Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):2150-2163. doi:10.7150/thno.18185 This issue Cite
Research Paper
Epigenomic and Functional Characterization of Junctophilin 3 (JPH3) as a Novel Tumor Suppressor Being Frequently Inactivated by Promoter CpG Methylation in Digestive Cancers
Xiaotong Hu1 ![]() , Yeye Kuang1, Lili Li2, Haimei Tang1, Qinglan Shi1, Xingsheng Shu2, Yanjiao Zhang2, Francis KL Chan3, Qian Tao2
, Yeye Kuang1, Lili Li2, Haimei Tang1, Qinglan Shi1, Xingsheng Shu2, Yanjiao Zhang2, Francis KL Chan3, Qian Tao2 ![]() , Chao He1
, Chao He1
1. Biomedical Research Center and Key Laboratory of Biotherapy of Zhejiang Province, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou 310016, China;
2. Cancer Epigenetics Laboratory, Department of Clinical Oncology, State Key Laboratory of Oncology in South China, Sir YK Pao Center for Cancer and Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong;
3. Institute of Digestive Disease and State Key Laboratory of Digestive Diseases, Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Hong Kong.
Received 2016-11-2; Accepted 2017-4-4; Published 2017-5-30
Citation:
Hu X, Kuang Y, Li L, Tang H, Shi Q, Shu X, Zhang Y, Chan FKL, Tao Q, He C. Epigenomic and Functional Characterization of Junctophilin 3 (JPH3) as a Novel Tumor Suppressor Being Frequently Inactivated by Promoter CpG Methylation in Digestive Cancers. Theranostics 2017; 7(7):2150-2163. doi:10.7150/thno.18185. https://www.thno.org/v07p2150.htm
Other stylesAbstract

Junctophilin (JPH) proteins stabilize junctional membrane complexes between plasma membrane and endoplasmic reticulum, also implicated in some human diseases. JPH3 mutations are linked to Huntington's disease-like 2 syndrome. Through epigenomic study of a colon cancer cell line pair (HCT116 and DKO), we identified JPH3 as a methylated novel tumor suppressor gene (TSG) candidate at 16q24. We further studied its epigenetic alterations and functions in digestive tumorigenesis. JPH3 expression at the RNA level was found to be frequently silenced or reduced in colorectal and gastric cancers due to its promoter CpG methylation, which is associated with tumor progression and poor survival of digestive cancer patients. Ectopic expression of JPH3 inhibited tumor cell growth in vitro and in vivo. JPH3 expression upregulated the cytosolic Ca2+ levels, and unfolded protein response gene expression upon endoplasmic reticulum stress. JPH3 also induced calpain activation and subsequent mitochondrial membrane depolarization and cell apoptosis. Thus, JPH3 was identified as a novel TSG methylated in colorectal and gastric tumors which promotes mitochondrial-mediated apoptosis, also as a potential metastasis and survival biomarker for digestive cancers.
Keywords: junctophilin, tumor suppressor gene, methylation, metastasis, 16q24.
Introduction
Gastric and colorectal cancers remain the third and fourth most common cancers and the second and third most common causes of cancer mortality worldwide. The gradual accumulation of multiple genetic and epigenetic changes genome-wide is the molecular basis of their initiation and progression [1]. Epigenetic modifications, particularly promoter CpG methylation, play a key role in their morphological progression. DNA methylation often results in the hypermethylation of selected gene promoters and inactivation of tumor suppressor genes (TSGs) [2]. Epigenetic disruption of TSGs is a fundamental contributor to digestive tumor development. Thus, identification of novel hypermethylated genes offers molecular insights into potential screening, monitoring, and therapeutic strategies to digestive cancers [3-5].
16q24 is a frequently deleted TSG locus in multiple tumors, including cancers of digestive, nasopharyngeal, breast, prostate and liver tissues. Loss of heterozygosity (LOH) at 16q24 is also frequently present in multiple other solid tumors, such as breast, prostate, gastric, colorectal, hepatocellular and Wilm's tumor [6-10], suggesting the presence of critical TSG(s) at this locus. Several bona fide TSGs have been identified at this locus including CDH13 [11]. We previously also used integrative genomics/epigenetics to identify TSGs at this critical locus and have successfully identified IRF8 [12], CDH11 [13]. We propose that more critical undocumented TSGs may reside at this locus, which needs to be further investigated.
Junctophilins (JPHs) belong to members of the junctional membrane complex (JMC) protein family, serving to stabilize junctional membrane complexes between endoplasmic reticulum (ER) and plasma membrane (PM), as well as maintain cellular ultrastructure between the cell surface and intracellular ionic channels. JPHs are essential for proper Ca2+-induced Ca2+ release during the excitation-contraction coupling. Four JPH family members, JPH1-JPH4, have been identified [14, 15] with highly conserved across species and tissue-specific expression manner. Abnormalities associated with JPH proteins have been linked to human diseases [16], including cardiomyopathy, arrhythmia, heart failure, skeletal muscle myopathy and Huntington's disease (HD)-like pathology. For an example, in Huntington's disease-like 2 (HDL2), decreased JPH3 transcripts and proteins are detected in HDL2 brain, probably due to the abnormal CAG/CTG repeat expansions at the HDL2 locus involving a junctional complex protein JPH3, while JPH3 hemizygous or null mice display impaired motor function [16, 17]. However, the association of alterations and functions in JPH family proteins with human tumorigenesis has been rarely studied.
In this study, we searched for methylated TSG candidates for digestive cancers through epigenomic (CpG methylome) study and expression profiling of a paired colon cancer cell line (HCT116 and HCT116-DKO with double knock-out of DNMT1 and DNMT3B [18, 19]), and identified a candidate 16q24 TSG - Junctophilin-3 (JPH3) as a methylated target. We systematically evaluated the inactivation of JPH3 by promoter CpG methylation in colorectal and gastric cancers, further explored its functions and mechanisms in the development of these cancers.
Results
Epigenomic identification of JPH3 as a methylated target in digestive cancers
Through analyzing whole-genome CpG methylation profiles (methylomes) of a paired colon cell line, HCT116 and DKO (with double knock-out of DNMT1 and DNMT3B), we found a significant signal enrichment of CpG methylation (cut off = 2) at the JPH3 promoter and exon 1 region in HCT116, but not in DKO (Figure 1A), suggesting that JPH3 is a methylated target in colorectal cancer.
We next examined JPH3 mRNA and protein expression in human normal adult tissues by semi-quantitative RT-PCR and immunohistochemistry. Results showed that in addition to its expected abundant expression in brain, JPH3 was also expressed in most other normal tissues including digestive tissues such as stomach, colon and rectum (Figure 1B). JPH3 is expressed in the cytoplasm of normal colonic and gastric mucosa epithelium (Figure 1B). We then examined its expression in colorectal and gastric tumor cell lines and found that JPH3 was silenced or dramatically reduced in most cell lines (Figure 1D, Suppl. Figure S1, and S3).
As JPH3 has 4 splicing variants, with only variant 1 as the longest full-length transcript containing 5 coding exons and encoding a fully functional 748-amino acid protein; while variant 2 and 3 are much shorter isoforms with only two coding exons and distinct C-termini, and variant 4 is actually a non-coding RNA. We designed more RT-PCR primers to examine the expression of these variants in cell lines and some primary tumors (Figure 1, Suppl. Figure S1, Suppl. Table S1). We found that variant 1, 4, 2 were frequently downregulated or silenced in most colorectal (CRC) and gastric cancer cell lines and primary tumors, while no expression of variant 3 was detected in any CRC and gastric tumor cell line or tumor (Figure 1, Suppl. Figure S1).
