of Gastric Cancer. Theranostics. 2017;7:213-27
102. Gao W, Xu J, Wang F, Zhang L, Peng R, Zhu Y. et al. Mitochondrial Proteomics Approach Reveals Voltage-Dependent Anion Channel 1 (VDAC1) as a Potential Biomarker of Gastric Cancer. Cell Physiol Biochem. 2015;37:2339-54
103. Kreso A, Dick John E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell. 2014;14:275-91
104. Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin Cancer Biol. 2015;31:16-27
105. Yashiro M, Morisaki T, Hasegawa T, Kakehashi A, Kinoshita H, Fukuoka T. et al. Abstract 1405: Novel biomarkers for gastric cancer stem cells utilizing comparative proteomics analysis. Cancer Res. 2015;75:1405-1405
106. Kato K, Kamada H, Fujimori T, Aritomo Y, Ono M, Masaki T. Molecular Biologic Approach to the Diagnosis of Pancreatic Carcinoma Using Specimens Obtained by EUS-Guided Fine Needle Aspiration. Gastroenterol Res Pract. 2012;2012:7
107. Yadav D, Lowenfels AB. The Epidemiology of Pancreatitis and Pancreatic Cancer. Gastroenterology. 2013;144:1252-61
108. Lin C, Wu WC, Zhao GC, Wang DS, Lou WH, Jin DY. ITRAQ-based quantitative proteomics reveals apolipoprotein A-I and transferrin as potential serum markers in CA19-9 negative pancreatic ductal adenocarcinoma. Medicine. 2016;95:e4527
109. Humphris JL, Chang DK, Johns AL, Scarlett CJ, Pajic M, Jones MD. et al. The prognostic and predictive value of serum CA19.9 in pancreatic cancer. Ann Oncol. 2012;23:1713-22
110. Magnani JL, Steplewski Z, Koprowski H, Ginsburg V. Identification of the Gastrointestinal and Pancreatic Cancer-associated Antigen Detected by Monoclonal Antibody 19-9 in the Sera of Patients as a Mucin. Cancer Res. 1983;43:5489-92
111. Tempero MA, Uchida E, Takasaki H, Burnett DA, Steplewski Z, Pour PM. Relationship of Carbohydrate Antigen 19-9 and Lewis Antigens in Pancreatic Cancer. Cancer Res. 1987;47:5501-3
112. Yoneyama T, Ohtsuki S, Honda K, Kobayashi M, Iwasaki M, Uchida Y. et al. Identification of IGFBP2 and IGFBP3 As Compensatory Biomarkers for CA19-9 in Early-Stage Pancreatic Cancer Using a Combination of Antibody-Based and LC-MS/MS-Based Proteomics. PLoS One. 2016;11:e0161009
113. Zhong N, Cui Y, Zhou X, Li T, Han J. Identification of prohibitin 1 as a potential prognostic biomarker in human pancreatic carcinoma using modified aqueous two-phase partition system combined with 2D-MALDI-TOF-TOF-MS/MS. Tumor Biol. 2015;36:1221-31
114. Takadate T, Onogawa T, Fukuda T, Motoi F, Suzuki T, Fujii K. et al. Novel prognostic protein markers of resectable pancreatic cancer identified by coupled shotgun and targeted proteomics using formalin-fixed paraffin-embedded tissues. Int J Cancer. 2013;132:1368-82
115. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
116. Kimhofer T, Fye H, Taylor-Robinson S, Thursz M, Holmes E. Proteomic and metabonomic biomarkers for hepatocellular carcinoma: a comprehensive review. Br J Cancer. 2015;112:1141-56
117. Naboulsi W MDA, Bracht T, Naboulsi W, Megger D A, Bracht T. et al. Quantitative tissue proteomics analysis reveals versican as potential biomarker for early-stage hepatocellular carcinoma. J Proteome Res. 2015;15:38-47
118. D.A. Megger TB, M. Kohl, M. Ahrens, W. Naboulsi, F. Weber, A.C. Hoffmann, C. Stephan, K. Kuhlmann, M. Eisenacher, J.F. Schlaak, H.A. Baba, H.E. Meyer, B. Sitek. Proteomic differences between hepatocellular carcinoma and non-tumorous liver tissue investigated by a combined 2D-DIGE and label-free quantitative proteomics study. Mol Cell Proteomics. 2013;12:2006-20
