Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussions
Conclusions
Materials and methods
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):1990-2002. doi:10.7150/thno.18136 This issue Cite
Research Paper
Surface De-PEGylation Controls Nanoparticle-Mediated siRNA Delivery In Vitro and In Vivo
Xi Zhu1, 2*, Wei Tao1*, Danny Liu1, 3, Jun Wu1, 4, Zilei Guo1, Xiaoyuan Ji1, Zameer Bharwani1, Lili Zhao1, 5, Xiaoping Zhao2, Omid C. Farokhzad1, 6 ![]() , Jinjun Shi1
, Jinjun Shi1 ![]()
1. Center for Nanomedicine and Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, USA;
2. National Shanghai Center for New Drug Safety Evaluation and Research, Shanghai, 201203, China;
3. Department of Chemistry, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada;
4. Department of Biomedical Engineering, School of Engineering, Sun Yet-sen University, Guangzhou, 510006, China;
5. Department of Endoscopy, the First Affiliated Hospital of Nanjing Medical University, Jiangsu Province Hospital, Nanjing, 210029, China;
6. King Abdulaziz University, Jeddah 21589, Saudi Arabia.
* X.Z. and W.T. contributed equally to this work.
Received 2016-10-29; Accepted 2017-3-11; Published 2017-5-12
Abstract

The present work proposes a unique de-PEGylation strategy for controllable delivery of small interfering RNA (siRNA) using a robust lipid-polymer hybrid nanoparticle (NP) platform. The self-assembled hybrid NPs are composed of a lipid-poly(ethylene glycol) (lipid-PEG) shell and a polymer/cationic lipid solid core, wherein the lipid-PEG molecules can gradually dissociate from NP surface in the presence of serum albumin. The de-PEGylation kinetics of a series of different lipid-PEGs is measured with their respective NPs, and the NP performance is comprehensively investigated in vitro and in vivo. This systematic study reveals that the lipophilic tails of lipid-PEG dictate its dissociation rate from NP surface, determining the uptake by tumor cells and macrophages, pharmacokinetics, biodistribution, and gene silencing efficacy of these hybrid siRNA NPs. Based on our observations, we here propose that lipid-PEGs with long and saturated lipophilic tails might be required for effective siRNA delivery to tumor cells and gene silencing of the lipid-polymer hybrid NPs after systemic administration.
Keywords: de-PEGylation, nanoparticle, self-assembly, siRNA delivery, cancer therapy.
Introduction
Surface coating of poly(ethylene glycol) (PEG) has been extensively exploited to improve the pharmacokinetics (PK) of therapeutic nanoparticles (NPs) for cancer treatment, via significantly reducing the recognition of NPs by the mononuclear phagocyte system (MPS) [1-3]. With prolonged systemic circulation, PEGylated NPs have a higher chance to extravasate out of leaky tumor vasculature and accumulate in solid tumors through the enhanced permeability and retention (EPR) effect [4-6]. Notably, several PEGylated liposomes and polymeric NPs have been clinically approved for cancer therapy, and many are under clinical investigation [7-9]. Meanwhile, surface de-PEGylation might be equally important for effective NP delivery into tumor cells due to various reasons [10, 11]. For instance, the surface PEG layer could negatively interfere with intracellular delivery of NPs [1, 11-13], which is particularly essential for the delivery of biomacromolecular therapeutics such as small interfering RNA (siRNA) that cannot readily cross cell membrane and require cytosolic transport for bioactivity [14-16]. Therefore, a balance between PEGylation and de-PEGylation is critical for siRNA NPs to simultaneously achieve both long-term blood circulation and sufficient intracellular uptake in the systemic delivery to tumors.
To address this dilemma, stimuli-triggered de-PEGylation strategies have been developed by attaching PEG molecules on NP surface through labile bonds that are sensitive to the unique features of tumor microenvironment (TME) and/or intracellular environment, such as low pH, overexpressed enzymes, and altered redox potential [11, 17, 18]. Several pH-responsive linkers (e.g., orthoester [19], vinyl ether [20], and hydrazine [21-23]) were studied for the PEG shedding of NPs to enhance gene and drug delivery. Overexpressed enzymes in the tumor tissue (e.g., matrix metalloproteinase (MMP)) [24-27] and altered intracellular redox potential [28-32] were also explored as stimulating triggers. One example is the development of cleavable lipid-PEGs by conjugating PEG molecules and phospholipids together with a MMP substrate peptide [24, 25]. Lipid/siRNA NPs formulated with this cleavable lipid-PEG exhibited improved gene silencing efficacy in tumors as a result of enhanced cellular uptake and endosomal escape [25]. In addition, de-PEGylation can further be combined with other targeting or penetration-enhancing strategies for the development of multifunctional NPs [22, 23, 26, 32, 33]. In vitro proofs of concept have been well demonstrated for these PEG-shedding systems, and some also have been preliminarily tested in animal models [21, 23-25, 27, 32, 34-36]. For example, a recent work reported a pH-sensitive hydrazone linkage-mediated PEG shedding strategy for increased retention and high therapeutic efficacy of lipid/siRNA NPs in breast cancer mouse models [36]. Despite of all these advances, such stimuli-responsive de-PEGylation approaches might pose additional complexity in terms of design, synthesis, and scaling of therapeutic NPs [18].
Herein, we report a tunable and robust PEG shedding strategy mediated by serum albumin, and systematically investigate the effects of de-PEGylation on NP-mediated siRNA delivery in vitro and in vivo. The siRNA NP platform (Figure 1a) was formulated by self-assembling a lipid-PEG layer on the surface of a poly(lactide-co-glycolide) (PLGA) polymer core containing cationic lipid/siRNA complexes [37], via hydrophobic interactions between the lipophilic tails of lipid-PEG and the PLGA polymer. Albumin is the most abundant plasma protein and has been recognized as the main fatty acid transporter by providing different binding sites for fatty acids with moderate to high affinity, thus enhancing their solubility in aqueous solutions such as blood plasma and interstitial fluid [38, 39]. A recent work further demonstrated that albumin can bind avidly to diacyl lipids [40]. We thus hypothesized that the lipid-PEG molecules could dissociate from the hybrid NP surface in the presence of serum albumin, and the dissociation kinetics may depend on the lipid-PEG properties. Our recent study discovered that the lipid segment (ceramide vs. DSPE) of lipid-PEG had a substantial effect on the de-PEGylation rate and the systemic circulation of the hybrid NPs, while PEG molecular weight might not be critical [37]. This observation inspired us to further clearly and systematically study how the lipid-PEG properties (e.g., lipid tail length, lipid tail saturation, and lipid charge) control the dissociation of lipid-PEGs from NP surface and affect siRNA NP delivery in vitro and in vivo, which we expect will provide significant insights for better understanding and design of the lipid-polymer hybrid NP platform. In this work, the de-PEGylation kinetics of eight lipid-PEG molecules with different alkyl chain length, saturation, and charge were measured with respective hybrid NPs. Furthermore, the tumor cell and macrophage uptake, gene silencing efficacy, and in vivo performance of these siRNA NPs were comprehensively examined and compared with the lipid-PEG dissociation rate. Results suggest that the alkyl chains of lipid-PEG determine the optimal de-PEGylation rates, by which the hybrid siRNA NPs can remain “stealth” long enough for high tumor extravasation and accumulation, and once reaching tumor tissue, can facilitate intracellular delivery to tumor cells for effective gene silencing.