JPH3 contains a typical CpG island (CGI) spanning its promoter and exon 1 (Figure 1C). Methylation-specific PCR (MSP) was used to analyze its methylation status and found that the JPH3 CGI was methylated in all cell lines with silenced or reduced expression (Figure 1D). JPH3 expression was restored after demethylation treatment with 5-Aza and TSA in methylated and silenced cell lines (Figure 1E). We further examined the detailed methylation profiles by bisulfite genomic sequencing (BGS) analysis of 81 CpG sites of the CGI, including sites analyzed by MSP (Figure 1F). Densely methylated CpG sites were detected in silenced cell lines, while both MSP and BGS showed that the JPH3 CGI was demethylated after 5-Aza and TSA treatment (Figure 1E, 1F), revealing a link between CGI methylation and JPH3 silencing. We also examined JPH3 expression and methylation in DKO cells. Results showed that JPH3 expression was restored in DKO cells, together with completely demethylated promoter alleles, suggesting that JPH3 transcription is regulated by promoter CpG methylation (Figure 1E).
JPH3 inhibited the growth and migration of digestive tumor cells
Frequent JPH3 silencing by promoter methylation in colorectal and gastric cancers suggests that JPH3 is likely a tumor suppressor. We investigated the effect of ectopic JPH3 expression on digestive tumor cell growth and migration. JPH3 expression plasmid was stably transfected into methylated and silenced HCT116 colon and MKN-28 gastric tumor cells. Ectopic JPH3 expression significantly suppressed the colony formation efficiency in plate and soft agar of transfected cells, relative to both vector-transfected cells and control cells (p<0.05, Figure 2A, 2B), indicating that JPH3 possesses growth inhibitory activities in colorectal and gastric cancer cells and functions as a tumor suppressor. Re-expression of JPH3 in these cells was confirmed by RT-PCR and Western blot (Figure 2C, Suppl. Figure S3). We evaluated the effects of JPH3 on the growth of MKN-28 cells in nude mice in vivo. The growth curve of JPH3 stably transfected and vector-transfected MKN-28 cells in nude mice was shown in Figure 2D, with no tumor grown in the JPH3-expressing cell group.
Figure 1
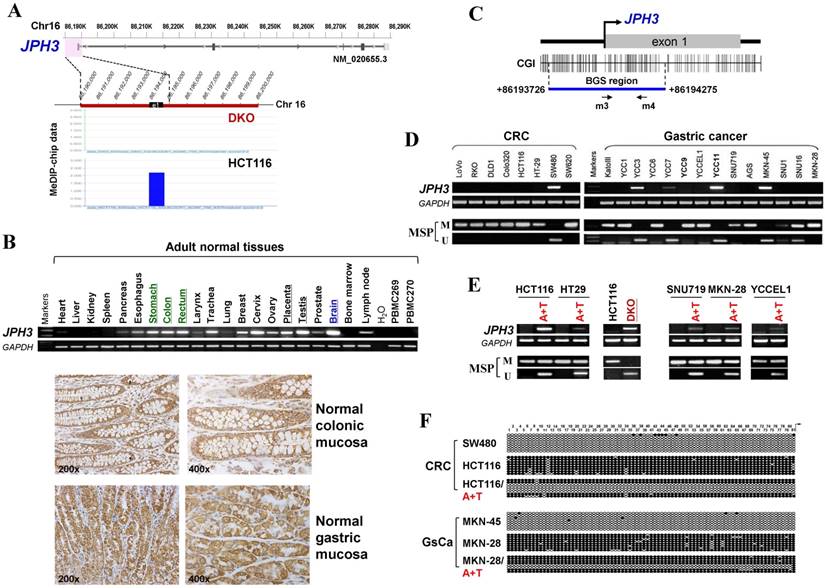
JPH3 expression and methylation in colorectal and gastric cancer cell lines and primary tumors. (A) CpG methylome study identified JPH3 as a methylated target in HCT116. JPH3 gene structure, promoter and exon 1 (UCSC Genome Browser NCBI36/hg18) are shown on the top panel. e1: exon 1. Positive methylation signal peak (blue) in HCT116 was identified by MeDIP-chip. (B) Broad expression of JPH3 in a normal adult tissue panel by semi-quantitative RT-PCR (primers detecting both variant 1 and 4), with GAPDH as a control (upper). Representative immunohistochemical staining for JPH3 in normal colonic and gastric mucosa. Original magnification: 200x, 400x (lower). (C) Structure of the JPH3 promoter CpG island (CGI). CpG sites are shown as short vertical lines. MSP primer sites and BGS region analyzed are also indicated. (D) Downregulation and silencing of JPH3 in colorectal and gastric cancer cell lines as a result of its CGI methylation, with RT-PCR primers detecting both variant 1 and 4. (E) Pharmacological demethylation with 5-Aza and TSA restored JPH3 expression in methylated and silenced cell lines. Representative results are shown. (F) Representative BGS results of cell lines. Vertical lines indicate individual CpG sites. Cloned BGS-PCR products were sequenced and each colony is shown as an individual row, representing a single allele of the CGI. Filled circles represent methylated and open circles unmethylated CpG sites. U: unmethylated; M: methylated. A: 5-Aza; T: TSA; CRC, colorectal cancer; GsCa, gastric cancer.
Figure 2
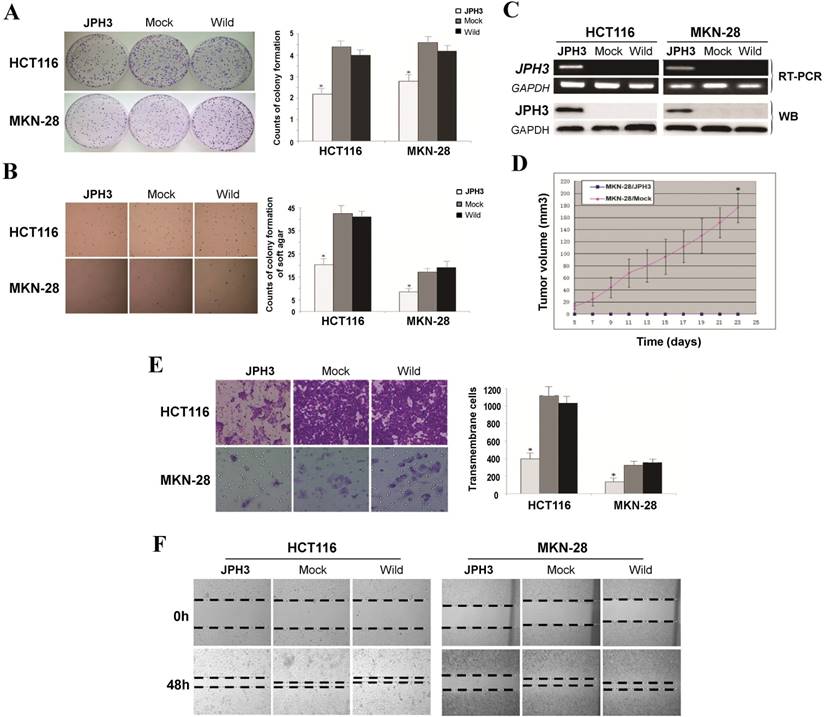
JPH3 inhibited the growth and migration of cancer cells. (A) Representative colony formation assay by monolayer culture and quantitative analysis. (B) Representative results of soft agar assays and quantitative analysis. (C) JPH3 expression in stably transfected cells confirmed by RT-PCR and Western blot. (D) Effect of JPH3 expression on tumor growth in nude mice. Result of tumor growth in nude mice inoculated subcutaneously with MKN-28/vector or MKN-28/JPH3 cells. *: p<0.05. (E) Migration assay with a 24-transwell system and quantitative analysis. The pictures were taken 24 h after seeding. Original magnification: 100x. (F) Wound-healing assay. Photos were taken every 24 h. Original magnification: 100x. All values are the mean ± s.d. of three independent experiments. *: p<0.05.
