Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(9):2477-2494. doi:10.7150/thno.18879 This issue Cite
Research Paper
PSMA-Targeted Theranostic Nanocarrier for Prostate Cancer
Orielyz Flores1, 2, Santimukul Santra1, Charalambos Kaittanis3, Rania Bassiouni2, Amr S Khaled4, Annette R. Khaled2, Jan Grimm3, J Manuel Perez5 ![]()
1. Nanoscience Technology Center and Chemistry Department, University of Central Florida, Orlando FL, 32827;
2. Burnett School of Biomedical Science, College of Medicine, University of Central Florida, Orlando FL, 32827;
3. Molecular Pharmacology Program and Department of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY, 10065;
4. Orlando VA Medical Center, Orlando, FL 32827;
5. Biomedical Imaging Research Institute, & Samuel Oschin Comprehensive Cancer Institute, Department of Biomedical Sciences and Department of Neurosurgery, Cedar Sinai Medical Center, Los Angeles CA, 90048.
Received 2016-12-22; Accepted 2017-4-12; Published 2017-6-24
Citation:
Flores O, Santra S, Kaittanis C, Bassiouni R, Khaled AS, Khaled AR, Grimm J, Perez JM. PSMA-Targeted Theranostic Nanocarrier for Prostate Cancer. Theranostics 2017; 7(9):2477-2494. doi:10.7150/thno.18879. https://www.thno.org/v07p2477.htm
Other stylesAbstract

Herein, we report the use of a theranostic nanocarrier (Folate-HBPE(CT20p)) to deliver a therapeutic peptide to prostate cancer tumors that express PSMA (folate hydrolase 1). The therapeutic peptide (CT20p) targets and inhibits the chaperonin-containing TCP-1 (CCT) protein-folding complex, is selectively cytotoxic to cancer cells, and is non-toxic to normal tissue. With the delivery of CT20p to prostate cancer cells via PSMA, a dual level of cancer specificity is achieved: (1) selective targeting to PSMA-expressing prostate tumors, and (2) specific cytotoxicity to cancer cells with minimal toxicity to normal cells. The PSMA-targeting theranostic nanocarrier can image PSMA-expressing cells and tumors when a near infrared dye is used as cargo. Meanwhile, it can be used to treat PSMA-expressing tumors when a therapeutic, such as the CT20p peptide, is encapsulated within the nanocarrier. Even when these PSMA-targeting nanocarriers are taken up by macrophages, minimal cell death is observed in these cells, in contrast with doxorubicin-based therapeutics that result in significant macrophage death. Incubation of PSMA-expressing prostate cancer cells with the Folate-HBPE(CT20p) nanocarriers induces considerable changes in cell morphology, reduction in the levels of integrin β1, and lower cell adhesion, eventually resulting in cell death. These results are relevant as integrin β1 plays a key role in prostate cancer invasion and metastatic potential. In addition, the use of the developed PSMA-targeting nanocarrier facilitates the selective in vivo delivery of CT20p to PSMA-positive tumor, inducing significant reduction in tumor size.
Keywords: polymeric nanoparticles, peptide, PSMA, prostate cancer.
Introduction
Current cancer chemotherapeutics are usually administered systemically, and affect not only the tumor but also healthy tissues, causing severe side effects. For this reason, targeted therapeutics have been designed to specifically localize in tumors [1-4]. This “magic bullet” approach typically involves the conjugation of a drug to a targeting ligand that specifically binds to a protein receptor overexpressed on cancer cells [5-8]. Most recently, the development of targeted nanotherapeutics promises to facilitate the delivery and accumulation of drugs in tumors that overexpress a particular cell-surface protein [9-12]. One of the main problems with targeted drug nanotherapeutics is that they can target the same cell surface protein on healthy cells or non-specifically accumulate in lymph nodes, liver and spleen, resulting in significant toxicity [13-15]. In addition, circulating macrophages and other cells of the immune system can uptake a significant amount of these nanotherapeutics, inducing cell death and affecting the immune system [16-19]. This non-selective cytotoxicity can be addressed by packaging a therapeutic drug that specifically affects the unique cancer biology of tumors, inducing selective killing only to tumor cells. For these reasons, a nanoparticle that specifically targets tumors via unique cancer cell-surface receptors, while encapsulating a cancer-specific therapeutic agent that is selectively cytotoxic only to cancer cells and non-toxic to normal tissue would result in an improved therapy for cancer. We hypothesized that this two-way tumor targeting approach on a nanotherapeutic agent would increase targeted tumor regression while minimizing size affects.
Recently, we reported the discovery of a cytotoxic peptide (CT20p; sequence VTIFVAGVLTASLTIWKKMG), based on the C-terminal α9 helix of Bax, which induced cell death by a different mechanism from the parent Bax protein [20]. Treatment of cells with CT20p lead to significant morphological changes associated with altered cytoskeletal dynamics, which included impairing mitochondrial movement and actin polymerization [21]. It was also found that the peptide has the potential to impair cancer cell invasiveness in a breast cancer model. This involved reduced cancer cell migration and loss of cell adhesion, leading to detachment and cell death in cancer cells [22]. These results were not observed with a control epithelial cell line, indicating that the lethal activity of the peptide was cancer-cell-specific. To explain the selective toxicity of CT20p, we recently identified chaperonin containing TCP-1 (CCT) as the intracellular target of CT20p [22]. CCT is a large macromolecular complex composed of eight subunits (CCTα, CCTβ, CCTγ, CCTδ, etc.) responsible for folding 5-10% of the cell proteome, and it is the essential chaperone for folding actin and tubulin into their native forms. CCT inhibition results in the changes in cytoskeletal dynamics and inhibition of actin and tubulin polymerization that we observed in CT20p-treated cells [21-22]. Our studies and that of others have reported that CCT is overexpressed in cancer cells and could be an indicator of cancer progression and metastasis [23-26], which makes it a novel target for cancer therapeutics. Interestingly, the cytotoxicity of CT20p in breast cancer cells correlated with the levels of CCT, which was decreased by comparison in normal breast epithelial cells [22].
The in vivo delivery of CT20p to tumor cells is challenging, due to the peptide's hydrophobicity, poor stability in serum, inefficient cancer cell uptake and unfavorable pharmacokinetics. Encapsulation of CT20p into a hyperbranched polymeric nanocarrier (HBPE) facilitated the delivery of the peptide to breast cancer tumors via the enhanced permeability and retention (EPR) effect.[20] HBPE nanocarriers protected CT20p while in circulation, releasing the peptide only in the acidic conditions of intracellular vesicles or by esterases found within cells. However, as EPR is not an efficient delivery approach for most primary tumors and even less for micro-metastasis, we reasoned that a specific tumor targeting of the HBPE(CT20p) nanocarrier would facilitate the specific delivery of CT20p in higher concentration to a tumor, resulting in an improved therapeutic effect. To test our hypothesis, we chose the prostate-specific membrane antigen (PSMA), a cell-membrane protein that is highly expressed in prostate cancer (PCa), as a target protein receptor to deliver CT20p. PSMA expression increases with PCa progression, providing an excellent target for treatment, especially for the more aggressive forms of the disease [27-31]. Although high levels of PSMA have also been found on PCa metastasis, no significant amounts were measured in accessible healthy tissues, making this target attractive for the treatment of metastatic PCa [27, 32, 33]. PSMA exhibits a dual enzymatic function as a glutamate carboxypeptidase and folate hydrolase, cleaving the amide bond of N-acetyl aspartylglutamate and hydrolyzing extracellular polyglutamated folate to mono-glutamic folic acid that can then be utilized by cells [28]. Furthermore, it has been proposed that upregulation of PSMA might provide PCa cells with a growth advantage, and implicate PSMA in the metabolism of polyglutamated folates and the subsequent uptake of folates [34, 35]. Folic acid, a high affinity ligand for the folate receptor (FR), retains its receptor binding and endocytosis properties when covalently linked to wide variety of molecules and nanoparticles [8, 36-40]. Therefore, we reasoned that folate-conjugating nanoparticles could be used as PSMA-targeted nanocarriers for the delivery of a therapeutic peptide to PSMA-positive PCa cells, inducing selective cancer cell death. Furthermore, as the nanocarrier can also be encapsulated with a near infrared fluorescent dye, a theranostic agent that reports on its tumor localization by fluorescence imaging can be achieved.
Herein we report the use of folate-conjugated polymeric nanoparticles as nanocarriers for the targeted delivery of a cancer-specific therapeutic peptide to prostate cancer via PSMA. The resulting PSMA-targeting nanocarrier is perfectly suited to facilitate the specific delivery of CT20p as the peptide can be encapsulated within the HBPE nanoparticles hydrophobic cavities. Furthermore, the free carboxylic groups on the nanocarrier's surface can be conjugated with folic acid, producing a multivalent agent that incorporates both therapy and targeting. First, we validated the use of folic acid as a PSMA-targeting ligand using an activatable fluorescent folate conjugate recently described by us. Upon incubation with PSMA-expressing PCa cells, the probe entered the cells, fluorescently labeling them. The use of folate to target PSMA was further validated using folate-conjugated nanoparticles that encapsulate a fluorescent dye Folate-HBPE(DiI). These PSMA-targeted nanocarriers were able to fluorescently label PSMA-expressing cells. When these nanocarriers were encapsulated with CT20p [Folate-HBPE (CT20p)], selective cytotoxicity was observed in cells that express PSMA. The Folate-HBPE(CT20p) nanocarrier did not cause cell death to macrophages, further indicating the cancer-specific therapeutic value of CT20p. In addition, treatment of the PSMA-expressing cells with the Folate-HBPE(CT20p) NPs induced cell detachment and decreased expression of β1 integrin, a surface protein that has been implicated in the potential of PCa to metastasize. Finally, studies using mice tumor xenografts showed that Folate-HBPE nanocarriers can specifically deliver CT20p to PSMA expressing prostate cancer tumors, achieving increased tumor regression compared non-targeted nanocarriers. Taken together, our data showed the feasibility of delivering a therapeutic, cancer-specific, peptide to prostate cancer via PSMA, achieving specific cancer cell detachment, cell death and tumor regression.