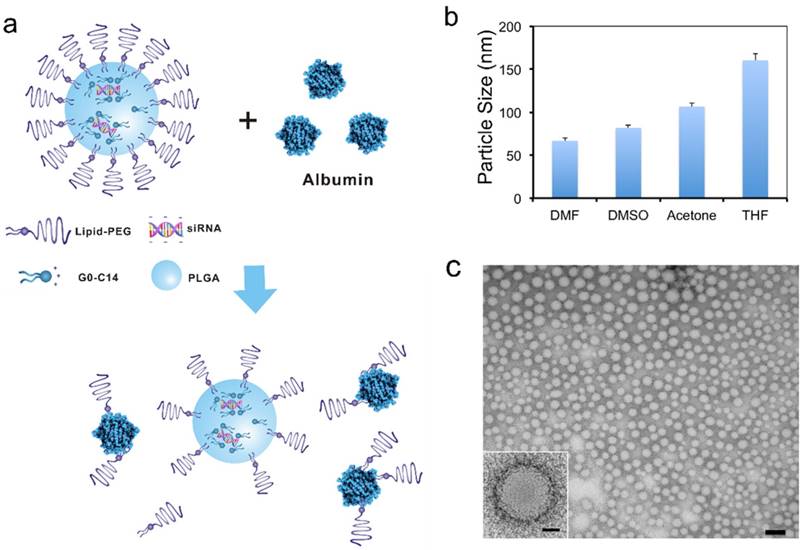
Lipid-polymer hybrid NPs for siRNA delivery. (a) Schematic of the NP structure and the lipid-PEG dissociation in the presence of serum albumin. (b) Size of the hybrid NPs prepared using different water-miscible organic solvents, as measured by DLS. (c) TEM image of the NPs prepared using DMF (scale bar: 100 nm). The scale bar is 20 nm for the insert image.
Results and Discussions
Self-assembled lipid-polymer hybrid NPs
The lipid-polymer hybrid NPs loaded with siRNA were prepared through a robust and simple self-assembly nanoprecipitation method [37]. Aqueous siRNA was first mixed with water-miscible organic solution containing cationic lipid-like compound G0-C14 and PLGA polymer. The negatively charged siRNA molecules could then be self-assembled quickly with G0-C14 into lipid-siRNA nanocomplexes. Subsequently, the organic solution was rapidly added into an aqueous solution of lipid-PEG, in which the PLGA polymer and cationic lipid/siRNA complex were co-precipitated to form a solid polymeric NP core surrounded by a lipid-PEG shell (Figure 1a). Four polar aprotic solvents with different dielectric constants were tested for preparing the hybrid siRNA NPs, including dimethylformamide (DMF), dimethylsulfoxide (DMSO), acetone, and tetrahydrofuran (THF). As shown by the dynamic light scattering (DLS) results (Figure 1b), sub-100 nm NPs were obtained with DMF and DMSO, which possess relatively high dielectric constants. Larger NPs were formed when using solvents with lower dielectric constants (i.e., acetone and THF). This observation is consistent with a previous study of developing similar lipid-polymer hybrid NPs for the delivery of chemotherapeutic drugs [41]. Low dielectric constant of a solvent implies its low polarity and poor water miscibility. Thus, when a low dielectric constant solvent is used, the organic solution of PLGA/nanocomplexes would have less efficient dispersion in the lipid-PEG aqueous solution. In the following studies, DMF was chosen for NP formulation, as the resulting smaller NPs may facilitate better tumor tissue accumulation through the EPR effect [42]. The hybrid siRNA NPs prepared with DMF also exhibited a spherical structure with a narrow size distribution as observed by TEM (Figure 1c).
Kinetics of lipid-PEG dissociation
During the nanoprecipitation process, lipid-PEG would self-assemble on the NP surface through hydrophobic interactions between the lipophilic alkyl chains and the PLGA core. Different from covalently conjugated PEG layer, physically adsorbed lipid-PEG molecules on NP surface are diffusible, which could thus result in de-PEGylation of the NPs over time. We hypothesized that both the assembly and the dissociation processes might be predominantly determined by the physicochemical properties of the lipid-PEG molecules. In this work, a series of eight lipid-PEGs with different alkyl chains and charges (Figure 2a) were used to evaluate their effect on the behaviors of the hybrid NPs for siRNA delivery, while keeping the PEG molecular weight (2 kDa) consistent. They are divided into two categories, neutral and anionic lipid-PEGs, according to the linkage (glycerol/serine vs. glycero-3-phosphoethanolamine) between the lipophilic tails and the PEG chain. Both categories include three lipid-PEGs with saturated alkyl chains of different length (C14, C16 and C18) and one lipid-PEG with unsaturated chain(s) (ceramide or DOPE).
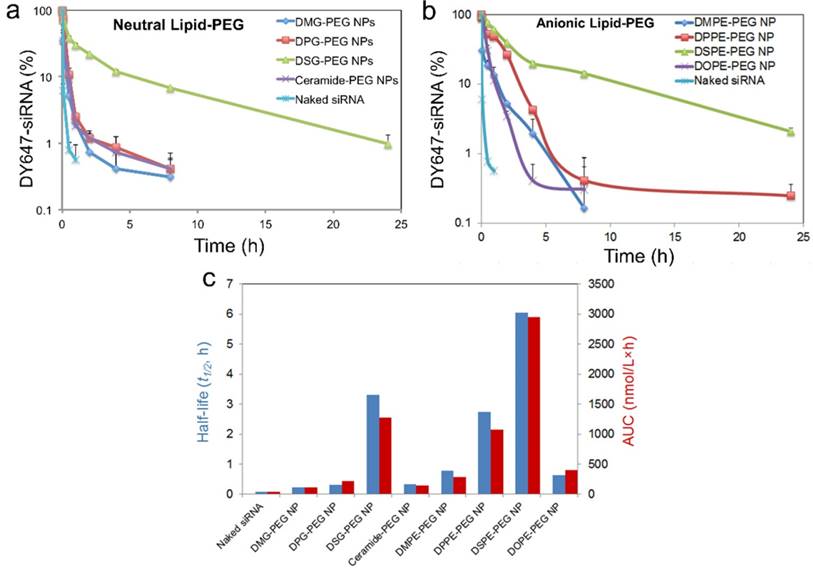
Effect of lipid-PEG on the properties and de-PEGylation of the hybrid siRNA NPs. (a) Chemical structure of eight different lipid-PEGs. (b) The amount of surface lipid-PEG relative to the PLGA polymer weight and the surface charge (zeta potential) on different NPs. (c) siRNA encapsulation efficiency of the NPs with different lipid-PEGs. Dissociation kinetics of (d) neutral lipid-PEGs and (e) anionic lipid-PEGs from respective NPs in the presence of serum albumin.
