Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):2092-2107. doi:10.7150/thno.19884 This issue Cite
Research Paper
c-Myc is Required for BRAFV600E-Induced Epigenetic Silencing by H3K27me3 in Tumorigenesis
Yiping Qu1, Qi Yang1, Juan Liu1, Bingyin Shi1, 2, Meiju Ji3, Gang Li4 ![]() , Peng Hou1, 2
, Peng Hou1, 2 ![]()
1. Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China;
2. Key Laboratory for Tumor Precision Medicine of Shaanxi Province, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China;
3. Center for Translational Medicine, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China;
4. Department of Neurosurgery, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, P.R. China.
Received 2017-3-1; Accepted 2017-3-31; Published 2017-5-26
Citation:
Qu Y, Yang Q, Liu J, Shi B, Ji M, Li G, Hou P. c-Myc is Required for BRAFV600E-Induced Epigenetic Silencing by H3K27me3 in Tumorigenesis. Theranostics 2017; 7(7):2092-2107. doi:10.7150/thno.19884. https://www.thno.org/v07p2092.htm
Other stylesAbstract

BRAFV600E mutation is frequently found in human cancers particularly thyroid cancer and melanoma, and is involved in the regulation of gene expression through activating MAPK/Erk signaling. Trimethylation of histone 3 lysine 27 (H3K27me3) is a critical epigenetic mark for the maintenance of gene silencing in tumorigenesis. However, molecular mechanism underlying the complex interplay between these two molecular events remains to be explored. In the present study, we conducted chromatin immunoprecipitation combined with next-generation sequencing (ChIP-Seq) and expression microarray analysis in NIH3T3 cells to explore the relationship between H3K27me3 and transcriptional regulation by BRAFV600E mutation. Our results showed that activated MAPK/Erk signaling by BRAFV600E mutation was a trigger of this epigenetic processing at many downstream target genes in cancer cell lines and BrafV600E-induced thyroid cancer of transgenetic mice. By integrating ChIP-Seq and gene expression microarray data, we identified 150 down-regulated loci with increased levels of H3K27me3 in BRAF-mutant cells relative to BRAF wild-type cells. Our data also demonstrated that c-Myc, a downstream key effector of BRAFV600E signaling, was required for BRAFV600E-induced changes in H3K27me3 through regulating the components of the polycomb repressive complex 2 (PRC2) genes Ezh2, Suz12 and Jarid2 at both transcriptional levels via direct binding to their regulatory elements and post-transcriptional levels via repressing the miR-26a, miR-200b and miR-155. In addition, BRAFV600E also caused gene silencing through Erk1/2-induced RNA polymerase II (RNAPII) poising and chromatin architecture. Collectively, our data uncover a previously unknown epigenetic mechanism in the tumorigenesis of BRAFV600E-driven cancers.
Keywords: BRAFV600E, c-Myc, H3K27me3, epigenetic silencing, tumorigenesis.
Introduction
Epigenetic modification of chromatin is a major mechanism in the regulation of gene expression (1, 2). Trimethylation of histone 3 lysine 27 (H3K27me3) is a repressive histone mark and can be regulated by Polycomb repressive complex 2 (PRC2), a component of the Polycomb machinery (3). Canonical PRC2 is composed of four core components, Ezh1/2, Suz12, Eed and RbAp46/48, and is responsible for the methylation of histone H3K27 such as H3K27me2/3 to initiate the formation of closed chromatin structures, thereby repressing transcription (4,5). In addition to the core components of PRC2, several cofactors such as Jarid2 have been demonstrated to interact with this complex and to regulate both its binding to chromatin and its enzymatic activity (6,7). In general, H3K27me3 is essential for normal development (8). However, there is a growing body of evidence suggesting that dysregulation of H3K27me3 is also frequently found in different types of cancer and is regarded as a hallmark of cancer (9).
Aberrant activation of Ras/Raf/Mek/Erk (MAPK/Erk) pathway promoting cell proliferation, survival and metastasis is a common mechanism in the genesis and progression of human cancers (10). The most common genetic alteration BRAFV600E mutation in this pathway is frequently found in many types of cancer particularly thyroid cancer and melanoma (11, 12). Through driving the MAPK/Erk signaling, BRAFV600E mutation plays a fundamental role in the tumorigenesis of thyroid and melanoma, and is also associated with poorer clinical outcomes of patients (13-15). Thus, it has become a major therapeutic target for these cancers in recent years.
It is the fact that activation of MAPK/Erk signaling by BRAFV600E mutation controls cell growth and survival in part through epigenetic modification including DNA methylation and histone modification. For example, our previous genome-wide methylation studies have demonstrated that a number of genes with important biological functions are altered in their methylation and, hence, expression in thyroid cancer and melanoma cells (16, 17). In addition, BRAFV600E mutation can cause the silencing of sodium iodide symporter (NIS) in thyroid cancer cells through promoting histone deacetylation at its promoter (18). However, it has remained to be determined whether genome-wide reprogramming of histone methylation such as H3K27me3 is instructed by BRAFV600E mutation during tumorigenesis.
We thus conducted chromatin immunoprecipitation combined with next-generation sequencing (ChIP-Seq) and expression microarray analysis in NIH3T3 cells expressing BRAFV600E mutant or wild-type BRAF. By integrating ChIP-Seq and gene expression microarray data, we identified 150 down-regulated loci with increased levels of H3K27me3 in BRAF-mutant cells relative to BRAF wild-type cells. Further studies demonstrated that BRAFV600E caused H3K27me3-induced gene silencing through regulating the expression of PRC2 complex components such as Ezh2, Suz12 and Jarid2 via c-Myc in vitro and in vivo. Therefore, the experimental evidence presented in the present study has important implications for a better understanding of the role of BRAFV600E mutation in tumorigenesis.
Materials and methods
Cell lines and drug treatments
Human thyroid cancer cell lines BCPAP and K1 were provided by Dr. Haixia Guan (The First Affiliated Hospital of China Medical University, Shenyang, China). Human melanoma cell lines M14 and A375, and mouse fibroblast cell line NIH3T3 were obtained from ATCC (Rockville, MD). BCPAP, K1 and M14 cells were routinely cultured at 37°C in RPMI 1640 medium with 10% fetal bovine serum (FBS), while A375 and NIH3T3 cells were cultured at 37°C in DMEM medium with 10% FBS. Cells were treated with a selective BRAF kinase inhibitor PLX4032 (S1267, Selleck), PLX4720 (S1152, Selleck), MEK inhibitor GSK1120212 (S2673, Selleck), AKT inhibitor MK-2206 (S1078, Selleck), and c-Myc inhibitor 10058-F4 (S7153, Selleck) at the indicated concentrations, individually or in different combinations. RNA and protein were then extracted after 48 h of continuous treatment and stored at -80°C until use.
Transfection of BRAF expression plasmids and RNA interference
The plasmid expressing wild-type BRAF and BRAFV600E mutant, and empty vector (pEFm6) were provided by Dr. Richard Marials (Cancer Research UK Manchester Institute, Manchester, UK). The NIH3T3 cells were transfected with different plasmid constructs using lipofectamine 2000 according to the instructions of the manufacturer as previously described (11). The siRNAs specifically targeting c-Myc and control siRNA were synthesized by GenePharma (Shanghai, China) and the sequences were presented in Table S1. Cells were transfected at 50% confluence using Lipofectamine 2000 (Invitrogen, Grand Island, NY), with a final siRNA concentration of 50 nM. Specific oligonucleotides with maximal knockdown efficiency were selected among three different sequences until use.
RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from the indicated cells using TRIzol reagent (Takara Inc., Dalian, P.R. China). Total RNA (~500 ng) was converted to cDNA using PrimeScript RT reagent Kit (Takara Inc., Dalian, P.R. China) according to the instructions of the manufacturer. MicroRNAs were converted using NCode™ VILO™ miRNA cDNA Synthesis Kit and EXPRESS SYBR® Green ER™ miRNA qRT-PCR Kits (Life, Inc., MA, USA). qRT-PCR assay was performed on a CFX96 Thermal Cycler DiceTM real-time PCR system (Bio-Rad Laboratories, Inc., CA, USA) using SYBR Premix Ex TaqTM (Takara Inc., Dalian, P.R. China) according to the instructions of manufacturer. The mRNA levels of the indicated genes were normalized to 18S rRNA. The expression of microRNAs was normalized to U6 rRNA. Each sample was analyzed in triplicate. The primer sequences were presented in Table S2.