119. Reis H, Pütter C, Megger DA, Bracht T, Weber F, Hoffmann A-C. et al. A structured proteomic approach identifies 14-3-3Sigma as a novel and reliable protein biomarker in panel based differential diagnostics of liver tumors. Biochim Biophys Acta. 2015;1854:641-50
120. Wang RC, Huang CY, Pan TL, Chen WY, Ho CT, Liu TZ. et al. Proteomic Characterization of Annexin l (ANX1) and Heat Shock Protein 27 (HSP27) as Biomarkers for Invasive Hepatocellular Carcinoma Cells. PLoS One. 2015;10:e0139232
121. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10-29
122. Parente F, Vailati C, Boemo C, Bonoldi E, Ardizzoia A, Ilardo A. et al. Improved 5-year survival of patients with immunochemical faecal blood test-screen-detected colorectal cancer versus non-screening cancers in northern Italy. Dig Liver Dis. 2015;47:68-72
123. Wang Y, Shan Q, Hou G, Zhang J, Bai J, Lv X. et al. Discovery of potential colorectal cancer serum biomarkers through quantitative proteomics on the colonic tissue interstitial fluids from the AOM-DSS mouse model. J Proteomics. 2016;132:31-40
124. Surinova S, Radová L, Choi M, Srovnal J, Brenner H, Vitek O. et al. Non-invasive prognostic protein biomarker signatures associated with colorectal cancer. EMBO Mol Med. 2015;7:1153-65
125. Tomonaga T, Kume H. Biomarker Discovery of Colorectal Cancer Using Membrane Proteins Extracted from Cancer Tissues. Rinsho byori. 2015;63:322-7
126. Bosch LJ, de Wit M, Hiemstra AC, Piersma S, Pham T, Oudgenoeg G. et al. Abstract 1563: Stool proteomics reveals novel candidate biomarkers for colorectal cancer screening. Cancer Res. 2015;75:1563-1563
127. Peltier J, Roperch JP, Audebert S, Borg JP, Camoin L. Quantitative proteomic analysis exploring progression of colorectal cancer: Modulation of the serpin family. J Proteomics. 2016;148:139-48
128. Sun Y, Yang Y, Shen H, Huang M, Wang Z, Liu Y. et al. iTRAQ-based chromatin proteomic screen reveals CHD4-dependent recruitment of MBD2 to sites of DNA damage. Biochem Biophys Res Commun. 2016;471:142-8
129. Smith SJ, Kroon JTM, Simon WJ, Slabas AR, Chivasa S. A novel function for Arabidopsis CYCLASE1 in programmed cell death revealed by iTRAQ analysis of extracellular matrix proteins. Mol Cell Proteomics. 2015
130. Evans C, Noirel J, Ow SY, Salim M, Pereira-Medrano AG, Couto N. et al. An insight into iTRAQ: where do we stand now? Anal Bioanal Chem. 2012;404:1011-27
131. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540-55
132. Miwa HE, Koba WR, Fine EJ, Giricz O, Kenny PA, Stanley P. Bisected, complex N-glycans and galectins in mouse mammary tumor progression and human breast cancer. Glycobiology. 2013
133. Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell surface protein glycosylation in cancer. Proteomics. 2014;14:525-46
134. Zhang Y, Jiao J, Yang P, Lu H. Mass spectrometry-based N-glycoproteomics for cancer biomarker discovery. Clin Proteomics. 2014;11:18
135. Sethi M K HWS, Fanayan S. Identifying N-Glycan Biomarkers in Colorectal Cancer by Mass Spectrometry. Acc Chem Res. 2016;49:2099-106
136. Koshiyama A, Ichibangase T, Imai K. Comprehensive fluorogenic derivatization-liquid chromatography/tandem mass spectrometry proteomic analysis of colorectal cancer cell to identify biomarker candidate. Biomed Chromatogr. 2013;27:440-50
137. Reck M, Popat S, Reinmuth N, De Ruysscher D, Kerr KM, Peters S. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014
138. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30