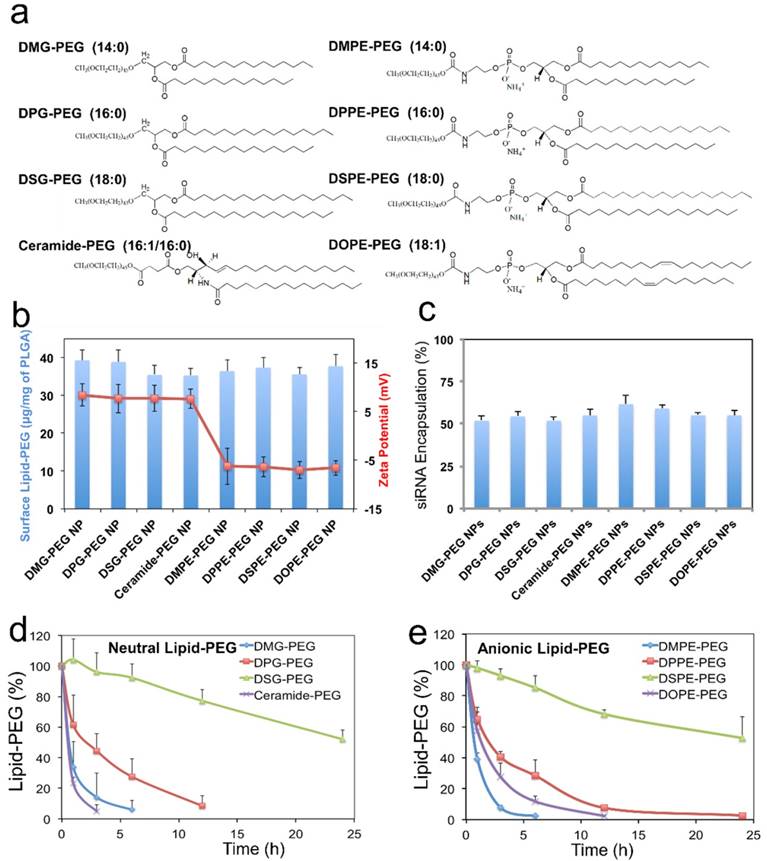
We first quantified the amount of lipid-PEG that self-assembled on the NPs using a method based on the spectrophotometric measurement of complexes formed by PEG and barium iodide [43, 44]. All NPs carried a similar amount of lipid-PEGs on the surface, which was ~ 35-40 μg/mg of the PLGA polymer (Figure 2b). It is worth noting that this self-assembly nanoprecipitation method requires no surfactant/stabilizer (e.g., polyvinyl alcohol) in the hybrid NP formulation, which was reported to significantly impede lipid-PEG association on the polymeric particle surface as a steric barrier [45]. This high surface lipid-PEG density may thus increase the NP stability in serum and reduce MPS recognition. The hybrid NPs prepared with neutral vs. anionic lipid-PEGs also carried different surface charges (Figure 2b). The NPs coated with neutral lipid-PEGs showed mildly positive zeta potential (~ 8 mV), while those with anionic lipid-PEGs exhibited mildly negative charges (~ -7 mV). The choice of lipid-PEGs also had no significant influence on the particle size (~ 60-70 nm) and the encapsulation efficiency of siRNA, which was ~ 50-60% for all the NPs (Figure 2c) as measured using fluorophore labeled siRNA (DY547-siRNA).
Considering that albumin is the main plasma protein in blood and has a strong binding with diacyl lipids [40], we utilized 4 w/v% serum albumin in phosphate buffered saline (PBS) to investigate the role of albumin on the de-PEGylation of the hybrid siRNA NPs. While all lipid-PEGs dissociated from NPs over time as shown in Figure 2d-e, the rate of dissociation was drastically different. For lipid-PEGs with saturated tails in both neutral and anionic categories, shorter alkyl chains resulted in faster dissociation. For example, after 24 h incubation with albumin, over 50% of DSG-PEG and DSPE-PEG (with C18 alkyl chains) still remained on NP surfaces. As a comparison, the amount of DMG-PEG and DMPE-PEG (with C14 alkyl chains) dwindled below the detection limit after 6-12 h. This phenomenon might be explained by the higher phase transition temperature and lower fluidity of the lipid tails with longer saturated alkyl chains, which could result in stronger hydrophobic interactions with the PLGA core. Interestingly, much faster dissociation was observed for the unsaturated lipid-PEGs than the saturated ones with the same length of alkyl chain (ceramide vs. DPG, or DOPE vs. DSPE). DOPE-PEG, which carries two unsaturated C18 alkyl chains, was released at a rate even faster than DPPE-PEG with two saturated C16 alkyl chains. Similarly, ceramide-PEG (with one unsaturated C16) was released faster than DMG-PEG (saturated C14). This may be attributed to the fact that the unsaturation on alkyl chain can lower the chain melting transition temperature, and thus increase the lipid fluidity [46]. On the other hand, neutral and anionic lipid-PEGs with the same alkyl chains showed similar dissociation kinetics, suggesting that the charge in the linkage group between PEG and alkyl chains had negligible effect on the de-PEGylation of these NPs. We also calculated the dissociation rate constants (kd) and half-lives (t1/2) of the eight lipid-PEGs from respective NPs by fitting the dissociation profiles in Figure 2d-e to a first-order dissociation model (Table 1).
The dissociation rate constant (kd) and dissociation half-life (t1/2) of different lipid-PEGs from respective NPs
| Neutral Lipid-PEG | DMG-PEG | DPG-PEG | DSG-PEG | Ceramide-PEG | |
| kd (h-1) | 0.52 | 0.21 | 0.025 | 1.05 | |
| t1/2 (h) | 1.33 | 3.30 | 27.73 | 0.66 | |
| Anionic Lipid-PEG | DMPE-PEG | DPPE-PEG | DSPE-PEG | DOPE-PEG | |
| kd (h-1) | 0.68 | 0.17 | 0.028 | 0.33 | |
| t1/2 (h) | 1.02 | 4.08 | 24.76 | 2.10 |
To further clarify the important role of albumin in de-PEGylation of the hybrid NPs, two anionic lipid-PEG NPs with the fastest vs. slowest dissociation rate (i.e. DMPE and DSPE) were chosen as models for further de-PEGylation study in PBS without albumin. As shown in Figure S1a, the de-PEGylation rates in the PBS control group were drastically reduced as compared to that in PBS containing 4 w/v% albumin (Figure 2e), suggesting that the de-PEGylation effect for our hybrid NPs could be closely related with the presence of albumin. In addition, the de-PEGylation kinetics in FBS, which contains albumin and a number of other proteins and can more closely mimic the blood, were determined with DMPE-PEG and DSPE-PEG NPs (Figure S1b). The similar lipid-PEG dissociation rates to those in PBS containing albumin (Figure 2e) further provide evidences for the strong effect of albumin on de-PEGylation. It is also worth mentioning that other factors such as the sheer force of blood flow may also have potential effect on the dissociation of lipid-PEG.