Western blot analysis
Cells were lysed in prechilled RIPA buffer containing protease inhibitors. Equal amounts of protein lysates were separated by SDS-PAGE and transferred onto PVDF membranes (Roche Diagnostics, Mannheim, Germany). The membranes were then incubated overnight with specific primary antibodies. Anti-B-raf (sc-9002), Anti-β-actin (sc-1616), and anti-c-Myc (sc-764) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-Ezh2 (ab3748), Anti-Suz12 (ab12073) and anti-H3 (ab1791) antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-pErk1/2 (#4370), anti-Erk1/2 (#9102), and anti-H3K27me3 (#9733) antibodies were purchased from Cell Signaling Technology (CST, MA, USA). Anti-pAKT473 (BS4007), anti-AKT (BS1379) and anti-GAPDH (AP0063) antibodies were purchased from Bioworld Technology (Bioworld, Inc., MN, USA). Anti-Jarid2 (NB100-2214) antibody was purchased from Novus (Novus, CO, USA). This was followed by incubation with species-specific HRP-conjugated secondary antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and antigen-antibody complexes were visualized using the Western Bright ECL detection system (Advansta, CA, USA).
ChIP-Seq and ChIP-quantitative PCR (qPCR) assays
The indicated cells were fixed with 1.0% formaldehyde at room temperature for 15 min, after which glycine was added to the medium. Cells were then lysed and stored at -80°C until use. The rabbit polyclonal antibodies against H3K27me3 (#9733, CST), H3K4me3 (#9727, CST) and c-Myc (sc-764, Santa Cruz) were used for the ChIP assay using the Pierce™ Magnetic ChIP Kit (Thermo, Inc., MA, USA) according to the instructions of manufacturer. Immunoprecipitated DNA (20 ng) was subjected to size selection of fragments around 200-bp in length. Sequencing libraries were consequently constructed by adaptor ligation and PCR. Sequencing was carried out with an Illumina HiSeq2500 system according to the manufacturer's protocol. Raw sequencing reads, with length in 50-bp per read, were aligned to UCSC mouse mm9 assembly and mapped on unique genomic locations. Model-based Analysis of ChIP-Seq (MACS) was used as described previously to map peaks between BRAF wild-type and BRAFV600E samples (19). All the raw datasets have been deposited in GEO and accession number is GSE93033. Motif analysis was performed using online software MEME-ChIP (http://meme-suite.org/tools/meme-chip) as described previously (20).
Input and ChIP products were used as templates for qPCR analysis. The primer sequences and positions were presented in Table S2. Data were calculated by 2-(Ct of IP sample -Ct of input sample) method and were presented as a percentage of input.
Gene expression profiling and data analysis
Microarray hybridization was performed using the Whole Human Genome Oligo Microarray (4×44K, Agilent Technologies, CA, USA) according to the instructions of manufacturer. Upon hybridization and washing, the arrays were scanned using a dual-laser DNA microarray scanner (Agilent Technologies, CA, USA). These data were extracted from images by the Feature Extraction software 6.1 (Agilent Technologies, CA, USA). Raw data were normalized by Lowess algorithm (Gene Spring Software 11.0, Agilent Technologies, CA, USA), and were deposited in GEO (accession number: GSE90589). DAVID (http://david.abcc.ncifcrf.gov/home.jsp) functional annotation cluster analysis was performed with a fold change of ≥2 (21). Only those terms that reported a value of ≤0.05 and count number ≥5 genes were selected for analysis. The gene ontology (GO) term biological process (BP) in DAVID was used to categorize the enriched biological themes. The pathway mapping analysis was performed using the KEGG Automatic Annotation Server (KAAS) (http://www.genome.jp/kegg/kaas/).
Animal studies
Transgenic mouse lines TPO-Cre and BrafCA were kindly provided by Dr. Kimura Shioko (National Institutes of Health, USA) and Prof. Martin McMahon (University of California, USA), respectively. TPO-Cre mice express Cre recombinase under control of human thyroid peroxidase (TPO) gene promoter. BrafCA mice express normal Braf prior to Cre-mediated recombination after mutationally activated BrafV600E (BrafVE) is expressed at physiological levels (22). Endogenous Braf alleles were replaced by BrafV600E and mice spontaneously develop PTC at 6-12 weeks of age by crossing with TPO-Cre mice. Six week-old TPO-Cre; BrafCA mice were randomly divided into three groups (five mice per group), and were treated with a combination of PLX4720 (30 mg/kg) with GSK1120212 (1mg/kg), 10058-F4 (30 mg/kg), and vehicle once a day for 2 weeks by gastric gavage and intraperitoneal injection. All mice were sacrificed and thyroids were collected and weighted 6 h after the final dose of drugs. The tissues were then fixed in 4% paraformaldehyde, embedded in paraffin and sectioned for hematoxylin & eosin (H&E) and immunohistochemical staining.
Immunohistochemical staining
Sections were cut at 5 µm thickness, deparaffinized and rehydrated. Endogenous peroxidase activity was blocked with hydrogen peroxide/methanol, and antigen retrieval was performed in a pH 9.0 TE (Tris-EDTA) buffer by autoclave for 10 min. The resultant tissue sections were then incubated with the antibodies against p-Erk1/2, c-Myc, Ezh2, Suz12, Jarid2 and H3K27me3 at 4°C overnight. After incubation with labeled streptavidin-biotin (LSAB) complex for 15 min, the slides were stained and visualized by using the iView DAB detection system (ZSGB-BIO, P.R. China). Each stained section was evaluated by a minimum of 10 randomly selected ×40 high-power fields for further statistical analysis.
Statistical analysis
Unless otherwise specified, the Student's t test was conducted to analyze significance of data using SPSS statistical package (16.0, Chicago, IL). The data are expressed as mean ± standard deviation (SD) or standard error of the mean (SEM) as indicated. P <0.05 was considered statistically significant.
Results
H3K27me3 is an epigenetic modification regulated by BrafV600E signaling
To explore the relationship between BrafV600E signaling and H3K27me3, we established mouse NIH3T3 cells expressing wild-type and active mutant (V600E) of human BRAF or that were transfected with empty vector (referred to hereafter as BrafWT cells, BrafV600E cells and VEC cells). As shown in Figure 1A, phosphorylation of Erk1/2 was significantly increased in BrafV600E cells as compared to BrafWT and VEC cells. Moreover, BrafV600E cells exhibited a dramatic increase in H3K27me3 levels relative to BrafWT and VEC cells. These results suggest that BrafV600E signaling induces trimethylation of H3K27 as an epigenetic modification.
Figure 1
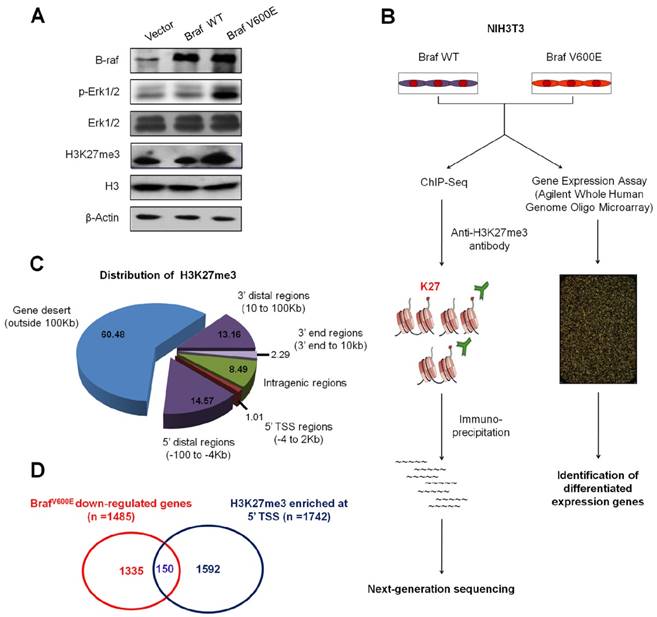
BrafV600E signaling induces epigenetic silencing by increasing H3K27me3 abundance in NIH3T3 cells. (A) Western blot analysis of B-raf, phosphorylated (p-) and total forms of Erk1/2 and H3K27me3. GAPDH and histone H3 were used as loading controls. (B) Flow chart shows the experimental design. (C) Pie chart shows distribution of H3K27me3 enrichment across whole genome in BrafV600E cells relative to BrafWT cells. (D) Venn diagram illustrates that 150 loci were marked by H3K27me3 and downregulated by BrafV600E signaling.
