Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(14):3415-3431. doi:10.7150/thno.20861 This issue Cite
Research Paper
Brusatol-Mediated Inhibition of c-Myc Increases HIF-1α Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia
Eun-Taex Oh1,2*, Chan Woo Kim3*, Ha Gyeong Kim3, Jae-Seon Lee4, Heon Joo Park2,3 ![]()
1. Department of Biomedical Sciences, College of Medicine, Inha University, Incheon 22212, Republic of Korea;
2. Hypoxia-related Disease Research Center, College of Medicine, Inha University, Incheon 22212, Republic of Korea;
3. Department of Microbiology, College of Medicine, Inha University, Incheon 22212, Republic of Korea;
4. Department of Molecular Medicine, College of Medicine, Inha University, Incheon 22212, Republic of Korea.
* E.-T. Oh and C.W. Kim contributed equally to this article
Received 2017-5-4; Accepted 2017-6-20; Published 2017-8-11
Citation:
Oh ET, Kim CW, Kim HG, Lee JS, Park HJ. Brusatol-Mediated Inhibition of c-Myc Increases HIF-1α Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia. Theranostics 2017; 7(14):3415-3431. doi:10.7150/thno.20861. https://www.thno.org/v07p3415.htm
Other stylesAbstract

HIF-1 (hypoxia-inducible factor-1) regulates the expression of ~100 genes involved in angiogenesis, metastasis, tumor growth, chemoresistance and radioresistance, underscoring the growing interest in targeting HIF-1 for cancer control. In the present study, we investigated the molecular mechanisms underlying brusatol-induced HIF-1α degradation and cell death in colorectal cancer under hypoxia (0.5% O2). Under hypoxia, pretreatment of cancer cells with brusatol increased HIF-1α degradation and cancer cell death in a dose-dependent manner. This effect was mediated by activation of prolyl hydroxylases (PHDs), as evidenced by the block of brusatol-induced HIF-1α degradation and cancer cell death by both pharmacological inhibition and siRNA-mediated knockdown of PHDs. In addition, a ferrous iron chelator (2,2'-bypyridyl) blocked brusatol-induced degradation of HIF-1α and cancer cell death in hypoxia by inhibiting PHD activation. We further found that brusatol inhibited c-Myc expression, and showed that overexpression of c-Myc prevented brusatol-induced degradation of HIF-1α and cancer cell death by increasing mitochondrial ROS production and subsequent ROS-mediated transition of ferrous iron to ferric iron. Consistent with these results, treatment of tumor-bearing mice with brusatol significantly suppressed tumor growth by promoting PHD-mediated HIF-1α degradation. Collectively, our results suggest that brusatol-mediated inhibition of c-Myc/ROS signaling pathway increases HIF-1α degradation by promoting PHD activity and induces cell death in colorectal cancer under hypoxia
Keywords: Brusatol, Cell death, Colorectal cancer, HIF-1α, Hypoxia.
Introduction
Brusatol is a type of quassinoid obtained from Brucea javanica, an evergreen shrub grown in Southeast Asia (1). It is known that brusatol and structurally related molecules, including bruceantin, bruceoside and bruceins, are capable of inducing an array of biological responses, exerting anti-inflammatory, antileukemia, and anticancer activities (1,2). The major mechanism responsible for the anticancer activity of brusatol at high micromolar concentrations is inhibition of protein synthesis (1-4). Importantly, diverse protein targets of brusatol have recently been reported to be involved in its anticancer activity. For example, brusatol has been shown to inhibit c‑Myc expression in leukemic cells, thereby increasing cytotoxicity of the cells (2), and increase the efficacy of chemotherapy by inhibiting NRF2 (nuclear factor-erythroid 2-related factor-2) expression in cancer cells (5). Additionally, it has been reported that brusatol regulates HIF-1α (hypoxia-inducible factor-1) protein, but not mRNA expression (6). However, the mechanism by which brusatol downregulates HIF‑1α protein expression and induces cancer cell death under hypoxic conditions is unclear.
Hypoxia is a common characteristic of tumor microenvironment that activates HIF signaling pathway in cancer cells (7). It has been reported that, under hypoxia, HIFs are overexpressed in various cancer cells (7), where they regulate the expression of a number of genes involved in angiogenesis, tumor growth, metastasis, metabolic reprogramming, chemoresistance, and radioresistance (7-12). HIFs are heterodimeric transcription factors that consist of one of three oxygen-regulated α-subunits, HIF-1α, HIF-2α and HIF-3α, and a constitutively expressed hydrocarbon receptor/nuclear translocator β subunit, HIF-1β (7, 13-17). HIF-1 function requires formation of a HIF-1α/HIF-1β heterodimer, in which HIF-1α serves as the major regulatory subunit responsible for the transcriptional function of the complex (8). HIF-1α expression is rapidly induced by hypoxia; upon reoxygenation of hypoxic cells, the protein rapidly degrades, with a half-life of less than 10 min (18). In well-oxygenated cells, the oxygen-dependent degradation (ODD) domain of HIF-1α is hydroxylated by three prolyl hydroxylases (PHD1-3), which utilize O2, ferrous iron, and α-ketoglutarate as substrates (18,19). The von Hippel-Lindau (pVHL) protein, which functions as an E3 ligase, binds to hydroxylated HIF-1α and recruits an E3 ubiquitin ligase complex that contains elongin-B, elongin-C, and cullin 2, thereby promoting ubiquitination and 26S proteasome-mediated degradation of HIF-1α (16,20,21). In hypoxic conditions, HIF-1α is not hydroxylated, preventing its interaction with pVHL and subsequent ubiquitination and proteasomal degradation (22). Following hypoxia-induced stabilization, HIF-1α is translocated into the nucleus with HIF‑1β and binds to hypoxia-response elements (HREs), thereby increasing the transcription of approximately 100 genes involved in cellular adaptation and survival under hypoxic conditions (7, 13, 22).
As mentioned, HIF-1 is a major regulator protein in the tumor survival and progression, as well as chemoreisistance and radioresistance (13). Therefore, HIF-1 has been regarded as an attractive target for cancer therapy over the past several years (6, 13). Recently, specific inhibitors targeting HIF-1 by inhibiting its mRNA expression, protein synthesis, protein stability and transactivation have been identified (6). In this present study, we investigated the mechanisms by which brusatol regulates HIF-1α expression and induces cancer cell death under hypoxic conditions. We show that brusatol induces a dose-dependent inhibition of HIF-1α expression through a mechanism that involves decreased c-Myc and hypoxia-mediated mitochondrial reactive oxygen species (ROS) production. The reduction in mitochondrial ROS, in turn, decreases HIF-1α stability in hypoxic cancer cells by inhibiting hypoxia-mediated transition of ferrous iron to ferric iron, thereby inducing cancer cell death. Collectively, these findings reveal a novel mechanism by which brusatol downregulates HIF-1α, thereby killing cancer cells in hypoxia.
Materials and Methods
Cell lines and culture conditions
The human colorectal cancer cell lines RKO, HCT116, SW480, CoLo205, DLD-1, HT29, and HCT15 were obtained from American Type Culture Collection (ATCC) and cultured in Dulbecco's Modified Eagle Medium (DMEM; Invitrogen) or RPMI-1640 (Invitrogen), as recommended by the supplier. Cells were incubated at 37°C in a humidified 5% CO2 incubator unless otherwise noted. Hypoxic conditions were achieved by incubating cells in an InvivO2 500 hypoxia workstation (The Baker Company). Oxygen concentrations were fixed using a gas mixture containing 5% CO2 with a balance of nitrogen.
Chemicals and antibodies
Brusatol was purchased from Tauto Biotech (Shanghai, China); cycloheximide (CHX) and MG132 were purchased from Calbiochem; 2,2'-bipyridiyl was purchased from Sigma-Aldrich; and dimethyloxalylglycine (DMOG) was purchased from Santa Cruz Biotechnology. Primary antibodies against the following proteins were used: HIF-1α (R&D Systems), HIF-1β (Cell Signaling Technology), PHD1 (R&D Systems), PHD2 (Cell Signaling Technology), PHD3 (Novus Biologicals), and c-Myc (Cell Signaling Technology). Secondary antibodies used for immunoblotting include horseradish peroxidase (HRP)-conjugated anti-mouse (Cell Signaling Technology), anti-rabbit (Cell Signaling Technology), and anti-goat (Upstate).
Immunoblot analysis
Cells were lysed using ice-cold RIPA buffer containing a protease inhibitor cocktail (Roche Applied Science), sodium orthovanadate (Sigma-Aldrich), and sodium fluoride (Sigma-Aldrich). Proteins in whole-cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by immunoblotting. Signals were detected using enhanced chemiluminescence reagents (Pierce).