Effect of lipid-PEG on in vitro siRNA delivery
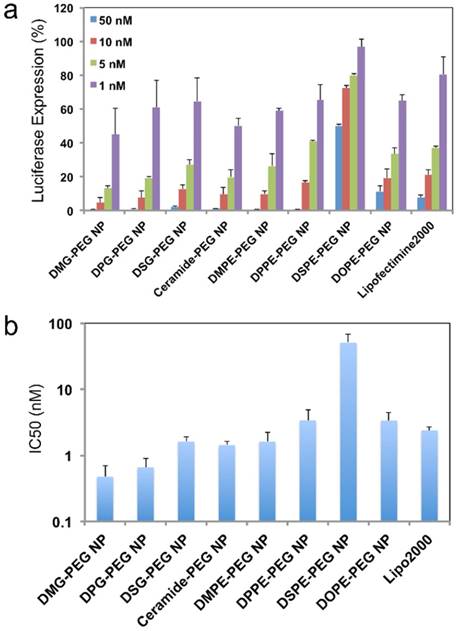
For in vitro evaluation of the hybrid siRNA NPs with different surface lipid-PEGs, a firefly luciferase-expressed HeLa cell line (Luc-HeLa) was used. The hybrid siRNA NPs without lipid-PEG coating was also used as a control. The cells were first transfected with NPs loaded with luciferase siRNA (siLuc). The reduction of luciferase expression was then measured to evaluate the siLuc NP-mediated silencing efficiency. Figure 3a show that the lipid-PEG plays a vital role in determining the silencing performance in vitro. All NPs exhibited silencing activity in a dose-dependent manner. At a siRNA concentration of 50 nM, most of the NPs demonstrated a nearly complete (> 95%) luciferase knockdown and they were significantly more effective than the commercial transfection reagent lipofectamine 2000 (Lipo2000), except the DSPE-PEG and DOPE-PEG NPs. The IC50 for in vitro silencing mediated by different NPs was calculated using the Phoenix® WinNonlin software (Figure 3b). In both neutral and anionic lipid-PEG categories, the NPs with faster lipid-PEG dissociation generally exhibited lower IC50. It is also noteworthy that neutral lipid-PEG NPs exhibited higher silencing efficacy compared with their anionic counterparts, although similar lipid-PEG dissociation profiles were observed (Figure 2d-e). As a comparison, the PLGA/G0-C14 siRNA NPs (without PEGylation) showed slightly better or comparable in vitro knockdown efficiency than the neutral lipid-PEG NPs with fast de-PEGylation rates (Figure S2).
In vitro silencing efficacy of the hybrid NPs. (a) Luciferase expression in Luc-HeLa cells treated with NP (siLuc) composed of different lipid-PEGs. Lipo2000 was used as a positive control. (b) IC50 for the in vitro luciferase silencing from (a).
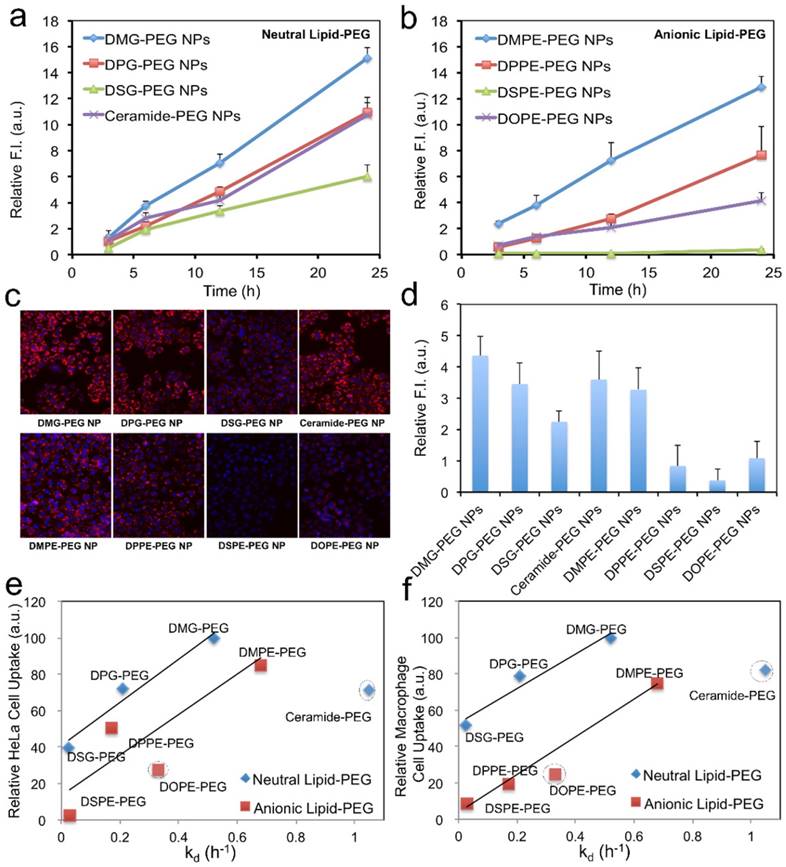
To understand the effect of lipid-PEG on in vitro silencing, we explored the uptake kinetics of the hybrid NPs loaded with DY547-siRNA in Luc-HeLa cells. Similar to the silencing results, the NPs with faster dissociation of saturated lipid-PEGs exhibited higher cellular internalization for both lipid-PEG categories, and the non-PEGylated PLGA/G0-C14 siRNA NPs also showed very high cellular internalization (Figure 4a-c and Figure S3). The neutral lipid-PEG NPs also showed higher cellular uptake compared to their anionic counterparts at 24 h. This could be explained by the surface charge difference of these NPs, as negatively charged NPs (with anionic lipid-PEGs) generally have less interaction with cell membranes than positively charged ones (with neutral lipid-PEGs). For the NPs coated with saturated lipid-PEGs, the cellular uptake result displayed similar trend with that of the silencing experiments, suggesting that the hybrid NPs with higher cellular uptake may potentially result in better luciferase silencing in Luc-HeLa cells. In addition to tumor cells, we also investigated the uptake of these NPs by macrophage cells (RAW264.7), which represent a major cell type in the MPS system for clearance of exogenous NPs. As shown in Figure 4d, the effect of lipid-PEGs on the macrophage uptake of NPs was consistent with that on Luc-HeLa cells. To further understand the correlation between the lipid-PEG dissociation and cellular uptake, we plotted the kd of different lipid-PEGs against the corresponding amount of NPs taken up by Luc-HeLa or macrophage cells (Figure 4e-f). Positive correlation was observed between the kd and the cellular uptake for lipid-PEGs with saturated alkyl tails in both neutral and anionic categories, but not for unsaturated ceramide-PEG and DOPE-PEG. This study also indicates that a compromise between high tumor cell uptake (for efficient gene silencing) and low macrophage recognition (for long blood circulation) may be needed especially for the non-targeted hybrid siRNA NPs to achieve optimal in vivo silencing.
Effect of lipid-PEG on NP uptake by tumor cells and macrophage cells. Uptake kinetics of siRNA NPs with (a) neutral and (b) anionic lipid-PEGs on Luc-HeLa cells. (c) Representative fluorescence images for the tumor cell uptake of NP (DY547-siRNA) with different lipid-PEGs after 24 h incubation. (d) NP uptake on macrophage cells (RAW264.7) after 12 h incubation. The correlation of (e) Luc-HeLa cell uptake and (f) macrophage cell uptake with the dissociation rate constant (kd) of different lipid-PEGs.