Identification of H3K27me3-enriched genomic regions associated with transcriptional silencing
ChIP-Seq and expression microarray were then employed to determine altered profiles of H3K27me3 in BrafV600E target genes in BrafV600E cells versus BrafWT cells (Figure 1B). A total of 25.1 and 22.2 million reads mapped to unique genomic locations in BrafV600E and BrafWT cells, respectively (data not shown). A ChIP-Seq peak calling program, MACS (19), was used to identify H3K27me3-enriched targets in BrafV600E cells relative to BrafWT cells. To characterize the pattern of H3K27me3 within these genes, using TSS (transcriptional start site) and gene body as separation points, we divided each gene into 5' distal regions (-100 to -4 Kb upstream of TSS), 5' TSS regions (-4 to 2 Kb around TSS), intragenic regions (2 Kb downstream of TSS to 3' end), 3' end regions (3' end to 10 Kb downstream of 3' end), 3' distal regions (10 to 100 Kb downstream of 3' end) and gene desert (outside 100 Kb from gene body). As shown in Figure 1C, H3K27me3 was mostly enriched in gene desert region (60.48%), which was consistent with a previous study (23). Moreover, we found enrichments in and around the 5´ TSSs of 1742 protein-coding genes in BrafV600E cells. Depletion of H3K27me3 was observed in 1135 genes whereas the levels remained unchanged for other 654 genes in both BrafV600E and BrafWT cells (Table S3). We subsequently identified 1485 downregulated and 1108 upregulated genes by microarray profiling (Table S3). We further integrated these gene expression data with ChIP-Seq results, and found that 150 downregulated gene loci had higher enrichments of H3K27me3 around TSS regions in BrafV600E cells relative to BrafWT cells (Figure 1D and Table S3).
Next, we used the Functional Annotation Cluster (FAC) tool available in the Database for Annotation, Visualization, and Integrated Discovery (DAVID) to classify biological processes of these downregulated genes, and found that they were mostly participated in extracellular matrix structural constituent (Figure S1). We then focused on the integrated 150 genes by using the GO term “Biological Process” (BP) analysis. The results with significantly P values from the resulting functional analysis for each group were listed in Table S3. We noted that these genes had a broad range of important molecular and cellular functions including various aspects of cell fate, suggesting that BrafV600E is involved in numerous essential biological functions through H3K27me3-mediated epigenetic silencing.
Validation of expression and H3K27me3 of BrafV600E-repressed genes
To validate the results of ChIP-Seq and microarray, 8 genes Abi3bp, Gimap6, Hopx, Pdzd2, Ptprd, Rspo2, Mtss1 and Nrxn3 from the 150 overlapped genes were randomly selected and verified by using ChIP-qPCR and qRT-PCR assays. As shown in Figure 2A, the expression of most genes, except for Nrxn3, was significantly decreased in BrafV600E cells as compared to BrafWT cells. To determine whether these BrafV600E-regulated genes can be epigenetically silenced in NIH3T3 cells, we tested the status of H3K27me3 in the promoters of these genes by ChIP-qPCR analysis. As expected, the fragments within their promoters were correspondingly enriched by anti-H3K27me3 antibody (Figure 2B).
To test the effect of BrafV600E on the expression and H3K27me3 status of these 8 genes in vivo, we established a transgenic mouse model of thyroid cancer (TPO-Cre/LSL-BrafV600E). The levels of phospho-Erk (p-Erk) and H3K27me3 were significantly increased in TPO-Cre/LSL-Braf V600E mice in comparison with Braf wild-type mice (Figure 2C). Next, we analyzed the expression levels of the above 8 genes in these mice. As shown in Figure 2D, most of these genes particularly Gimap6, Pdzd2, Ptprd and Nrxn3 were downregulated in thyroid cancer from TPO-Cre/LSL-BrafV600E mice as compared to Braf wild-type mice. To further determine the role of BrafV600E in the regulation of these genes, we treated TPO-Cre/LSL-Braf V600E mice with a combination of BRAF kinase inhibitor PLX4720 and MEK inhibitor GSK1120212. Compared with vehicle treatment, combined treatment of two inhibitors caused a significant reduction in tumor volume and weight (Figure 3A and B). Moreover, the levels of phospho-Erk (p-Erk) and H3K27me3 were dramatically inhibited by combined treatment of PLX4720 and GSK1120212 in TPO-Cre/LSL-Braf V600E mice (Figure 3C). Importantly, we found that combined treatment significantly restored the expression of these downregulated genes as compared to vehicle treatment (Figure 3D). Altogether, these findings suggest that BrafV600E-induced changes in H3K27me3 in gene promoters contribute to their transcription silencing in tumorigenesis.
Figure 2
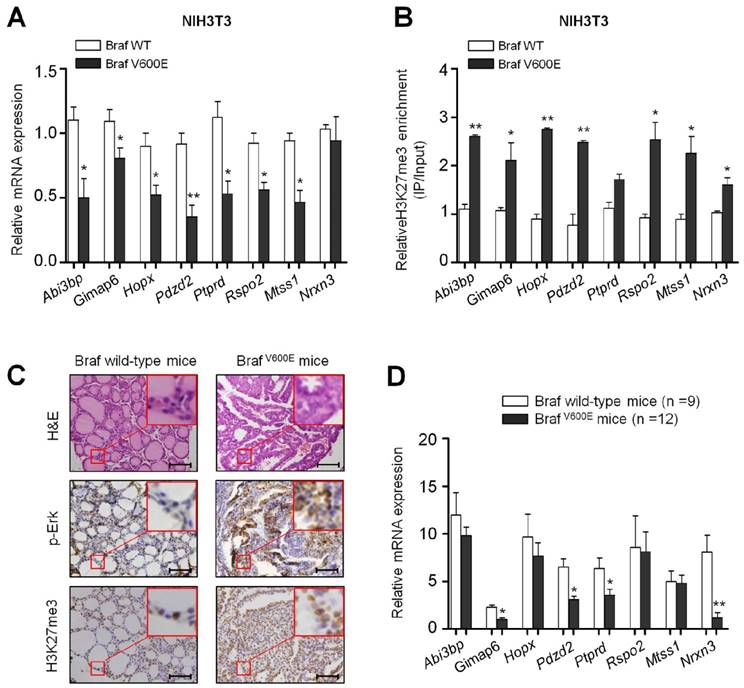
Validation of mRNA expression and H3K27me3 of BrafV600E-repressed genes in vitro and in vivo. (A) qRT-PCR analysis of Abi3bp, Gimap6, Hopx, Pdzd2, Ptprd, Rspo2, Mtss1 and Nrxn3 expression in BrafV600E cells relative to that in BrafWT cells. 18S rRNA was used as an endogenous control. Data were presented as mean ± SD from three independent experiments. (B) ChIP-qPCR analysis of promoter regions of the indicated genes in BrafV600E cells relative to that in BrafWT cells using anti-H3K27me3 antibody. Data were presented as mean ± SD from three independent assays. (C) Comparison of the levels of p-Erk1/2 and H3K27me3 between TPO-Cre/LSL-BrafV600E (BrafV600E) and Braf wild-type mice by IHC analysis. The inserts show the magnified image of the area indicated by the red square. Scale bars, 200 μm. (D) qRT-PCR analysis of Abi3bp, Gimap6, Hopx, Pdzd2, Ptprd, Rspo2, Mtss1 and Nrxn3 expression in BrafV600E mice relative to that in Braf wild-type mice. 18S rRNA was used as an endogenous control. Data were presented as mean ± SD. Statistically significant differences were indicated: *, P <0.05; **, P <0.01.