To further evaluate the effects of JPH3 expression on the migration and invasion of colorectal and gastric cancer cells, transwell migration and wound-healing assays were performed. JPH3 expression also inhibited cancer cell migration and invasion significantly (p<0.05, Figure 2E, 2F). These results suggested that JPH3 indeed acts as a tumor suppressor in colorectal and gastric cancer cells through inhibiting cell growth and migration.
JPH3 promoted ER stress-induced apoptosis and unfolded protein response
To further determine the mechanisms of JPH3-mediated tumor inhibition, we quantified cell cycle distribution using flow cytometry. There were no significant differences in cell cycle phase distribution between JPH3-transfected and control cells. We further checked spontaneous, 5-fluorouracil and ER stress-induced apoptosis by inducer brefeldin A (BFA) treatment. JPH3 expression increased ER stress-induced apoptosis (Figure 3A) but not the spontaneous and 5-fluorouracil induced apoptosis. JPH3 expression also increased caspase 3, 9 and PARP cleavage which was further enhanced by BFA treatment (Figure 3B). Although caspase 4 activation has been reported to be responsible for ER stress-mediated apoptosis, our data indicate that JPH3 expression-elicited apoptosis might be caspase 4-independent (Figure 3B).
Figure 3
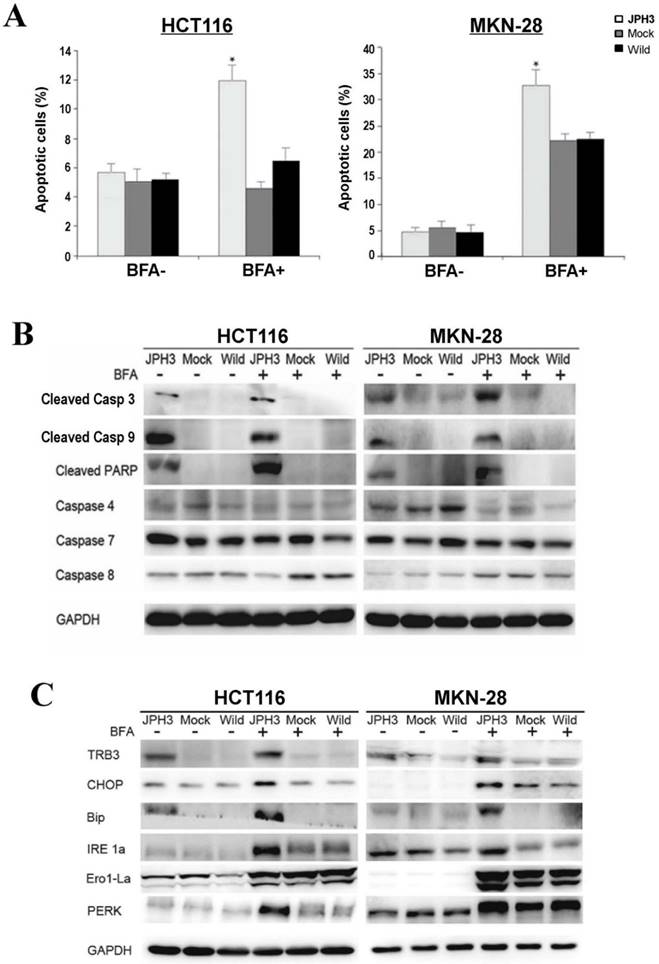
Effects of ectopic JPH3 expression on the apoptosis and UPR of tumor cells with or without BFA treatment. (A) Quantitative analysis results of annexin V-FITC/PI staining before and after BFA treatment in HCT116 and MKN-28 cells. Values are the mean ± s.d. of three independent experiments. *: p<0.05. (B) Cleaved Caspase 3, Caspase 9 and PARP were significantly induced by JPH3 expression. (C) Expression of several key UPR markers examined by Western blot.
In addition, we assessed the effects of JPH3 on tumor suppressor p53, a key regulation of cell cycle arrest and apoptosis. Immunofluorescence and WB results showed no difference in p53 protein levels between JPH3 expression cells and their controls (Suppl. Figure S4).
To determine the effect of JPH3 expression on unfolded protein response (UPR), the expression of some UPR-related proteins was examined. As shown in Figure 3C, JPH3 expression upregulated several UPR-related proteins, including TRB3, CHOP, Bip, IRE1a, Ero1-La, and PERK, especially after BFA treatment. These results suggested that JPH3 could increase ER stress-induced apoptosis and UPR in colorectal and gastric cancer cells.
JPH3 regulated the mitochondrial apoptotic signaling pathway
We next investigated whether JPH3 affects mitochondrial apoptotic signaling pathway. Our results show that JPH3 expression could induce collapse of the mitochondrial transmembrane potential (MMP) even before BFA treatment (Figure 4A). To determine whether the MMP change led to release of mitochondrial apoptogenic factor Cyto c and Smac, the cytosolic and mitochondrial fractions were analyzed by immunoblotting. Results showed that JPH3 expression increased the release of Cyto C and Smac into the cytosol (Figure 4B).
Figure 4
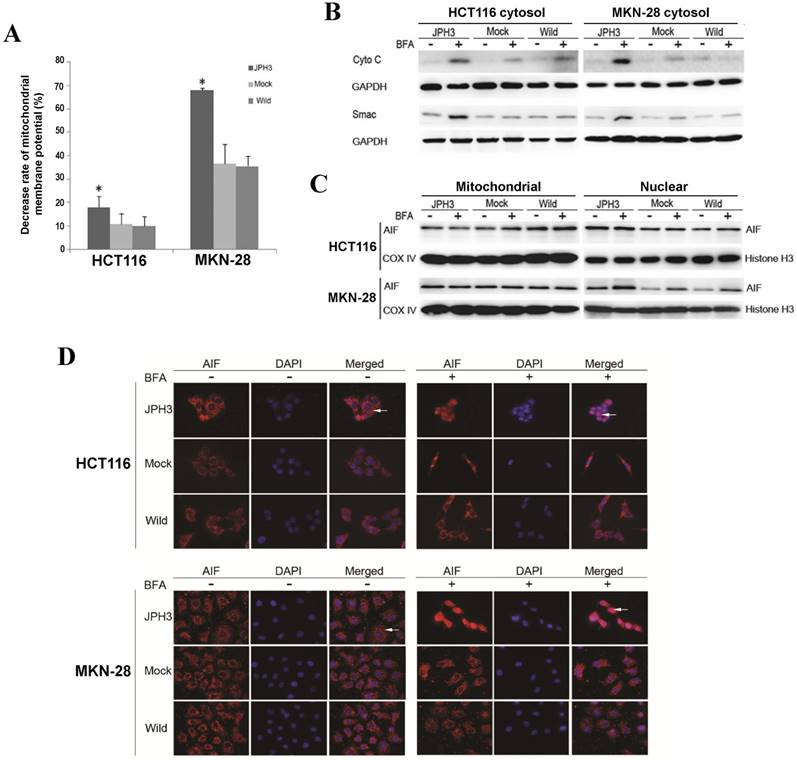
Effects of ectopic JPH3 expression induced digestive cancer cell apoptosis through mitochondria-mediated apoptosis pathway. (A) Mitochondrial membrane potential was significantly decreased by JPH3 expression. (B) JPH3 expression increased the release of Cyto C and Smac into the cytosol. (C) Western blot and (D) immunofluorescence results showed that JPH3 expression induced translocation of mitochondrial AIF into the nucleus significantly after BFA treatment.