Results
Validation of folate as a ligand to target PSMA in PCa cells using an activatable Folate-S-S-Doxorubicin probe
To validate the use of folate as a potential ligand to target PSMA, we used an activatable Folate-S-S-Doxorubicin fluorescent probe, which has been previously described by us.[8] The probe consists of folic acid conjugated to doxorubicin (Doxo) via a disulfide bond (Figure 1A), and was designed to have the fluorescence of Doxo quenched (OFF-state) by the proximity of folate. Upon binding and internalization, the disulfide bond within the Folate-S-S-Doxo probe is cleaved by intracellular glutathione and the fluorescence of Doxo is regained (ON-state), fluorescently labeling the cell's cytoplasm and indicating successful internalization of the probe (Figure 1B). We therefore hypothesized that if PSMA facilitates the internalization of a folate-containing probes, it would facilitate the binding, internalization and activation of Folate-S-S-Doxo on PSMA expressing PCa cells. To test this hypothesis, we incubated LNCaP, a PCa cell line that expresses PSMA with the Folate-S-S-Doxo probe. Results showed a significant amount of fluorescence in the cytoplasm of the cells within 12 hours of incubation (Figure 1C). In contrast, when similar experiments were performed using PC3 cells, a PCa cell line that does not express PSMA, no fluorescence was observed in the cytoplasm of these cells (Figure 1D). Even after 48 hours of incubation, no cell-associated fluorescence was observed due to the lack of PSMA expression. When a PC3 cell line that stably expresses PSMA was used (PSMA(+) PC3 cells), internalization of the Folate-S-S-Doxo probe was regained. As in the case of LNCaP cells, internalization resulted in activation of the probe and fluorescent labeling of the cell cytoplasm within 12 hours (Figure 1E). As internalization was not observed in PC3 cells, but was observed in the PSMA-transfected PC3 cells, this suggested that the internalization and activation of the Folate-S-S-Doxo probe was mediated by the presence of transmembrane PSMA. This was further validated by the fact that the internalization of the probe in the PSMA(+) PC3 cells, was blocked by 2-PMPA (data not shown). 2-PMPA is a high affinity PSMA ligand that strongly binds to the active side of PSMA where the polyglutamated folate is cleaved.[34, 41] Within 24 hours of incubation, the activated doxorubicin was able to migrate to the nucleus where it induced cell death. As expected, cell death of the prostate cancer cells that express PSMA is observed within 24 hours indicating successful migration of the free doxorubicin into the nucleus of these PCa cells (Figure S1A and S1B). When these cells were pre-incubated with 2-PMPA, and then incubated with Folate-S-S-Doxo, no cell-associated fluorescence or cytotoxicity was observed, even after a 48 hour of incubation (Figure S1C and S1D). These results clearly indicated that the Folate-S-S-Doxo probe was internalized by LNCaP and PSMA(+) PC3 cells and became activated upon internalization. The observed internalization seems to be mediated by PSMA, as pre-incubation of the cells with the non-cleavable PSMA ligand (2-PMPA) blocked the internalization of the probe. Most importantly, as neither LNCaP nor PC3 cells express significant amounts of the folate receptor,[42] the observed internalization of the Folate-S-S-Doxo probe must have occurred via PSMA. The lack of cell surface expression of the folate receptor on the LNCaP and PC3 cells was further validated by flow cytometry (Figure S2) using an anti-folate receptor antibody. Taken together, these results showed that a folate ligand can be used to specifically target the binding and internalization of a folate-containing probe to PCa cells via PSMA.
Fabrication of the folate-conjugated HBPE nanocarriers
The HBPE nanocarriers were synthesized as described before and encapsulation of the therapeutic CT20p peptide was achieved as described in Material and Methods. In a typical experiment, a peptide loading of 3.5 nM CT20p per mL of nanoparticle suspension is achieved. To facilitate aqueous stability and to minimize non-specific binding, a short polyethylene glycol moiety (PEG) was conjugated to the nanocarrier's surface to facilitate aqueous stability. The folic acid was conjugated using ethylene diamine as a linker (Figure 2A) and typical water-based EDC/NHS bioconjugation chemistry. Our synthetic procedure yielded spherical Folate-HBPE(CT20p) nanocarriers of approximately 80 nm in size as determined by STEM (Figure 2B). The resulting suspension was stable in PBS (pH = 7.4) for months even after the folate ligand was conjugated to the nanocarrier's surface. The hydrophobic character of the CT20p peptide and DiI dye allows for a stable encapsulation into the hydrophobic pockets of the nanocarrier at physiological pH (7.4). However, at pH 5.0, the typical pH of the lysosome, the CT20p cargo is released in a time-dependent manner, starting to reach a plateau in 15 hours (Figure 2C). Release at acidic pH is likely facilitated by changes in the protonation of the polymer's carboxylic acid group within the nanoparticle that disturb hydrogen bonding and van der Waals interactions that stabilize the encapsulation of the CT20p peptide cargo, thus triggering release at lower pH. This is important because after endosomal uptake of Folate-HBPE(CT20p), CT20p would need to be released within the cells' late endosome or lysosome to be effective and induce cell death. For the assessment of nanocarrier internalization by fluorescence microscopy and FACS, a fluorescent dye (DiI) was encapsulated into the HBPE nanocarrier during their synthesis, yielding Folate-HBPE(DiI). No significant change in nanoparticle size or aqueous stability was observed when DiI was encapsulated instead of CT20p.
Figure 1
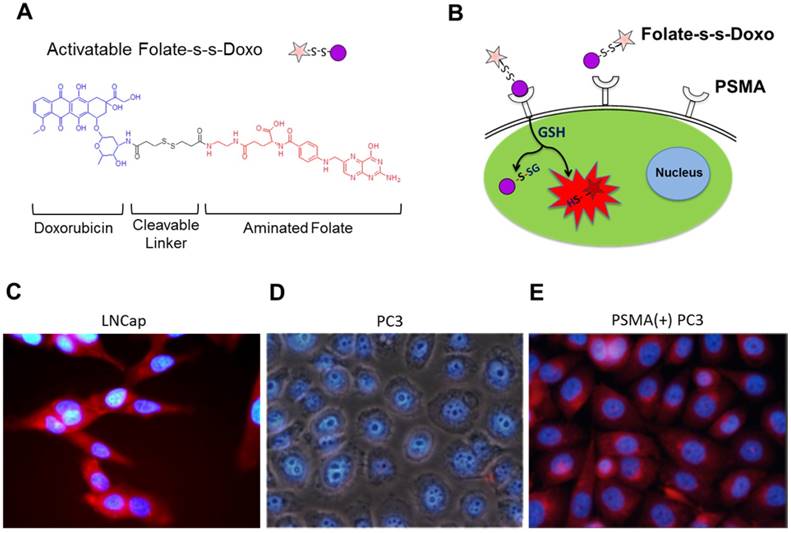
(A) Chemical structure of a cleavable Folate-Doxorubicin probe. (B) Mechanism of fluorescent activation upon PSMA-mediated cell internalization. The fluorescence of Doxorubicin (Doxo) is quenched by the close proximity of the folic acid (folate) ligand that act as both quencher and targeting ligand. Upon PSMA-mediated internalization of the probe, the disulfide bond that links Doxo and Folate is cleaved by reduced glutathione in the cytoplasm, activating the fluorescence of doxorubicin. Fluorescence microscopy images of (C) LNCaP, (D) PC3 and (E) PSMA(+) PC3 prostate cancer cell lines upon a 12 hour incubation with the activated probe Folate-s-s-Doxo (1.2 uM at 37C). Internalization and fluorescence activation of the probe is observed in the PSMA expressing PCa cells (LNCaP and PSMA(+) PC3), but not in wild type PC3, that does not express PSMA.
Figure 2
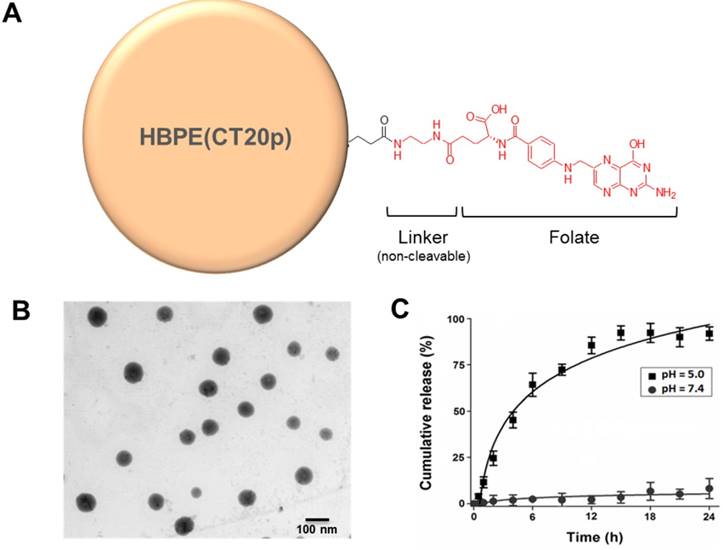
(A) Diagram depicting the conjugation of folate to HBPE(CT20p) NPs using ethylene diamine as a non-cleavable linker. For clarity, only one ligand is depicted attached to the nanoparticle, but multiple folate ligands are attached, yielding a multivalent folate conjugated nanoparticle (B). Scanning transmission electron micrograph (STEM) image of the Folate-HBPE(CT20p) NPs. (C) Release profile of the CT20p peptide from the HBPE nanoparticle at pH 5.0 and 7.4. The stability of CT20p encapsulation at physiological pH is shown by the lack of peptide release at pH 7.4. However, at pH 5, the typical lysosomal pH, a time-dependent release of the peptide is observed.
Validation of a folate-conjugated nanocarrier [Folate-HBPE(DiI)] to target PSMA in PCa cells
Next, we investigated if the HBPE nanocarrier conjugated with folate can bind and internalize into PCa cells via PSMA, similarly to the activatable Folate-S-S-Doxo fluorescent probe. In these experiments, the Folate-HBPE(DiI) nanocarriers were incubated with the prostate cancer cell lines to facilitate assessment of nanocarrier internalization by fluorescence microscopy and flow cytometry. Minimal fluorescence was observed in non-treated (control) LNCaP and PC3 cells, as expected (Figure 3A). In contrast, upon incubation of LNCaP cells with the Folate-HBPE(DiI), marked cell-associated fluorescence was observed by flow cytometry and fluorescence microscopy (Figure 3B). This cell-associated fluorescence was abrogated when the LNcaP cells were pre-treated with 2-PMPA (Figure 3C). Furthermore, minimal fluorescence was observed in PC3 cells upon incubation with Folate-HBPE(DiI) (Figure 3D) as these PCa cells do not express PSMA. However, when the PSMA transfected PC3 cells were incubated with the Folate-HBPE(DiI), significant cell associated fluorescence was observed (Figure 3E) that was abrogated by pre-incubation of the cells with 2-PMPA (Figure 3F). Next, a panel of cancer cell lines was tested for binding to Folate-HBPE(DiI) using flow cytometry and results were compared to those obtained using J591-fluorescein, a fluorescent labeled anti-PSMA antibody.[43] Results showed a correlation between the level of PSMA cell surface expression, judged by the levels of J591 antibody binding (Figure 4A) to the levels detected with the Folate-HBPE(DiI) NPs (Figure 4B). Both the LNCaP and PSMA(+) PC3 cell lines are fluorescently labeled by both the J591 antibody and the Folate-HBPE(DiI) nanoparticles. However, PC3, did not interact with either the folate-conjugated nanocarrier or the anti-PSMA antibody. Other non-prostatic cancer cell lines that do not express PSMA did not bind to either the Folate-HBPE(DiI) or the J591 antibody. These results demonstrate that targeting PSMA with Folate-HBPE(DiI) NPs allowed for discrimination between PSMA-positive and PSMA-negative cells similar to the J591 antibody, which binds outside of the catalytic side of the protein.[43-46] Taken together, these results validate the use of a folate ligand to address the binding and internalization of a polymeric nanocarrier to PSMA expressing PCa cells, releasing cargos inside the cell for imaging and/or therapeutic purposes.