RNA isolation and quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from RKO and HCT116 cells using the TRIzol reagent (Invitrogen) and treated with DNase I (New England Biolabs). cDNA was synthesized from (1 μg) of total RNA using AccuPower RT PreMix (Bioneer, Daejeon, Republic of Korea), and then amplified by PCR using the appropriate primers for Glut1 (glucose transporter 1), PDK1 (pyruvate dehydrogenase kinase 1), PGK1 (phosphoglycerate kinase 1), CA9 (carbonic anhydrase 9), HIF-1α, and 18S rDNA (Bioneer). qPCR and analyses were performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad).
Plasmid construction
The c-Myc expression plasmid was constructed by first obtaining total RNA from RKO cells using the TRIzol reagent (Invitrogen) and then generating cDNA using SuperScriptIII Reverse Transcriptase (Invitrogen). The open reading frame of c-Myc was subsequently amplified by PCR using the appropriate primers, and the resulting PCR products were digested with restriction enzymes and directly ligated into the pCDNA3.1 (Invitrogen) vector. The cloned plasmids were analyzed by restriction enzyme digestion and DNA sequencing (Bionics, Seoul, Republic of Korea).
Transfection assays
Cells (5 × 104) were seeded in 25-cm2 flasks, incubated overnight, and co-transfected with a 50-μl mixture containing 1 μg pODD-luc (Addgene) or p3×HRE‑luc (Addgene) plasmid, 0.1 μg pCMV-β-galactosidase plasmid (transfection control; Stratagene), and TurboFect in vitro transfection reagent (Fermentas). After 16 h, cells were exposed to 20% or 0.5% O2 for 8 h. Luciferase activity was determined using a Luciferase assay system (Promega) and normalized with respect to β-galactosidase activity, assessed using a β-galactosidase enzyme assay system (Promega), according to the manufacturer's instructions. Three independent transfections were performed in each trial.
Measurement of O2 consumption
Cells (5 × 104), seeded in 96-well plates and incubated overnight, were incubated with or without 100 nM brusatol and exposed to 0.5% O2 for 8 h. O2 consumption was determined using a Mito-ID O2 Extracellular Sensor Kit (Enzo), as described by the manufacturer, and normalized to the protein concentration.
siRNA transfection
PHD1, PHD2, and PHD3 were knocked down by RNA interference (RNAi) using the following 19-bp (including a 2-deoxynucleotide overhang) small interfering RNAs (siRNAs; Bioneer Corporation): PHD1, 5ʹ-GACAAGUAUCAGCUAGCAUdTdT; PHD2, 5ʹ-GAGUAGAGCAUAUAGAGAUdTdT; and PHD3, 5ʹ-CGUGUAUCGUUCCCUCUdTdT. Stealth RNAi (Invitrogen) was used as a negative control (siCont). For transfection, cells were seeded in 25-cm2 flasks, grown to ~80% confluence, and transfected with siRNA duplexes using LipofectAMINE 2000 (Invitrogen) following the manufacturer's instructions. After a 48-h incubation, cells were processed as indicated for analysis.
Measurement of iron
Cells were harvested from confluent 75-cm2 flasks for each analysis. Levels of ferrous iron and total iron were analyzed in cell lines using a ferrous iron measurement kit from Abcam as described by the manufacturer.
Measurement of mitochondrial ROS production
Cells were seeded onto an 8-well chamber slide and treated with MitoTracker Green (Invitrogen) for 30 min. The cells were then washed with phosphate-buffered saline (PBS), incubated with 100 nM brusatol and MitoSOX Red (Invitrogen) for 1 h, and exposed to 20% or 0.5% O2 for 4 h. The resulting fluorescence was detected with a Nikon confocal laser-scanning microscope.
Quantification of clonogenic death
Various numbers of cells were plated on 60-mm dishes and treated with a range of concentrations of brusatol (0-100 nM), DMOG (1 mM), 2,2'-bipyridyl (200 μM), or FeCl2 (200 μM) for 1 h. The cells were further incubated under hypoxia for 4 h, gently washed three times with medium, and cultured for 14 d with the standard DMEM at 37°C in a 5% CO2 incubator to allow colonies to form. Cells in colonies were fixed in 95% methanol, stained with 0.5% crystal violet, and the numbers of colonies (≥50 cells/colony) from triplicate dishes were counted. Mean colony numbers were plotted relative to those formed by untreated cells.
Xenograft tumor model
All procedures were carried out according to the Institutional Animal Care and Use Committee protocol approved for this study by Inha University (INHA 150605-363). Eight-week-old, male nude mice (BALB/c-nu) were purchased from Orient Bio Laboratory Animal Inc. (Seoul, Korea) and maintained in a room at 25°C with a 12-h light/12-h dark cycle with ad libitum access to sterile water and food. Tumor xenografts were generated by injecting RKO or HCT116 cells (5 × 106 cells/mouse) subcutaneously into the right flank of male nude mice. Mice were randomized into three groups (n = 7 mice/group), and administered brusatol (2 and 4 mg/kg) by intraperitoneal injection three times a week for 32 d. Tumor sizes were measured every 3 to 4 d using a digital caliper, and tumor volume was calculated using the following formula: V = length × width2/2. Mice were monitored daily for evidence of disease or death. Mice were killed after 32 d and tumors were harvested.
Immunohistochemistry and immunofluorescence staining
Tissues were fixed in a buffered formalin solution and embedded in paraffin. Sections (4 μm thickness) were dewaxed and rehydrated with a graded ethanol series. Antigen was retried by heating the slides for 10 min in a microwave oven in 10 mM citrate buffer (pH 6.0). After blocking endogenous peroxidase activity by incubation in 3% hydrogen peroxide in PBS for 10 min, sections were incubated overnight at 4°C with anti-HIF-1α antibody (Sigma-Aldrich). HIF-1α immunohistochemistry was performed using an indirect immunoperoxidase method. For immunofluorescence staining, sections were incubated with an anti-c-Myc antibody (Abcam), and confocal images were captured on a Zeiss LSM 510 Confocal System (Carl Zeiss Inc.) using Zeiss AIM software. Sections were examined microscopically, and representative fields of view were imaged under 400× magnification.
TUNEL assay
Apoptosis in xenograft tumors was measured by TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) assay using a kit (Roche Life Science) according to the manufacturer's instructions. Briefly, tissue sections were incubated with proteinase K (20 µg/ml in 10 mM Tris-HCl, pH 7.4) for 15 min at 37°C. DNA breaks were then labeled with terminal deoxytransferase (TdT) and biotinylated deoxy UTP. Endogenous peroxidase activity was quenched by incubation with 3% hydrogen peroxide.
Statistical analysis
All immunoblots are representative of at least three separate experiments. All grouped data are presented as means ± SD. Differences between groups were analyzed by analysis of variance (ANOVA) or Student's t-test, as appropriate, using GraphPad Prism software.
Results
Effects of brusatol on HIF-1α expression in colorectal cancer cells under hypoxia
To investigate the effects of brusatol on HIF expression in colorectal cancer cells, we incubated RKO and HCT116 cells with brusatol (0-100 nM) for 1 h, then further exposed the cells to 20% or 0.5% O2 for 4 h, and assessed HIF-1α and HIF-1β expression by immunoblot analysis. Brusatol caused a concentration-dependent decrease in HIF-1α expression in RKO cells under hypoxia, but did not alter HIF-1β levels (Figure 1A and 1B). To confirm these findings, we incubated RKO and HCT116 cells with or without 100 nM brusatol for 1 h, then further exposed the cells to 20% or 0.5% O2 for 8 h, and assessed HIF-1α and HIF-1β expression by immunoblot analysis. HIF-1α expression was markedly increased in RKO (Figure 1C) and HCT116 (Figure 1D) cells exposed to 0.5% O2, exhibiting a peak increase at 4 h, followed by a gradual decline in expression (Figure 1C and 1D). Brusatol (100 nM) also effectively inhibited HIF-1α expression in both cell lines under hypoxic conditions; by contrast, HIF-1β remained unaffected regardless of oxygen concentration (Figure 1C and 1D). Similar results were observed in SW480, CoLo205, DLD-1, HT29, and HCT15 colorectal cancer cell lines (Figure S1).
We next evaluated the effects of brusatol on the transcriptional activity of HIF-1α, which regulates numerous hypoxia-adaptive genes that contain HREs in their promoters (7, 13). We treated RKO and HCT116 cells with 100 nM brusatol, and analyzed the expression of HIF-1α target genes using qPCR. As shown in Figure 1E and 1F, brusatol pretreatment under hypoxia decreased the expression of HIF-1α target genes, including Glut1, PGK1, PDK1, and CA9. To confirm these results, we transfected RKO and HCT116 cells with a reporter plasmid (p3×HRE-luc) and pCMV-β-galactosidase (transfection control), treated them with brusatol, and then exposed them to 20% or 0.5% O2 for 8 h. Brusatol reduced luciferase activity by ~90% under hypoxia (Figure 1G and 1H). Taken together, these data indicate that brusatol treatment decreases HIF-1α expression, thereby decreasing HIF-1α-mediated transcription and expression of downstream target genes.