To further determine whether another mitochondrial apoptogenic factor, AIF, was also involved in JPH3 expression induced apoptosis, Western blot and immunofluorescence experiments were performed to confirm the nuclear translocation of AIF (Figure 4C, 4D). After BFA treatment, AIF colocalized with DAPI nuclear stain in most JPH3 expressing cells, while in control cells AIF staining was always observed in the cytosol (Figure 4D). Meanwhile, Western blot showed that AIF expression increased in nuclear fraction but decreased in mitochondrial fraction, especially after BFA treatment (Figure 4C). These data suggested that JPH3 is involved in modulation of the mitochondrial apoptotic signaling pathway in colorectal and gastric tumor cells.
JPH3 expression increased cytosolic Ca2+ level and calpain activation
As previous studies reported that JPHs are essential for the maintenance of JMC structures and thus participate in the maintenance of intracellular Ca2+ homeostasis and regulation of intracellular calcium signaling pathways. We also analyzed the effect of JPH3 expression on cytosolic Ca2+ levels. JPH3 expression increased the cytosolic Ca2+ level to about twice that of the controls (Figure 5A, 5B).
Figure 5
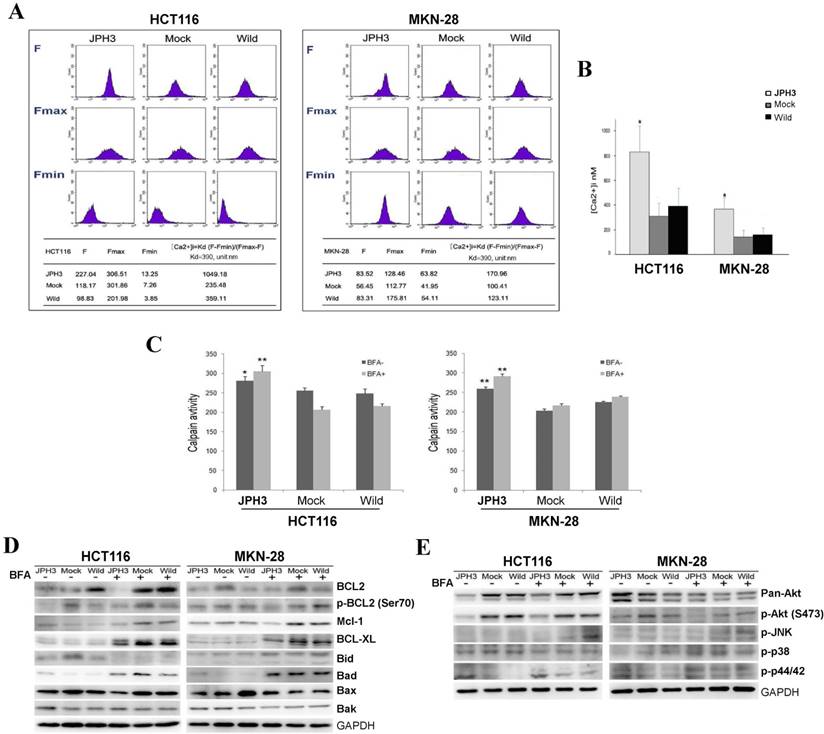
Effects of ectopic JPH3 expression on the level of cytosolic Ca2+, Calpain activity, several important protein kinases and BCL2 family protein expression. (A-B) JPH3 expression significantly increased the level of cytosolic Ca2+ and (C) Calpain activity. (D) JPH3 expression significantly decreased the pro-survival Bcl2 family proteins including Bcl2, P-Bcl2 (Ser70), Mcl-1 and Bcl-xL, but had no apparent effect on the pro-apoptosis Bcl2 family proteins. (E) JPH3 expression significantly decreased p-Akt (S473) expression and had no apparent effects on the others.
To further define the Ca2+-mediated signaling pathway, we examined whether calpain was activated by JPH3 expression. Calpain activity was significantly higher in JPH3 expressing cells than in the controls especially after BFA treatment (Figure 5C). These results suggested that JPH3 increases the level of cytosolic Ca2+ and calpain activation in colorectal and gastric cancer cells.
JPH3 suppressed pro-survival Bcl2 family members and Akt phosphorylation
Most stimuli that induce mitochondria-dependent apoptosis promote the permeabilization of the outer mitochondrial membrane, which is controlled by proteins of the Bcl2 family. So we checked pro-survival members Bcl2, p-Bcl2 (Ser70), p-Bcl2 (Thr56), Mcl-1, Bcl-xL and the pro-apoptotic proteins Bid, Bad, Bax, and Bak by Western blot. JPH3 expression suppressed Bcl2 and p-Bcl2 (Ser70) and the other pro-survival members Mcl-1 and Bcl-xL but did not affect the pro-apoptotic proteins (Figure 5D). We also checked some important protein kinases which play a critical role in controlling cell survival and mitochondrial apoptosis. JPH3 expression only significantly suppressed Akt phosphorylation (p-Akt at S473) (Figure 5E). These data showed that JPH3 induces mitochondrial apoptosis through suppressing pro-survival Bcl2 family members and Akt phosphorylation.
Correlation of JPH3 expression with clinicopathological features of digestive cancer patients
To evaluate JPH3 expression and methylation in digestive primary tumors, we analyzed JPH3 methylation by MSP in 53 colorectal and 32 gastric tumor samples with paired normal tissues. JPH3 methylation was higher in tumors than paired normal tissues (Figure 6A). BGS confirmed the MSP results in primary tumors (Figure 6B). Moreover, JPH3 methylation is significantly correlated with its low expression (Figure 6E, Table 3).
JPH3 protein expression was next investigated by immunohistochemistry in normal and cancer tissues, including 10 normal colonic and 10 normal gastric mucosa biopsy samples, 137 colorectal and 192 gastric cancer paraffin blocks. Representative immunohistochemical staining results are shown in Figure 6C. JPH3 was found to be highly expressed in the cytoplasm of epithelial and some stromal cells in normal colonic and gastric mucosa. In contrast, JPH3 expression was absent in most primary tumor samples. 56.9% (78/137) of colorectal and 39.6% (76/192) of gastric tumors showed weak or moderate expression, relative to its expression in normal mucosa.
The correlation between clinicopathological parameters of colorectal and gastric cancer patients and JPH3 protein expression are summarized in Table 1 and Table 2. In colorectal cancer, low JPH3 protein expression was significantly correlated with high T, N and Duke stages (p=0.023, 0.007 and 0.023 respectively). In gastric cancer, low JPH3 protein expression was significantly correlated with poor differentiation (p=0.000), high clinical stage (p=0.018), more lymph node metastases (p=0.008), more liver metastases (p=0.025) and invasion (p=0.019). More importantly, the difference of 5-year overall survival rates for colorectal and gastric patients between JPH3 high and low expression were both statistically significant (Figure 6D).
In addition, we also retrieved JPH3 methylation and expression data of digestive tumors in Cancer Genome Atlas (TCGA) database (Figure 6F). JPH3 expression levels in colorectal (n=379) and gastric (n=415) tumors are lower than that in normal controls, while JPH3 methylation levels were significantly higher in colorectal (n=233) and gastric (n=48) tumors than normal controls. Moreover in colorectal cancer, JPH3 expression and methylation have significant correlation with clinical stages (rho=0.2898852, p=2.322e-08) including t stages (rho=0.2548168, p=5.709e-07) and N stages (rho=0.251135, p=8.387e-07). These results suggest that JPH3 downregulation by promoter CpG methylation is correlated with disease progression and poor survival of colorectal and gastric cancer patients.