Figure 3
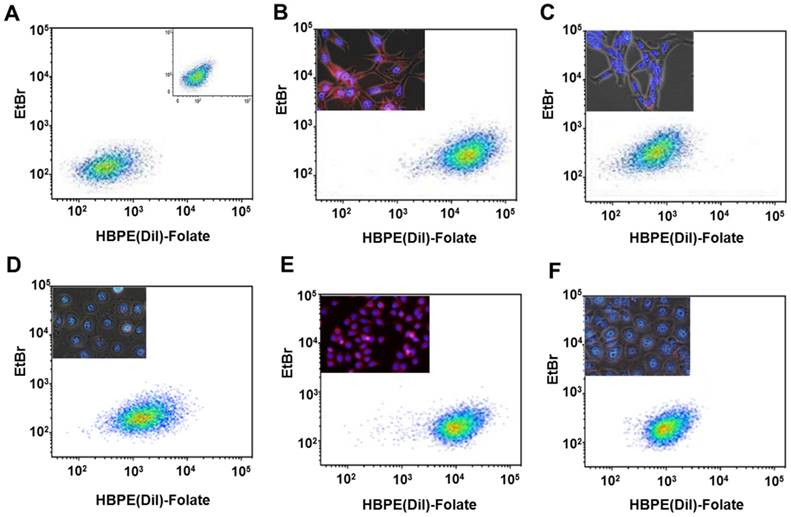
Assessment of targeting and PSMA-mediated cell internalization of Folate-HBPE(DiI)-NPs by flow cytometry analysis. (A) Control, non -treated LNCaP and PC-3 (inset) cell lines, (B) LNCaP, (C) LNCaP pre-treated with PMPA. (D) PC-3, (E) PSMA(+) PC-3, (F) PSMA(+) PC-3 pre-treated with PMPA. Inset in B-E: Fluorescence microscopy images of the corresponding cells. Internalization of Folate-HBPE(DiI)-NPs and fluorescence labeling is observed in the PSMA expressing PCa cells (LNCaP and PSMA(+) PC3), but not in wild type PC3, that does not express PSMA.
Figure 4
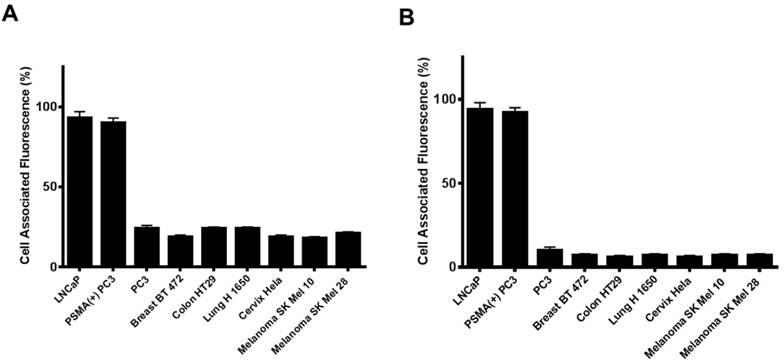
Cell associated fluorescence of various tumor cell lines upon incubation with an anti-PSMA antibody (Fluorescein-J591) (A) or Folate-HBPE(DiI) (B). Note the strong cell associated fluorescence in the cell lines treated with either the anti-PSMA antibody or the folate nanoparticle, indicating that the folate nanoparticle can discriminate between cells that express PSMA from those that do not.
PSMA-targeted and cancer-specific cytotoxicity of Folate-HBPE(CT20p) in PCa cell lines
To test the efficacy of the Folate-HBPE(CT20p) nanocarriers in LNCaP and PC3 prostate cancer cells, the endogenous levels of CCT, the cellular target of CT20p, were examined by detecting the CCT-β subunit. Results show that the prostate cancer cell lines, LNCaP, PC3 and PSMA transfected PC3 cell lines contain similar levels of the CCT-β protein by Western blot (Figure 5A). In addition, genomic analysis through the TCGA and cBioPortal databases indicate that the mRNA levels of CCT-β are essentially the same between the prostate cancer cell lines LNCaP and PC3 (Figure 5B), and that the gene for CCT-β (cct2) is amplified in patients with prostate cancer (Figure 5C). These results indicate that both LNCaP and PC3 cell lines should be responsive to treatment with a CT20p therapy. However, if selective PSMA internalization is achieved using the Folate-HBPE(CT20p) nanocarriers, the PSMA-expressing cell lines (e.g.,LNCaP) would be more responsive that the cell line that do not express PSMA (e.g., PC3). Our results show that upon incubation of LNCaP cells with Folate-HBPE(CT20p), dose- (based on CT20p peptide) and time-dependent responses were observed, achieving cell death within 48 hours with only a nanomolar (IC50 = 6 nM) concentration of the peptide (Figure 6A, 6D). Similar results were obtained with PSMA (+) PC3 cells (Figure 6B, 6E). As expected, no cytotoxicity was observed in PC3 cells due to the lack of PSMA expression on these cells (Figure 6C, 6F). Fluorescence microscopy studies of LNCaP and PSMA(+) PC3 cells incubated with Folate-HBPE(CT20p/DiI) nanocarrier that contains both CT20p and DiI, showed a significant amount of cell associated fluorescence and cell death that was abrogated by pre-incubation with 2-PMPA (Supplementary Figure S3). These results indicated that the Folate-HBPE(CT20p) can be used to specifically deliver CT20p to PCa cells via PSMA, achieving target-specific cell death that can be blocked by 2-PMPA. In addition, cell viability was measured by determining changes in cell membrane asymmetry and permeability using flow cytometry.[21] In this assay, the corresponding prostate cancer cells were first incubated with the Folate-HBPE(CT20p) at the IC50 dose (6.0 nM) and the cell's permeability was determined by the internalization and DNA-binding of a Sytox AADvanced dye. This dye is cell impermeable and non-fluorescent, but upon permeabilization of the cell (during late apoptosis or necrosis), the dye enters the dying cell and binds to DNA with an enhancement in the dye's fluorescence. Meanwhile, cell membrane asymmetry (apoptosis) was determine by incubation with the ratiometric probe (4'-N,N-diethylamino-6-(N,N,N-dodecyl-methylamino-sulfopropyl)-methyl-3-hydroxyflavone, F2N12S). In combination, these two dyes were capable of distinguishing live from apoptotic or necrotic cells as indicated in Figure 7A. Results showed that in the untreated LNCaP cells 88% are alive, with only 12% of the cells undergoing cell death (Figure 7B). The percent of LNCaP live cells were dramatically reduced to 28%, upon incubation with the Folate-HBPE(CT20p) NPs for 48 hours (Figure 7C). Most of the treated LNCaP cells (69%) were apoptotic. Pre-incubation of the cells with 2-PMPA increased the levels of live cells to 77%, as internalization of Folate-HBPE(CT20p) was reduced by 2-PMPA binding to PSMA on the surface of LNCaP cells (Figure 7D).
Current cancer chemotherapeutics, such as doxorubicin and cisplatin, although highly effective, often affect not only cancer cells but also normal tissues inducing problematic sites effects.[47-49] In addition, cancer cells often develop resistance to these drugs rendering them ineffective.[48, 50] With the goal of reducing systemic, non-specific toxicity to normal tissues and drug resistance, these drugs have been either modified with targeting ligands or encapsulated into nanoparticles.[8, 36, 51-53] However, as cells of the immune system, such as macrophages, can uptake these nanoparticles, cell death to these cells often occurred upon uptake of nanoparticles carrying these drugs. In a representative experiment using RAW mouse macrophages, the viability of untreated cells (92%) was minimally reduced to 84% upon incubation of these cells with Folate-HBPE(CT20p) (Figure 8A and 8B, respectively). In contrast, when macrophages were incubated with Doxorubicin, a dramatic reduction in the number of viable cells was observed with less than 1% of the cells being still alive, and most of the cells being apoptotic (24%) or necrotic (76%) (Figure 8C). Interestingly, when macrophages were incubated with Folate-S-S-Doxo, only 32% of the cells remained alive with most of them (64%) being necrotic (Figure 8D). These results are significant as they show that CT20p is cytotoxic to prostate cancer cells but not macrophages in contrast to doxorubicin which is cytotoxic to these cells in either preparation (with or without folate).
PSMA-specific reduction in the levels of cell surface integrin β1 (CD29) and cell adhesion
Previously, we reported that in a breast cancer cell model, CT20p induced a dramatic reduction in the levels of polymerized tubulin [22] and F-actin that triggers changes in the cell cytoskeleton and a reduction in the levels of cell surface integrin and cell adhesion, contributing to cell death.[21] Since cytoskeletal proteins interact with integrins and in prostate cancer integrins support metastasis and angiogenesis, mediating signals between the extracellular matrix and the actin cytoskeleton that enable invasiveness, we investigated if incubation of LNCaP cells with Folate-HBPE(CT20p) affected the levels of integrin β1. We selected this integrin as it has been found that integrin β1 (CD29) plays a key role in the metastatic progression of prostate cancer.[54] Assessing surface integrin levels by flow cytometry, we found no significant change in the levels of integrin β1 on PC3 cells incubated with Folate-HBPE(CT20p) as expected (Figure 9A). However, a significant decrease within 24 hours and an even larger decrease at 48 hours in the levels of integrin β1 is observed in PC3-PSMA(+) (Figure 9B) and LNCaP (Figure 9C) cells, respectively. These results correlate with decrease in the percentages of cell adhesion in the PSMA-expressing cells, but not in the PSMA-negative cells (Figure 9D-9F) incubated with Folate-HBPE(CT20p) for 24 and 48 hours. These results also suggested that Folate-HBPE(CT20p), by inhibiting the protein-folding activity of CCT, could reduce the pool of cytoskeletal proteins and reduce integrin levels in prostate cancer cells to impair key aspects of metastasis, specifically cell adhesion and invasion in cancer cells that express PSMA. As PSMA expression has been detected not only in primary prostate cancer tumors, but also in prostate cancer metastatic lesions [31, 55], Folate-HBPE(CT20p) could have a promising use for the treatment of metastatic PCa. Taken together, these results showed that Folate-HBPE(CT20p) can selectively target PSMA expressing PCa cells, causing a reduction of CCT client proteins, among which are actin and tubulin, inducing cell detachment that is in large part mediated by a reduction in the levels of β1 integrin. These effects lead to targeted cell death (e.g. anoikis), potentially minimizing the cancer cell's ability to metastasize in vivo, while not being toxic to macrophages and other non-cancerous tissues.