Figure 1
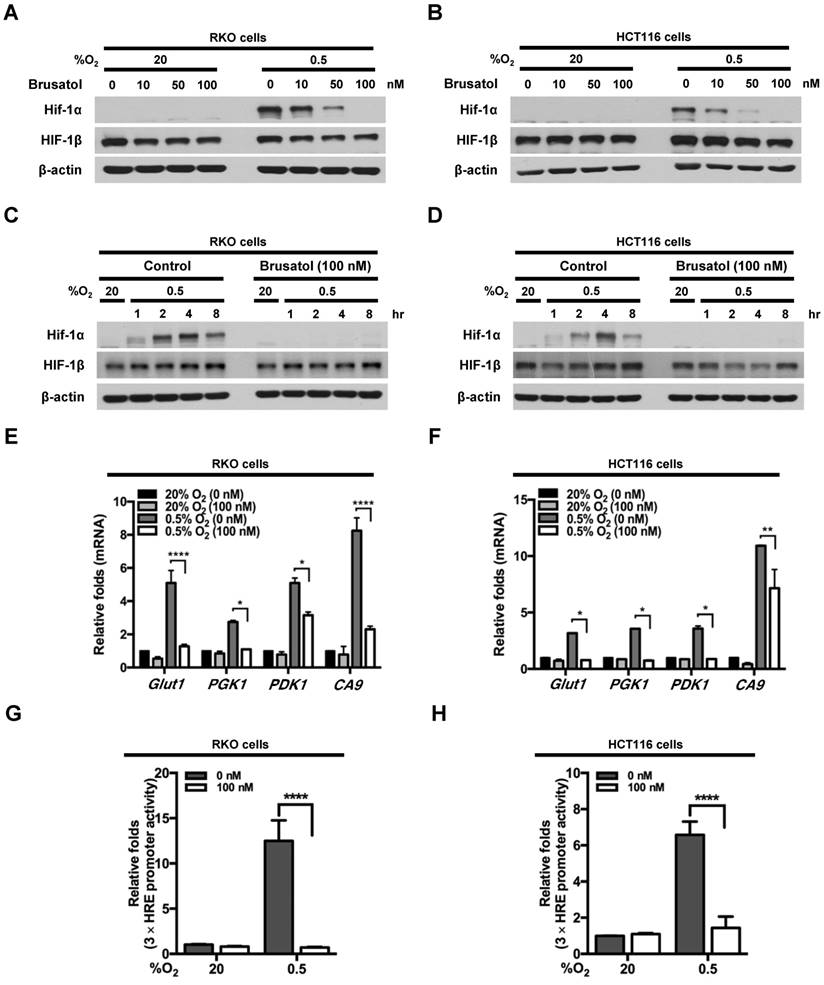
Effects of brusatol on HIF-1α expression in colorectal cancer cells under hypoxia. (A and B) RKO (A) and HCT116 (B) cells were treated with different concentrations of brusatol (0-100 nM) for 1 h and exposed to 20% or 0.5% O2. After a 4-h incubation, cells were harvested and whole-cell lysates were analyzed by immunoblotting for the indicated proteins. (C and D) RKO (C) and HCT116 (D) cells were incubated with or without 100 nM brusatol. After an 8 h exposure to 20% or 0.5% O2, cells were harvested at the indicated times. (E and F) RKO (E) and HCT116 (F) cells were incubated with or without 100 nM brusatol for 1 h, exposed to 20% or 0.5% O2 for 8 h, and then harvested. qPCR was used to amplify Glut1, PGK1, PDK1, and CA9; 18S rRNA was used as an internal control. Data are presented as means ± SD (*P < 0.05, **P < 0.01, ****P < 0.0001; ANOVA). (G and H) RKO (G) and HCT116 (H) cells were co-transfected with p3×HRE-luc and pCMV-β-galactosidase, cultured for 16 h, and then incubated with or without 100 nM brusatol for 1 h. Cells were then exposed to 20% or 0.5% O2 for 8 h. Luciferase activity measured in whole-cell lysates was normalized to that of β-galactosidase. Data are presented as means ± SD (****P < 0.0001; ANOVA).
Brusatol decreases HIF-1α protein stability in colorectal cancer cells under hypoxia
To determine how brusatol regulates HIF-1α expression, we quantified HIF-1α mRNA levels in RKO and HCT116 cells following treatment with brusatol. As shown in Figure 2A and 2B, HIF-1α mRNA levels were unaffected by brusatol treatment under either normoxic or hypoxic conditions. Therefore, we investigated whether brusatol affects the accumulation and stability of HIF-1α protein. We first exposed RKO and HCT116 cells to 0.5% O2 for 4 h to obtain maximum HIF-1α expression. Then, we treated the cells with or without 10 μg/ml CHX, the protein synthesis inhibitor, or 100 nM brusatol, and further incubated them for 3 h under hypoxia. The cells were harvested at the indicated times, and the decay in HIF-1α protein over time was measured by immunoblot analysis. HIF-1α was degraded in a time-dependent (1-3 h) manner in the presence of 10 μg/ml CHX and 100 nM brusatol, with a more rapid increase in degradation observed after 2 h (Figure 2C and 2D). A previous report found that HIF-1α degradation is regulated by the proteasome system (13). To determine whether brusatol increases proteasome-mediated degradation of HIF-1α under hypoxia, we treated RKO and HCT116 cells with or without the proteasome inhibitor MG132. After 1 h, we treated with or without the 100 nM brusatol and exposed them to 0.5% O2 for 4 h. Then, the level of HIF-1α was assessed by immunoblot analysis. As shown in Figure S2, MG132 treatment inhibited brusatol-induced degradation of HIF-1α in colorectal cancer cells under hypoxia. Collectively, these results demonstrate that brusatol increases proteasome-mediated HIF-1α degradation in hypoxic cells.
Figure 2
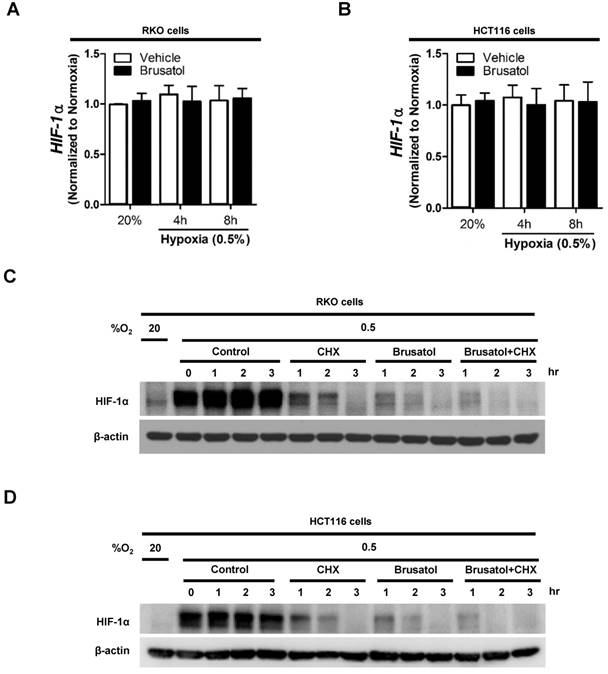
Brusatol decreases HIF-1α protein stability in colorectal cancer cells under hypoxia. (A and B) RKO (A) and HCT116 (B) cells were incubated with or without 100 nM brusatol and exposed to 20% or 0.5% O2. After an 8 h incubation, cells were harvested at the indicated times. qPCR was used to amplify HIF-1α; 18S rRNA was used as the internal control. (C and D) RKO (C) and HCT116 (D) cells were exposed to 0.5% O2 for 2 h, incubated with 10 μg/ml CHX for 3 h, and then harvested at the indicated times. HIF-1α protein levels were examined by immunoblot analysis; β-actin was used as an internal control.
PHDs are required for brusatol-induced HIF-1α degradation in colorectal cancer cells under hypoxia
In our previous report, we reported that the hydroxylation activity of PHDs is maintained in cancer cells under hypoxia (7, 13). Therefore, we investigated whether brusatol-induced HIF-1α degradation in hypoxic cancer cells was mediated by activation of PHDs. To this end, we treated RKO and HCT116 cells with DMOG, a PHD inhibitor, in the presence or absence of 100 nM brusatol, and exposed these cells to 0.5% O2 for 4 h. As shown in Figure 3A and 3B, DMOG prevented the brusatol-induced decrease in HIF1α expression in colorectal cancer cells under hypoxia. To define the potential contribution of individual PHDs to the regulation of HIF-1α in brusatol-treated cells under hypoxic conditions, we transfected RKO and HCT116 cells with siRNAs targeting PHD1 (siPHD1), PHD2 (siPHD2), or PHD3 (siPHD3). Next, the cells were incubated with or without 100 nM brusatol and exposed to 0.5% O2 for 4 h. siRNAs targeting PHDs prevented the brustol-induced HIF-1α degradation in hypoxic cancer cells (Figure 3C and 3D). In particular, the restoration of HIF-1α in siPHD2 treated cells was significantly higher than that in siPHD1 or siPHD3 treated cells (Figure 3C and 3D). To further validate this, we transfected RKO and HCT116 cells with siPHD1, siPHD2, or siPHD3. We then transfected these cells with p3×HRE-luc or pODD-luc reporter constructs together with pCMV-β-galactosidase, treated them with brusatol, and exposed them to 20% or 0.5% O2 for 8 h. Luciferase activity increased mainly following treatment with siRNAs under hypoxia (Figure 3C and 3D), confirming the immunoblot analysis results.