c-Myc is a major determinant of BrafV600E-induced changes in H3K27me3 through regulating the PRC2 components
Given that the PRC2 is responsible for H3K27me3, a histone mark that closely correlates with transcriptional silencing (8,9), we thus examined the effect of BrafV600E on the expression of PRC2 components including two core components Ezh2 and Suz12, and a cofactor Jarid2 (7, 24-26) in NIH3T3 cells. As shown in Figure 4A, BrafV600E transgene significantly activated the MAPK/Erk signaling, characterized by increased phosphorylation of Erk1/2 (p-Erk1/2). Compared to BrafWT and VEC cells, protein levels of Ezh2, Suz12 and Jarid2 were increased in BrafV600E cells. Accordingly, H3K27me3 levels were also upregulated in BrafV600E cells. Our data also showed that c-Myc, a downstream effector of MAPK/Erk cascade, was upregulated in BrafV600E cells as compared to BrafWT and VEC cells (Figure 4A). In addition, we found that basal expression of Ezh2, Suz12 and Jarid2 was inhibited in NIH3T3 cells when c-Myc was knocked down by two different small interfering RNAs (si-Myc1 and si-Myc2) (Figure 4B). Meanwhile, basal levels of H3K27me3 were decreased upon c-Myc knockdown (Figure 4B). As expected, our data demonstrated that the regulatory role of BrafV600E on PRC2 components and H3K27me3 was significantly attenuated upon c-Myc depletion (Figure 4C). These findings suggest that c-Myc is essential for BrafV600E-induced changes in H3K27me3 through modulating the expression of PRC2 components in tumorigenesis. In addition, we also tested the effect of BrafV600E transgene on the expression of c-Myc and PRC2 components in vivo by using a mouse model of thyroid cancer. As shown in Figure 4D, the levels of c-Myc, Ezh2, Suz12, Jarid2 and H3K27me3 were significantly increased in TPO-Cre/LSL-Braf V600E mice as compared to Braf wild-type mice. When the TPO-Cre/LSL-Braf V600E mice were treated with a combination of PLX4720 and GSK1120212, we found that these two inhibitors dramatically decreased the levels of c-Myc, these three PRC2 components and H3K27me3 as compared to vehicle treatment (Figure 4D). Next, we attempted to determine the role of c-Myc in regulating H3K27me3 in vivo using TPO-Cre/LSL-Braf V600E mice. As shown in Figure 5A and B, c-Myc inhibitor 10058-F4 caused a significant reduction in tumor volume and weigh as compared to the control. Moreover, 10058-F4 dramatically downregulated the expression of Ncl, a downstream target of c-Myc (27), suggesting that transcriptional activity of c-Myc was inhibited by 10058-F4. Similar to the in vitro findings, in vivo data showed that 10058-F4 dramatically decreased the levels of PRC2 components and H3K27me3 in TPO-Cre/LSL-Braf V600E mice (Figure 5C). As a result, the expression of the above 8 downregulated genes was restored by 10058-F4 particularly Abi3bp, Gimap6, Hopx, Ptprd, and Rspo2 as compared to vehicle treatment (Figure 5D). These results suggest the importance of c-Myc in the regulation of PRC2-dependent H3K27me3 in the BrafV600E-driven cancers.
Figure 3
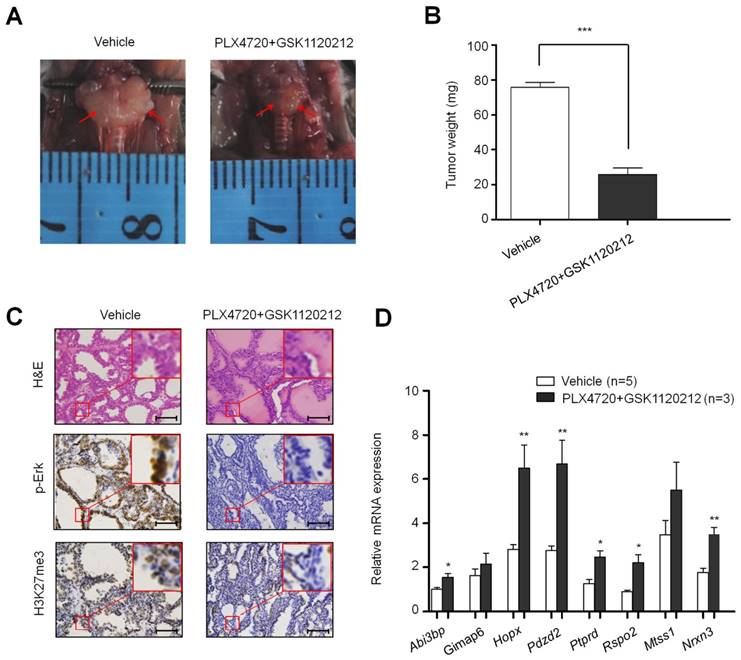
The effect of inhibition of BrafV600E signaling on H3K27me3 levels and the expression of BrafV600E-repressed genes in BrafV600E mice. Comparison of tumor volume (A), weight (B), and the levels of p-Erk1/2 and H3K27me3 (C) between BrafV600E mice treated with a combination of PLX4720 and GSK1120212 (n=3) and vehicle (n=5), respectively. Scale bars, 200 μm. (D) qRT-PCR analysis of Abi3bp, Gimap6, Hopx, Pdzd2, Ptprd, Rspo2, Mtss1 and Nrxn3 expression in combination-treated mice relative to that in vehicle-treated mice. DMSO was used as the vehicle control. 18S rRNA was used as an endogenous control. Data were presented as mean ± SD. Statistically significant differences were indicated: *, P<0.05; **, P<0.01; ***, P<0.001.
Figure 4
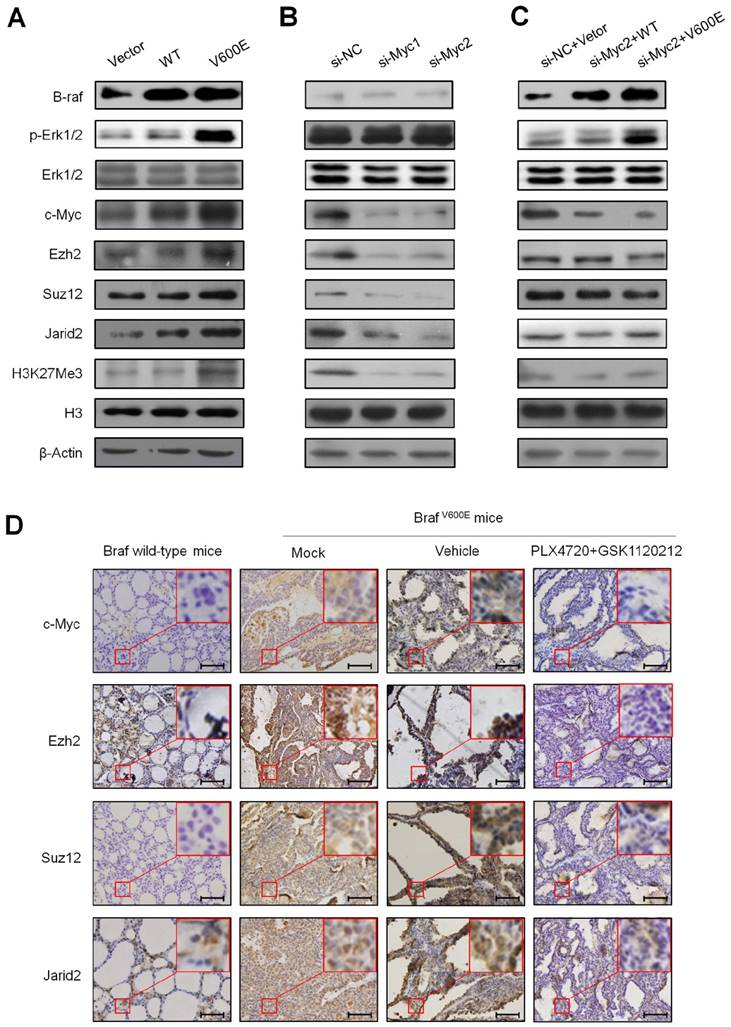
BrafV600E increases H3K27me3 levels through regulating the PRC2 components via c-Myc. (A) Western blot analysis of B-raf, p-Erk1/2, total Erk1/2, c-Myc, Ezh2, Suz12, Jarid2 and H3K27me3 in VEC, BrafWT and BrafV600E cells, respectively. β-Actin and histone H3 were used as loading controls. (B) Western blot analysis of Ezh2, Suz12, Jarid2 and H3K27me3 upon c-Myc depletion in NIH3T3 cells. β-Actin and histone H3 were used as loading controls. si-NC, si-Myc1 and 2 represent control and specific c-Myc siRNAs, respectively. (C) Western blot analysis of Ezh2, Suz12, Jarid2 and H3K27me3 in BrafV600E cells relative to that in BrafWT cells. β-Actin and histone H3 were used as loading controls. (D) A comparison of the levels of c-Myc, Ezh2, Suz12, Jarid2 and H3K27me3 in the indicated mice by IHC staining. DMSO was used as the vehicle control. The insert shows the magnified image of the area indicated by the red square. Scale bars, 200 μm.