Figure 5
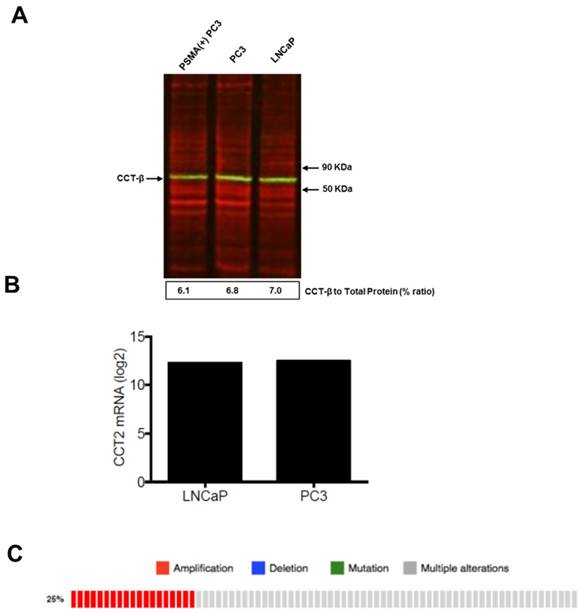
(A) Levels of CCTβ protein in LNCaP, PC3 and PSMA(+) PC3 cell lines by Western Blot analysis. Green bands indicate the presence of CCT-β in the samples. Red bands indicate total protein stain, loading control. (B) Genomic analysis of CCTβ mRNA levels in LNCaP, PC3 cell lines and (C) patients with neuroendocrine prostate cancer. Genomic analysis was performed using a Prostate Cancer TCGA database.
Figure 6
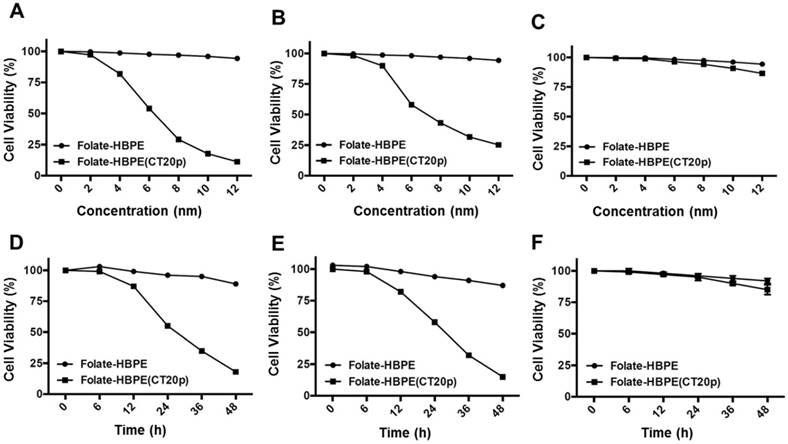
Dose- (a - c) and time- (d - f) dependent cytotoxicity assay (MTT) of prostate cancer cells treated with Folate HBPE(CT20p)-NPs and Folate-HBPE. LNCaP (A,D), PSMA(+) PC3 (B,E) and PC3 (C,F). A reduction in cell viability is observed in the cells that express PSMA when treated with Folate HBPE(CT20p)-NPs.
Figure 7
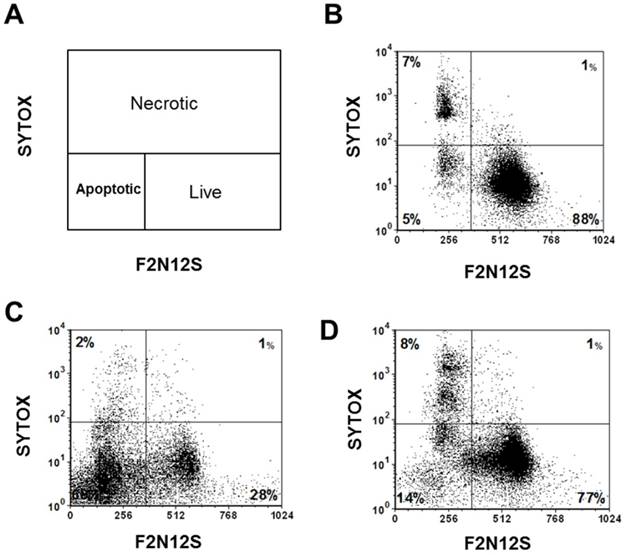
Cell viability assay via the assessment of cell membrane permeability (SYTOX AADvanced staining) and cell membrane asymmetry (F2N12S dyes staining). (A) Diagram showing the quadrants representing the population of viable (live), apoptotic and necrotic cells based on the relative uptake of Sytox and F2N12S. As cells undergo apoptosis and necrosis, they will become more permeable and lose membrane symmetry, resulting in shifting of the cell population to the left and up. (B) Untreated LNCaP cells, control. (C) LNCaP cells treated with Folate-HBPE(CT20p). (D) LNCaP cells pre-incubated with PMPA and then treated with Folate-HBPE(CT20p). Incubation time is 48h in all experiments.
Figure 8
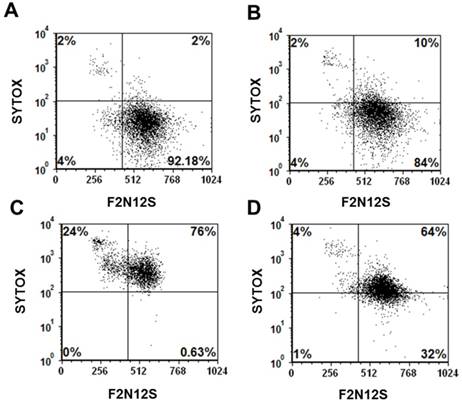
Cell viability (Sytox AADvanced/F2N12S) assay of: (A) untreated mouse macrophages (RAW), control, (B) RAW cells treated with Folate-HBPE(CT20p), (C) RAW cells treated with doxorubicin (D) RAW cells treated with Folate s-s-Doxo. Incubation time is 48h in all experiments.
Figure 9
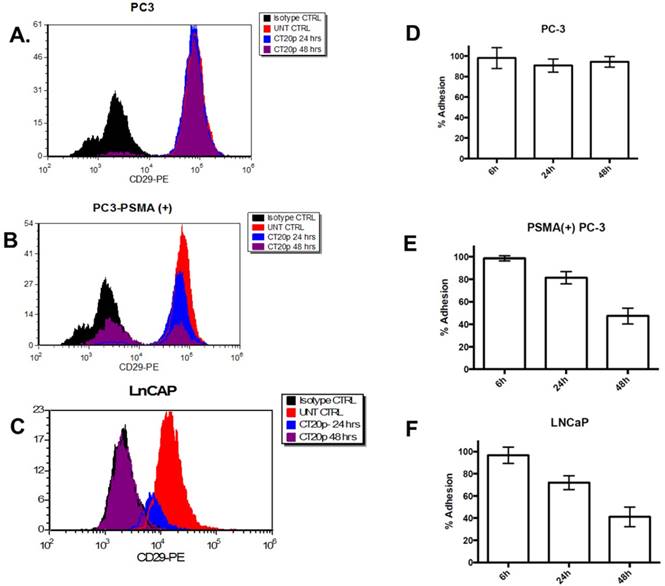
Levels of integrin β1 (CD29) in: (A) PC3, (B) PSMA(+) PC3 and (C) LnCaP cells treated with Folate-HBPE(CT20p). Corresponding cells were incubated with an anti-integrin β1 antibody conjugated with a phycoerythrin fluorescent dye. Binding to the corresponding cells was determined by flow cytometry to assess the levels of cell surface integrin β1 in the corresponding cells. UNT CTRL (red lines) represent the level of fluorescence of the corresponding to the untreated cells and correspond to the basal levels of integrin β1 on the cells surface. Isotype CTRL (black lines) represent the level of fluorescence corresponding to the non-specific binding of an antibody that does not bind to integrin β1. Upon incubation of the corresponding cells with Folate-HBPE(CT20p), a decrease in the levels of cell surface integrin β1 was determined by a movement to the left of the fluorescence peak, toward the Isotype CTRL peak. Corresponding percentages of cell adhesion in (D) PC3, (E) PSMA(+) PC3 and (F) LnCaP cells treated with Folate-HBPE(CT20p), determined using a crystal violet assay.
In vivo targeting of Folate-HBPE(CT20p) and specific tumor regression of PSMA expressing prostate cancer tumor xenographs
The PSMA-specific targeting of the Folate-HBPE(CT20p) nanocarrier was tested in vivo using mice bearing PSMA(+) PC3 tumors. First, we studied the PSMA-targeting ability of HBPE nanocarriers containing a near infrared DiR dye (Folate-HBPE(DiR)) to assess for specific tumor targeting via PSMA. For these experiments, PSMA(+) PC3 cells (1 × 106) were injected into the right flank of a nude male mice, while the same amount of wild type PC3 cells were injected into the left flank. Tumors were allowed to grow for a week. Then, an intravenous (IV) injection of Folate-HBPE(DiR) (2 mg/kg/dose), was administered to the mice. After 24 hours, mouse fluorescence imaging showed a strong fluorescence signal in the PSMA(+) PCa tumors, indicating selective delivery of the nanocarriers to the PSMA-expressing tumors (Figure 10A). No fluorescence was observed in wild type PC3 tumors, due to their lack of PSMA expression. This experiment was repeated twice to confirm that the fluorescent signal was restricted to the PSMA+ tumors obtaining similar results (Figure S4A). In addition, when mice were injected with HBPE(DiR) NPs with no folate conjugated on its surface, no tumor associated fluorescence was observed by mouse fluorescent imaging. (Figure S4B). These results suggested that the folate-conjugated HBPE nanocarrier can be used to selectively target PSMA-expressing PCa tumors in vivo.