Table 1
Association between JPH3 protein expression and clinicopathological parameters in 137 primary colorectal tumor cases
| Clinicopathological parameters | n | JPH3 protein expression | P-value | |
|---|---|---|---|---|
| Low expression n=87 (63.5%) | High expression n=50 (36.5%) | |||
| Sex | ||||
| Male | 79 | 46 (58.2%) | 33 (41.8%) | |
| Female | 58 | 41 (70.7%) | 17 (29.3%) | 0.134 |
| Age | ||||
| <63 | 70 | 46 (65.7%) | 24 (34.3%) | |
| ≥63 | 67 | 41 (61.2%) | 26 (38.8%) | 0.583 |
| Differentiation | ||||
| Well Moderate | 72 46 | 41 (56.9%) 31(67.4%) | 31 (43.1%) 15 (32.6%) | |
| Poor | 19 | 15 (78.9%) | 4 (21.1%) | 0.166 |
| pT categories | ||||
| T1/T2 | 31 | 15 (48.4%) | 16 (51.6%) | |
| T3/T4 | 106 | 72 (67.9%) | 34 (32.1%) | 0.047 |
| pN categories | ||||
| pN0 | 69 | 36 (52.2%) | 33 (47.8%) | |
| pN1/2 | 68 | 51 (75%) | 17 (25%) | 0.006 |
| pM categories | ||||
| pM0 | 122 | 74 (60.7%) | 48 (39.3%) | |
| pM1 | 15 | 13 (86.7%) | 2 (13.3%) | 0.048 |
| Duke stage | ||||
| A/B | 66 | 34 (51.5%) | 32 (48.5%) | |
| C/D | 71 | 53 (74.6%) | 18 (25.4%) | 0.005 |
Figure 6
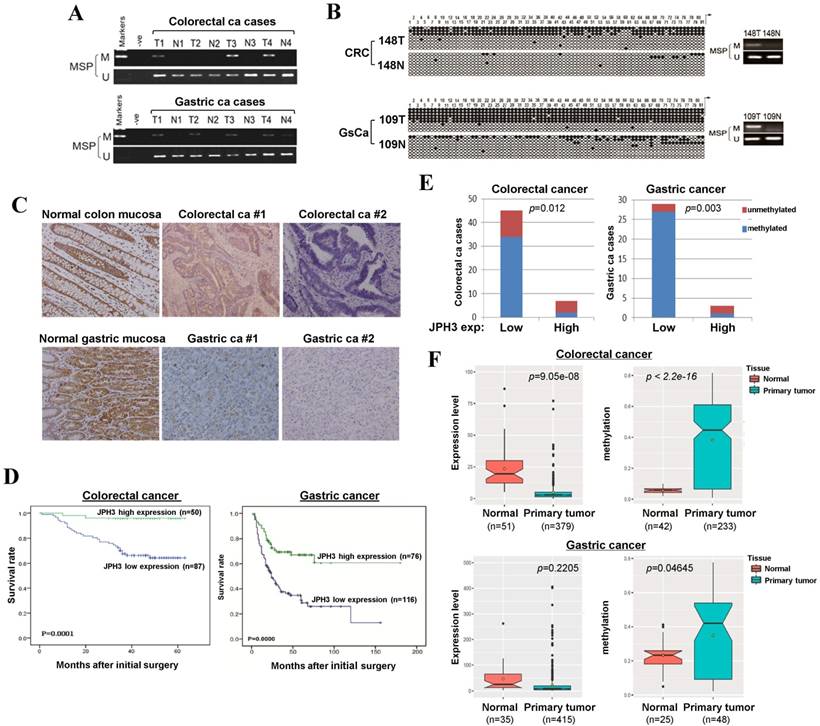
JPH3 reduction by promoter methylation in colorectal and gastric primary tumors and its correlation with clinicopathological features. Representative analysis of JPH3 methylation in primary tumors (T) and normal tissues (N) by (A) MSP and (B) BGS. (C) Representative immunohistochemical staining for JPH3 in normal gastric and colonic mucosa, as well as their primary carcinoma tissues. Original magnification: 200x. (D) Overall survival analysis results in colorectal and gastric cancer patients. (E) Correlation of JPH3 methylation and expression in primary colorectal and gastric tumors. (F) Analysis of JPH3 expression and methylation in colorectal and gastric cancer patients from the TCGA database, as well as their correlation with clinicopathological parameters.
Discussion
JPHs stabilize the junctional membrane complexes (JMC) between PM and ER, which are the structure for cell surface and ion channel cross-talking [20, 21]. JPH proteins contain eight N-terminal “membrane occupation and recognition nexus” (MORN) domains, a space-spanning alpha helix, and a C-terminal “transmembrane domain” (TMD). The MORN domains mediate binding to the plasma membrane, while the hydrophobic TMD is anchored into the SR membrane [14]. JPHs are found to be highly conserved in evolution through analysis of over 60 JPH proteins from over 40 species, in particular the MORN motifs found in all species [22].
JPHs family contains four members, JPH1-4. JPH1 is predominantly expressed in skeletal muscle; JPH2 is distributed in both skeletal muscle and heart; JPH3 and JPH4 are abundantly expressed in brain. JPH1 or JPH2 knock-out in mice induces disorganized JMC and disrupted Ca2+ homeostasis in skeletal muscle, and finally neonatal or embryonic death, respectively [23, 24]. JPH1 and JPH2 knockdown study in mouse skeletal muscle fibers also showed decreases in intracellular Ca2+ release and the disorganization of JMC [25]. In the case of the neuronal JPH isoforms (JPH3 and JPH4), JPH3 null mice exhibit adult onset, progressive motor dysfunction, whereas JPH3 hemizygous mice have a similar but milder phenotype. The only information that has been reported about JPH3 involves a mouse model for neurodegenerative HDL2 disorder that showed a decrease in JPH3 expression due to changes at the genomic level [26, 27]. Loss of JPH3 protein expression contributes to HDL2 pathogenesis perhaps through disruption of calcium flux or ER dysregulation [17]. No information is available so far for the possible roles of JPHs during human carcinogenesis yet.