Figure 5
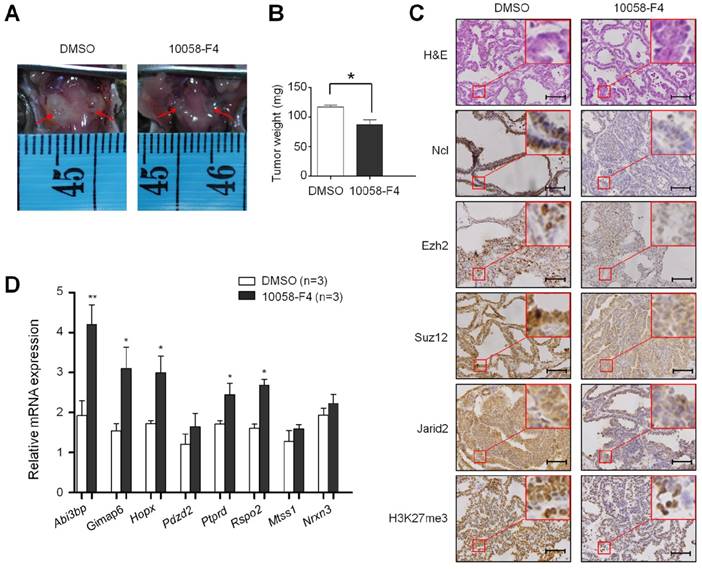
The effect of inhibition of c-Myc by 10058-F4 on the levels of the PRC2 components and H3K27me3 in BrafV600E mice. Comparison of tumor volume (A), weight (B), and the levels of Ncl, Ezh2, Suz12, Jarid2 and H3K27me3 (C) between BrafV600E mice treated with 10058-F4 (n=3) and vehicle (n=3), respectively. DMSO was used as the vehicle control. The insert shows the magnified image of the area indicated by the red square. Scale bars, 200 μm. (D) qRT-PCR analysis of Abi3bp, Gimap6, Hopx, Pdzd2, Ptprd, Rspo2, Mtss1 and Nrxn3 expression in 10058-F4-treated mice relative to that in vehicle-treated mice. DMSO was used as the vehicle control. 18S rRNA was used as an endogenous control. Data were presented as mean ± SD. Statistically significant differences were indicated: *, P<0.05; **, P<0.01; ***, P<0.001.
Next, we made efforts to confirm the above conclusions in human cancer cells. Thus, we chose two BRAFV600E-mutant thyroid cancer cell lines BCPAP and K1, and two BRAFV600E-mutant melanoma cell lines M14 and A375 in the present study. For BCPAP and K1 cells, we treated these cells with PLX4032, GSK1120212, MK-2206 and 10058-F4 individually or in different combinations. As shown in Figure 6A, these inhibitors exhibited an inhibitory effect on c-Myc, and downregulated the levels of some of PRC2 components and H3K27me3 particularly PLX4032, GSK1120212 and 10058-F4 regardless of individual or combinations. Our data also showed that, of these PRC2 components, Jarid2 expression was not significantly inhibited by each treatment alone. However, combinations of PLX4032 or GSK1120212 and 10058-F4 dramatically downregulated Jarid2 expression in both BCPAP and K1 cells. For M14 and A375 cells, we treated these cells with PLX4032, GSK1120212 and 10058-F4 individually or in combinations. Similar to the findings in thyroid cancer cells, blockade of the MAPK/Erk cascade or c-Myc by these inhibitors downregulated the expression of some of PRC2 components and decreased the levels of H3K27me3 in melanoma cells (Figure 6B).
c-Myc regulates the expression of Ezh2, Suz12 and Jarid2 at both transcriptional and post-transcriptional levels
There is evidence showing that c-Myc directly upregulates the transcription of core components of the PRC2 through binding to their regulatory elements (28). Thus, we next determine the role of c-Myc in the regulation of PRC2 components by BrafV600E. As shown in Figure 7A (upper-left panel), we found that mRNA levels of Ezh2, Suz12 and Jarid2 were upregulated in BrafV600E cells as compared to BrafWT cells. Basal mRNA levels of these three genes were significantly inhibited upon c-Myc knockdown in NIH3T3 cells (Figure 7A, upper-middle panel). Similar to the findings in Figure 4C, our data showed that c-Myc depletion dramatically inhibited the regulatory effect of BrafV600E on the PRC2 components in NIH3T3 cells (Figure 7A, upper-right panel). In addition, a previous study has demonstrated that a number of microRNAs (miRNAs) were transcriptionally suppressed by c-Myc (29), and some of them such as miR-26a, miR-200b and miR-155 have been reported to regulate the expression of PRC2 components at post-transcriptional levels (30-32). Thus, we attempted to test whether BrafV600E increased the expression of PRC2 components through regulating miR-26a, miR-200b and miR-155 via c-Myc. As expected, we found that BrafV600E transgene significantly inhibited the expression of these miRNAs in NIH3T3 cells (Figure 7A, lower-left panel), and knocking down c-Myc in NIH3T3 cells upregulated the expression of miR-26a, miR-200b and miR-155 (Figure 7A, lower-middle panel). Crucially, c-Myc depletion completely abolished the inhibitory effect of BrafV600E on the expression of these three microRNAs (Figure 7A, lower-right panel). Collectively, these data suggest that BrafV600E regulates the expression of the PRC2 components to cause redistribution of H3K27me3 through c-Myc at both transcriptional and post-transcriptional levels, contributing to gene silencing in tumorigenesis.
Next, we attempted to explore the mechanism underlying the regulation of PRC2 components by c-Myc. Given that c-Myc has been reported to transcriptionally regulate the expression of the PRC2 components (28), we thus performed chromatin immunoprecipitation (ChIP-qPCR) assay to show direct binding of c-Myc to their promoter regions. As shown in Figure 7B, using anti-c-Myc antibody, we observed 5.2 to 17.6-fold enrichment of promoter amplicons of Ezh2, Suz12 and Jarid2 in BrafV600E cells relative to BrafWT cells. A recent study has indicated that c-Myc interacts with MAX and WDR5 to upregulate their expression through recruiting H3K4me3 deposition in gene promoters (33). We thus analyzed the H3K4me3 levels at gene promoters of Ezh2, Suz12 and Jarid2 in BrafV600E and BrafWT cells using ChIP-qPCR assay. However, the H3K4me3 levels were not significantly changed between these two cells (Figure 7C). These observations further support that c-Myc regulates the transcription of the PRC2 components through direct binding to their regulatory elements. In addition, we similarly found that promoter amplicons of miR-26a, miR-200b and miR-155 were significantly enriched by anti-c-Myc antibody in BrafV600E cells relative to BrafWT cells (Figure 7D). Given that c-Myc recruits the PRC2 complex to cause the H3K27me3 reprogram, contributing to silencing of certain microRNAs through interacting with EZH2 (34), we thus performed the ChIP-qPCR assay to detect the H3K27me3 levels at the promoters of miR-26a, miR-200b and miR-155. As shown in Figure 7E, although the H3K27me3 levels were slightly higher in BrafV600E cells than BrafWT cells, the difference did not reach statistical significance, suggesting that c-Myc may inhibit the transcription of these microRNAs through an unknown mechanism, but not H3K27me3 redistribution.