Figure 10
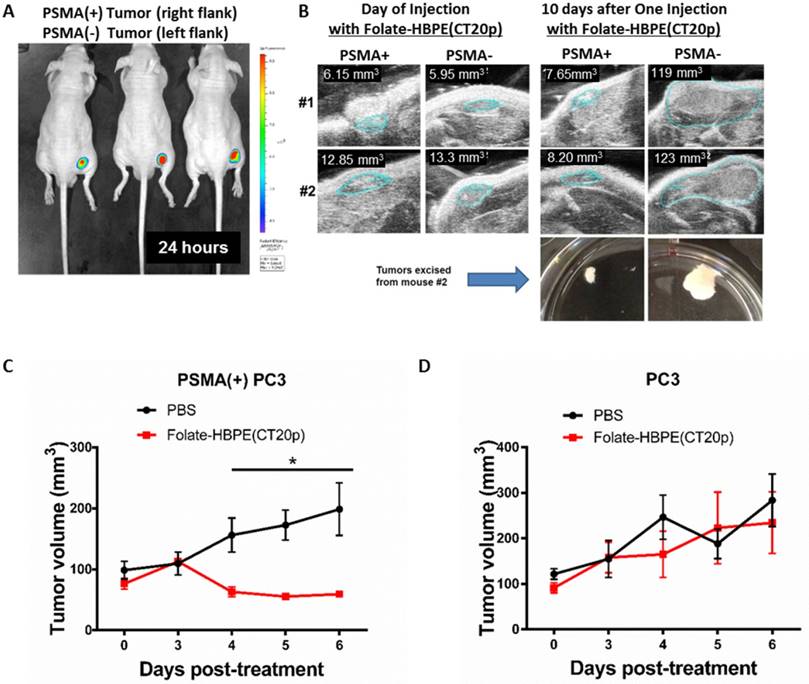
Targeting and treatment of prostate cancer tumors expressing PSMA with Folate-HBPE(CT20p). Mice (n=3) were injected subcutaneously (SC) with PSMA (+) (right flank) or PSMA(-) (left flank) prostate cancer cells. Upon tumor detection (~2 weeks), mice were injected intravenously (IV) with Folate-HBPE-NPs (2mg/kg/dose) containing (A) DiR, a near IR dye or (B) CT20p. Mice were imaged after 24 hours using a fluorescence in vivo imaging system (IVIS) to assess the specific targeting of the folate conjugated nanoparticles to PSMA expressing tumors (A). Ultrasound imaging was performed to assess tumor regression of mice treated with the Folate-HBPE(CT20p). Growth curves of (C) PSMA(+) PC3 or (D) wild type PC3 tumors with or without treatment with Folate-HBPE(CT20p).
Next, the PSMA-targeted anti-tumor effect of the Folate-HBPE(CT20p) was evaluated in mice bearing PSMA(+) and PSMA(-) PC3 tumors. A single intravenous (IV) treatment with Folate HBPE(CT20p) (2 mg/kg/dose or ~3.4 nM CT20p) caused significant reduction in the growth of the PSMA(+) PC3 but not the wild type PC3 tumors (Figure 10B), supporting the previous data in Figure 10A. A marked difference in the size of the excised tumors is observed with ultrasound imaging (Figure 10B). Histological examination of excised tumor tissues by a pathologist revealed fragmentation and areas of necrosis in the PSMA(+) tumors (Figure S5A), not evident in the PSMA(-) tumors (Figure S5B) or in the liver or spleen (Figure S5C and S5D), supporting that the Folate-HBPE(CT20p) delivered their toxic cargo (CT20p) specifically to the PSMA(+) tumors with minimal damage to liver and spleen. A growth-curve of tumors in mice treated with Folate-HBPE(CT20p) showed a suppression of growth in the PSMA(+) tumors (Figure 10C, black line), while the PSMA(-) tumors did not respond to the treatment and continued growing (Figure 10D). Similar results were also observed in mice with LNCaP tumor, where injection of Folate-HBPE(CT20p) was able to control tumor growth, as opposed to the PBS-control (Figure 11A). Treatment of the PSMA-expressing tumor-bearing mice with Folate-HBPE(CT20p) did not affect their weight (Figure 11B) or normal behavior. As also observed in the PSMA(+) PC3 tumors, a mark difference in the size of the excised LNCaP tumors from the Folate-HBPE(CT20p)-treated mice was observed (Figure 11C and 11D). Biodistribution experiments using Folate-HBPE(DiR) in the LNCaP tumor model showed accumulation of nanoparticles at 24 hours in tumors, as well as in the liver, with less accumulation in other tissues (Figure S6).
Taken together, we have shown that folate conjugated polymeric nanocarrier can be used to target PSMA in PCa cells, delivering a cytotoxic peptide (CT20p) selective for cells that highly express CCT to induce PCa cell death.
Figures 11
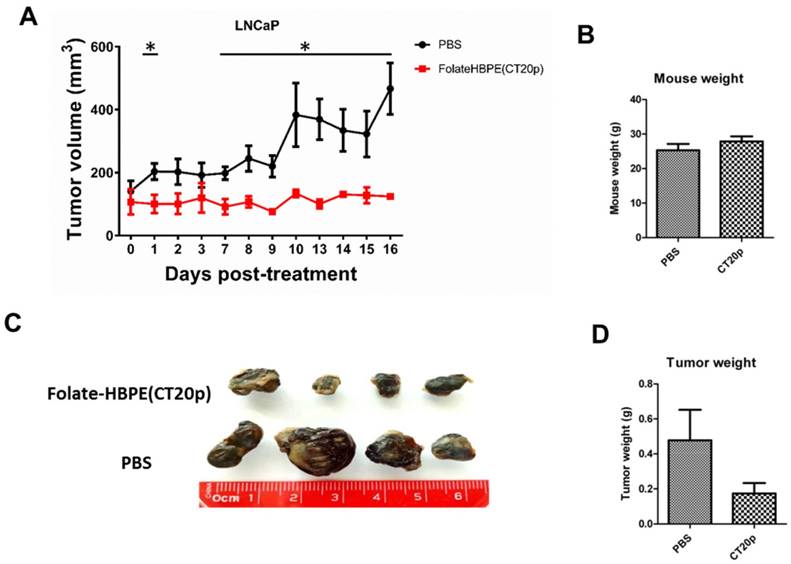
(A) Growth curves of LNCaP tumors treated with Folate-HBPE(CT20p) or PBS. (B) Average weight of the PBS-treated and Folate-HBPE(CT20p) treated mice (n = 4) at the end of the experiment. (C) Tumor size comparison after necropsy. (D) Average weight of the PBS-treated and Folate-HBPE(CT20p) tumors at the end of the experiment.
Discussion
PSMA is a unique prostate cancer specific target that has been largely investigated for the delivery of imaging and therapeutic agents to treat prostate cancer. Its overexpression in prostate cancer tissue and in the neovasculature of most solid tumors makes it an ideal target for cancer [31]. Its importance as a potential clinical therapeutic target is recapitulated by multiple clinical trials that use PSMA targeting to deliver small molecules or nanoparticle-based therapeutics to treat prostate cancer [56-59]. However, targeting PSMA has been facilitated mostly by anti-PSMA antibodies (e.g. J591 or 7E11) [59-63], PSMA aptamers [64, 65], glutamate ureas [66-69] and a few studies that reported the use of folic acid to target PSMA [70-73].
Herein, we engineered a multivalent folate conjugated hyperbranched polymeric nanoparticles as theranostic nanocarriers to deliver near infrared fluorescent dyes and a therapeutic peptide, CT20p, to prostate cancer cells via PSMA. CT20p is our therapeutic cargo that has been shown to selectively kill breast cancer cells by reducing the pool of CCT client proteins, leading to loss of actin and tubulin polymerization and changes in cytoskeletal architecture that in combination cause dramatic alterations in cancer cell morphology [20, 21]. Subsequently, these changes cause a decrease in the levels of cell surface integrins and a reduction in the degree of cell adhesion and migration, resulting in cell death. This cell death is independent of conventional apoptotic modulators, such as caspases or Bcl-2. [20] In this report, we showed that a multivalent folate-conjugated nanocarrier that encapsulated CT20p [Folate-HBPE(CT20p)] can induce PSMA-specific prostate cancer cell killing. The ability of PSMA to facilitate the binding and internalization of the folate conjugated nanoparticles is due to the ability of PSMA to bind and internalize folate-conjugated compounds, such as polyglutamated folates [74, 75]. This targeting approach to deliver therapeutics to prostate cancer is advantageous, as PSMA is not only expressed in the primary PCa tissue but also its metastatic lesions [27, 32]. Furthermore, CT20p is cytotoxic to cancer cells that highly express its target, CCT, and less toxic to healthy tissues with lower basal levels of CCT, including macrophages, spleen and liver tissue. This is important as most of the toxicity associated with current chemotherapeutics (e.g., doxorubicin, among others) is due to their uptake and killing in healthy cells.[47] As CT20p only affects cells highly expressing CCT, non-specific binding of Folate-HBPE(CT20p) to normal tissue and circulating blood cells that express the folate receptor would cause minimal damage. This contrasts with Doxorubicin, which we showed that kills macrophages at comparable concentration. The depletion of cells of the immune system such as macrophages, white and red cells has been implicated to some of its toxic side effects.[47, 76] As our folate-nanocarriers can be taken up by these immune cells, the use of a cancer-specific agent like CT20p and a therapeutic anticancer cargo is highly advantageous.
Our results show that by targeting the delivery of a cancer-specific therapeutic peptide (CT20p) to prostate cancer cells using PSMA, a dual level of cancer specificity is achieved. First, by using a nanoparticle delivery system that targets PSMA, selective targeting to prostate tumors is achieved with minimal accumulation in healthy tissue. Second, yet another level of cancer specificity is achieved by selecting a therapeutic cargo that is toxic only to cancer cells with elevated levels of CCT. In this way, even if non-specific uptake by healthy cells occurs, minimal damage will occur as the cargo (CT20p) is specific only to cells with increased CCT. In combination, this dual level of cancer killing-specificity could result in fewer side effects. Animal studies show that the Folate-HBPE(CT20p) can target PSMA and in vitro cell culture studies corroborate the specificity of the PSMA targeting as 2-PMPA, a high affinity PSMA ligand, blocks the internalization of these nanocarriers. In addition, uptake of these nanocarriers is observed in LNCaP cells that highly express PSMA, but not in PC3 that lack expression of this surface protein. Most importantly, when PC3 cells that were genetically modified to express PSMA are used, uptake of the Folate-HBPE(CT20p) is abrogated by pre-incubation of 2-PMPA. Internalization of the folate-conjugating nanocarrier is not facilitated by the folate receptor as the prostate cancer cell lines used in this study (LNCaP and PC3) has been reported to express low amounts of folate receptors (FOLR) [77]. Furthermore, comparison of the PSMA vs. FOLR gene expression levels in these cells using a public gene expression database (CAportal), corroborate a 3-fold higher expression of PSMA in LNCaP cells, compared to PC3 (Figure S7). The expression levels of the FOLR 1, 2, and 3 receptors are low in both LNCaP and PC3 cells. Taken together, with the fact that similar results are observed with a Folate-S-S-Doxo activatable probe, these results further indicate that uptake of the folate conjugated probes was facilitated by PSMA.
Incubation of the PSMA-positive prostate cancer cells with Folate-HBPE(CT20p) induces considerable changes in cell morphology, as evident by fluorescence microscopy images, triggering cell detachment and eventual cell death. Meanwhile, no changes are observed in PSMA-negative PC3 cells, even when these cells also expressed CCT-beta, the intracellular target of the therapeutic cargo CT20p. This is further proof of the PSMA-mediated internalization of our Folate nanocarrier that induces specific cell killing in PSMA-expressing. In addition, a reduction in the levels of integrin β1 is observed within 24 h. These results are important as integrin β1 plays a key role in prostate cancer invasion and metastatic potential [54]. Therefore, developing a targeted therapeutic agent that reduce the levels of this integrin in prostate cancer such as Folate-HBPE(CT20p) could not only reduce the viability of these cells but reduce their ability to metastasize in vivo. Studies are underway to test this hypothesis.