139. Wang CL, Wang CI, Liao PC, Chen CD, Liang Y, Chuang WY. et al. Discovery of retinoblastoma-associated binding protein 46 as a novel prognostic marker for distant metastasis in nonsmall cell lung cancer by combined analysis of cancer cell secretome and pleural effusion proteome. J Proteome Res. 2009;8:4428-40
140. Wang Z, Wang C, Huang X, Shen Y, Shen J, Ying K. Differential proteome profiling of pleural effusions from lung cancer and benign inflammatory disease patients. Biochim Biophys Acta. 2012;1824:692-700
141. Li Y, Lian H, Jia Q, Wan Y. Proteome screening of pleural effusions identifies IL1A as a diagnostic biomarker for non-small cell lung cancer. Biochem Biophys Res Commun. 2015;457:177-82
142. Na SS, Aldonza MB, Sung HJ, Kim YI, Son YS, Cho S. et al. Stanniocalcin-2 (STC2): A potential lung cancer biomarker promotes lung cancer metastasis and progression. Biochim Biophys Acta. 2015;1854:668-76
143. An J, Tang C, Wang N, Liu Y, Guo W, Li X. et al. Preliminary study of MALDI-TOF mass spectrometry-based screening of patients with the NSCLC serum-specific peptides. Zhongguo Fei Ai Za Zhi. 2013;16:233-9
144. Chouaib S, Kieda C, Benlalam H, Noman MZ, Mami-Chouaib F, Ruegg C. Endothelial Cells as Key Determinants of the Tumor Microenvironment: Interaction with Tumor Cells, Extracellular Matrix and Immune Killer Cells. Crit Rev Immunol. 2010;30:529-45
145. Reymond N, d'Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858-70
146. Matsuda K, Ohga N, Hida Y, Muraki C, Tsuchiya K, Kurosu T. et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem Biophys Res Commun. 2010;394:947-54
147. Zhuo H, Lyu Z, Su J, He J, Pei Y, Cheng X. et al. Effect of Lung Squamous Cell Carcinoma Tumor Microenvironment on the CD105+ Endothelial Cell Proteome. J Proteome Res. 2014;13:4717-29
148. Jin H, Cheng X, Pei Y, Fu J, Lyu Z, Peng H. et al. Identification and verification of transgelin-2 as a potential biomarker of tumor-derived lung-cancer endothelial cells by comparative proteomics. J Proteomics. 2016;136:77-88
149. Rovithi M, Lind JSW, Pham TV, Voortman J, Knol JC, Verheul HMW. et al. Response and toxicity prediction by MALDI-TOF-MS serum peptide profiling in patients with non-small cell lung cancer. Proteomics Clin Appl. 2016;10:743-9
150. Walker MJ, Zhou C, Backen A, Pernemalm M, Williamson AJK, Priest LJC. et al. Discovery and Validation of Predictive Biomarkers of Survival for Non-small Cell Lung Cancer Patients Undergoing Radical Radiotherapy: Two Proteins With Predictive Value. EBioMedicine. 2015;2:841-50
151. Birse CE, Lagier RJ, FitzHugh W, Pass HI, Rom WN, Edell ES. et al. Blood-based lung cancer biomarkers identified through proteomic discovery in cancer tissues, cell lines and conditioned medium. Clin Proteomics. 2015;12:18
152. Xiao H, Zhang L, Zhou H, Lee JM, Garon EB, Wong DTW. Proteomic analysis of human saliva from lung cancer patients using two-dimensional difference gel electrophoresis and mass spectrometry. Mol Cell Proteomics. 2012;11:M111.012112
153. Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer. 2016;16:525-37
154. Kim YJ, Sertamo K, Pierrard M-A, Mesmin C, Kim SY, Schlesser M. et al. Verification of the biomarker candidates for non-small-cell lung cancer using a targeted proteomics approach. J Proteome Res. 2015;14:1412-9
155. Hong S, Kang YA, Cho BC, Kim DJ. Elevated Serum C-Reactive Protein as a Prognostic Marker in Small Cell Lung Cancer. Yonsei Med J. 2012;53:111-7