To determine the effect of the MAPK/Erk signaling and c-Myc on the expression of these PRC2 components and microRNAs in human cancer cells, we treated the BCPAP, K1, M14 and A375 cells with PLX4032 and 10058-F4, respectively. As expected, blockade of the MAPK/Erk signaling by PLX4032 significantly decreased mRNA expression of Ezh2, Suz12 and Jarid2 in these cell lines. On the other hand, PLX4032 increased the expression of miR-26a, miR-200b and miR-155 in the same cell lines (Figure 8A). Similarly, we also found that 10058-F4 increased the expression of Ezh2, Suz12 and Jarid2 and decreased the expression of miR-26a, miR-200b and miR-155 in these four cell lines (Figure 8B). These data further support the importance of the MAPK/ Erk/c-Myc axis in the regulation of PRC2-dependent H3K27me3 in tumorigenesis.
Notably, a previous study has indicated that activated Erk2 itself can suppress gene expression by directly binding to a specific enriched GA/GC motif and then phosphate RNA polymerase II (RNAPII) on Ser-5 residues (S5P). RNAPII (S5P) will lose its extending ability, causing a poised transcription status of genes (35). Moreover, this binding effect also regulates nucleosome disruption and H3K27me3 deposition through recruiting Jarid2 to the specific GA/GC motif DNA elements (35). This was supported by our data that promoter amplicons containing GA/GC motif of Gimap6, Rspo2 and Hopx were significantly enriched by anti-p-Erk antibody in BrafV600E cells. Meanwhile, promoter amplicons of Rspo2 and Hopx were also enriched by anti-Jarid2 antibody in BrafV600E cells (Figure S2). Taken together, these observations suggest that BrafV600E contributes to epigenetic silencing through multiple distinct mechanisms.
Discussion
Oncogenic signaling pathways regulate the transcription of tumour-related genes in part through epigenetic mechanisms including DNA methylation, histone acetylation and methylation (16-18, 36, 37). For example, the activation of the PI3K/Akt pathway may be a trigger of gene silencing by H3K27me3 redistribution in breast cancer (36). In addition, a previous study has demonstrated that H3K27me3 levels and transcription are both regulated by the activated H-Ras (G12V) (37); however, Ras-induced changes in H3K27me3 are not a trigger for, but rather a consequence of, changes in transcriptional activity. One possibility is that the activated Ras signaling may affect the transcription of epigenetic chromatin modification enzymes such as the PRC2 complex, further leading to changes in H3K27me3. In the present study, we suggest that the activation of MAPK/Erk cascade by BRAFV600E is a trigger of epigenetic silencing in tumorigenesis. This oncogenic mutation is a high-frequent alteration in human cancers particularly in thyroid cancer and melanoma (11, 12), and has been demonstrated to induce epigenetic silencing of tumor-related genes through DNA methylation and histone deacetylation in these cancers (16-18). We further revealed that BRAFV600E caused changes in H3K27me3, thereby leading to epigenetic silencing of multiple loci. This new line of evidence suggests an interesting mechanism that BRAFV600E induces malignant transformation through its epigenetic effects in tumorigenesis.
Figure 6
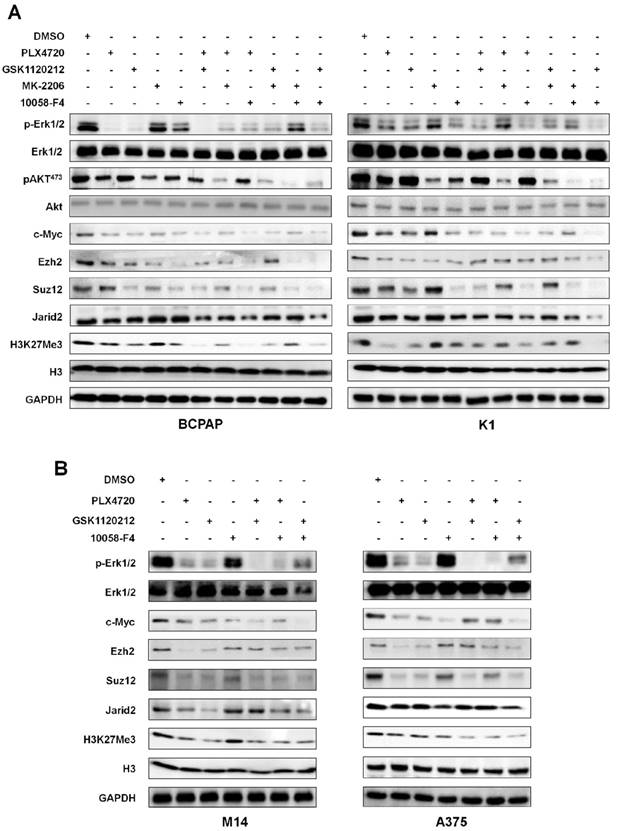
The effect of inhibition of major signaling pathways on the levels of the PRC2 components and H3K27me3 in thyroid cancer and melanoma cells. Western blot was performed to evaluate the effect of the indicated inhibitors individually or in combinations on the levels of the PRC2 components and H3K27me3 in thyroid cancer cell lines BCPAP and K1 (A) and melanoma cell lines M14 and A375 (B). GAPDH and histone H3 were used as loading controls. DMSO was used as the vehicle control.
Figure 7
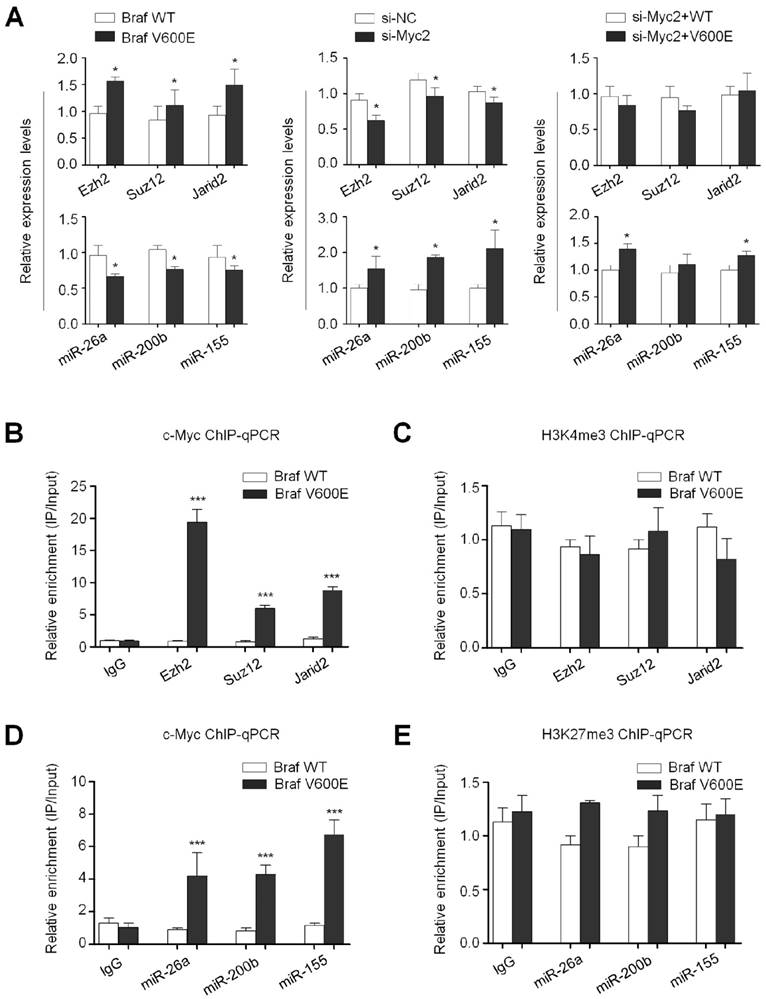
BrafV600E regulates the expression of the PRC2 components through c-Myc at both transcriptional and post-transcriptional levels. (A) qRT-PCR analysis of Ezh2, Suz12, Jarid2, miR-26a, miR-200b and miR-155 in BrafV600E cells relative to that in BrafWT cells (left panel). qRT-PCR analysis of Ezh2, Suz12, Jarid2, miR-26a, miR-200b and miR-155 upon c-Myc depletion in NIH3T3 cells (middle panel). qRT-PCR analysis of Ezh2, Suz12, Jarid2, miR-26a, miR-200b and miR-155 upon c-Myc depletion in BrafV600E cells relative to that in BrafWT cells (right panel). 18S rRNA was used as an endogenous control. Data were presented as mean ± SD from three independent experiments. (B-E) ChIP-qPCR analysis of the promoter regions of the indicated genes including Ezh2, Suz12, Jarid2, miR-26a, miR-200b and miR-155 in BrafV600E cells relative to that in BrafWT cells using the antibodies indicated in each panel. Data were presented as mean ± SD from three independent experiments. Statistically significant differences were indicated: *, P<0.05; ***, P<0.001.
