Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(7):1890-1900. doi:10.7150/thno.19135 This issue Cite
Research Paper
Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia
Lifeng Feng1*, Jiaqiu Li2*, Lixian Yang1, Libo Zhu3, Xiufeng Huang3, Shuzheng Zhang4, Likang Luo5, Zhinong Jiang6, Tingting Jiang1, Wenxia Xu1, Xian Wang2, Hongchuan Jin1 ![]()
1. Laboratory of Cancer Biology, Key Lab of Biotherapy in Zhejiang, Sir Run Run Shaw Hospital, Medical school of Zhejiang University;
2. Department of Medical Oncology, Sir Run Run Shaw Hospital, Medical school of Zhejiang University;
3. Department of Gynecology, Women's Hospital, Medical school of Zhejiang University;
4. Department of Gynecology, Xiaoshan Women's Hospital, Hangzhou;
5. Department of Pathology, Xiaoshan Women's Hospital, Hangzhou;
6. Department of Pathology, Sir Run Run Shaw Hospital, Medical school of Zhejiang University.
* Equal contribution
Received 2017-1-10; Accepted 2017-2-20; Published 2017-4-10
Citation:
Feng L, Li J, Yang L, Zhu L, Huang X, Zhang S, Luo L, Jiang Z, Jiang T, Xu W, Wang X, Jin H. Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics 2017; 7(7):1890-1900. doi:10.7150/thno.19135. https://www.thno.org/v07p1890.htm
Other stylesAbstract

Long-term application of Tamoxifen (TAM) is usually recommended for hormone receptor positive breast cancer patients. Unfortunately, TAM will inevitably increase the incidence of endometrial hyperplasia even endometrial cancer. Despite of substantial investigations, no effective approaches to prevent TAM-induced endometrial carcinogenesis have been acknowledged. In this study, we found that inhibition of Nrf2 could be valuable to prevent TAM-induced endometrial hyperplasia. Upon TAM treatment, the mRNA and protein expression of autophagy adaptor SQSTM1 was specifically increased in endometrial cells but not breast cancer cells. Knocking-down of SQSTM1 expression retarded TAM-promoted growth of endometrial cancer cells. TAM stimulated SQSTM1 transcription specifically in endometrial cells by enhancing phosphorylation and nuclear translocation of Nrf2. Indeed, the expression of Nrf2 and SQSTM1 were positively correlated in primary endometrial tissues. In rats with TAM-induced endometrial hyperplasia, both Nrf2 and SQSTM1 expression were increased. Nrf2 inhibitor brusatol effectively attenuated TAM-induced SQSTM1 upregulation and endometrial hyperplasia. The kinase of Nrf2, PRKCD, was activated by TAM. Once PRKCD was depleted, TAM failed to promote Nrf2 phosphorylation and SQSTM1 expression. In summary, TAM stimulated Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia by activating PRKCD. Therefore, blocking PRKCD-Nrf2-SQSTM1 signaling could be useful to prevent TAM-induced endometrial hyperplasia.
Introduction
As the first generation of selective estrogen receptor modulators (SERMs), tamoxifen (TAM) is a triphenylethylene derivative widely used for the treatment of HR (Hormone receptor) positive breast cancer [1, 2]. Though new generations of SERMs such as toremifen, raloxifene and anastrozole have been developed, TAM is still the dominant endocrine therapy drug recommended for the long-term management of premenopausal breast cancer patients [3, 4]. However, the long-term use of TAM will increase the incidence of endometrial hyperplasia, even endometrial cancer [2, 5, 6]. Previous reports revealed that TAM could act as weak estrogen in uterus to activate the transcription of ER (estrogen receptor) target genes, such as PAX2 [7], NR5A [8] and Syncytin-1 [9]. However, effective clinical approaches to prevent TAM-induced endometrial carcinogenesis are still lacking.
SQSTM1/p62 (Sequestosome 1), is an autophagy-related adaptor protein to mediate the degradation of ubiquitinated cargo, via its LC3 interacting region (LIR) and C terminal ubiquitin association (UBA) domain [10-12]. In addition, it can interact with KEAP1 (Kelch-like ECH-Associated protein 1) to disrupt Keap1-Nrf2 interaction, thus stabilizing Nrf2 (nuclear factor erythroid 2-related factor 2, NFE2L2) to trigger the antioxidant response [13, 14]. SQSTM1 can also interact with many other signaling molecules such as TRAF-6 (TNF receptor-associated factor 6) [15, 16] and Raptor [17, 18], leading to the activation of NFκB and mTORC1 (mammalian target of rapamycin complex 1) pathway, respectively. As a proof-of-principle, SQSTM1 was highly expressed to promote the pathogenesis of many tumors. For example, autophagy deficiency promoted tumorigenesis by activating NFκB and Nrf2 signaling in a SQSTM1 dependent manner [19-21]. SQSTM1 expression can also be induced by oncogenic Ras signaling to sustain active NFκB signaling in lung adenocarcinoma and pancreatic ductal adenocarcinoma [22, 23]. Yin Yang 1 (YY1) inhibited miR372 transcription to elevate SQSTM1 expression in breast cancer [24]. Moreover, SQSTM1 can be activated by phosphorylation at serine 351 in hepatocellular carcinoma [25]. However, it remains unknown whether SQSTM1 is relevant to TAM-induced endometrial carcinogenesis.