156. Ryan BM, Pine SR, Chaturvedi AK, Caporaso N, Harris CC. A Combined Prognostic Serum Interleukin-8 and Interleukin-6 Classifier for Stage 1 Lung Cancer in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. J Thorac Oncol. 2014;9:1494-503
157. Song G, Liu Y, Wang Y, Ren G, Guo S, Ren J. et al. Personalized biomarkers to monitor disease progression in advanced non-small-cell lung cancer patients treated with icotinib. Clin Chim Acta. 2015;440:44-8
158. Barrera L, Montes-Servín E, Barrera A, Ramírez-Tirado LA, Salinas-Parra F, Bañales-Méndez JL. et al. Cytokine profile determined by data-mining analysis set into clusters of non-small-cell lung cancer patients according to prognosis. Ann Oncol. 2015;26:428-35
159. Sengupta D, Tackett AJ. Proteomic Findings in Melanoma. J Proteomics Bioinform. 2016;9:e29
160. Byrum SD, Larson SK, Avaritt NL, Moreland LE, Mackintosh SG, Cheung WL. et al. Quantitative Proteomics Identifies Activation of Hallmark Pathways of Cancer in Patient Melanoma. J Proteomics Bioinform. 2013;6:043-50
161. Qendro V, Lundgren DH, Rezaul K, Mahony F, Ferrell N, Bi A. et al. Large-Scale Proteomic Characterization of Melanoma Expressed Proteins Reveals Nestin and Vimentin as Biomarkers That Can Potentially Distinguish Melanoma Subtypes. J Proteome Res. 2014;13:5031-40
162. Yuana Y, Sturk A, Nieuwland R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013;27:31-9
163. Riches A, Campbell E, Borger E, Powis S. Regulation of exosome release from mammary epithelial and breast cancer cells - A new regulatory pathway. Eur J Cancer. 2014;50:1025-34
164. Ekström EJ, Bergenfelz C, von Bülow V, Serifler F, Carlemalm E, Jönsson G. et al. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol Cancer. 2014;13:88
165. Hood JL, San RS, Wickline SA. Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis. Cancer Res. 2011;71:3792-801
166. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G. et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883-91
167. Lazar I, Clement E, Ducoux-Petit M, Denat L, Soldan V, Dauvillier S. et al. Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigment Cell Melanoma Res. 2015;28:464-75
168. Angi M, Kalirai H, Coupland S. Proteomic analysis of the uveal melanoma (UM) secretome reveals novel insights and potential biomarkers. Acta Ophthalmol (Copenh). 2015:93
169. Crabb JW, Hu B, Crabb JS, Triozzi P, Saunthararajah Y, Tubbs R. et al. iTRAQ Quantitative Proteomic Comparison of Metastatic and Non-Metastatic Uveal Melanoma Tumors. PLoS One. 2015;10:e0135543
170. Mittal P JM. Proteomics: An Indispensable Tool for Novel Biomarker Identification in Melanoma. J Data Mining Genomics Proteomics. 2016;7:204
171. Mahfoud OK, Rakovich TY, Prina-Mello A, Movia D, Alves F, Volkov Y. Detection of ErbB2: nanotechnological solutions for clinical diagnostics. RSC Adv. 2014;4:3422-42
172. Varga D, Koenig J, Kuhr K, Strunz K, Geyer V, Kurzeder C. et al. Comparison of early onset breast cancer patients to older premenopausal breast cancer patients. Arch Gynecol Obstet. 2010;282:427-32
173. Patris S, De Pauw P, Vandeput M, Huet J, Van Antwerpen P, Muyldermans S. et al. Nanoimmunoassay onto a screen printed electrode for HER2 breast cancer biomarker determination. Talanta. 2014;130:164-70
174. Aslebagh R, Ngounou A, Channaveerappa D, Arcaro K, Darie C. Proteomics Study of Human Breast Milk for Breast Cancer Biomarkers Discovery. FASEB J. 2015:29
175. Dowling P, Henry M, Meleady P, Clarke C, Gately K, O'Byrne K. et al. Metabolomic and proteomic analysis of breast cancer patient samples suggests that glutamate and 12-HETE in combination with CA15-3 may be useful biomarkers reflecting tumour burden. Metabolomics. 2015;11:620-35