Table 2
Association between JPH3 protein expression and clinicopathological parameters in 192 primary gastric tumor cases
| Clinicopathological parameters | n | JPH3 protein expression | P-value | |
|---|---|---|---|---|
| Low expression n=116 (60.4%) | High expression n=76 (39.6%) | |||
| Sex | ||||
| Male | 138 | 83 (60.1%) | 55 (39.9%) | |
| Female | 54 | 33 (61.1%) | 21 (38.9%) | 0.946 |
| Age | ||||
| >62 | 95 | 57 (60%) | 38 (40%) | |
| ≤62 | 97 | 59 (60.8%) | 38 (39.2%) | 0.907 |
| Differentiation | ||||
| Well and Moderate | 75 | 33 (44%) | 42 (56%) | |
| Poor | 117 | 83 (70.9%) | 34 (29.1%) | 0.000 |
| Clinical stage | ||||
| I | 43 | 21 (48.8%) | 22 (51.2%) | |
| II | 40 | 18 (45%) | 22 (55%) | |
| III | 76 | 52 (68.4%) | 24 (31.6%) | |
| IV | 33 | 25 (75.8%) | 8 (24.2%) | 0.018 |
| Lymph node metastasis | ||||
| Absent | 86 | 43 (50%) | 43 (50%) | |
| Present | 106 | 73 (68.9%) | 33 (31.1%) | 0.008 |
| Liver metastasis | ||||
| Absent | 157 | 89 (56.7%) | 68 (43.3%) | |
| Present | 35 | 27 (77.1%) | 8 (22.9%) | 0.025 |
| Invasion | ||||
| T1-2 | 62 | 30 (48.4%) | 32 (51.6%) | |
| T3-4 | 130 | 86 (66.2%) | 44 (33.8%) | 0.019 |
Table 3
Correlation between JPH3 expression and methylation in colorectal and gastric cancers
| JPH3 promoter | No. | JPH3 protein expression | P-value | ||
|---|---|---|---|---|---|
| Low expression | High expression | ||||
| CRC | M | 36 | 34 | 2 | 0.012 |
| U | 16 | 11 | 5 | ||
| GsCa | M U | 28 4 | 27 2 | 1 2 | 0.003 |
JPH3 has 4 splicing variants, with variant 1 as the full-length transcript encoding a fully functional 748-amino acid protein; while variant 2 and 3 are much shorter isoforms and variant 4 is a non-coding RNA. We found that JPH3 variant 1,2,4 were frequently downregulated or silenced in CRC and gastric tumor cell lines and tumors, while variant 3 is never expressed. Thus in this study, we mainly studied JPH3 variant 1 in digestive system tumorigenesis. Whether other JPH3 variants also play some roles in tumorigenesis need further investigation. We also checked the expression of a related member JPH4 in tumor cell lines and primary cases (Suppl. Figure S1). JPH4 was frequently downregulated in tumor cells and primary tumors, while no correlation of JPH4 and JPH3 expression was detected. The functions of JPH4 in cancer pathogenesis need further investigations.
DNMT1 and DNMT3B are two major DNA methyltransferases responsible for the maintenance and de novo CpG methylation, and their disruption results in >95% loss of overall genomic methylation as well as CpG island demethylation [28]. The colon cell line pair (HCT116 and DNMT1/DNMT3B double knock-out (DKO)) provides us with a useful model system to identify key methylated cancer genes involved in colon tumorigenesis, which has been widely used by other researchers [18, 29, 30]. Through methylome analysis of HCT116 and DKO pair cell line, we identified JPH3 as a methylated TSG candidate in colon cancer. We found that in addition to its abundant expression in neuronal cells, JPH3 is also readily expressed in some other normal tissues including digestive systems. For the first time, we report that JPH3 was frequently silenced or reduced in digestive tumors due to its promoter CpG methylation, indicating its potential value as a biomarker for these cancers in future. As clinical primary tumor tissue samples always contain infiltrating non-malignant cells, it is well known that unmethylated TSG promoter alleles are always detected in primary tumor tissue samples, which actually acts as a good positive control for the integrity of bisulfited tumor genomic DNA. Thus, we also successfully detected unmethylated JPH3 alleles in all the primary tumor tissue samples (Figure 6).
Meanwhile, restoring JPH3 expression inhibits tumor cell growth in vitro and in vivo, indicating that JPH3 acts as a tumor suppressor in colorectal and gastric cancers. We also demonstrated that its tumor suppressor function is, at least partially, due to its induction of mitochondrial-mediated apoptosis through unfolded protein response involving perturbed calcium homeostasis. Previous studies show that JPHs are essential for the maintenance of JMC structures and thus participates in the maintenance of intracellular Ca2+ homeostasis and regulation of intracellular calcium signaling pathways [14]. We also found that JPH3 expression increased the cytosolic Ca2+ level to about twice that of the controls. So as in neuronal cells, JPH3 expression could also participates in the maintenance of intracellular Ca2+ homeostasis in colorectal and gastric tumor cells.
Endoplasmic reticulum serves two major functions in the cells. The newly synthesized proteins destined for secretion, cell surface or intracellular organelles are properly folded in this organelle and ER also provides the cells with a Ca2+ reservoir and controls the free and bound Ca2+ levels in the cells [31-33]. Though cancer cells always reprogramme their metabolism to support the rapid proliferation, eventually loss of nutrients/energy leads to accumulation of unfolded and misfolded proteins in ER and disruption of Ca2+ homeostasis. So tumor cells often suffer from more ER stress than do normal cells. Since ER is also an important sensor for cellular stress, a group of signal transduction pathways will be activated to maintain ER homeostasis upon ER stress in human cancers. Two important pathways are the unfolded protein response (UPR) and autophagy [34, 35].
We found that in colorectal and gastric cancer cells, JPH3 expression significantly induced UPR but not autophagy. The initial purpose of UPR is to stop the unpropor protein translation and reestablish homeostasis and normal ER function and meanwhile increase production of molecular chaperones involved in correct protein folding [36, 37]. However, when the ER stress is extensive or sustained, the adaptive mechanisms fail to reestablish homeostasis and the normal function of ER, the UPR pathways may launch apoptotic pathways to remove the stressed cells [38]. This is consistent with our results that JPH3 expression significantly induced apoptosis after ER stress induction, but did not significantly alter spontaneous or 5-fluorouracil induced apoptosis. Additionally, JPH3 expression significantly elevated the expression level of CHOP and TRB3 which both function to mediate programmed cell death.
In addition to activation of UPR, JPH3 expression also induce calpain activation and subsequent mitochondrial membrane depolarization, followed by mitochondrial release of Cyto C, Smac and consequent activation of caspase -9 and -3. Moreover, JPH3 expression increased PARP cleavage and induced AIF nuclear translocation. Both pro- and anti-apoptotic Bcl2 family members play an important role in the regulation of outer mitochondrial membrane permeability [39]. However, our results indicated that JPH3 expression mainly suppresses Bcl2 and p-Bcl2 (Ser70) and the pro-survival members Mcl-1 and Bcl-xL, but does not affect pro-apoptotic proteins. As 16q24 LOH is frequently present in multiple other solid tumors, 16q24 is also a TSG locus for common tumors other than digestive cancers. Whether epigenetic disruption of JPH3 is involved in the pathogenesis of other solid tumors needs further investigation.
Figure 7
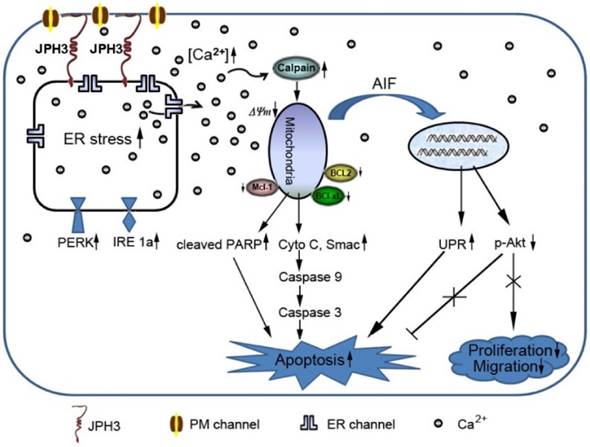
Diagram showing the mechanisms with which JPH3 expression induced cancer cell apoptosis by unfolded protein response involving perturbed calcium homeostasis.
In conclusion, JPH3 was found to be a novel tumor suppressor downregulated and methylated in colorectal and gastric cancers, which increases mitochondrial- mediated apoptosis through UPR involving perturbed calcium homeostasis (Figure 7). This appears to be the first report that a junctional complex junctophilin family protein is involved in tumor pathogenesis. JPH3 may potentially serve as a biomarker colorectal and gastric cancer. Further studies on the detailed intracellular Ca2+ signaling pathways affected by JPH3 expression could lead to a better understanding of the complex regulation of calcium homeostasis in normal and cancer cells, as well as identify new therapeutic strategies for gastrointestinal cancer.