Transcription factor Nrf2 promotes the transcription of its target genes to regulate the antioxidant response through binding to the antioxidant response element (ARE) [26, 27]. Meanwhile, activation of Nrf2 signaling was relevant to cancer development [26, 28, 29]. For example, oncogenic Ras signaling promoted drug resistance by increasing Nrf2 transcription [30]. In addition, phosphorylation of Nrf2 at serine 40 by protein kinase C delta (PRKCD) promoted the disassociation of Nrf2 from KEAP1 and facilitated the nuclear translocation of Nrf2 [31]. On the other hand, serine 568 phosphorylation of Nrf2 by Fyn promoted its nuclear export and subsequent degradation [32].
In this study, we found that TAM stimulated Nrf2-dependent SQSTM1 transcription to promote cellular growth specifically in endometrial but not breast cancer cells. Upon TAM treatment, Nrf2 translocated to nucleus to activate SQSTM1 transcription, as a consequence of PRKCD-mediated phosphorylation. Inhibition of Nrf2 significantly attenuated TAM-induced endometrial hyperplasia, indicating that Nrf2 inhibitor could be valuable for the prevention of TAM-induced endometrial carcinogenesis.
Materials and Methods
Antibodies, Plasmids and Chemicals
Cells were cultured in DMEM or RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum at 37℃ with 5% CO2 and 95% humidity. Anti-SQSTM1/p62 antibody and SQSTM1/p62 plasmid were used as previous reported [24]. Anti-Nrf2 antibody was purchased from Proteintech (Wuhan, China), anti-Nrf2 (phospho S40) antibody and anti-Histone H3 were purchased from Abcam (Shanghai, China), anti-GLS antibody was purchased from Abgent (Suzhou, China), anti-PKC delta antibody was purchased from BD biosciences (Shanghai, China), anti-phospho PKC delta (Thr505) and anti-Actin antibody were purchased from Cell Signaling Technology (Shanghai, China). TAM and 4-OH-Tamoxifen (4-OH-TAM) were purchased from Sigma Aldrich (Shanghai, China). Brusatol was purchased from Tauto Biotech (Shanghai, China).
Plasmids and siRNA Transfection
For plasmid transfection, cells were seeded overnight in 6 or 12 well plates and 1-2 μg of plasmids were transfected with X-tremeGENE HP DNA Transfection Reagent (Roche Applied Science, Shanghai, China). Cells were harvested for RNA and protein extraction after 48-72 hours of transfection. For siRNA transfection, cells were seeded overnight in 6 or 12 well plates and transfected using LipofectamineTM RNAiMAX transfection reagent (Thermofisher Scientific) according to the manufacturer's instructions. After 24h transfection, 4-OH-TAM or inhibitors were added for another 24 or 48 hours. All siRNAs were synthesized by Genepharma (Shanghai, China) and the detail sequences were listed in Supplementary Table 1.
Cell growth assay
The CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay kit (Promega, Beijing, China) was applied to measure cell viability. Briefly, 72 hours after 4-OH-TAM treatment, the MTS reagents were added to each well, and cell viability was measured following the manufacturer's instruction.
Western Blotting
Cell lysates harvested were quantitated by BCA protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Lysates were resolved by SDS-PAGE, transferred to PVDF membranes and probed with the indicated primary antibodies, then washed with TBS-T (TBS with 0.1% of Tween-20) and incubated with suitable HRP-conjugated secondary antibodies (Dako, Glostrup, Denmark). The membranes were visualized with enhanced chemiluminescence (EMD Millipore, Billerica, MA, USA) in Amersham Imager 600 system (GE Healthcare Life Sciences, Shanghai, China).
RNA extraction and Quantitative real-time RT-PCR
Total RNA was extracted with Trizol reagent (Invitrogen) according to the manufacturer's instructions. RNA concentrations were quantified by NanoDrop 2000 (Nanodrop, Wilmington, Del., USA). The High Capacity cDNA Reverse Transcription Kit (Thermofisher Scientific Inc., Shanghai, China) was used for reverse transcription according to the manual provided. Briefly, 2μg total RNA was used for each reaction. The mRNA expression level was determined by quantitative real-time PCR using SYBR Green Master Mix Kit and Light Cycler 480 II system (Roche, Shanghai, China). Beta-actin was used as an internal control of RNA integrity. All primers used were listed in Supplementary Table 2.