In a previous report, inhibition of mitochondrial function was shown to allow PHDs to remain active in low-oxygen environments (13), supporting the possibility that brusatol treatment reduces mitochondrial function and thereby decreases oxygen consumption. To investigate whether mitochondrial function is impaired in brusatol-treated cells, we exposed RKO and HCT116 cells to 0.5% O2 for 8 h in the absence or presence of 100 nM brusatol, and monitored oxygen consumption. Brusatol treatment reduced oxygen consumption by as much as 70% (Figure 3G and 3H). These results indicate that brusatol decreases HIF-1α protein stability in hypoxic colorectal cancer cells by activating PHDs.
Brusatol increases PHD-mediated HIF-1α degradation by inhibiting hypoxia-induced transition of ferrous iron to its ferric state
Mitochondrial ROS are essential for O2 sensing and subsequent HIF-1α stabilization in hypoxic cells (23), and increased ROS production increases the hypoxia-mediated transition of ferrous iron to its ferric state (24). The increase of ferric iron facilitates the rapid inhibition of PHD hydroxylase activity (25). As shown above (Figure 3), brusatol increased the activation of PHDs and the degradation of HIF-1α. Therefore, we investigated whether brusatol affects mitochondrial ROS production and cellular Fenton chemistry. To this end, we treated RKO and HCT116 cells with 2,2'-bipyridyl, a chelator of ferrous iron, in the presence or absence of 100 nM brusatol, and exposed these cells to 0.5% O2 for 4 h. As shown in Figure 4A and 4B, 2,2'-bipyridyl effectively inhibited the brusatol-induced degradation of HIF-1α under hypoxia. To validate this observation, we treated RKO and HCT116 cells with 200 μM FeCl2 in the presence of 100 nM brusatol and 200 μM 2,2'-bipyridyl, and exposed these cells to 0.5% O2 for 4 h. As shown in Figure 4C and 4D, FeCl2 blunted the 2,2'-bipyridyl-mediated restoration of HIF-1α in brusatol-treated hypoxic cells. To further confirm this, we transfected RKO and HCT116 cells with p3×HRE‑luc or pODD-luc reporter constructs together with pCMV-β-galactosidase, and then treated cells with 100 nM brusatol, 200 μM 2,2'-bipyridyl, or 200 μM FeCl2, and exposed them to 0.5% O2 for 8 h (Figure 4E and 4F). Consistent with the results of immunoblot analyses, brusatol inhibited transactivation of HIF-1α and increased its hydroxylation. Next, we investigated whether brusatol affects mitochondrial ROS production in hypoxic cells. We first treated RKO and HCT116 cells with MitoTracker Green and MitoSOX Red, and then with 100 nM brusatol. Next, the cells were exposed to 0.5% O2 for 4 h. As shown in Figure 4G and 4H, brusatol decreased the hypoxia-induced production of mitochondrial ROS. To define the potential contribution of brusatol-induced inhibition of ROS production to the transition of ferrous iron to its ferric state, we treated RKO and HCT116 cells with 100 nM brusatol and exposed them to 0.5% O2 for 4 h. As shown in Figure 4I and 4J, brusatol treatment increased intracellular ferrous iron by inhibiting ROS production in hypoxic cells. These results indicate that brusatol decreases HIF-1α protein stability in hypoxic cancer cells through activation of PHDs by inhibiting mitochondrial ROS production and the subsequent transition of ferrous iron to ferric iron.
Figure 3
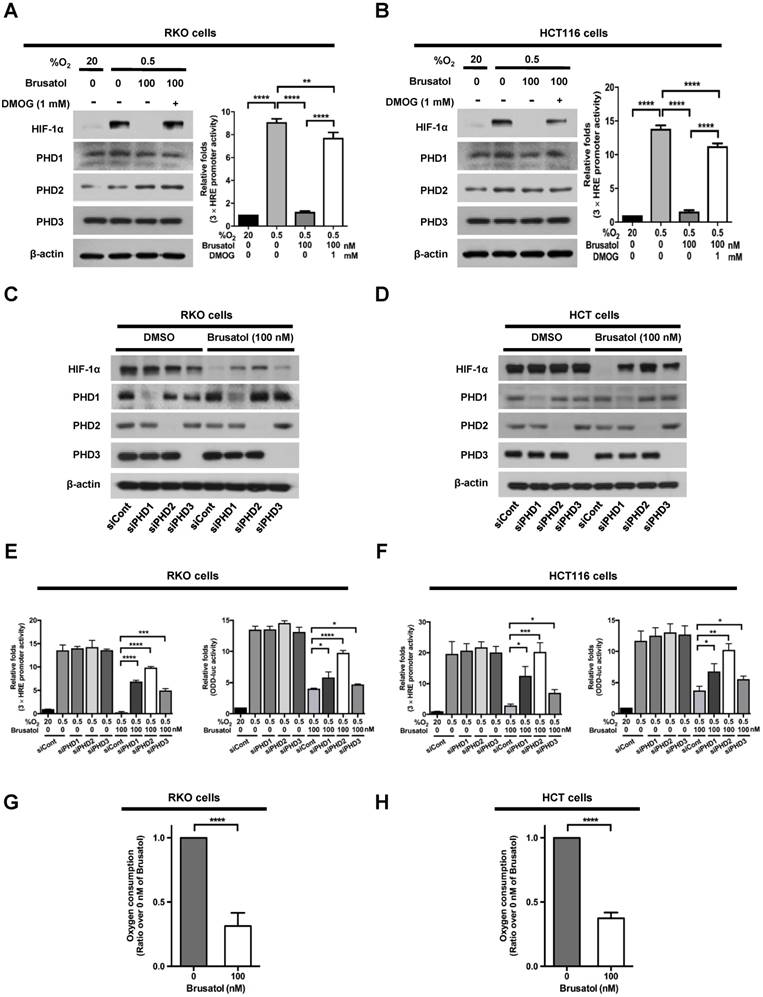
PHDs are required for brusatol-induced HIF-1α degradation in colorectal cancer cells under hypoxia. (A and B) Left panels and right panels depict immunoblot analysis and HRE-luciferase promoter assay, respectively. RKO (A) and HCT116 (B) were incubated with or without 1 mM DMOG. After a 1-h incubation, cells were incubated with or without 100 nM brusatol for 1 h, and then exposed to 20% or 0.5% O2 for 4 h and harvested. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. RKO (A) and HCT116 (B) cells co-transfected with p3×HRE-luc and pCMV-β-galactosidase were cultured for 16 h, then incubated with or without 100 nM brusatol for 1 h and exposed to 20% or 0.5% O2 for 8 h. Luciferase activity in whole-cell lysates was normalized to that of β-galactosidase. Data are presented as means ± SD (**P < 0.01, ****P < 0.0001; ANOVA). (C and D) RKO (C) and HCT116 (D) cells were transfected with siRNAs targeting PHD1, PHD2, or PHD3 under hypoxia. After a 48-h incubation, cells were treated with 100 nM brusatol or DMSO (vehicle control), exposed to 0.5% O2 for 4 h, and then harvested. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. (E and F) Left panels and right panels depict HRE-luciferase promoter assay and hydroxylation assay, respectively. RKO (E) and HCT116 (F) cells transfected with p3×HRE-luc (left panels) or pODD-luc (right panels) and pCMV-β-galactosidase were cultured for 16 h, then incubated with or without 100 nM brusatol for 1 h, and exposed to 20% or 0.5% O2 for 8 h. Luciferase activity in whole-cell lysates was normalized to that of β-galactosidase. Data are presented as means ± SD (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ANOVA). (G and H) Effects of brusatol on O2 consumption in RKO (G) and HCT116 (H) cells under hypoxia. Values are presented as means ± SD from three experiments (****P < 0.0001; ANOVA).