Effect of lipid-PEG on systemic in vivo siRNA delivery
To assess the in vivo performance of these hybrid NPs for siRNA delivery, we first examined the PK by injecting NP (DY647-siRNA) to normal C57BL/6 mice through the tail vein. NPs with different lipid-PEGs were evaluated and compared with naked siRNA. Figure 5a-b shows that naked siRNA was rapidly cleared from the systemic circulation within 30 min, while siRNA NPs stayed longer in blood. Drastically different PK behaviors were observed among these NPs and between the neutral and anionic categories. For the NPs coated with neutral lipid-PEGs, only the DSG-PEG NP exhibited a long circulation half-life (t1/2) ~ 3.3 h. All the other three NPs showed a t1/2 less than 20 min (Figure 5c). The NPs with anionic lipid-PEGs demonstrated much longer circulation than the neutral counterparts. The trend found in the PK study for anionic lipid-PEG NPs analogously resembled that observed in the dissociation study. DSPE-PEG NPs exhibited the most impressive circulation property with t1/2 ~ 6 h, and the area under the curve (AUC) is ~ 68-fold of that for naked siRNA (Figure 5c). Besides, NPs with unsaturated ceramide-PEG and DOPE-PEG exhibited relatively rapid blood clearance, as well as the bare PLGA NPs in the control group which were cleared more easily (Figure S4).
Effect of lipid-PEG on PK of the hybrid siRNA NPs. (a, b) Circulation profile of naked siRNA and different siRNA NPs composed of neutral or anionic lipid-PEGs in normal C57BL/6 mice after IV injection. siRNA was labeled with fluorophore DY647. (c) Circulation half-life and AUC of naked siRNA and siRNA NPs composed of different lipid-PEGs.
Next we checked the biodistribution (BioD) and tumor accumulation of the hybrid siRNA NPs upon intravenous (IV) injection into mice bearing subcutaneous Luc-HeLa tumor xenograft. As we have learned that, due to the EPR effect, NPs with long circulation lives could have higher chance to accumulate in tumor tissues than NPs with short PK [47-50]. Therefore, we herein selected all the three NPs with relatively long blood circulation (DOPE-PEG, DPPE-PEG, and DSPE-PEG NPs), and two NPs with short circulation (ceramide-PEG and DSG-PEG) and naked siRNA as controls. For fluorescence imaging, the siRNA was labeled with near infrared dye DY677. Twenty-four hours post-IV injection, major organs and tumors were harvested and imaged (Figure 6a). It was observed that naked siRNA had low accumulation in all tissues except kidney. The NPs with longer circulating t1/2 generally led to higher tumor accumulation. This can be explained by the fact that only the NPs that were not rapidly cleared from the circulation will have a chance to encounter the leaky tumor vasculature. NPs prepared with DSG-PEG, DOPE-PEG, and DPPE-PEG achieved similar tumor accumulation, which is ~ 4.7-6.1-fold higher than that of naked siRNA (Figure 6b). DSPE-PEG NPs with the longest circulating t1/2 exhibited a 14.5-fold higher tumor accumulation than naked siRNA. In comparison, the short-circulating ceramide-PEG NPs only increased the siRNA accumulation by a 2.2-fold. Interestingly, the NPs coated with neutral ceramide-PEG or DSG-PEG demonstrated a higher lung accumulation vs. other NPs. Further investigation on this observation might lead to other potential applications of the hybrid NPs.
Biodistribution and in vivo gene-silencing efficacy. (a) Ex vivo fluorescence image of the tumor and major organs from athymic nude mice bearing Luc-HeLa tumor treated with naked DY677-siRNA vs. NP (DY677-siRNA) with different lipid-PEGs. (b) Quantitative analysis of biodistribution of NP (siRNA) in different tissues and the Luc-HeLa tumor (n=3). (c) In vivo bioluminescence imaging of mice bearing Luc-HeLa tumor following IV injection of free siLuc or NP (siLuc) for 3 consecutive days. Images were taken at 5 min after intraperitoneal injection of 2 mg of D-Luciferin per mouse. (d) Luciferase expression in the protein extracts of tumor tissue treated with NP (siLuc) vs. naked siLuc (n=4, * p<0.05 vs. naked siRNA).
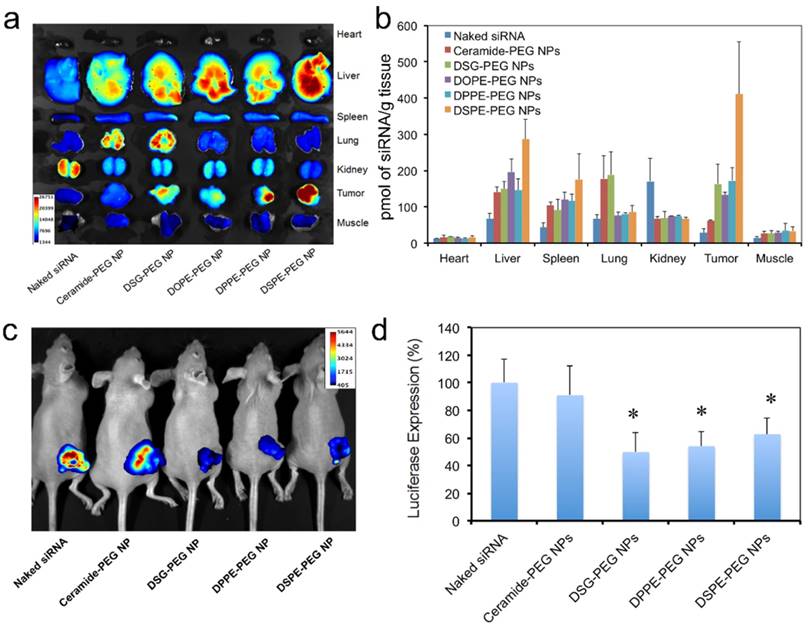
The in vivo silencing efficacy of these hybrid siRNA NPs was also evaluated on athymic nude mice bearing subcutaneous Luc-HeLa xenograft tumor. The mice received daily IV injection of naked siLuc or NP (siLuc) for three consecutive days. Three NPs (DSG-PEG, DPPE-PEG, and DSPE-PEG) with relatively long blood circulation and high tumor accumulation, and one NP (ceramide-PEG) with low tumor accumulation, were selected in this study. Two days after the final injection, each mouse was injected intraperitoneally with 2 mg of D-Luciferin, and 5 min later, the animal was imaged for bioluminescence (Figure 6c). For quantification analysis, the animals were sacrificed, and then the protein extracts of tumor tissue were prepared using lysis buffer to measure the firefly luciferase level (Figure 6d). As can be seen, the animals treated with the rapidly cleared ceramide-PEG NPs exhibited an insignificant reduction of the luciferase level compared to those treated with naked siRNA. DSG-PEG and DPPE-PEG NPs, which exhibited excellent in vitro silencing effect and relatively long blood circulation, were both effective in reducing bioluminescence intensity, and led to ~ 50% luciferase silencing in vivo. Interestingly, for DSPE-PEG NP, despite its low in vitro silencing efficiency (Figure 3a), ~ 40% knockdown of luciferase expression was achieved in tumor tissues. We speculated that the in vivo dissociation of lipid-PEG from NP surface might be faster than that in vitro owing to the more complicated environment and the blood flow. Our observations suggest that the in vivo gene silencing efficacy may depend on different aspects of the NPs, and neither in vitro data nor in vivo PK/BioD alone could be used to predict the in vivo efficacy. From these results, we postulate that lipid-PEGs with long and saturated lipophilic tails may be required for the lipid-polymer hybrid siRNA NPs to achieve effective gene silencing in tumor tissues after systemic administration.