176. Maurizio E, Wiśniewski JR, Ciani Y, Amato A, Arnoldo L, Penzo C. et al. Translating Proteomic Into Functional Data: An High Mobility Group A1 (HMGA1) Proteomic Signature Has Prognostic Value in Breast Cancer. Mol Cell Proteomics. 2016;15:109-23
177. Liu NQ, Dekker LJM, Stingl C, Güzel C, De Marchi T, Martens JWM. et al. Quantitative proteomic analysis of microdissected breast cancer tissues: comparison of label-free and SILAC-based quantification with shotgun, directed, and targeted MS approaches. J Proteome Res. 2013;12:4627-41
178. Yang J, Xiong X, Liu S, Zhu J, Luo M, Liu L. et al. Identification of novel serum peptides biomarkers for female breast cancer patients in Western China. Proteomics. 2016;16:925-34
179. Bouchal P, Dvorakova M, Roumeliotis T, Bortlicek Z, Ihnatova I, Struharova I. et al. Combined proteomics and transcriptomics identifies carboxypeptidase B1 and NF-κB associated proteins as putative biomarkers of metastasis in low grade breast cancer. Mol Cell Proteomics. 2015
180. De Marchi T, Liu NQ, Stingl C, Timmermans MA, Smid M, Look MP. et al. 4-protein signature predicting tamoxifen treatment outcome in recurrent breast cancer. Mol Oncol. 2016;10:24-39
181. Mangé A, Dimitrakopoulos L, Soosaipillai A, Coopman P, Diamandis EP, Solassol J. An integrated cell line-based discovery strategy identified follistatin and kallikrein 6 as serum biomarker candidates of breast carcinoma. J Proteomics. 2016;142:114-21
182. Henderson MC, Hollingsworth AB, Gordon K, Silver M, Mulpuri R, Letsios E. et al. Integration of Serum Protein Biomarker and Tumor Associated Autoantibody Expression Data Increases the Ability of a Blood-Based Proteomic Assay to Identify Breast Cancer. PLoS One. 2016;11:e0157692
183. Beretov J, Wasinger VC, Millar EKA, Schwartz P, Graham PH, Li Y. Proteomic Analysis of Urine to Identify Breast Cancer Biomarker Candidates Using a Label-Free LC-MS/MS Approach. PLoS One. 2015;10:e0141876
184. Romero I, Bast RC. Minireview: human ovarian cancer: biology, current management, and paths to personalizing therapy. Endocrinology. 2012;153:1593-602
185. Ezzati M, Abdullah A, Shariftabrizi A, Hou J, Kopf M, Stedman JK. et al. Recent Advancements in Prognostic Factors of Epithelial Ovarian Carcinoma. Int Sch Res Notices. 2014;2014:10
186. Leung F, Bernardini MQ, Clarke B, Rouzbahman M, Diamandis EP, Kulasingam V. Abstract B13: Discovery of novel subtype-specific ovarian cancer biomarkers via integrated tissue proteomics. Clin Cancer Res. 2016;22:B13-B
187. Nepomuceno AI, Shao H, Jing K, Ma Y, Petitte JN, Idowu MO. et al. In-depth LC-MS/MS analysis of the chicken ovarian cancer proteome reveals conserved and novel differentially regulated proteins in humans. Anal Bioanal Chem. 2015;407:6851-63
188. Poersch A, Grassi ML, Carvalho VPd, Lanfredi GP, Palma CdS, Greene LJ. et al. A proteomic signature of ovarian cancer tumor fluid identified by highthroughput and verified by targeted proteomics. J Proteomics. 2016;145:226-36
189. Deng J, Wang L, Ni J, Beretov J, Wasinger V, Wu D. et al. Proteomics discovery of chemoresistant biomarkers for ovarian cancer therapy. Expert Rev Proteomics. 2016;13:905-15
190. Chappell NP, Teng P-N, Hood BL, Wang G, Darcy KM, Hamilton CA. et al. Mitochondrial proteomic analysis of cisplatin resistance in ovarian cancer. J Proteome Res. 2012;11:4605-14
191. Zhang SF, Wang XY, Fu ZQ, Peng QH, Zhang JY, Ye F. et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy. 2015;11:225-38