Figure 8
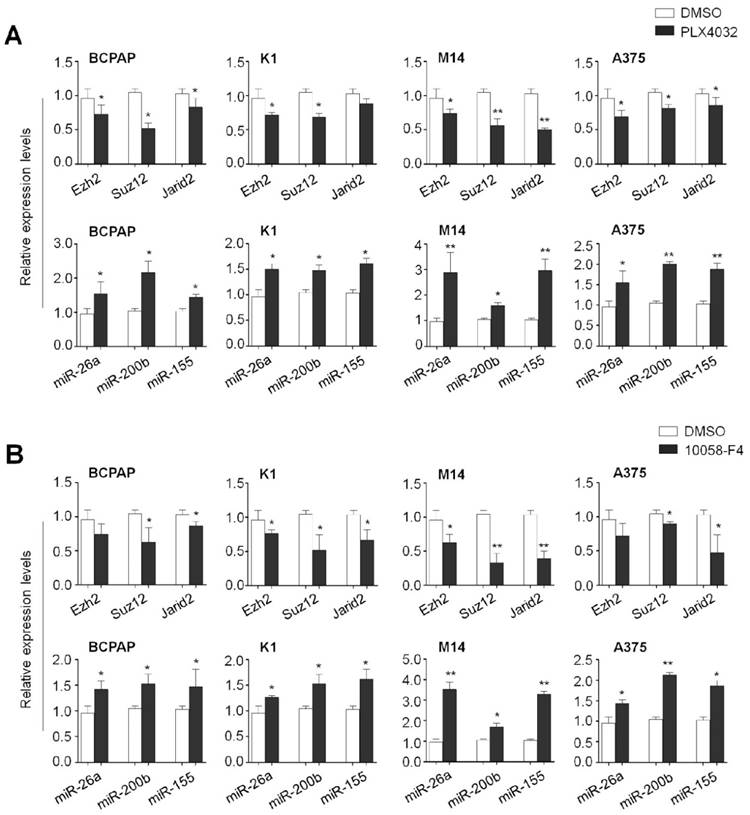
The effect of BrafV600E signaling and c-Myc on the expression of Ezh2, Suz12, Jarid2, miR-26a, miR-200b and miR-155 in thyroid cancer and melanoma cells. qRT-PCR analysis was performed to evaluate the effect of BRAF kinase inhibitor PLX4032 and c-Myc inhibitor 10058-F4 on the expression of Ezh2, Suz12, Jarid2, miR-26a, miR-200b and miR-155 in thyroid cancer cell lines BCPAP and K1 (A) and melanoma cell lines M14 and A375 (B). DMSO was used as the vehicle control. 18S rRNA was used as an endogenous control. Data were presented as mean ± SD from three independent experiments. Statistically significant differences were indicated: *, P<0.05; **, P<0.01.
It is the fact that H3K27me3, a repressive histone mark, is regulated by the PRC2 complex including core components such as Ezh1/2, Suz12 and Eed, and several cofactors such as Jarid2 (4,6,7). These cofactors interact with core components to modulate both their binding to chromatin and their enzymatic activity (6). In the present study, we found that BrafV600E transgene increased the levels of the H3K27me3 and PRC2 components including Ezh2, Suz12 and Jarid2 in vitro and in vivo, and blockade of MAPK/Erk signaling by PLX4032 decreased their levels, suggesting that BrafV600E may induce redistribution of H3K27me3 through regulating the PRC2 components. Next, we attempted to explore molecular mechanisms underlying the regulation of PRC2 components by BrafV600E through a series of in vitro and in vivo experiments. It is well-known that c-Myc, a downstream key effector of BRAFV600E signaling, functions as an oncogenic transcription factor, and is overexpressed in the majority of human cancers (38). c-Myc is able to regulate the expression of thousands of target genes through binding to regulatory elements in their promoters (38). However, this effect depends on interaction of c-Myc with its obligate partner MAX (39). There is evidence showing that c-Myc can regulate the transcription of the PRC2 components in embryonic stem cells (28). Indeed, we found that BrafV600E upregulated the expression of c-Myc in BrafV600E cancer cells or BrafV600E-driven mouse thyroid cancer, and c-Myc depletion expectedly abolished the regulatory role of BrafV600E on these PRC2 components and H3K27me3 in vitro and in vivo. Altogether, these observations suggest that c-Myc is required for BrafV600E-induced epigenetic silencing by H3K27me3 in tumorigenesis.
Recent efforts have been undertaken to explore molecular mechanisms responsible for the regulation of PRC2 components by c-Myc. First, there is evidence demonstrating that direct binding of c-Myc to the regulatory elements of the PRC2 components activates their transcription by recruiting chromatin modifier enzymes to their E-box elements, ultimately leading to the increase of active RNA polymerase II on their promoters (28). This was supported by our data that promoter amplicons of Ezh2, Suz12 and Jarid2 were significantly enriched by anti-c-Myc antibody in BrafV600E cells relative to BrafWT cells. In addition, it has been proposed that binding of c-Myc to chromatin is enriched in active histone modifications such as H3K4me3 (33, 40). However, we did not find significant changes in H3K4me3 between BrafV600E and BrafWT cells. These findings suggest that c-Myc activates the transcription of the PRC2 components by direct binding to their regulatory elements independent of H3K4me3 reprogram in tumorigenesis. In addition to transcription levels, c-Myc may also regulate the expression of its downstream targets including the PRC2 components at post-transcriptional levels. There is increasing evidence showing that c-Myc inhibits the expression of a series of microRNAs through directly binding to their promoters or forming complexes with their indispensability transcription factors to result in transcriptional repression (29). Some of these microRNAs such as miR-26a, miR-200b and miR-155 have been demonstrated to target the PRC2 components including Ezh2, Suz12 and Jarid2 (30-32). In the present study, we observed that BrafV600E significantly decreased the expression of miR-26a, miR-200b and miR-155, and c-Myc depletion completely reversed the inhibitory effect of BrafV600E on these miRNAs, suggesting that c-Myc is essential for this regulatory process. In addition, we found that anti-c-Myc antibody significantly enriched promoter amplicons of these miRNAs in BrafV600E cells relative to BrafWT cells, further supporting that c-Myc represses the transcription of these miRNAs through directly binding to their promoters. As a result, c-Myc may upregulate the levels of the PRC2 components at post-transcriptional levels through repressing the above miRNAs, contributing epigenetic silencing in BRAFV600E-driven cancers. We also demonstrated that inhibition of MAPK/Erk cascade or c-Myc effectively increased the expression of miR-26a, miR-200b and miR-155, and decreased the expression of the PRC2 components in human cancer cell lines, further supporting the above conclusions.
In summary, we demonstrate that BRAFV600E signaling induces epigenetic silencing by H3K27me3, and c-Myc is required in this process. Our findings are consistent with a model (Figure 9) in which BRAFV600E signaling induces epigenetic silencing not only through upregulating the PRC2 components via c-Myc at both transcriptional and post-transcriptional levels, but also through Erk1/2-induced RNAPII poising and chromatin architecture. Thus, the present study reveals a previously unrecognized epigenetic mechanism in BRAFV600E-driven tumorigenesis.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We would like to thank Dr. Haixia Guan (The First Affiliated Hospital of China Medical University, Shenyang, China) for kindly providing human thyroid cancer cell lines BCPAP and K1, and Dr. Kimura Shioko (National Institutes of Health, USA) and Prof. Martin McMahon (University of California, USA) for kindly providing transgenic mouse lines TPO-Cre and BrafCA.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81372217, 81402340, 81572627 and 81672645), and Science and Technology Project of Shaanxi Province (No. 2013SF2-13).