Conclusions
Surface PEGylation can facilitate the long systemic circulation of therapeutic NPs by lowering MPS clearance and thus enhance NP extravasation into tumor tissues through the EPR effect, while de-PEGylation favors the intracellular transport of NPs, a critical step in the delivery of biomacromolecules such as siRNA to tumor cells. Therefore, when taking these factors into consideration, the PEG dissociation from NP surface needs to be carefully explored for optimal gene silencing in tumors. With the hybrid siRNA NP platform, we systematically investigated the effects of de-PEGylation on siRNA delivery. The lipid-PEG dissociation, which was controlled by the physicochemical characters of its lipid tails, correlated with the cellular uptake, gene silencing, and in vivo performance of these siRNA NPs. By selecting different lipid-PEGs in the NP formulation, the in vitro and in vivo behaviors of the hybrid siRNA NP system could be easily manipulated.
Different from the stimuli-triggered de-PEGylation strategies, our alternative approach avoids the design of TME stimuli-responsive chemistry, which may introduce additional complexities in the synthesis and scale-up of therapeutic NPs. Notably, the self-assembly of lipid-PEGs on the hybrid NP surface is also very robust, thus offering a convenient approach for high-throughput screening of lipid-PEG molecules. The optimization of other factors, such as the PEG length and the combination of multiple different lipid-PEGs, may further lead to better in vivo efficacy of the hybrid RNAi NPs. By enabling effective silencing of specific genes in tumor tissues following systemic administration, this NP platform could serve as a robust toolkit for fundamental cancer research and rapid in vivo validation of potential therapeutic targets in cancer pathogenesis, in particular those considered as 'undruggable'. In addition, the hybrid siRNA NPs can also simultaneously encapsulate small molecular drugs (e.g., taxanes and cisplatin prodrugs) within the PLGA polymer core[51-53], and the co-delivery of siRNA and drug combinations is expected to have a synergistic anti-tumor effect[33, 54]. In summary, we successfully demonstrated that surface lipid-PEG dissociation controls the in vitro and in vivo performance of the lipid-polymer hybrid siRNA NPs, and we expect this robust NP platform to be highly useful in cancer research and treatment.
Materials and methods
Materials
Methoxy-terminated DMPE-PEG, DPPE-PEG, DSPE-PEG, DOPE-PEG, and ceramide-PEG were obtained from Avanti Polar Lipids, and methoxy-terminated DMG-PEG, DPG-PEG, and DSG-PEG were from NOF American. The PEG molecular weight is 2 kDa. Ethylenediamine core-poly (amidoamine) (PAMAM) generation 0 dendrimer (G0), D-Luciferin, and bovine serum albumin were purchased from Sigma-Aldrich. Ester-terminated poly(D,L-lactide-co-glycolide) (PLGA, viscosity of 0.26-0.54 dL/g) was obtained from Durect Corporation. Iodine solution was obtained from Alfa Aesar. Transfection agent lipofectamine2000 (Lipo2000) was purchased from Invitrogen. Steady-Glo luciferase assay system was purchased from Promega. Luciferase siRNA (siLuc) and fluorophore-labeled siRNA (DY547-siRNA, DY647-siRNA, and DY677-siRNA) were acquired from Dharmacon. The siLuc sequence is: 5'-CUU ACG CUG AGU ACU UCG AdTdT-3' (sense) and 5'-UCG AAG UAC UCA GCG UAA GdTdT-3' (antisense). DY547 and DY647 were labeled at the 5'-end of the sense strand of siLuc. DY677 was labeled at the 5'-end of both the sense and antisense strands of siLuc.
Synthesis of G0-C14 compound
Cationic lipid-like G0-C14 compound was synthesized by the ring opening reaction of alkyl epoxides with PAMAM, according to a previously described procedure.[52, 55] PAMAM G0 and 1,2-epoxytetradecane were mixed at a molar ratio of 1:7, and reacted under vigorous stirring at 90 °C for 48 h. The crude reaction mixture was purified by silica chromatography with a gradient elution from CH2Cl2 to 75:22:3 CH2Cl2/MeOH/NH4OH.
Preparation and characterization of lipid-polymer hybrid siRNA NPs
The lipid-polymer hybrid siRNA NPs were formulated by a self-assembly nanoprecipitation method we recently developed with modifications [37]. In brief, 5 mg PLGA and 0.5 mg G0-C14 are dissolved in 1 mL organic solvent (e.g., DMF). A 50 µL siRNA (4 nmoles) aqueous solution is mixed with the organic solution by pipetting to form siRNA/G0-C14 nanocomplexes. Next, the organic solution with the nanocomplexes and polymers was added into a 20 mL aqueous solution containing 2 mg lipid-PEG (e.g., DSG-PEG). 0.2 mL of phosphate buffered saline (PBS) was subsequently added to stabilize the formed NPs. The suspension was then stirred at room temperature for 30 min. The hybrid NPs were finally washed in Amicon tubes (MWCO 100kDa; Millipore) to remove remaining organic solvents and free compounds with ice-cold water, and concentrated in 1 mL PBS solution. The PLGA/G0-C14 siRNA NPs without PEGylation were prepared with the same method except that lipid-PEGs were replaced with stabilizer polyvinyl alcohol (0.5%, w/v). The particle size and surface charge (zeta potential) were determined by dynamic light scattering (DLS; 15-mW laser incident beam of 676 nm; Brookhaven Instruments Corporation). NP samples for transmission electron microscopy (TEM) imaging were stained with 1% uranyl acetate and imaged by a Tecnai G2 Spirit BioTWIN microscope (FEI Company) operating at 80 kV.
Lipid-PEG quantification and dissociation kinetics
The amount of lipid-PEG molecules on NP surface and the dissociation of lipid-PEG from NPs in PBS (with 4 w/v% bovine serum albumin), PBS without albumin, or FBS were quantified based on the spectrophotometric measurement of complexes formed between PEG and barium iodide [44]. For quantification of surface lipid-PEG amount, the freshly prepared NP solution after 30 min stirring was directly ultra-centrifuged and the pellet was re-suspended in water for reaction with the barium iodide solution (100 μL DMSO, 20 µL BaCl2 (5%) and 20 µL I2 solution (0.1 N)). For the dissociation kinetics study, NPs were first incubated with PBS (with 4 w/v% bovine serum albumin), PBS without albumin, or FBS at 37 ºC. At predetermined time points, NP suspension was ultra-centrifuged to remove supernatant, and the NP pellet was re-suspended in water. Then 100 μL of NP solution (1.25 mg/mL at PLGA polymer weight) was mixed with 100 μL DMSO, 20 µL BaCl2 (5 %) and 20 µl I2 solution (0.1 N). Calibration curve was prepared with corresponding lipid-PEG solution with same concentration of PLGA (1.25 mg/mL) and G0-C14 (0.125 mg/mL). After incubation at room temperature for 15 min, absorbance at 535 nm was measured on the microplate reader.
Cell culture
Firefly luciferase-expressing HeLa (Luc-HeLa) and RAW264.7 macrophage were maintained in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen) with high glucose, 10% (v/v) fetal bovine serum (FBS; Gibco). Both cells were incubated at 37ºC in 5% CO2.