Brusatol-mediated inhibition of c-Myc expression increases HIF-1α degradation by inhibiting hypoxia-induced mitochondrial ROS production
In tumor microenvironment, hypoxia-induced mitochondrial ROS oxidizes and inactivates the ferrous iron within the active sites of the PHDs (23-25). c-Myc has been shown to play an important role in mitochondrial ROS production, and its overexpression increases HIF-1α expression by inhibiting activation of PHDs in hypoxic cancer cells (26). Thus, inhibition of c-Myc is tightly linked to diminished HIF-1α levels in a prolyl hydroxylase and von Hippel-Lindau protein-dependent manner (27). Therefore, treatment of antioxidants vitamin C and N-acetylcysteine reduces the HIF-1α levels in cancer cells (27). Due to effect of brusatol on decrease of HIF-1α protein stability in hypoxic cancer cells through activation of PHDs by inhibiting mitochondrial ROS production and the transition of ferrous iron to ferric iron as shown in Figure 4. Therefore, we next investigate the potential involvement of c-Myc in brusatol-induced degradation of HIF-1α in hypoxic cancer cells using immunoblot analysis to examine the expression of c-Myc before and after brusatol treatment. As shown in Figure 5A and 5B, brusatol treatment effectively inhibited c-Myc expression in hypoxic RKO and HCT116 cells. Furthermore, as shown in Figure 5C and 5D, overexpression of c-Myc restored brusatol-induced degradation of HIF-1α. To define the potential contribution of c-Myc to HIF-1α regulation in brusatol-treated hypoxic cells, we transfected RKO and HCT116 cells with a pc-Myc expression plasmid or pCont (empty vector), treated the cells with 100 nM brusatol for 1 h, and then exposed them to 0.5% O2 for 4 h. Consistent with the results of immunoblot analyses, c-Myc overexpression effectively restored degradation of HIF-1α by increasing ferrous iron in hypoxic cancer cells (Figure 5E-5H). Taken together, these results indicate that brusatol-induced degradation of c-Myc increases degradation of HIF-1α by inhibiting mitochondrial ROS production and the transition of ferrous iron to its ferric state.
Brusatol induces cancer cell death in vitro under hypoxic conditions
To investigate the effect of brusatol on hypoxic cancer cell survival, we treated RKO and HCT116 cells with brusatol (0-100 nM) for 1 h and exposed them to 0.5% O2 for 4 h at 37°C. The cells were then washed three times with PBS and cultured for 14 d. Cell survival was assessed by counting the numbers of colonies formed. As shown in Figure 6A and 6B, brusatol induced a concentration-dependent decrease in clonogenic survival in RKO and HCT116 cells under hypoxia. Because brusatol induced PHD activation and HIF-1α degradation (Figure 3), we tested whether blocking PHDs prevents brusatol-induced cancer cell death under hypoxic conditions through inhibition of HIF-1α degradation. As shown in Figure 6C and 6D, DMOG reduced brusatol-induced clonogenic cell death in hypoxic cancer cells by as much as ~30%. Next, we investigated whether brusatol-induced accumulation of ferrous iron increases cancer cell death under hypoxia. As shown in Figure 6E and 6F, 2,2'-bipyridyl prevented brusatol-induced cancer cell death, and FeCl2 increased brusatol-induced cancer cell death in hypoxic cancer cells treated with 2,2'-bipyridyl and brusatol. Finally, we investigated whether c-Myc decreases brusatol-induced cancer cell death under hypoxic conditions, and found that overexpression of c-Myc effectively inhibited brusatol-induced cancer cell death (Figure 6G and 6H). Collectively, these findings indicate that brusatol induces clonogenic cell death in hypoxic cancer cells by inhibiting the c-Myc/ROS/ferric iron signaling pathway.
Brusatol suppresses growth of xenograft tumors
Finally, to evaluate the in vivo antitumor activity of brusatol, we established a mouse colorectal cancer xenograft model by subcutaneously inoculating nude mice with RKO or HCT116 cells, and then administering brusatol (2 and 4 mg/kg) by intraperitoneal injection three times per week for 32 d. Tumor size was measured every 3 to 4 d, and all mice were sacrificed at the end of experiment, at which point tumors were dissected and weighed. The body weight of xenograft mice was also monitored throughout the treatment period to evaluate systemic toxicity. Body weight was not significantly affected by brusatol, indicating that the dose of brusatol did not cause overt toxic effects (Figure S3). Representative images of mice harboring RKO or HCT116 xenografts at 32 d are presented in Figure 7A and 7B. As shown in Figure 7A-D, brusatol treatment significantly decreased tumor growth in both RKO and HCT116 tumors. Specifically, the volume of RKO tumors of mice treated with 2 and 4 mg/kg brusatol was 49.02 ± 6.28 % and 65.9 ± 3.94 % of the volume of control tumors, respectively (*P < 0.05 for both). Likewise, the volume of HCT116 tumor of mice treated with 2 and 4 mg/kg brusatol was 36.2 ± 13.84 % and 64.9 ± 5.39 % of the volume of control tumors, respectively (*P < 0.05 for both). Brusatol treatment also significantly decreased tumor weight in RKO and HCT116 xenograft models compared with controls (Figure 7E and 7F; *P < 0.05 for both). To elucidate whether treatment of tumor-bearing host mice with brusatol causes degradation of c-Myc and HIF-1α, we evaluated HIF-1α and c-Myc expression in RKO xenografts using immunohistochemical and immunofluorescence staining. As shown in Figure 7G and 7H, brusatol treatment (2 and 4 mg/kg) of tumor-bearing mice significantly suppressed the expression of HIF-1α and c-Myc. We also evaluated the induction of apoptosis in RKO xenograft tumors by TUNEL assay. The incidence of apoptotic bodies was significantly higher in tumor-bearing mice treated with brusatol than that in controls (Figure 7I). These results demonstrate that brusatol induces cell death by promoting PHD-mediated degradation of HIF-1α in hypoxic tumors, which in turn suppresses tumor growth.
Figure 4
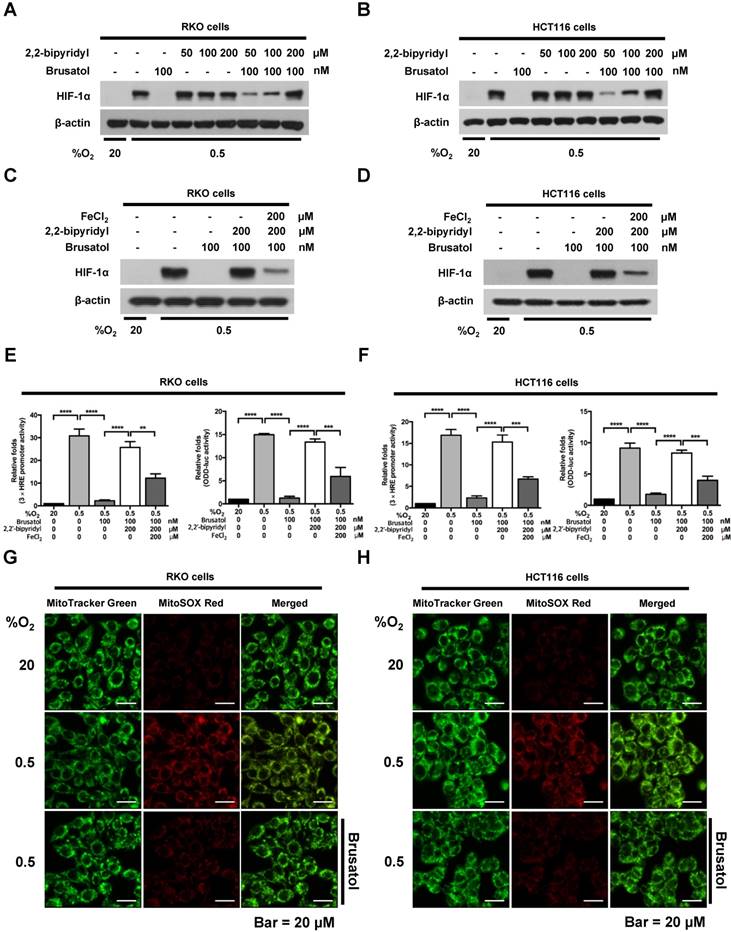
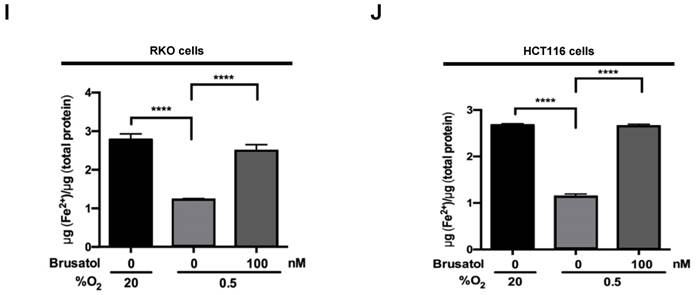
Brusatol increases PHD-mediated HIF-1α degradation by inhibiting hypoxia-induced transition of ferrous iron to its ferric state. (A and B) RKO (A) and HCT116 (B) cells were incubated with or without 100 nM brusatol in the presence and absence of different concentrations of 2,2'-bipyridyl (50-200 μM). After a 1 h incubation, cells were exposed to 20% or 0.5% O2 for 4 h and then harvested. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. (C and D) RKO (C) and HCT116 (D) cells were incubated with or without 200 μM 2,2'-bipyridyl or 200 μM FeCl2 in the presence and absence of 100 nM brusatol. After a 1-h incubation, cells were exposed to 20% or 0.5% O2 for 4 h and then harvested. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. (E and F) Left panels and right panels depict HRE-luciferase promoter assay and hydroxylation assay, respectively. RKO (E) and HCT116 (F) cells transfected with p3×HRE-luc (left panels) or pODD-luc (right panels) and pCMV-β-galactosidase were cultured for 16 h, then incubated with or without 200 μM 2,2'-bipyridyl or 200 μM FeCl2 in the presence and absence of 100 nM brusatol. After a 1-h incubation, cells were exposed to 20% or 0.5% O2 for 8 h and then harvested. Luciferase activity in whole-cell lysates was normalized to that of β-galactosidase. Data are presented as means ± SD (**P < 0.01, ***P < 0.001, ****P < 0.0001; ANOVA). (G and H) RKO (G) and HCT116 (H) cells were treated with MitoTracker and MitoSOX for 1 h, washed three times with pre-warmed PBS, and exposed to 0.5% O2 for 4 h. Fluorescence was detected with a Nikon confocal laser-scanning microscope. (I and J) Measurement of intracellular ferrous iron concentrations in RKO (I) and HCT116 (J) cells incubated with or without 100 nM brusatol under normoxia and hypoxia.