192. Shetty V, Nickens Z, Testa J, Hafner J, Sinnathamby G, Philip R. Quantitative immunoproteomics analysis reveals novel MHC class I presented peptides in cisplatin-resistant ovarian cancer cells. J Proteomics. 2012;75:3270-90
193. Zhang H, Liu T, Zhang Z, Payne Samuel H, Zhang B, McDermott Jason E. et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell. 2016;166:755-65
194. Lin SN, Taylor J, Alperstein S, Hoda R, Holcomb K. Does speculum lubricant affect liquid-based Papanicolaou test adequacy? Cancer Cytopathol. 2014;122:221-6
195. Bakkum-Gamez J, Dowdy S. Retooling the Pap Smear for Ovarian and Endometrial Cancer Detection. Clin Chem. 2014;60:22-4
196. Boylan KL, Afiuni-Zadeh S, Geller MA, Hickey K, Griffin TJ, Pambuccian SE. et al. A feasibility study to identify proteins in the residual Pap test fluid of women with normal cytology by mass spectrometry-based proteomics. Clin Proteomics. 2014;11:30
197. Skubitz APN, Afiuni S, Boylan KLM, Geller M, Argenta P, Hoffman S. et al. Abstract B34: Tandem Mass Tag 10-plex isobaric labeling of Pap test proteins: A novel method for the identification of ovarian cancer protein biomarkers by mass spectrometry. Clin Cancer Res. 2016;22:B34-B
198. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
199. Tanase CP, Codrici E, Popescu ID, Mihai S, Enciu A-M, Necula LG. et al. Prostate cancer proteomics: Current trends and future perspectives for biomarker discovery. Oncotarget. 2017;8:18497
200. Zhao Z, Wu F, Ding S, Sun L, Liu Z, Ding K. et al. Label-free quantitative proteomic analysis reveals potential biomarkers and pathways in renal cell carcinoma. Tumor Biol. 2015;36:939-51
201. Øvrehus MA, Zürbig P, Vikse BE, Hallan SI. Urinary proteomics in chronic kidney disease: diagnosis and risk of progression beyond albuminuria. Clin Proteomics. 2015;12:21
202. Sandim V, Pereira DdA, Kalume DE, Oliveira-Carvalho AL, Ornellas AA, Soares MR. et al. Proteomic analysis reveals differentially secreted proteins in the urine from patients with clear cell renal cell carcinoma. Urol Oncol. 2016;34:5.e11-5.e25
203. Neely BA, Wilkins CE, Marlow LA, Malyarenko D, Kim Y, Ignatchenko A. et al. Proteotranscriptomic Analysis Reveals Stage Specific Changes in the Molecular Landscape of Clear-Cell Renal Cell Carcinoma. PLoS One. 2016;11:e0154074
204. Robinson D, Van Allen Eliezer M, Wu YM, Schultz N, Lonigro Robert J, Mosquera J-M. et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;161:1215-28
205. Øverbye A, Skotland T, Koehler CJ, Thiede B, Seierstad T, Berge V. et al. Identification of prostate cancer biomarkers in urinary exosomes. Oncotarget. 2015;6:30357-76
206. Kim Y, Jeon J, Mejia S, Yao CQ, Ignatchenko V, Nyalwidhe JO. et al. Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nat Commun. 2016;7:11906
207. Street JM, Koritzinsky EH, Glispie DM, Star RA, Yuen PST. Urine Exosomes: An Emerging Trove of Biomarkers. Advances in Clinical Chemistry: Elsevier. 2016
208. Frantzi M, Latosinska A, Fluhe L, Hupe MC, Critselis E, Kramer MW. et al. Developing proteomic biomarkers for bladder cancer: towards clinical application. Nat Rev Urol. 2015;12:317-30
209. Guo J, Ren Y, Hou G, Wen B, Xian F, Chen Z. et al. A Comprehensive Investigation toward the Indicative Proteins of Bladder Cancer in Urine: From Surveying Cell Secretomes to Verifying Urine Proteins. J Proteome Res. 2016;15:2164-77
210. Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25-41