In vitro luciferase silencing
Luc-HeLa cells were seeded into 96-well plates (5,000 cells per well) and allowed to attach in cell culture medium at 37ºC in a 5% CO2 incubator overnight. Cells were then transfected with NPs or Lipo2000-siLuc complexes at siRNA concentration of 50 nM, 10 nM, 5 nM, and 1 nM for 24 h. Lipo2000-siLuc complexes were prepared according to the manufacturer's protocol. Luc-HeLa cells were then washed with fresh medium and further cultured in medium for another 48 h. The expression of luciferase in HeLa cells was determined using Steady-Glo luciferase assay kits. Cell number was measured using AlamarBlue assay. The luminescence/fluorescence intensity was measured using a microplate reader. All the in vitro transfection experiments were performed in triplicate.
NP uptake
For cellular uptake kinetics study, HeLa or RAW264.7 cells were seeded into 96-well plates and allowed to attach for 24 h. Next, the cells were incubated with NP (DY547-siRNA) at the siRNA concentration of 50 nM for different periods of time (3, 6, 12, and 24 h), and then washed with PBS, fixed with 4% paraformaldehyde, and stained with Hoechst 33342 (2 µg/mL) for nuclei identification. Images were acquired on an inverted Fluorescence Microscope (Zeiss Axiovert 200) and analyzed using Fiji/Image-J software.
Animals
All in vivo studies were performed in accordance with National Institutes of Health animal care guidelines and in strict pathogen-free conditions in the animal facility of Brigham and Women's Hospital. Animal protocol was approved by the Institutional Animal Care and Use Committees on animal care (Harvard Medical School). Mice were obtained from Charles River Laboratories. The animals were allowed free access to sterile food pellets and water.
Pharmacokinetic study
For pharmacokinetic study, normal C57BL/6 mice were intravenously injected with fluorophore (DY647)-labeled siRNA NPs or naked DY647-siRNA through the tail vein. At different time points, blood was drawn retro-orbitally and siRNA fluorescence was measured using the BioTek microplate reader. Standard curve was generated by measuring the fluorescence intensity of different amounts of NP (DY647-siRNA) or free DY647-siRNA added in blood from untreated mice. The total blood volume was estimated as 58.5 mL blood per kg of bodyweight.
Luc-HeLa tumor xenograft model
For the establishment of Luc-HeLa tumor xenograft model, 3×106 cells were suspended in 1:1 (v/v %) serum free media and Matrigel (BD Biosciences), and implanted subcutaneously into the flank of 5-week-old female athymic nude mice.
Biodistribution
For biodistribution study, female athymic nude mice bearing Luc-HeLa tumor were administered with (DY677)-labeled siRNA NPs or naked DY677-siRNA through the lateral tail vein injection. The mice were sacrificed 24 h after administration, and organs and tumors were harvested and imaged using the Syngene PXi imaging system (Synoptics Ltd). To quantify the accumulation of siRNA, organs and tumors harvested from mice were weighted and homogenized. Fluorescent intensity of the homogenate was measured using the Syngene PXi imaging system and quantified by Image-J [56].
In vivo luciferase silencing
To evaluate in vivo silencing efficacy, female athymic nude mice bearing Luc-HeLa tumor were administered daily by IV injection of free siLuc or NP (siLuc) at a dose of 1.2 mg siRNA per kg of animal weight, for three consecutive days. Two days after the final injection, the animals were anesthetized and injected intraperitoneally with of 2 mg of D-Luciferin per mouse. Five minutes later, the animals were imaged for the bioluminescence using the Syngene PXi imaging system. For quantification analysis, the animals were sacrificed, and then the protein extracts of tumor tissue were prepared using lysis buffer. The luciferase level was measured using the Steady-Glo luciferase assay kit, and the total protein was quantified using BCA assay.
Statistical analysis
Data are presented as mean ± S.D., unless otherwise stated. Statistical analysis was performed using two-tailed student's t test. A p-value < 0.05 is considered statistically significant.
Supplementary Material
Supplementary figures.
Abbreviations
siRNA: small interfering RNA
NP: nanoparticle
PEG: poly(ethylene glycol)
PK: pharmacokinetics
MPS: mononuclear phagocyte system
EPR: enhanced permeability and retention
TME: tumor microenvironment
MMP: matrix metalloproteinase
PLGA: poly(lactide-co-glycolide)
DMF: dimethylformamide
DMSO: dimethylsulfoxide
THF: tetrahydrofuran
DLS: dynamic light scattering
siLuc: luciferase siRNA
PBS: phosphate buffered saline
FBS: fetal bovine serum
DMEM: Dulbecco's Modified Eagle's Medium
kd: dissociation rate constant
t1/2: half-life
Lipo2000: lipofectamine 2000
DMPE: 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamine
DPPE: 1,2-Dipalmitoryl-sn-glycero-3-phosphoethanolamine
DSPE: 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine
DOPE: 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine
DMG: 1,2-Dimyristoyl-sn-glycerol, methoxypolyethylene glycol
DPG: 1,2-Dipalmitoyl-sn-glycerol, methoxypolyethylene glycol
DSG: 1,2-Distearoyl-sn-glycerol, methoxypolyethylene glycol
PAMAM: Ethylenediamine core-poly (amidoamine)
G0-C14: Epoxytetradecane-modified PAMAM generation 0 dendrimer
TEM: Transmission Electron Microscopy
Acknowledgements
This work was in part supported by the DoD grant (W81XWH-15-1-0728), the NIH grants (R00CA160350 and CA200900), the Movember-Prostate Cancer Foundation (PCF) Challenge Award, the David Koch-PCF program in nanotherapeutics, the J. Eustace Wolfington-PCF Young Investigator Award, the Shanghai Sailing Program (16YF1411300), and the Technology Development Grant of Pudong New Area (PKF2014-C01).
Competing Interests
O.C.F. has financial interests in Selecta Biosciences, Tarveda Therapeutics and Placon Therapeutics. Other authors declare no competing financial interests.