Discussion
Here, we investigated the mechanisms underlying brusatol-induced colorectal cancer cell death under hypoxic conditions. We found that brusatol decreases mitochondrial ROS, thereby inhibiting hypoxia-induced transition of ferrous iron to its ferric state. The resulting accumulation of ferrous iron activated PHDs, which in turn induces HIF-1α degradation, leading to colorectal cancer cell death.
Brusatol and structurally similar compounds are known to improve the response of cancer cells to various chemotherapeutic and radiotherapeutic treatments by inhibiting NRF2 defense mechanisms (5, 28). It has also been suggested that the inhibitory effect of brusatol against NRF2 is independent of KEAP1, a component of the proteasomal and autophagic protein-degradation system in A549 human lung adenocarcinoma cells (29). However, a recent mass-spectrometry study demonstrated that brusatol was not a specific NRF2 inhibitor because it rapidly and markedly decreased the expression of a great number of proteins (30, 31). In the present study, brusatol did not alter NRF2 expression in RKO or HCT116 colon cancer cells under normoxic or hypoxic conditions (data not shown). This difference between colorectal and lung cancer cell lines may reflect differences inherent to the specific cell types.
Hypoxia-induced accumulation of HIF-1α allows cancer cells to survive oxygen deprivation, and induces angiogenesis, tumor growth, metastasis, chemoresistance, and radioresistance (13-16). Thus, there is growing interest in treating cancers with HIF-1α inhibitors (13-16). Brusatol was previously described to induce HIF-1α degradation in a microenvironment that mimics hypoxia (6). Vartanian et al. demonstrated that inhibition of overall protein synthesis was observed in some cell lines derived from leukemia, lung adenocarcinoma, Ehrlich carcinoma, and hepatoma when the concentration of brusatol is higher than 500 nM (31). However, Lu et al. showed that brusatol inhibited HIF-1α expression at low concentration without affecting protein synthesis (6). However, how brusatol regulates HIF‑1α protein stability and induces cancer cell death under hypoxic conditions still remains unclear. Consistent with this previous report, we observed that, at low concentation, brusatol effectively induced HIF-1α degradation without affecting protein synthesis in the present study (Figure 1A-1D, Figure S1 and Figure S2). HIF-1α induces more than 100 target genes that regulate hypoxic adaptation and survival by binding to HREs in the promoter region of these genes (13). In the present study, brusatol-induced degradation of HIF-1α reduced HRE promoter activity by 8-10-fold (Figure 1G and 1H), thereby decreasing the expression of its target genes, including Glut1, PGK1, PDK1, and CA9 (Figure 1E and 1F). As a consequence, brusatol-induced inhibition of HIF-1α increased cancer cell death under hypoxia (Figure 6A and 6B).
Brusatol was shown to increase HIF-1α degradation without altering its mRNA levels (6). Consistent with this report, brusatol did not change HIF-1α mRNA expression in RKO cells and HCT116 cells in our study (Figure 2A and 2B). After confirming that brusatol regulates HIF-1α at the protein level, we investigated the underlying mechanism. Using CHX to block de novo protein synthesis in colorectal cancer cells, we confirmed that brusatol decreased HIF-1α protein stability under hypoxia (Figure 2).
Figure 5
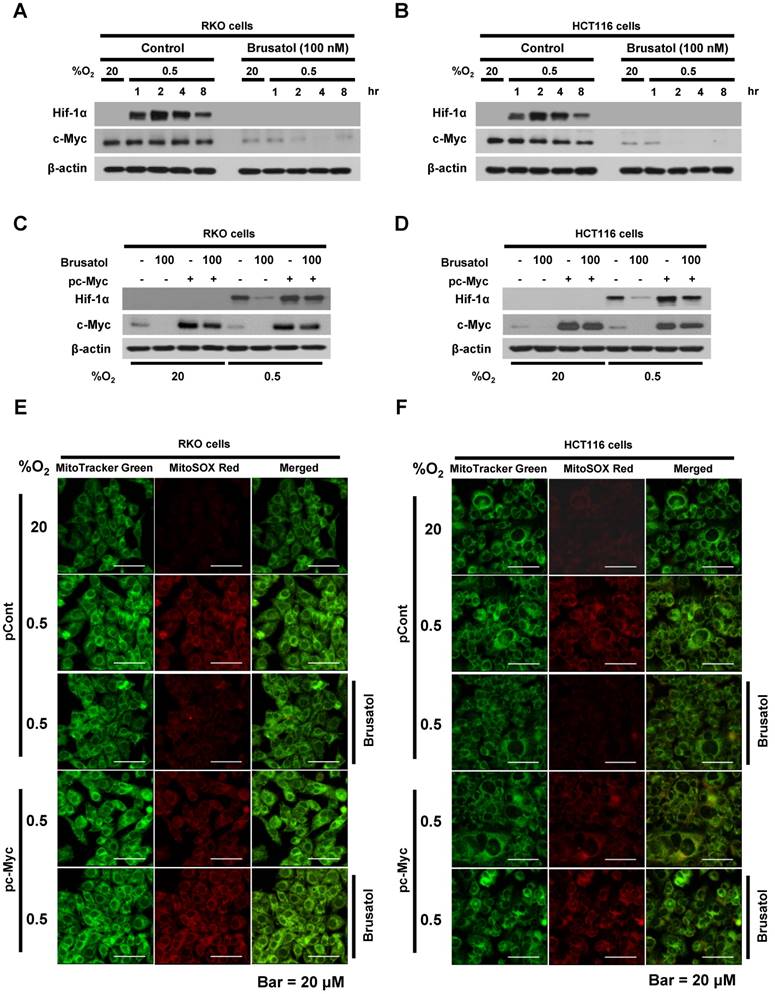
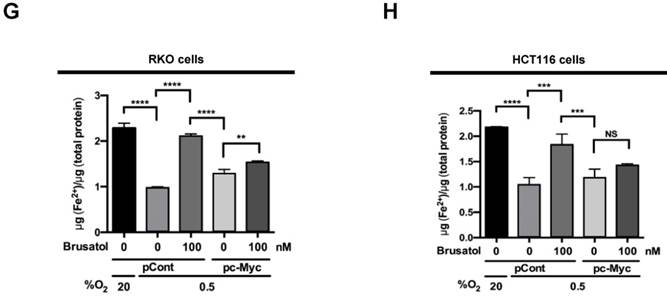
Brusatol-mediated inhibition of c-Myc expression increases HIF-1α degradation by inhibiting hypoxia-induced mitochondrial ROS production. (A and B) RKO (A) and HCT116 (B) cells were incubated with or without 100 nM brusatol. After a 1-h incubation, cells were exposed to 20% or 0.5% O2 for 8 h and then harvested at the indicated time. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. (C and D) RKO (C) and HCT116 (D) cells were transfected with pc-Myc or pCont (empty vector) for 48 h, and then incubated with or without 100 nM brusatol. After a 1-h incubation, cells were exposed to 20% or 0.5% O2 for 4 h and harvested. Whole-cell lysates were analyzed by immunoblotting for the indicated proteins. (E and F) RKO (E) and HCT116 (F) cells were treated with MitoTracker and MitoSOX for 1 h, washed three times with pre-warmed PBS, and exposed to 0.5% O2 for 4 h. Fluorescence was detected with a Nikon confocal laser-scanning microscope. (G and H) Effect of c-Myc expression on intracellular ferrous iron concentrations in RKO (G) and HCT116 (H) cells incubated with or without 100 nM brusatol under hypoxia. Data are presented as means ± SD (**P < 0.01, ***P < 0.001, ****P < 0.0001; ANOVA).