Materials and Methods
Cell lines, tumor samples, and normal tissues
A panel of gastric (KatoIII, YCC1, YCC3, YCC6, YCC7, YCC9, YCCEL1, YCC11, SNU719, AGS, MKN-45, SNU1, SNU16, MKN-28) and colon cancer cell lines (SW480, SW620, DLD1, Colo320, HT-29, LoVo, RKO, HCT116, HCT116-DKO with double knock-out of DNMT1 and DNMT3B) were used. Cell lines were maintained in RPMI or DMEM medium (Gibco BRL, Rockville, MD) with 10% fetal bovine serum. Human normal adult tissue RNA samples were purchased commercially (Stratagene, La Jolla, CA, or Millipore Chemicon, Billerica, MA).
A total of 192 gastric cancer patients and 137 colorectal cancer patients who underwent surgery between Feb 2004 and June 2006 at the Sir Run Run Shaw Hospital (Hangzhou, Zhejiang, China) were investigated. Patients who received pre-operative chemotherapy were excluded. 10 normal gastric and 10 normal colon mucosa biopsy samples were used as normal controls. This study was approved and monitored by the ethics committee of Sir Run Run Shaw Hospital, Zhejiang University.
Establishment of paired colon cancer cell line methylomes by MeDIP-chip
Methylated DNA immunoprecipitation (MeDIP) coupled with promoter microarray hybridization (MeDIP-chip) was performed as previously [19]. Briefly, methylated HCT116 and DKO cell DNA was immunoprecipited by monoclonal antibody against 5-methylcytidine (33D3, Diagenode, Seraing, Belgium), and then hybridized to NimbleGen™ HG18 Meth (385K CGI plus) promoter arrays (Array Star, Inc., MD). Bioinformatics analysis of methylome data was performed as previously [19].
5-aza-2′-deoxycytidine (5-Aza) and trichostatin A (TSA) treatment
Cells with silenced JPH3 expression were seeded at a density of 1×106 cells/ml. After overnight culture, cells were treated with the demethylation agent 5-aza-2'-deoxycytidine (5-Aza; Sigma-Aldrich, St Louis, MO) at a final concentration of 10 µM for 72 hours and then further treated with 100 nM trichostatin A (TSA; Cayman Chemical Co, Ann Arbor, MI) for 16 hours. Cells were subsequently harvested for DNA and RNA extraction.
Bisulfite treatment and promoter methylation analysis
Bisulfite modification of DNA, methylation specific PCR (MSP) and bisulfite genome sequencing (BGS) were carried out as described previously [40, 41]. MSP and BGS primers are listed in Suppl. Table S1. Genomic positions of MSP and BGS primers are shown in Suppl. Figure S2.
JPH3-expressing plasmid and cell transfection
The JPH3-expressing plasmid was kindly provided by Prof. Keith Robertson (Mayo Clinic, Rochester, MN) and sequence verified. Colon cell line HCT116 and gastric cell line MKN-28 were transfected with pCMV-Tag2A-JPH3 plasmid (Agilent, Santa Clara, California) or empty vector as a control using MegaTran 1.0 transfection reagent (Origene, Maryland, MD). Stable JPH3-expressing clones were selected for further studies.
Determination of mitochondrial transmembrane potential
The cells were trypsinized, washed in ice-cold phosphate buffered saline, and stained with JC-1 (5ug/ml; Molecular Probes) for 30 min at 37℃ in 5% CO2 atmosphere. Then, the changes in the mitochondrial transmembrane potential (MMP) were monitored and analyzed. The percentage of red and green fluorescence was estimated by flow cytometry.
Measurement of intracellular Ca2+
Change in the intracellular Ca2+ level was analyzed using Fluo-3 AM, a cell permeant, Ca2+-sensitive fluorescent dye. Cells were loaded with 2µM Fluo-3 AM diluted in Krebs-Ringer buffer [KRB; 0.7mM Na2HPO4, 1.5mM NaH2PO4 (pH 7.4), 10mM d-glucose, 120mM NaCl, 4.5mM KCl, 0.5mM MgCl2] for 30min in a CO2 incubator. After washing with KRB to remove the residual dye, the cells were harvested, washed with Ca2+-free PBS and analyzed by flow cytometry.
Calpain activity assay
Calpain activity was measured using the cell-permeable fluorogenic calpain substrate, S-LLVY-AMC. In brief, cells were harvested, washed twice with HEPES-buffered Hank's balanced salt solution (HBSS; pH 7.4) without phenol red, resuspended in HEPES-HBSS at 3×105 cells/ml, and prewarmed at 37℃ in a CO2 incubator for 10 min. The substrate S-LLVY-AMC (25 µM) was then added, and the AMC fluorescence intensity was measured using a spectrofluorometer (BIO-TEK ELX800) at excitation and emission wavelengths of 400 and 500 nm, respectively.
Subcellular fractionation
Nuclear and cytoplasmic protein extractions were extracted using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Biotech, Peking, China) and mitochondrial fractions were isolated using the Cell Mitochondria Isolation Kit (Beyotime Biotech, Peking, China).
In vivo subcutaneous tumor model
All in vivo experimental protocols were approved by the animal care committee of Sir Run Run Shaw Hospital, Zhejiang University. Viable MKN-28 cells (3.5×106 cells in 0.1ml phosphate buffer saline) were injected subcutaneously into the right dorsal flank of 6-week-old female BALB/c nude mice (six mice per group). Tumor volume was assessed every 2 days for 4 weeks. Tumor volume was calculated using the following formula: (short diameter)2×(long diameter)/2.
TCGA database analysis
JPH3 expression and methylation of were analyzed using The Cancer Genome Atlas (TCGA) database. JPH3 expression and processed clinical data of gastric and colorectal patients in TCGA were obtained from Fire browse database (http://www.firebrowse.org). For colorectal cancer, a total of 379 tumor samples and 51 normal samples were studied. For gastric cancer, a total of 415 tumor samples and 35 normal samples were studied. Gene expression and methylation levels were measured using normalized log2 RSEM index from FireBrowse database. Wilcox rank sum test was used to analyze the significance in differential gene expression. Pearson method was applied as a correction to multiple comparisons.
Statistical analysis
The results in the figures are all expressed as mean values ± s.d. Statistical analysis was performed in SPSS 11.0 for Windows (SPSS Inc., Chicago, IL, U.S.). The two-tailed Chi-square test was used to analyze the association of JPH3 protein expression with the different clinicopathological parameters. For all tests, p<0.05 was considered to be of statistical significance.