Author Contributions
PH and YQ conceived and designed the experiments. YQ, QY and JL performed the experiments. YQ, MJ and PH analyzed the data. BS, GL and PH contributed reagents/materials/analysis tools. YQ and PH wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
Figure 9
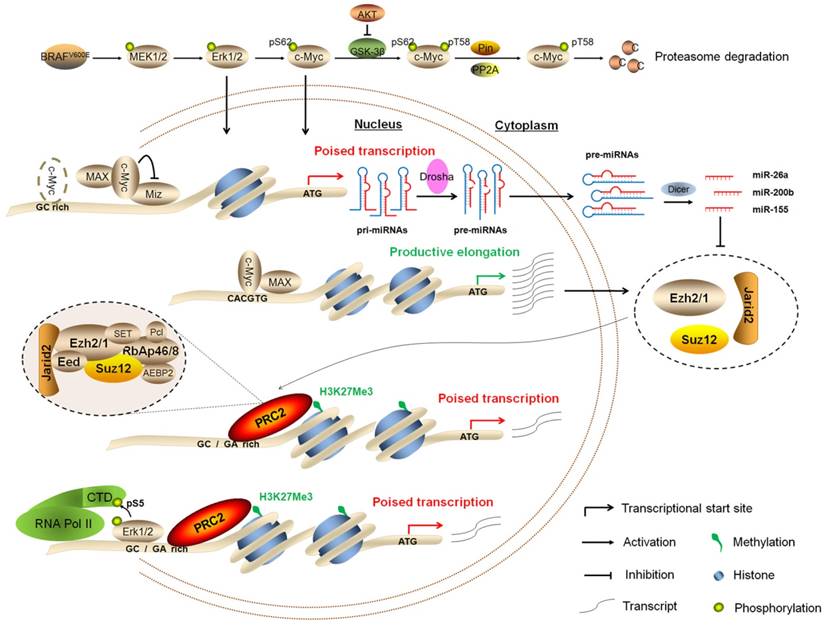
Schematic model of BRAFV600E signaling-induced epigenetic silencing by H3K27me3 in tumorigenesis. BRAFV600E mutation constitutively activates the MAPK/Erk signaling, resulting in upregulation of its downstream effector c-Myc in many types of cancer particularly thyroid cancer and melanoma. In general, c-Myc is degraded via the ubiquitin-proteasome system. However, this process can be inhibited by activated PI3K/Akt pathway in cancer cells. When c-Myc translocates into the nucleus, it regulates the expression of its target genes through binding to their regulatory elements via interaction with its obligate partner MAX or other unknown mechanisms. In the present study, c-Myc regulates the expression of the PRC2 components, contributing to epigenetic silencing through two distinct mechanisms. First, c-Myc transcriptionally regulates the expression of the PRC2 components through binding to the regulatory elements within their promoters. Second, c-Myc transcriptionally represses the expression of a panel of miRNAs, whereas some of them target and regulate the expression of the PRC2 components at post-transcriptional levels. In addition, BRAFV600E signaling also induces epigenetic silencing through Erk1/2-induced RNAPII poising and chromatin architecture.
References
1. Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148-1159
2. Taby R, Issa J.P. Cancer epigenetics. CA Cancer J Clin. 2010;60:376-392
3. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039-1043
4. Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306:1574-1577
5. Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D. et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell. 2010;38:452-464
6. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343-349
7. Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M. et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol Cell. 2015;57:769-783
8. Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697-708
9. Conway E, Healy E, Bracken AP. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol. 2015;37:42-48
10. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120:3446-3456
11. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S. et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-954
12. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676-690
13. Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ. et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246
14. Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role molecular bases and clinical implications. Endocr Rev. 2007;28:742-762
15. Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B. et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA. 2013;309:1493-1501
16. Hou P, Liu D, Xing M. Genome-wide alterations in gene methylation by the BRAF V600E mutation in papillary thyroid cancer cells. Endocr Relat Cancer. 2011;18:687-697
17. Hou P, Liu D, Dong J, Xing M. The BRAF V600E causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle. 2012;11:286-295
18. Zhang Z, Liu D, Murugan AK, Liu Z, Xing M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr Relat. Cancer. 2014;21:161-173
19. Wang L, Chen J, Wang C, Uuskula-Reimand L, Chen K, Medina-Rivera A. et al. MACE: model based analysis of ChIP-exo. Nucleic Acids Res. 2014;42:e156
20. Ma W, Noble WS, Bailey TL. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat Protoc. 2014;9:1428-1450
21. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44-57
22. Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation progression and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379-384
23. Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I. et al. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res. 2009;19:221-233
24. Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV. et al. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. 2010;464:306-310
25. Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. Jarid2 and PRC2 partners in regulating gene expression. Genes Dev. 2010;24:368-380
26. Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jørgensen HF. et al. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat Cell Biol. 2010;12:618-624
27. Greasley PJ, Bonnard C, Amati B. Myc induces the nucleolin and BN51 genes: possible implications in ribosome biogenesis. Nucleic Acids Res. 2000;28:446-453
28. Neri F, Zippo A, Krepelova A, Cherubini A, Rocchigiani M, Oliviero S. Myc regulates the transcription of the PRC2 gene to control the expression of developmental genes in embryonic stem cells. Mol Cell Biol. 2012;32:840-851
29. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM. et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43-50
30. Lu J, He ML, Wang L, Chen Y, Liu X, Dong Q. et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011;71:225-233
31. Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39:761-772
32. Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V. et al. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity. 2014;40:865-879
33. Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q. et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell. 2015;58:440-452
34. Wang L, Zhang X, Jia LT, Hu SJ, Zhao J, Yang JD. et al. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology. 2014;59:1850-1863
35. Tee WW, Shen Steven S, Oksuz O, Narendra V, Reinberg D. Erk1/2 Activity Promotes Chromatin Features and RNAPII Phosphorylation at Developmental Promoters in Mouse ESCs. Cell. 2014;156:678-690
36. Zuo T, Liu TM, Lan X, Weng YI, Shen R, Gu F. et al. Epigenetic silencing mediated through activated PI3K/AKT signaling in breast cancer. Cancer Res. 2011;71:1752-1762
37. Hosogane M, Funayama R, Nishida Y, Nagashima T, Nakayama K. Ras-induced changes in H3K27me3 occur after those in transcriptional activity. PLoS Genet. 2013;9:e1003698
38. Dang CV. MYC on the path to cancer. Cell. 2012;149:22-35
39. Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipperprotein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211-1217
40. Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall'Olio V. et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764-770
Author contact
![]() Corresponding authors: Peng Hou, Ph.D., Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China Tel/Fax: 86-298-532-4039; E-mail: phouedu.cn; Dr. Gang Li, Department of Neurosurgery, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, P.R. China Tel/Fax: +86-29-85323259; E-mail: liliangxbytcom
Corresponding authors: Peng Hou, Ph.D., Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, P.R. China Tel/Fax: 86-298-532-4039; E-mail: phouedu.cn; Dr. Gang Li, Department of Neurosurgery, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, P.R. China Tel/Fax: +86-29-85323259; E-mail: liliangxbytcom
Citation styles
APA
Qu, Y., Yang, Q., Liu, J., Shi, B., Ji, M., Li, G., Hou, P. (2017). c-Myc is Required for BRAFV600E-Induced Epigenetic Silencing by H3K27me3 in Tumorigenesis. Theranostics, 7(7), 2092-2107. https://doi.org/10.7150/thno.19884.
ACS
Qu, Y.; Yang, Q.; Liu, J.; Shi, B.; Ji, M.; Li, G.; Hou, P. c-Myc is Required for BRAFV600E-Induced Epigenetic Silencing by H3K27me3 in Tumorigenesis. Theranostics 2017, 7 (7), 2092-2107. DOI: 10.7150/thno.19884.
NLM
Qu Y, Yang Q, Liu J, Shi B, Ji M, Li G, Hou P. c-Myc is Required for BRAFV600E-Induced Epigenetic Silencing by H3K27me3 in Tumorigenesis. Theranostics 2017; 7(7):2092-2107. doi:10.7150/thno.19884. https://www.thno.org/v07p2092.htm
CSE
Qu Y, Yang Q, Liu J, Shi B, Ji M, Li G, Hou P. 2017. c-Myc is Required for BRAFV600E-Induced Epigenetic Silencing by H3K27me3 in Tumorigenesis. Theranostics. 7(7):2092-2107.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.