Materials and Methods
All chemical and reagents were analytical grade and used as supplied without further purification. Acetonitrile, dimethyl sulfoxide, dimethylformamide, N-hydroxysuccinimide (NHS) and doxorubicin hydrochloride (Doxo), poly-D-lysine hydrobromide were purchased from Sigma-Aldrich. 1-ethyl-3-[3-(dimethy- lamino)propyl] carbodiimide hydrochloride (EDC) was obtained from Pierce Biotechnology. Near Infrared dyes (DiI or DiR), 6-diamidino-2-phenylindole (DAPI), puromycin, Sytox AAdvanced and F2N12S Violet Ratiometric Apoptosis kit were purchased from Invitrogen. Dialysis membranes (MWCO 6-8K, 23 mm Flat-width) were obtained from Spectrum Laboratories. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from 2-PMPA was obtained from Tocris. Invitrogen. PC3 cells and LNCaP cells and RAW 264.7 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). PSMA (+) PC3 cells were obtained from Dr. Jam Grimm (Memorial-Sloan Kettering Cancer Center, MSKCC, New York, NY). Dulbecco's Modified Eagle's Medium (DMEM), Ham's F-12K (Kaighn's), RPMI-1640 basal media, trypsin, penicillin-streptomycin, were acquired from Cellgro and FBS, fetal bovine serum, from American Type Culture Collection. The PE-labeled mouse antihuman CD29 was from BD Bioscience, the Folate Receptor Polyclonal antibody was from Thermo Fisher, and the anti CCTβ monoclonal antibody was from Millipore. The peptide CT20p (Ac-VTIFVAGVLTASLTIWKKMG-NH2), was commercially synthesized (Biopeptide Co., Inc, San Diego, CA, USA) at > 98% purity. Foxn1nu/Foxn1nu nude mice purchased form Charles River. Holey carbon-coated copper 400-mesh grid was acquired from SPI supplies.
Synthesis and Characterization of Folate-Conjugated HBPE Nanoparticles
The HBPE polymer was synthesized as previously described.[52, 20] The HBPE nanoparticle encapsulating either a fluorescent dye (DiI or DiR) or the therapeutic peptide CT20p were fabricated by the solvent diffusion method.[52, 20] In brief, 5 μL of a 10 μg/μL of CT20p or fluorescent dye solution in DMF were mixed with a solution containing 15 mg of HBPE polymers in 250 μL of DMF. The resulting solution of HBPE polymer and cargo (dye or peptide) in DMF was added drop-wise to deionized water (5 mL) under continuous stirring at room temperature. The resulting HBPE nanoparticles were dialyzed (MWCO 6-8K) in ultra-pure water first and then PBS solution (pH = 7.4), before sterilized using 0.22 µm pore size filter.
To conjugate the nanoparticles with folate, folic acid was first conjugated with ethylene diamine to yield aminated folate following a procedure previously described [78]. Then, the carboxylated nanoparticles were PEGylated with Carboxy-PEG4-Amine (10mmol) via EDC/NHS chemistry. Specifically, a 200 μL solution of 10 mmol of EDC in MES buffer (pH=6.1) was added dropwise to an aqueous solution containing 1.0 mol of carboxylated HBPE nanoparticles. Followed by, the dropwise addition of 10 mmol NHS in 200 μL MES buffer. After 3 min incubation at room temperature, 200 μL of a solution of 10 mmol of Carboxy-PEG4-Amine in water was added dropwise and followed by continuous stirring for 3 h. The resulting PEGylated-HBPE nanoparticles were purified by dialysis (MWCO 6-8K) against ultrapure water and a PBS solution (pH = 7.4), before subsequent conjugation with aminated folate (10 mmol) via EDC/NHS chemistry, following a procedure similar to the one followed for the conjugation of Carboxy-PEG4-Amine. The Folate-Peg4-HBPE nanoparticles encapsulating either CT20p or a fluorescent dye were dialyzed in PBS and sterilized using a 0.22 µm pore size filter (Millipore). Preparations were stored at 4°C. Dynamic light scattering and zeta potential analysis of the folate decorated nanoparticles preparations were performed using a Zetasizer Nano ZS from Malvern Instruments. In addition, the shape of the nanoparticles was analyzed by Scanning Transmission Electron Microscopy (STEM) using a Zeiss ULTRA-55 FEG SEM instrument with STEM detector samples for STEM were prepared on a copper grid.
In Vitro Drug Release
The stability of encapsulation at physiological pH and the assessment of the CT20p release from the nanoparticle at low pH were analyzed using a dynamic dialysis technique describe before.[51] Briefly, 100 μL of HBPE nanoparticles were placed in a dialysis bag (MWCO 6-8 K) and dialyzed against a PBS solution (pH 7.4 or 5). The amount of released CT20p was determined at different time points within a period of 24 hours by analyzing 1 mL aliquots by absorbance at 280 nm due to the presence of tryptophan (W) in the peptide. The concentration of the CT20p was estimated using a calibration curve. The percentage of cumulative release was calculated applying the following equation:
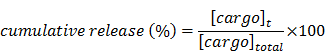
where [cargo]t is the amount of CT20p released at time t, and [cargo]total is the total CT20p present in the HBPE nanoparticles.
Cell Culture
PC3 cells were grown in Ham's F-12K (Kaighn's) medium (F12K) supplemented with 10% FBS and 1% penicillin-streptomycin, whereas PSMA (+) PC3 cells were maintained in F12K basal medium supplemented with 10% FBS, 1% penicillin-streptomycin and 0.05% puromycin was added to ensure the selection of the cell line. LNCaP cells were maintained in RPMI-1640 basal medium supplemented with 10% FBS and 1% penicillin-streptomycin. RAW 264.7 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) modified to contain 4 mM L-glutamine, 4500 mg/L glucose, 1 mM sodium pyruvate, and 1500 mg/L sodium bicarbonate, 10% FBS and 1% penicillin-streptomycin. All cell lines were maintained at 37 °C, 5% CO2 in a humidified incubator.
Cellular Uptake of Folate-S-S-Doxo
For these experiments, fifty thousand cells (PC3 cells, PSMA(+) PC3 cells or LNCaP cells) were seeded in a 24 well plate (200 mm2) and grown overnight, before treatment. The cells were incubated with Folate-S-S-Doxo (1.2 μM) at 37 °C, 5% CO2 in a humidified incubator for 12 h and 24 h. Subsequently, the cells were washed three times with 1X PBS and fixed with 4% paraformaldehyde in 1X PBS and stained with DAPI (1 mg/mL) for 10 min. The cells were washed and examined by fluorescence microscopy at the corresponding time points.
Inhibition of Cellular Uptake of Folate-HBPE Nanoparticles
To corroborate receptor-mediated uptake of the Folate-HBPE(DiI) NPs, PC3, PSMA(+) PC3 and LNCaP cell lines were seeded on a 35-mm culture plate. When the cells reached ~50% confluence, the plates were treated Folate-HBPE(DiI) (0.07 mg of HBPE nanoparticles/1.5 mL) and incubated at 37 °C in 5% CO2 for 24 h. The PSMA receptor inhibitor, PMPA, was added an hour prior treatment in excess, 100X treatment amount (~1 μM PMPA), if applicable. After incubation, the cells were washed twice with 1X PBS and fixed with a 4% paraformaldehyde solution followed by nuclei staining with DAPI. The uptake of the cells was examined with a florescence microscope equipped with a 10x or 40x objective. Likewise, PSMA-mediated cell internalization of Folate-HBPE(DiI) NPs was assessed by FACS. PC3, PSMA(+) PC3 or LNCaP cells were grown and treated as described above. After incubation, the media was collected and 500 μL of 0.05% trypsin were added and incubated until cell detachment was observed. Both fractions were combined and centrifuged at 1000 rpm; the cell pellets were collected and suspended in 1X PBS. All cellular suspensions (1x106 cells/ml) were examined by flow cytometry and assayed using the Folate-HBPE(DiI) and Ethidium bromide (EtBr). Fluorescence was measured at 616 nm for EtBr and at 565 nm for Dil. The acquisition number of the cells was set at 10,000. Analysis of data was done using FSC Express software (DeNovo).
Immunoblotting
To screen for the presence of CCT-β in the studies cell lines. Cell lysate were obtained by mechanical homogenation in a 210 mM sucrose, 70 mM mannitol, 10 mM HEPES, 1 mM EDTA buffer, pH 7.4. Cell lysates were centrifuged at 1,000xg for 10 minutes and the supernatant were runby SDS-PAGE before transferred to an immunobilon-FL membrane (Millipore). The blot was first probed with a primary antibody against CCT-β (Millipore), followed by a IRDye 800CW secondary antibody (LI-COR). The blot was then imaged on an Oddysey detection system, 800 nm channel (LI-COR). For total protein quantification, the REVERT Total Protein Stain kit (LI-COR) was used following manufacturer protocol and imaged on an Oddysey detection system, 700nm channel (LI-COR).
MTT Assay
MTT cell viability assay was used to test the effect of Folate-HBPE(CT20p) nanoparticles on the cell proliferation of PC3, PSMA(+) PC3 and LNCaP cell lines. Specifically, all cell lines (2,500 cells/well) were seeded in 96-well plate and incubated with the nanoparticles at indicated concentrations for 24 h at 37°C, 5% CO2. Afterwards, each well was washed three times with 1X PBS and treated with 20 μL of MTT solution (5 μg/μL) for 4 h at 37°C, 5% CO2. The resulting formazan crystals were dissolved in acidified isopropanol (10:1 isopropanol:0.1 N HCl) at room temperature and the absorbance was measured at 570 nm using a Synergy HT multi-detection microplate reader. These experiments were performed in triplicates and the control experiments were carried out in the same way as described above, except that no treatment was added. The percentage of cell survival as a function of CT20p concentration was later plotted to determine the relative IC50 value, which stands for the CT20p concentration needed to prevent cell proliferation by 50%.
Flow Cytometry
Short-term cell survival was assessed using the flow cytometry based SYTOX® AADvanced™ and Violet Ratiometric Membrane Asymmetry Probe, 4'-N,N-diethylamino-6-(N,N,N-dodecyl-methylamino-sulfopropyl)-methyl-3-hydroxyflavone (F2N12S) from ThermoFisher ,and used according to manufacturer's protocol. Briefly, RAW 264.7, PC3, PSMA(+) PC3 or LNCaP cells were seeded in on a 35-mm plates at a density 3 × 105 cells/well and grown until reaching confluence (~50%). Plates were treated with Folate-HBPE(CT20p) (7 nM), Doxo (1.2 μM), Doxo-S-S-Folate (1.2 μM) or controls and incubated at 37 °C in 5% CO2 for a 48 h. If applicable, an excess PMPA (~1 μM) was added an hour prior treatment. Media were collected and 500 μL of 0.05% trypsin was added and incubated until cell detachment. Both fractions were combined and centrifuged at 1000 rpm, the cell pellets were collected and suspended in 1X PBS. The cellular suspensions were assayed using the SYTOX® AADvanced™ assay, using an excitation wavelength of 488 nm and emissions of 695 nm. Membrane asymmetry was assessed using the Violet Ratiometric Membrane Asymmetry Probe/Dead Cell Apoptosis Kit. The acquisition number of the cells was set at 10,000. Data was collected using the BD FACS Canto flow cytometer and the analysis of data was done using FSC Express software (DeNovo).