Previous studies have shown that the PHD1, PHD2, and PHD3 decrease HIF-1α stability in cancer cells under hypoxia (7, 13). To investigate how brusatol induces HIF-1α degradation in colorectal cancer cells under hypoxia, we assessed the expression of PHDs, but observed no change in the levels of these regulatory proteins (data not shown). We then investigated whether brusatol affected the activity of PHDs by treating colorectal cancer cells with DMOG, a pharmacological inhibitor of PHDs, and siRNAs targeting individual PHD isoforms. We observed that inhibition of PHD restored HIF-1α expression and transactivation in hypoxic brusatol-treated cancer cells (Figure 3A-3F) and reduced brusatol-induced cell death (Figure 6C and 6D). The canonical pathway of PHD-mediated HIF-1α degradation requires oxygen, and it has been previously reported that inhibition of mitochondrial function allows PHDs to remain active in cancer cells under hypoxia (13). Consistent with this, we observed that PHDs play an important role in brusatol-induced HIF-1α degradation (Figure 3A-3F), and further showed that brusatol decreases mitochondrial function, thereby reducing oxygen consumption (Figure 3G and 3H). Therefore, our results suggest that brusatol-induced inhibition of oxygen consumption leads to activation of PHDs, which in turn increases HIF-1α degradation (Figure 3).
Under hypoxia, mitochondrial ROS are required for O2-sensing and subsequent HIF-1α stabilization in hypoxic cells (23). Hypoxia-induced mitochondrial ROS production promotes the transition of intracellular ferrous iron to its ferric state (24), and the subsequent increase in intracellular ferric iron facilitates the rapid inhibition of hydroxylase-mediated activation of PHDs (26). We investigated the effect of brusatol and the hypoxia-mediated reduction in mitochondrial ROS production in cancer cells (Figure 4G-4J). 2,2'-bipyridyl, a ferrous iron chelator, effectively inhibited brusatol-induced HIF-1α degradation (Figure 4A-4D) and reduced brusatol-induced cell death under hypoxia (Figure 6E and 6F). Collectively, these results demonstrate that the brusatol-induced reduction in mitochondrial ROS increases HIF-1α degradation through the accumulation of intracellular ferrous iron (Figure 4) and causes cancer cell death under hypoxic conditions (Figure 6E and 6F).
Previous reports have identified an important role for c-Myc in mitochondrial ROS production, showing that upregulation of c-Myc increases HIF-1α expression by inhibiting PHD activation in cells under hypoxia (3, 23-25, 32). Thus, antioxidants reduce the HIF-1α levels in cancer cells (27). In addition, another report describes that brusatol reduces generation of mitochondrial ROS in HCT116 (6). Therefore, we investigated the effect of brusatol on HIF-1α degradation by inhibiting expression of c-Myc-mediated generation of mitochondrial ROS. We found that brusatol inhibits c-Myc expression in colorectal hypoxic cancer cells and mouse xenograft tumors (Figure 5A, 5B and Figure 7H), and showed that overexpression of c-Myc restores brusatol-induced HIF-1α degradation in brusatol-treated colorectal cancer cells under hypoxia (Figure 5C and 5D) through the production of mitochondrial ROS (Figure 5E and 5F).
Figure 6
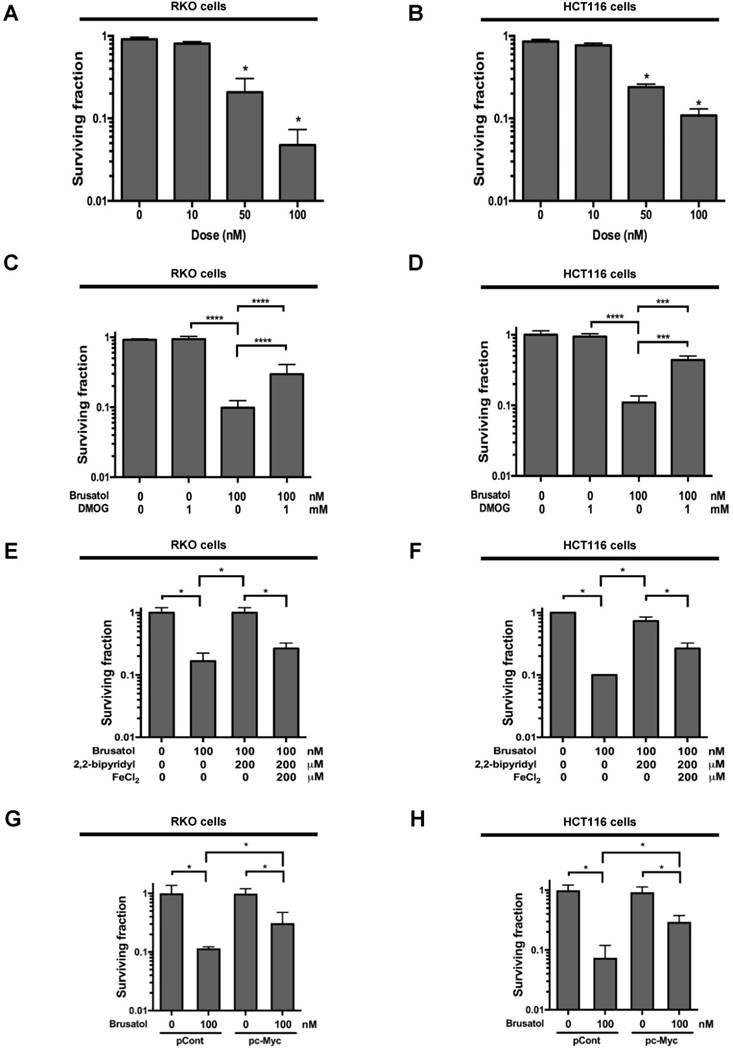
Brusatol induces cancer cell death in vitro under hypoxic conditions. (A and B) Effects of brusatol on clonogenic cell death in RKO (A) and HCT116 (B) cells under hypoxia. (C and D) Effects of PHDs on brusatol-induced clonogenic cell death in RKO (C) and HCT116 (D) cells under hypoxia. (E and F) Effects of intracellular ferrous iron on brusatol-induced clonogenic cell death in RKO (E) and HCT116 (F) cells under hypoxia. (G and H) Effects of c-Myc on brusatol-induced clonogenic cell death in RKO (G) and HCT116 (H) cells under hypoxia. Data are presented as means ± SD (*P < 0.05, ***P < 0.001, ****P < 0.0001; ANOVA).
Figure 7
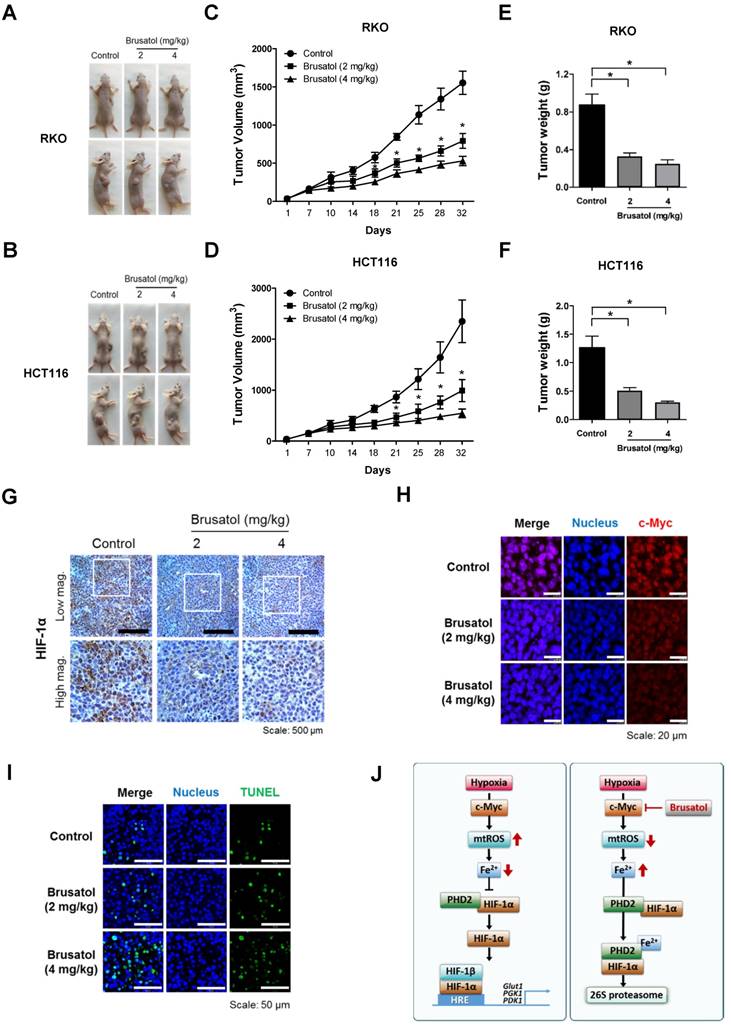
Brusatol suppresses colorectal cancer growth in vivo. (A and B) Representative images captured 32 d post-inoculation of RKO and HCT116 cells. (C and D) Measurement of tumor volume. Nude mice were subcutaneously implanted with RKO or HCT116 cells, and then intraperitoneally injected with brusatol (2, 4 mg/kg) or normal saline three times per week. Tumor volumes were calculated according to the formula, width2 × length/2. Data represent means ± S.E (n = 7 mice/group; *P < 0.05; ANOVA). (E and F) Tumor weight was measured after sacrificing mice at the end of the experiment. Data represent means ± S.E (n = 7 mice/group; *P < 0.05; ANOVA). (G) Representative photographs of HIF-1α immunohistochemical staining in tumors derived from RKO xenografts. Scale bar: 500 μm. Top: low magnification; bottom: high magnification. (H) Immunofluorescence staining of c-Myc in paraffin-embedded tissue specimens from mouse xenograft tumors. Scale bar: 20 μm. Blue, nuclei; red, c-Myc. (I) Apoptosis in tumor sections of the indicated experimental groups was determined using an in situ cell death detection kit. Scale bar: 50 μm. Blue, nuclei; green, TUNEL staining. (J) Schematic model showing how brusatol induces cancer cell death under hypoxia by activation of PHDs.