Abbreviations
TSG, tumor suppressor gene; LOH, Loss of heterozygosity; HD, Huntington's disease; HDL2, Huntington's disease-like 2; JPH, Junctophilin; JPH3, Junctophilin 3; JMC, junctional membrane complex; 5-Aza, 5-aza-2′-deoxycytidine; TSA, trichostatin A; MSP, methylation-specific PCR; BGS, bisulfite genome sequencing; HBSS, Hank's balanced salt solution; CGI, CpG island; BFA, brefeldin A; EMT, epithelial-mesenchymal transition; MMP, mitochondrial transmembrane potential; UPR, unfolded protein response; PM, plasma membrane; ER, endoplasmic reticulum; SR, sarcoplasmic reticulum; MORN, membrane occupation and recognition nexus; TMD, transmembrane domain; HDL2, Huntington disease-like 2.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Profs. Bert Vogelstein for the DKO cell line, Sun Young Rha for some gastric cell lines, and Keith D Robertson for the JPH3 full-length cDNA. This work was supported by National Natural Science Foundation (NSFC) of China (grant no. 81372622, 81472211 and 81572327), Major Projects in Zhejiang Province (grant no. 2012C13014-1), Hong Kong HMRF (#13120082), and VC special research fund from the Chinese University of Hong Kong.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Arnold CN, Goel A, Blum HE, Boland CR. Molecular pathogenesis of colorectal cancer: implications for molecular diagnosis. Cancer. 2005;104:2035-47
2. Palii SS, Robertson KD. Epigenetic control of tumor suppression. Crit Rev Eukaryot Gene Expr. 2007;17:295-316
3. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107-16
4. Duffy MJ, Napieralski R, Martens JW, Span PN, Spyratos F, Sweep FC. et al. Methylated genes as new cancer biomarkers. Eur J Cancer. 2009;45:335-46
5. Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330-9
6. Jin H, Wang X, Ying J, Wong AH, Li H, Lee KY. et al. Epigenetic identification of ADAMTS18 as a novel 16q23.1 tumor suppressor frequently silenced in esophageal, nasopharyngeal and multiple other carcinomas. Oncogene. 2007;26:7490-8
7. Lo KW, Teo PM, Hui AB, To KF, Tsang YS, Chan SY. et al. High resolution allelotype of microdissected primary nasopharyngeal carcinoma. Cancer Res. 2000;60:3348-53
8. Paige AJ, Taylor KJ, Stewart A, Sgouros JG, Gabra H, Sellar GC. et al. A 700-kb physical map of a region of 16q23.2 homozygously deleted in multiple cancers and spanning the common fragile site FRA16D. Cancer Res. 2000;60:1690-7
9. Mori Y, Matsunaga M, Abe T, Fukushige S, Miura K, Sunamura M. et al. Chromosome band 16q24 is frequently deleted in human gastric cancer. Br J Cancer. 1999;80:556-62
10. Al-Tassan NA, Whiffin N, Hosking FJ, Palles C, Farrington SM, Dobbins SE. et al. A new GWAS and meta-analysis with 1000Genomes imputation identifies novel risk variants for colorectal cancer. Sci Rep. 2015;5:10442
11. Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M. et al. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res. 2001;61:4556-60
12. Lee KY, Geng H, Ng KM, Yu J, van Hasselt A, Cao Y. et al. Epigenetic disruption of interferon-gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene. 2008;27:5267-76
13. Li L, Ying J, Li H, Zhang Y, Shu X, Fan Y. et al. The human cadherin 11 is a pro-apoptotic tumor suppressor modulating cell stemness through Wnt/beta-catenin signaling and silenced in common carcinomas. Oncogene. 2012;31:3901-12
14. Nishi M, Mizushima A, Nakagawara K, Takeshima H. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 2000;273:920-7
15. Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11-22
16. Landstrom AP, Beavers DL, Wehrens XH. The junctophilin family of proteins: from bench to bedside. Trends Mol Med. 2014;20:353-62
17. Seixas AI, Holmes SE, Takeshima H, Pavlovich A, Sachs N, Pruitt JL. et al. Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Ann Neurol. 2012;71:245-57
18. Wang J, Xia Y, Li L, Gong D, Yao Y, Luo H. et al. Double restriction-enzyme digestion improves the coverage and accuracy of genome-wide CpG methylation profiling by reduced representation bisulfite sequencing. BMC Genomics. 2013;14:11
19. Li L, Zhang Y, Fan Y, Sun K, Su X, Du Z. et al. Characterization of the nasopharyngeal carcinoma methylome identifies aberrant disruption of key signaling pathways and methylated tumor suppressor genes. Epigenomics. 2015;7:155-73
20. Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595-636
21. Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13-26
22. Garbino A, van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, Wehrens XH. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175-86
23. Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K. et al. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol. 2001;154:1059-67
24. van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N. et al. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979-88
25. Hirata Y, Brotto M, Weisleder N, Chu Y, Lin P, Zhao X. et al. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J. 2006;90:4418-27
26. Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X. et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70:427-40
27. Holmes SE, O'Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG. et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377-8
28. Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE. et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552-6
29. Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM. et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3:1709-23
30. McGarvey KM, Van Neste L, Cope L, Ohm JE, Herman JG, Van Criekinge W. et al. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008;68:5753-9
31. Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235-49
32. Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487-508
33. Momoi T. Conformational diseases and ER stress-mediated cell death: apoptotic cell death and autophagic cell death. Curr Mol Med. 2006;6:111-8
34. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519-29
35. Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299-304
36. Dorner AJ, Wasley LC, Kaufman RJ. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J Biol Chem. 1989;264:20602-7
37. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739-89
38. Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36:329-37
39. Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:63-7
40. Murray PG, Qiu GH, Fu L, Waites ER, Srivastava G, Heys D. et al. Frequent epigenetic inactivation of the RASSF1A tumor suppressor gene in Hodgkin's lymphoma. Oncogene. 2004;23:1326-31
41. Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW. et al. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006;25:1070-80
Author contact
![]() Corresponding authors: X Hu, Biomedical Research Center, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang 310016, China. Tel: (86) 571-86006363; Fax: (86) 571-86006363; E-mail: huxtcom; or Q Tao, Rm 315, Cancer Center, PWH, The Chinese University of Hong Kong, Shatin, Hong Kong. Tel: (852) 2632-1340; Fax: (852) 2648-8842; E-mail: qtaoedu.hk
Corresponding authors: X Hu, Biomedical Research Center, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, Zhejiang 310016, China. Tel: (86) 571-86006363; Fax: (86) 571-86006363; E-mail: huxtcom; or Q Tao, Rm 315, Cancer Center, PWH, The Chinese University of Hong Kong, Shatin, Hong Kong. Tel: (852) 2632-1340; Fax: (852) 2648-8842; E-mail: qtaoedu.hk
Citation styles
APA
Hu, X., Kuang, Y., Li, L., Tang, H., Shi, Q., Shu, X., Zhang, Y., Chan, F.KL., Tao, Q., He, C. (2017). Epigenomic and Functional Characterization of Junctophilin 3 (JPH3) as a Novel Tumor Suppressor Being Frequently Inactivated by Promoter CpG Methylation in Digestive Cancers. Theranostics, 7(7), 2150-2163. https://doi.org/10.7150/thno.18185.
ACS
Hu, X.; Kuang, Y.; Li, L.; Tang, H.; Shi, Q.; Shu, X.; Zhang, Y.; Chan, F.KL.; Tao, Q.; He, C. Epigenomic and Functional Characterization of Junctophilin 3 (JPH3) as a Novel Tumor Suppressor Being Frequently Inactivated by Promoter CpG Methylation in Digestive Cancers. Theranostics 2017, 7 (7), 2150-2163. DOI: 10.7150/thno.18185.
NLM
Hu X, Kuang Y, Li L, Tang H, Shi Q, Shu X, Zhang Y, Chan FKL, Tao Q, He C. Epigenomic and Functional Characterization of Junctophilin 3 (JPH3) as a Novel Tumor Suppressor Being Frequently Inactivated by Promoter CpG Methylation in Digestive Cancers. Theranostics 2017; 7(7):2150-2163. doi:10.7150/thno.18185. https://www.thno.org/v07p2150.htm
CSE
Hu X, Kuang Y, Li L, Tang H, Shi Q, Shu X, Zhang Y, Chan FKL, Tao Q, He C. 2017. Epigenomic and Functional Characterization of Junctophilin 3 (JPH3) as a Novel Tumor Suppressor Being Frequently Inactivated by Promoter CpG Methylation in Digestive Cancers. Theranostics. 7(7):2150-2163.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.