References
1. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49:6288-308
2. Guo X, Huang L. Recent advances in nonviral vectors for gene delivery. Acc Chem Res. 2012;45:971-9
3. Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134:2139-47
4. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
5. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliver Rev. 2014;66:2-25
6. Tao W, Zhang J, Zeng X, Liu D, Liu G, Zhu X. et al. Blended nanoparticle system based on miscible structurally similar polymers: a safe, simple, targeted, and surprisingly high efficiency vehicle for cancer therapy. Adv Healthc Mater. 2015;4:1203-14
7. Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83:761-9
8. Shi J, Xiao Z, Kamaly N, Farokhzad OC. Self-assembled targeted nanoparticles: evolution of technologies and bench to bedside translation. Acc Chem Res. 2011;44:1123-34
9. Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41:2971-3010
10. Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178-81
11. Romberg B, Hennink WE, Storm G. Sheddable coatings for long-circulating nanoparticles. Pharm Res. 2008;25:55-71
12. Remaut K, Lucas B, Braeckmans K, Demeester J, De Smedt SC. Pegylation of liposomes favours the endosomal degradation of the delivered phosphodiester oligonucleotides. J Control Release. 2007;117:256-66
13. Peeters L, Sanders NN, Jones A, Demeester J, De Smedt SC. Post-pegylated lipoplexes are promising vehicles for gene delivery in RPE cells. J Control Release. 2007;121:208-17
14. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129-38
15. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
16. Islam MA, Reesor EK, Xu Y, Zope HR, Zetter BR, Shi J. Biomaterials for mRNA delivery. Biomater Sci. 2015;3:1519-33
17. Zhang Y, Satterlee A, Huang L. In vivo gene delivery by nonviral vectors: overcoming hurdles? Mol Ther. 2012;20:1298-304
18. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12:991-1003
19. Li W, Huang Z, MacKay JA, Grube S, Szoka FC Jr. Low-pH-sensitive poly(ethylene glycol) (PEG)-stabilized plasmid nanolipoparticles: effects of PEG chain length, lipid composition and assembly conditions on gene delivery. J Gene Med. 2005;7:67-79
20. Shin J, Shum P, Thompson DH. Acid-triggered release via dePEGylation of DOPE liposomes containing acid-labile vinyl ether PEG-lipids. J Control Release. 2003;91:187-200
21. Walker GF, Fella C, Pelisek J, Fahrmeir J, Boeckle S, Ogris M. et al. Toward synthetic viruses: endosomal pH-triggered deshielding of targeted polyplexes greatly enhances gene transfer in vitro and in vivo. Mol Ther. 2005;11:418-25
22. Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264-73
23. Niu M, Naguib YW, Aldayel AM, Shi YC, Hursting SD, Hersh MA. et al. Biodistribution and in vivo activities of tumor-associated macrophage-targeting nanoparticles incorporated with doxorubicin. Mol Pharm. 2014;11:4425-36
24. Hatakeyama H, Akita H, Kogure K, Oishi M, Nagasaki Y, Kihira Y. et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14:68-77
25. Hatakeyama H, Akita H, Ito E, Hayashi Y, Oishi M, Nagasaki Y. et al. Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials. 2011;32:4306-16
26. Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS nano. 2012;6:3491-8
27. Zou Z, He X, He D, Wang K, Qing Z, Yang X. et al. Programmed packaging of mesoporous silica nanocarriers for matrix metalloprotease 2-triggered tumor targeting and release. Biomaterials. 2015;58:35-45
28. Takae S, Miyata K, Oba M, Ishii T, Nishiyama N, Itaka K. et al. PEG-detachable polyplex micelles based on disulfide-linked block catiomers as bioresponsive nonviral gene vectors. J Am Chem Soc. 2008;130:6001-9
29. Wang K, Liu Y, Yi W-J, Li C, Li Y-Y, Zhuo R-X. et al. Novel shell-cross-linked micelles with detachable PEG corona for glutathione-mediated intracellular drug delivery. Soft Matter. 2013;9:692-9
30. Cerritelli S, Velluto D, Hubbell JA. PEG-SS-PPS: reduction-sensitive disulfide block copolymer vesicles for intracellular drug delivery. Biomacromolecules. 2007;8:1966-72
31. Gao W, Langer R, Farokhzad OC. Poly (ethylene glycol) with observable shedding. Angew Chem Int Ed Engl. 2010;49:6567-71
32. Suma T, Miyata K, Anraku Y, Watanabe S, Christie RJ, Takemoto H. et al. Smart multilayered assembly for biocompatible siRNA delivery featuring dissolvable silica, endosome-disrupting polycation, and detachable PEG. ACS nano. 2012;6:6693-705
33. Salzano G, Costa DF, Sarisozen C, Luther E, Mattheolabakis G, Dhargalkar PP. et al. Mixed Nanosized Polymeric Micelles as Promoter of Doxorubicin and miRNA-34a Co-Delivery Triggered by Dual Stimuli in Tumor Tissue. Small. 2016:10 1002/smll.201600925
34. Yang X-Z, Du J-Z, Dou S, Mao C-Q, Long H-Y, Wang J. Sheddable Ternary Nanoparticles for Tumor Acidity-Targeted siRNA Delivery. ACS Nano. 2012;6:771-81
35. Wang H-X, Yang X-Z, Sun C-Y, Mao C-Q, Zhu Y-H, Wang J. Matrix metalloproteinase 2-responsive micelle for siRNA delivery. Biomaterials. 2014;35:7622-34
36. Gujrati M, Vaidya AM, Mack M, Snyder D, Malamas A, Lu ZR. Targeted Dual pH-Sensitive Lipid ECO/siRNA Self-Assembly Nanoparticles Facilitate In Vivo Cytosolic sieIF4E Delivery and Overcome Paclitaxel Resistance in Breast Cancer Therapy. Adv Healthc Mater. 2016;5:2882-95
37. Zhu X, Xu Y, Solis LM, Tao W, Wang L, Behrens C. et al. Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc Natl Acad Sci USA. 2015;112:7779-84
38. van der Vusse GJ. Albumin as Fatty Acid Transporter. Drug Metabolism. Pharmacokinetics. 2009;24:300-7
39. Liu Z, Chen X. Simple bioconjugate chemistry serves great clinical advances: albumin as a versatile platform for diagnosis and precision therapy. Chem Soc Rev. 2016;45:1432-56
40. Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519-22
41. Chan JM, Zhang L, Yuet KP, Liao G, Rhee J-W, Langer R. et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30:1627-34
42. Stylianopoulos T. EPR-effect: utilizing size-dependent nanoparticle delivery to solid tumors. Ther Deliv. 2013;4:421-3
43. Simone E, Dziubla T, Shuvaev V, Muzykantov VR. Synthesis and characterization of polymer nanocarriers for the targeted delivery of therapeutic enzymes. Free Radicals and Antioxidant Protocols. 2010:145-64
44. Cheng TL, Chuang KH, Chen BM, Roffler SR. Analytical measurement of PEGylated molecules. Bioconjug Chem. 2012;23:881-99
45. Chene CJ, Saltzman WM. Enhanced siRNA delivery into cells by exploiting the synergy between targeting ligands and cell-penetrating peptides. Biomaterials. 2011;32:6194-203
46. Koynova R, Caffrey M. Phases and phase transitions of the phosphatidylcholines. Biochim Biophys Acta. 1998;1376:91-145
47. Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505-15
48. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
49. Yu M, Zheng J. Clearance Pathways and Tumor Targeting of Imaging Nanoparticles. ACS Nano. 2015;9:6655-74
50. Zeng X, Liu G, Tao W, Ma Y, Zhang X, He F. et al. A Drug-Self-Gated Mesoporous Antitumor Nanoplatform Based on pH-Sensitive Dynamic Covalent Bond. Adv Funct Mater. 2017:10 1002/adfm.201605985
51. Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T. et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39
52. Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS. et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci USA. 2013;110:18638-43
53. Krishnamurthy S, Vaiyapuri R, Zhang L, Chan JM. Lipid-coated polymeric nanoparticles for cancer drug delivery. Biomater Sci. 2015;3:923-36
54. Creixell M, Peppas NA. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today. 2012;7:367-79
55. Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR. et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci USA. 2010;107:1864-9
56. Tao W, Zeng X, Wu J, Zhu X, Yu X, Zhang X. et al. Polydopamine-Based Surface Modification of Novel Nanoparticle-Aptamer Bioconjugates for In Vivo Breast Cancer Targeting and Enhanced Therapeutic Effects. Theranostics. 2016;6:470-84
Author contact
![]() Corresponding authors: Email: ofarokhzadharvard.edu (O.C.F.), Email: jshiharvard.edu (J.S.).
Corresponding authors: Email: ofarokhzadharvard.edu (O.C.F.), Email: jshiharvard.edu (J.S.).