Cellular Adhesion Assay
A standard crystal violet adhesion assay was done as previously described by MJ. Humphries [79]. Plates were used uncoated for PC3 and PSMA(+) PC3 cell lines and coated (1.0 mL/25 cm2 of a 10 μg/mL solution of poly-D-lysine) for LNCaP. All cell lines were seeded in 96 well plates at a density of 15,000 cells/well followed by incubation with Folate-HBPE(CT20p) (7 nM) for 6 h, 24 h and 48 h. Afterwards, cells were allowed to gently shake for 15 sec, fixed using 4% paraformaldehyde and stained using an aqueous solution of crystal violet (5 mg/ml). Absorbance at 595 nm was read on a Synergy HT multi-detection microplate reader (Biotek).
Measurement of Cell Surface Integrin Expression
PC3, PSMA(+) PC3 and LNCaP cells were treated with Folate-HBPE(CT20p) NPs (7 nM) for 24 h and 48 h. Next, the cells were trypsinized, washed with 5% FBS in PBS 1X and stained with PE labeled mouse anti-human CD29 and corresponding PE isotype control. Data was acquired with an Accuri C6 flow cytometer and analyzed using FCS Express software.
In vivo Studies
For in vivo experiments, 1.5 million cells in 100 μL matrigel/media (1:1) mixture were delivered subcutaneously into the flank region of 6-10 week-old males, Foxn1nu/Foxn1nu nude mice. Tumor volume and growth was evaluated by ultrasound (Visual Sonics Vevo 2100; Toronto, Canada) and caliper measurements. Tumor volume was determined as described by Tomayko et al. [80] For studies using PC3 tumor mouse models, the number of mice used was 3 per group (n=3). For studies using LNCaP tumor models, the number of mice per group was n=4. In all experiments, tumor bearing mice received 100 μL of 1mg/kg of either Folate-HBPE (CT20p) NPs, control HBPE (CT20p) NPs or 100 ul PBS intravenously. The 1-mg amount was calculated based on the amount of CT20p in the nanoparticle. For tissue staining, a standard Hematoxylin and Eosin (H&E) protocol was implemented. A board certified pathologist performed the histological examination. Mice were weighed at the completion of the experiment. Tumors were removed and weighted as well. This study was prepared in understanding with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Institutional Animal Care and Use Committee at the University of Central Florida approved the animal study protocol.
Statistical analysis
Representative experiments are shown with at least three technical replicates. For animal studies, statistical calculations were performed using GraphPad Prism software (GraphPad). Statistical significance was defined as P < 0.05. Interrogation of The Cancer Genome Atlas (TCGA) database was accomplished using the websource cBioPortal for Cancer Genomics (http://cbioportal.Org).[81, 82]
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by NIH/NIBIB grant R01 EB 019288 awarded to JMP and ARK. Partial funding was provided by grants from the Breast Cancer Research Foundation and the USA Department of Defense (DoD PC111667).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Freeman AI, Mayhew E. Targeted drug delivery. Cancer. 1986;58:573-83
2. FitzGerald D, Pastan I. Targeted toxin therapy for the treatment of cancer. J Natl Cancer Inst. 1989;81:1455-63
3. Jerjian TV, Glode AE, Thompson LA, O'Bryant CL. Antibody-Drug Conjugates: A Clinical Pharmacy Perspective on an Emerging Cancer Therapy. Pharmacotherapy. 2016;36:99-116
4. Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov. 2015;14:603-22
5. Arosio D, Casagrande C. Advancement in integrin facilitated drug delivery. Adv Drug Deliv Rev. 2016;97:111-43
6. Vergote I, Leamon CP. Vintafolide: a novel targeted therapy for the treatment of folate receptor expressing tumors. Ther Adv Med Oncol. 2015;7:206-18
7. Assaraf YG, Leamon CP, Reddy JA. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist Updat. 2014;17:89-95
8. Santra S, Kaittanis C, Santiesteban OJ, Perez JM. Cell-specific, activatable, and theranostic prodrug for dual-targeted cancer imaging and therapy. J Am Chem Soc. 2011;133:16680-8
9. Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903-10
10. Liu K, Jiang X, Hunziker P. Carbohydrate-based amphiphilic nano delivery systems for cancer therapy. Nanoscale. 2016;8:16091-156
11. Jo SD, Ku SH, Won Y-Y, Kim SH, Kwon IC. Targeted nanotheranostics for future personalized medicine: recent progress in cancer therapy. Theranostics. 2016;6:1362-77
12. Ryu JH, Koo H, Sun I-C, Yuk SH, Choi K, Kim K. et al. Tumor-targeting multi-functional nanoparticles for theragnosis: new paradigm for cancer therapy. Adv Drug Deliv Rev. 2012;64:1447-58
13. Maurer-Jones MA, Bantz KC, Love SA, Marquis BJ, Haynes CL. Toxicity of therapeutic nanoparticles. Nanomedicine. 2009;4:219-241
14. Liu S, Kurzrock R. Understanding Toxicities of Targeted Agents: Implications for Anti-tumor Activity and Management. Semin Oncology. 2015;42:863-75
15. Liu S, Kurzrock R. Toxicity of targeted therapy: Implications for response and impact of genetic polymorphisms. Cancer Treat Rev. 2014;40:883-91
16. Liu C-Y, Tsai T-H, Huang Y-C, Shieh H-R, Liao H-F, Chen Y-J. Differential immunomodulating effects of pegylated liposomal doxorubicin nanoparticles on human macrophages. J Nanosci Nanotechnol. 2012;12:7739-46
17. Tan Q, Liu X, Fu X, Li Q, Dou J, Zhai G. Current development in nanoformulations of docetaxel. Expert Opin Drug Delivery. 2012;9:975-90
18. Marcato P, Favaro W, Duran N. Cisplatin properties in a nanobiotechnological approach to cancer: a mini-review. Curr Cancer Drug Targets. 2014;14:458-76
19. Cadoo K, Lowery M, McCaffrey J. Re: Lymphopenia Associated With Adjuvant Anthracycline/Taxane Regimens. Clin Breast Cancer. 2009;9:262
20. Boohaker RJ, Zhang G, Lee MW, Nemec KN, Santra S, Perez JM. et al. Rational Development of a Cytotoxic Peptide to Trigger Cell Death. Mol Pharm. 2012;9:2080-93
21. Lee MW, Bassiouni R, Sparrow NA, Iketani A, Boohaker RJ, Moskowitz C. et al. The CT20 peptide causes detachment and death of metastatic breast cancer cells by promoting mitochondrial aggregation and cytoskeletal disruption. Cell Death Dis. 2014;5:e1249
22. Bassiouni R, Nemec KN, Iketani A, Flores O, Showalter A, Khaled AS. et al. Chaperonin Containing-TCP-1 Protein Level in Breast Cancer Cells Predicts Therapeutic Application of a Cytotoxic Peptide. Clinical Cancer Research. 2016 clincanres.2502.015
23. Boudiaf-Benmammar C, Cresteil T, Melki R. The cytosolic chaperonin CCT/TRiC and cancer cell proliferation. PloS One. 2013;8:e60895
24. Zhang Y, Wang Y, Wei Y, Wu J, Zhang P, Shen S. et al. Molecular chaperone CCT3 supports proper mitotic progression and cell proliferation in hepatocellular carcinoma cells. Cancer Lett. 2016;372:101-9
25. Chen L, Zhang Z, Qiu J, Zhang L, Luo X, Jang J. Chaperonin CCT-Mediated AIB1 Folding Promotes the Growth of ERα-Positive Breast Cancer Cells on Hard Substrates. PloS One. 2014;9:e96085
26. Guest ST, Kratche ZR, Bollig-Fischer A, Haddad R, Ethier SP. Two members of the TRiC chaperonin complex, CCT2 and TCP1 are essential for survival of breast cancer cells and are linked to driving oncogenes. Exp Cell Res. 2015;332:223-35
27. Ross JS, Sheehan CE, Fisher HA, Kaufman RP Jr, Kaur P, Gray K. et al. Correlation of primary tumor prostate-specific membrane antigen expression with disease recurrence in prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2003;9:6357-62
28. Ghosh A, Heston WD. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J Cell Biochem. 2004;91:528-39
29. Wright GL Jr, Grob BM, Haley C, Grossman K, Newhall K, Petrylak D. et al. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996;48:326-34
30. Murphy GP, Elgamal AA, Su SL, Bostwick DG, Holmes EH. Current evaluation of the tissue localization and diagnostic utility of prostate specific membrane antigen. Cancer. 1998;83:2259-69
31. Chang SS, Reuter VE, Heston WD, Gaudin PB. Comparison of anti-prostate-specific membrane antigen antibodies and other immunomarkers in metastatic prostate carcinoma. Urology. 2001;57:1179-83
32. Perner S, Hofer MD, Kim R, Shah RB, Li H, Moller P. et al. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum Pathol. 2007;38:696-701
33. Silver DA, Pellicer I, Fair WR, Heston W, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81-5
34. Yao V, Berkman CE, Choi JK. Expression of prostate-specific membrane antigen (PSMA), increases cell folate uptake and proliferation and suggests a novel role for PSMA in the uptake of the non-polyglutamated folate, folic acid. Prostate. 2010;70:305
35. Yao V, Bacich DJ. Prostate specific membrane antigen (PSMA) expression gives prostate cancer cells a growth advantage in a physiologically relevant folate environment in vitro. Prostate. 2006;66:867-75
36. Choi SK, Thomas T, Li M-H, Kotlyar A, Desai A, Baker JR. Light-controlled release of caged doxorubicin from folate receptor-targeting PAMAM dendrimer nanoconjugate. Chem Commun. 2010;46:2632-4
37. Low PS, Antony AC. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv Drug Deliv Rev. 2004;56:1055-8
38. Leamon CP, Low PS. Delivery of macromolecules into living cells: a method that exploits folate receptor endocytosis. Proc Natl Acad Sci USA. 1991;88:5572-6
39. Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg Med Chem Lett. 2006;16:5350-5
40. Leamon CP, Reddy JA, Vlahov IR, Vetzel M, Parker N, Nicoson JS. et al. Synthesis and biological evaluation of EC72: a new folate-targeted chemotherapeutic. Bioconjug Chemistry. 2005;16:803-11