Our results further showed that c-Myc overexpression also decreases accumulation of intracellular ferrous iron in brusatol-treated cancer cells under hypoxia (Figure 5G and 5H) by increasing mitochondrial ROS (Figure 5E and 5F), thereby reducing brusatol-induced colorectal cancer cell death under hypoxia (Figure 6G and 6H). Moreover, brusatol treatment induced cancer cell death by promoting PHD-mediated degradation of HIF-1α, which in turn significantly suppressed tumor growth in both RKO and HCT116 xenografts. (Figure 7A-I). These results strongly suggest that brusatol increases PHD-induced degradation of HIF-1α by inhibiting c-Myc expression under hypoxia, thereby decreasing mitochondrial ROS production and causing cancer cell death. Although the detailed mechanism by which brusatol decreases c-Myc expression remains unclear, our findings collectively demonstrate that brusatol induces cell death in colorectal cancer cells under hypoxia by promoting PHD-mediated degradation of HIF-1α (Figure 7J).
Abbreviations
HIF-1: hypoxia-inducible factor-1; PHDs: prolyl hydroxylases; NRF2: nuclear factor-erythroid 2-related factor-2; ODD: oxygen-dependent degradation; pVHL: von Hippel-Lindau; HREs: hypoxia-response elements; ROS: reactive oxygen species; ATCC: American Type Culture Collection; DMEM: Dulbecco's Modified Eagle Medium; CHX: cycloheximide; DMOG: dimethyloxalylglycine; HRP: horseradish peroxidase; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; RNAi: RNA interference; TUNEL: terminal deoxynucleotidyl transferase dUTP nick-end labeling; TdT: terminal deoxytransferase.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MISP) under NRF-2014R1A5A2009392, NRF-2015M2B2B1068599 (NRF-2015M2B2B1068623), NRF-2015R1C1A1A01054870, and NRF-2015R1D1A1A01056610.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Turpaev K, Welsh N. Brusatol inhibits the response of cultured beta-cells to pro-inflammatory cytokines in vitro. Biochem Biophys Res Commun. 2015;460:868-72
2. Mata-Greenwood E, Cuendet M, Sher D. et al. Brusatol-mediated induction of leukemin cell differentiation and G(1) arrest is associated with down-regulation of c-myc. Leukemia. 2002;16:2275-84
3. Liao LL, Kupchan SM, Horwitz SB. Mode of action of the antitumor compound bruceantin, an inhibitor of protein synthesis. Mol Pharmacol. 1976;12:167-176
4. Harder B, Tian W, La Clair JJ. et al. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol Carcinog. 2016 doi:10.1002/mc22609
5. Ren D, Villeneuve NF, Jiang T. et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defensemechanism. Proc Natl Acad Sci USA. 2011;108:1433-8
6. Lu Y, Wang B, Shi Q. et al. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci Rep. 2016;6:39123
7. Oh ET, Kim JW, Kim JM. et al. NQO1 inhibits proteasome-mediated degradation of HIF-1α. Nat Commun. 2016;7:13593
8. Wang GL, Jiang BH, Rue EA. et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510-4
9. Chan DA, Sutphin PD, Yen SE. et al. Coordinate reugulation of the oxygen-dependent degradation domain of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2005;25:6415-26
10. Keith B, Simo MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465-72
11. Sutphin PD, Chan Da, Giaccia AJ. Dead cells don's form tumors: HIF-dependent cytotoxins. Cell Cycle. 2004;3:160-3
12. Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Meastasis Rev. 2007;26:341-52
13. Oh ET, Kim CW, Kim SJ. et al. Docetaxel induced-JNK2/PHD1 signaling pathway increase degradation of HIF-1α and causes cancer cell death under hypoxia. Sci Rep. 2016;6:27382
14. Ema M, Taya S, Tokotani N. et al. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF express and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA. 1997;94:4273-8
15. Gu YZ, Moran SM, Hogenesch JB. et al. Molecular characterizaition and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998;7:205-13
16. Luo W, Zhong J, Chang R. et al. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but not HIF-2alpha. J Biol Chem. 2010;285:3651-63
17. Trisciuoglio D, Gabellini C, Desideri M. et al. Bcl-2 regulates HIF-1alpha protein stabilization in hypoxic melanoma cells via the molecular chaperone HSP90. PLoS One. 2010;5:e11772
18. Ivan M, Kondo K, Yang H. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464-8
19. Jaakkola P, Mole DR, Tian YM. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468-72
20. Liu YV, Baek JH, Zhang H. et al. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell. 2007;25:207-17
21. Luo YV, Semenza GL. RACK1 vs. HSP90: competition for HIF-1α degradation vs. stabilization. Cell Cycle. 2007;6:656-9
22. Escuin D, Kline ER, Giannakakou P. Both microtubule-stabilizing and microtubule-destabilizing drugs inhibit hypoxia-inducible factor-1α accumulation and activity by disrupting microtubule function. Cancer Res. 2005;65:9021-28
23. Simon MC. Mitochondrial reactive oxygen species are required for hypoxic HIF alpha stabilization. Adv Exp Med Biol. 2006;588:165-70
24. Tayor CT. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J. 2008;409:19-26
25. Dang CV, Kim JW, Gao P. et al. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51-6
26. Sabharwal SS, Schumacker PT. Mitochondiral ROS in cancer: initiators, amplifiers or an Achilles'heel? Nat Rev Cancer. 2014;14:709-21
27. Gao P, Zhang H, Dinavahi R. et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230-8
28. Sun X, Wang Q, Wang Y. et al. Brusatol enhances the radiosensitivity of A549 cells by promoting ROS production and enhancing DNA damage. Int J Mol Sci. 2016;17:doi 10.3390/ijms17070997
29. Olayanju A, Copple IM, Bryan HK. et al. Brusatol proves a rapid and transient inhibition of Nrf2 of signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med. 2015;78:202-12
30. Menegon S, Coumbano A, Giordano S. The dual roles of NRF2 in Cancer. Trends Mol Med. 2016;22:578-92
31. Vartanian S, Ma TP, Lee J. et al. Application of mass spectrometry profiling to establish brusatol as an inhibitor of global protein synthesis. Mol Cell Proteomics. 2016;15:1220-31
32. Ferber EC, Peck B, Delpuech O. et al. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012;19:968-79
Author contact
![]() Corresponding author: Heon Joo Park, MD, PhD., Inha University College of Medicine, Department of Microbiology, Hypoxia-related Disease Research Center, 100, Inha-ro, Nam-gu, Incheon 22212, Republic of Korea Tel: +82-32-860-9800 Fax: +82-32-885-8302 E-mail: park001ac.kr
Corresponding author: Heon Joo Park, MD, PhD., Inha University College of Medicine, Department of Microbiology, Hypoxia-related Disease Research Center, 100, Inha-ro, Nam-gu, Incheon 22212, Republic of Korea Tel: +82-32-860-9800 Fax: +82-32-885-8302 E-mail: park001ac.kr
Citation styles
APA
Oh, E.T., Kim, C.W., Kim, H.G., Lee, J.S., Park, H.J. (2017). Brusatol-Mediated Inhibition of c-Myc Increases HIF-1α Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia. Theranostics, 7(14), 3415-3431. https://doi.org/10.7150/thno.20861.
ACS
Oh, E.T.; Kim, C.W.; Kim, H.G.; Lee, J.S.; Park, H.J. Brusatol-Mediated Inhibition of c-Myc Increases HIF-1α Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia. Theranostics 2017, 7 (14), 3415-3431. DOI: 10.7150/thno.20861.
NLM
Oh ET, Kim CW, Kim HG, Lee JS, Park HJ. Brusatol-Mediated Inhibition of c-Myc Increases HIF-1α Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia. Theranostics 2017; 7(14):3415-3431. doi:10.7150/thno.20861. https://www.thno.org/v07p3415.htm
CSE
Oh ET, Kim CW, Kim HG, Lee JS, Park HJ. 2017. Brusatol-Mediated Inhibition of c-Myc Increases HIF-1α Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia. Theranostics. 7(14):3415-3431.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.