211. Lin SY, Chang CH, Wu HC, Lin CC, Chang KP, Yang CR. et al. Proteome Profiling of Urinary Exosomes Identifies Alpha 1-Antitrypsin and H2B1K as Diagnostic and Prognostic Biomarkers for Urothelial Carcinoma. Sci Rep. 2016;6:34446
212. Liu M, Hu Z, Qi L, Wang J, Zhou T, Guo Y. et al. Scanning of novel cancer/testis proteins by human testis proteomic analysis. Proteomics. 2013;13:1200-10
213. Maes E, Mertens I, Valkenborg D, Pauwels P, Rolfo C, Baggerman G. Proteomics in cancer research: Are we ready for clinical practice? Crit Rev Oncol Hematol. 2015;96:437-48
214. Kern SE. Why your new cancer biomarker may never work: recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012;72:6097-101
215. Pavlou MP, Diamandis EP, Blasutig IM. The Long Journey of Cancer Biomarkers from the Bench to the Clinic. Clin Chem. 2013;59:147-57
216. Diamandis EP. Cancer biomarkers: can we turn recent failures into success?. J Natl Cancer Inst. 2010:102
217. Diamandis EP. The failure of protein cancer biomarkers to reach the clinic: why, and what can be done to address the problem? BMC Med. 2012;10:87
218. Diamandis Eleftherios P. Present and future of cancer biomarkers. Clin Chem Lab Med. 2014;52:791
219. Mordente A, Meucci E, Martorana GE, Silvestrini A. Cancer Biomarkers Discovery and Validation: State of the Art, Problems and Future Perspectives. In: Scatena R, editor. Advances in Cancer Biomarkers: From biochemistry to clinic for a critical revision. Dordrecht: Springer Netherlands. 2015:9-26
220. Wan-Ibrahim WI, Singh VA, Hashim OH, Abdul-Rahman PS. Biomarkers for Bone Tumors: Discovery from Genomics and Proteomics Studies and Their Challenges. Mol Med. 2015;21:861-72
221. Cramer D W BRC, Berg C D. et al. Ovarian cancer biomarker performance in prostate, lung, colorectal, and ovarian cancer screening trial specimens. Cancer Prev Res. 2011;4:365-74
222. Molina R EJM, Augé J M. et al. HE4 a novel tumour marker for ovarian cancer: comparison with CA 125 and ROMA algorithm in patients with gynaecological diseases. Tumor Biol. 2011;32:1087-95
223. Hernández B, Parnell A, Pennington SR. Why have so few proteomic biomarkers “survived” validation? (Sample size and independent validation considerations). Proteomics. 2014;14:1587-92
224. Füzéry AK, Levin J, Chan MM, Chan DW. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics. 2013;10:13
Author contact
![]() Corresponding authors: nyheedu.cn (N. He); hndengyancom (Y. Deng); august555482com (X. Zeng).
Corresponding authors: nyheedu.cn (N. He); hndengyancom (Y. Deng); august555482com (X. Zeng).
Citation styles
APA
Huang, R., Chen, Z., He, L., He, N., Xi, Z., Li, Z., Deng, Y., Zeng, X. (2017). Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: approaches and application. Theranostics, 7(14), 3559-3572. https://doi.org/10.7150/thno.20797.
ACS
Huang, R.; Chen, Z.; He, L.; He, N.; Xi, Z.; Li, Z.; Deng, Y.; Zeng, X. Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: approaches and application. Theranostics 2017, 7 (14), 3559-3572. DOI: 10.7150/thno.20797.
NLM
Huang R, Chen Z, He L, He N, Xi Z, Li Z, Deng Y, Zeng X. Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: approaches and application. Theranostics 2017; 7(14):3559-3572. doi:10.7150/thno.20797. https://www.thno.org/v07p3559.htm
CSE
Huang R, Chen Z, He L, He N, Xi Z, Li Z, Deng Y, Zeng X. 2017. Mass spectrometry-assisted gel-based proteomics in cancer biomarker discovery: approaches and application. Theranostics. 7(14):3559-3572.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.