41. Kularatne SA, Wang K, Santhapuram HK, Low PS. Prostate-specific membrane antigen targeted imaging and therapy of prostate cancer using a PSMA inhibitor as a homing ligand. Mol Pharm. 2009;6:780-9
42. Ghosh A, Wang X, Klein E, Heston WD. Novel role of prostate-specific membrane antigen in suppressing prostate cancer invasiveness. Cancer Res. 2005;65:727-31
43. Smith-Jones PM, Vallabhajosula S, Navarro V, Bastidas D, Goldsmith SJ, Bander NH. Radiolabeled monoclonal antibodies specific to the extracellular domain of prostate-specific membrane antigen: preclinical studies in nude mice bearing LNCaP human prostate tumor. J Nucl Med. 2003;44:610-7
44. Bander NH, Trabulsi EJ, Kostakoglu L, Yao D, Vallabhajosula S, Smith-Jones P. et al. Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J Urology. 2003;170:1717-21
45. Tagawa ST, Beltran H, Vallabhajosula S, Goldsmith SJ, Osborne J, Matulich D. et al. Anti-Prostate Specific Membrane Antigen-based Radioimmunotherapy for Prostate Cancer. Cancer. 2010;116:1075-83
46. Pandit-Taskar N, O'Donoghue JA, Divgi CR, Wills EA, Schwartz L, Gönen M. et al. Indium 111-labeled J591 anti-PSMA antibody for vascular targeted imaging in progressive solid tumors. EJNMMI Res. 2015;5:28
47. El-Sayyad HI, Ismail MF, Shalaby FM, Abou-El-Magd RF, Gaur RL, Fernando A. et al. Histopathological effects of cisplatin, doxorubicin and 5-flurouracil (5-FU) on the liver of male albino rats. Int J Biol Sci. 2009;5:466-73
48. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265-79
49. Ajani JA. Optimizing docetaxel chemotherapy in patients with cancer of the gastric and gastroesophageal junction. Cancer. 2008;113:945-55
50. Hao XY, Bergh J, Brodin O, Hellman U, Mannervik B. Acquired resistance to cisplatin and doxorubicin in a small cell lung cancer cell line is correlated to elevated expression of glutathione-linked detoxification enzymes. Carcinogenesis. 1994;15:1167-73
51. Santra S, Kaittanis C, Grimm J, Perez JM. Drug/dye-loaded, multifunctional iron oxide nanoparticles for combined targeted cancer therapy and dual optical/magnetic resonance imaging. Small. 2009;5:1862-8
52. Santra S, Kaittanis C, Perez JM. Aliphatic Hyperbranched Polyester: A New Building Block in the Construction of Multifunctional Nanoparticles and Nanocomposites. Langmuir. 2010;26:5364-73
53. ElBayoumi TA, Torchilin VP. Tumor-Targeted Nanomedicines: Enhanced Anti-Tumor Efficacy In vivo of Doxorubicin-Loaded Long-Circulating Liposomes Modified with Cancer-Specific Monoclonal Antibody. Clin Cancer Res: an official journal of the American Association for Cancer Research. 2009;15:1973-80
54. Lee YC, Jin JK, Cheng CJ, Huang CF, Song JH, Huang M. et al. Targeting constitutively activated beta1 integrins inhibits prostate cancer metastasis. Mol Cancer Res. 2013;11:405-17
55. Bostwick DG, Pacelli A, Blute M, Roche P, Murphy GP. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: a study of 184 cases. Cancer. 1998;82:2256-61
56. Taneja SS. ProstaScint® Scan: Contemporary Use in Clinical Practice. Rev Urol. 2004;6:S19-S28
57. Bander NH, Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ. Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J Clin Onc. 2005;23:4591-601
58. Rowe SP, Macura KJ, Ciarallo A, Mena E, Blackford A, Nadal R. et al. Comparison of Prostate-Specific Membrane Antigen-Based 18F-DCFBC PET/CT to Conventional Imaging Modalities for Detection of Hormone-Naïve and Castration-Resistant Metastatic Prostate Cancer. J Nucl Med. 2016;57:46-53
59. Barrett JA, Coleman RE, Goldsmith SJ, Vallabhajosula S, Petry NA, Cho S. et al. First-in-man evaluation of 2 high-affinity PSMA-avid small molecules for imaging prostate cancer. J Nucl Med. 2013;54:380-7
60. Chevalier S, Moffett S, Turcotte E, Luz M, Chauvette L, Derbekyan V. et al. The dog prostate cancer (DPC-1) model: a reliable tool for molecular imaging of prostate tumors and metastases. EJNMMI Res. 2015;5:1
61. Cho H-S, Dong Z, Pauletti GM, Zhang J, Xu H, Gu H. et al. Fluorescent, Superparamagnetic Nanospheres for Drug Storage, Targeting, and Imaging: A Multifunctional Nanocarrier System for Cancer Diagnosis and Treatment. ACS Nano. 2010;4:5398-404
62. Tse BW-C, Cowin GJ, Soekmadji C, Jovanovic L, Vasireddy RS, Ling M-T. et al. PSMA-targeting iron oxide magnetic nanoparticles enhance MRI of preclinical prostate cancer. Nanomedicine. 2015;10:375-86
63. Viola-Villegas NT, Sevak KK, Carlin SD, Doran MG, Evans HW, Bartlett DW. et al. Noninvasive imaging of PSMA in prostate tumors with 89Zr-labeled huJ591 engineered antibody fragments: the faster alternatives. Mol Pharm. 2014;11:3965-73
64. Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW. et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sciences USA. 2006;103:6315-20
65. Hrkach J, Von Hoff D, Ali MM, Andrianova E, Auer J, Campbell T. et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128-39
66. Lütje S, Heskamp S, Cornelissen AS, Poeppel TD, van den Broek SA, Rosenbaum-Krumme S. et al. PSMA ligands for radionuclide imaging and therapy of prostate cancer: clinical status. Theranostics. 2015;5:1388
67. Shallal HM, Minn I, Banerjee SR, Lisok A, Mease RC, Pomper MG. Heterobivalent agents targeting PSMA and integrin-αvβ3. Bioconjug Chem. 2014;25:393-405
68. Ray Banerjee S, Chen Z, Pullambhatla M, Lisok A, Chen J, Mease RC. et al. Preclinical Comparative Study of 68Ga-Labeled DOTA, NOTA, and HBED-CC Chelated Radiotracers for Targeting PSMA. Bioconjug Chem. 2016;27:1447-55
69. Chen Y, Foss CA, Byun Y, Nimmagadda S, Pullambhatla M, Fox JJ. et al. Radiohalogenated prostate-specific membrane antigen (PSMA)-based ureas as imaging agents for prostate cancer. J Med Chem. 2008;51:7933-43
70. Jivrajani M, Nivsarkar M. Ligand-targeted bacterial minicells: Futuristic nano-sized drug delivery system for the efficient and cost effective delivery of shRNA to cancer cells. Nanomedicine. 2016;12:2485-98
71. Li X, McTaggart M, Malardier-Jugroot C. Synthesis and characterization of a pH responsive folic acid functionalized polymeric drug delivery system. Biophys Chem. 2016;214:17-26
72. Yoshizawa T, Hattori Y, Hakoshima M, Koga K, Maitani Y. Folate-linked lipid-based nanoparticles for synthetic siRNA delivery in KB tumor xenografts. Eur J Pharm Biopharm. 2008;70:718-25
73. Hattori Y, Maitani Y. Enhanced in vitro DNA transfection efficiency by novel folate-linked nanoparticles in human prostate cancer and oral cancer. J Control Release. 2004;97:173-83
74. Yao V, Berkman CE, Choi JK, O'Keefe DS, Bacich DJ. Expression of prostate-specific membrane antigen (PSMA), increases cell folate uptake and proliferation and suggests a novel role for PSMA in the uptake of the non-polyglutamated folate, folic acid. Prostate. 2010;70:305-16
75. Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc Natl Acad Sci U S A. 2005;102:5981-6
76. Daemen T, Hofstede G, Ten Kate MT, Bakker-Woudenberg IA, Scherphof GL. Liposomal doxorubicin-induced toxicity: Depletion and impairment of phagocytic activity of liver macrophages. Inter J Cancer. 1995;61:716-21
77. Hattori Y, Maitani Y. Folate-linked nanoparticle-mediated suicide gene therapy in human prostate cancer and nasopharyngeal cancer with herpes simplex virus thymidine kinase. Cancer Gene Ther. 2005;12:796-809
78. Zhang Z, Huey Lee S, Feng SS. Folate-decorated poly(lactide-co-glycolide)-vitamin E TPGS nanoparticles for targeted drug delivery. Biomaterials. 2007;28:1889-99
79. Humphries M. Cell adhesion assays. Mol Biotechnol. 2001;18:57-61
80. Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer chemotherapy and pharmacology. 1989;24:148-54
81. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
82. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
Author contact
![]() Corresponding author: J Manuel Perez, PhD, Professor, Department of Biomedical Sciences & Department of Neurosurgery, Biomedical Imaging Research Institute & Samuel Oschin Comprehensive Cancer Institute, Cedar Sinai Medical Center, 127 S. San Vicente Blvd, Suite A8113, Los Angeles CA, 90048 Email: jmanuel.perezorg
Corresponding author: J Manuel Perez, PhD, Professor, Department of Biomedical Sciences & Department of Neurosurgery, Biomedical Imaging Research Institute & Samuel Oschin Comprehensive Cancer Institute, Cedar Sinai Medical Center, 127 S. San Vicente Blvd, Suite A8113, Los Angeles CA, 90048 Email: jmanuel.perezorg
Citation styles
APA
Flores, O., Santra, S., Kaittanis, C., Bassiouni, R., Khaled, A.S., Khaled, A.R., Grimm, J., Perez, J.M. (2017). PSMA-Targeted Theranostic Nanocarrier for Prostate Cancer. Theranostics, 7(9), 2477-2494. https://doi.org/10.7150/thno.18879.
ACS
Flores, O.; Santra, S.; Kaittanis, C.; Bassiouni, R.; Khaled, A.S.; Khaled, A.R.; Grimm, J.; Perez, J.M. PSMA-Targeted Theranostic Nanocarrier for Prostate Cancer. Theranostics 2017, 7 (9), 2477-2494. DOI: 10.7150/thno.18879.
NLM
Flores O, Santra S, Kaittanis C, Bassiouni R, Khaled AS, Khaled AR, Grimm J, Perez JM. PSMA-Targeted Theranostic Nanocarrier for Prostate Cancer. Theranostics 2017; 7(9):2477-2494. doi:10.7150/thno.18879. https://www.thno.org/v07p2477.htm
CSE
Flores O, Santra S, Kaittanis C, Bassiouni R, Khaled AS, Khaled AR, Grimm J, Perez JM. 2017. PSMA-Targeted Theranostic Nanocarrier for Prostate Cancer. Theranostics. 7(9):2477-2494.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.