Nuclear and cytoplasmic protein extraction
The Thermo Scientific™ NE-PER™ Nuclear and Cytoplasmic Extraction Reagent was applied for the extraction of nuclear and cytoplasmic proteins. 5-8×105/well of cells were seeded in 6-well plates overnight, and incubated with 4-OH-TAM for various times as indicated. Cells were then collected by trypsin digestion, washed with chilled PBS. The extraction reagents were added to the pelleted cells step by step. The cytoplasmic and nuclear proteins were collected separately, following the manual provided.
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) was performed with the SimpleChIP™ Enzymatic Chromatin IP Kit (#9003, Cell Signaling Technology, Boston, MA, USA) according to the manufacturer's instructions. Briefly, cells were first fixed with 4% formaldehyde for crosslinking of protein and chromatins were harvested and fragmented using sonication. The chromatins were immunoprecipitated with antibody specific to Nrf2. After immunoprecipitation, the protein-DNA crosslinks were reversed and genomic DNAs were purified. The enriched DNA fragments were detected by quantitative real-time PCR with a pair of primers around the Nrf2 binding sites of SQSTM1 gene. The primers used were list in Supplementary Table 2.
Immunohistochemistry (IHC) staining
Formalin-fixed and paraffin-embedded human endometrial tissue sections were immunostained with anti-SQSTM1 or anti-Nrf2 antibodies, using microwave antigen retrieval in 0.01M pH6.0 citrate buffer. After washing, signal was detected using suitable HRP labeled secondary antibody with DAB as the chromagen (Dako, Denmark).
Animal Studies
Female SD rats about 8 weeks old were purchased from Slac Laboratory Animal Inc. (Shanghai China) and maintained under specific pathogen-free conditions. For ovariectomy, rats were anesthetized with 8% chloral hydrate (0.5ml/100g). The ovaries were ligatured from uterus with hemostat and surgical suture, and removed by cutting through tissues on the rostral side of each hemostat. The ovariectomized rats were randomly divided into three groups, DMSO group, DMSO+TAM group and TAM+Bru group. In DMSO group, mice were intraperitoneal injected with DMSO as the control. In DMSO+TAM group, mice were intraperitoneal injected with TAM (10mg/kg) in DMSO. In TAM+Bru group, mice were intraperitoneal injected with TAM plus brusatol (1mg/kg) twice per week. Forty days later, the rats were anesthetized with chloral hydrate and the uterus were collected for RNA and protein extraction, or fixed with 4% formaldehyde immediately for HE staining. The thickness of endometrium was measured from 3 random sites of each sample after HE staining.
Luciferase activity assay
SQSTM1/p62 promoter (p62-pro) fragment (from -2046 to +127) with Nrf2 binding site (from -1258 to -1245) was cloned by PCR. Primers used were included in Supplementary Table 2. PCR product was double digested with NheI/HindIII and inserted into pGL2 vector (Promega, Shanghai, China). Cells (1×105) seeded in 12-well plates were co-transfected with Flag-Nrf2 or its mutants, various luciferase reporters and pRL-TK Renilla control luciferase reporter (Promega). Forty-eight hours after transfection, the activities of firefly luciferase and renilla luciferase were measured using the Dual-GloTM luciferase assay system (Promega). Relative luciferase activity was normalized with renilla luciferase activity.
Statistical analysis
Unless specified, the Student's t tests were performed to analyze statistical difference. P values less than 0.05 were considered statistically significant.
Results
TAM promoted endometrial cell growth via upregulating SQSTM1 expression
4-OH-tamoxifen (4-OH-TAM, the active form of TAM) can effectively inhibit the growth of breast cancer MCF7 cells. However, it failed to inhibit even moderately promoted the growth of two independent endometrial cancer cell lines RL95-2 and AN3CA (Supplementary Figure 1). Interestingly, both 4-OH-TAM and TAM increased SQSTM1 protein level in a dose- or time-dependent manner, in both RL95-2 and AN3CA but not MCF7 cells (Figure 1A and B, Supplementary Figure 2A and B). High concentration (20µM) of 4-OH-TAM significantly increased SQSTM1 expression and caused no growth inhibition of either RL95-2 or AN3CA cells. However, 4-OH-TAM killed most of MCF cells at this concentration. Importantly, TAM-promoted growth of RL95-2 and AN3CA cells were attenuated upon SQSTM1 knock-down (Figure 1C). In contrast, TAM induced growth inhibition in MCF7 cells regardless of SQSTM1 expression (Figure 1C).
Figure 1
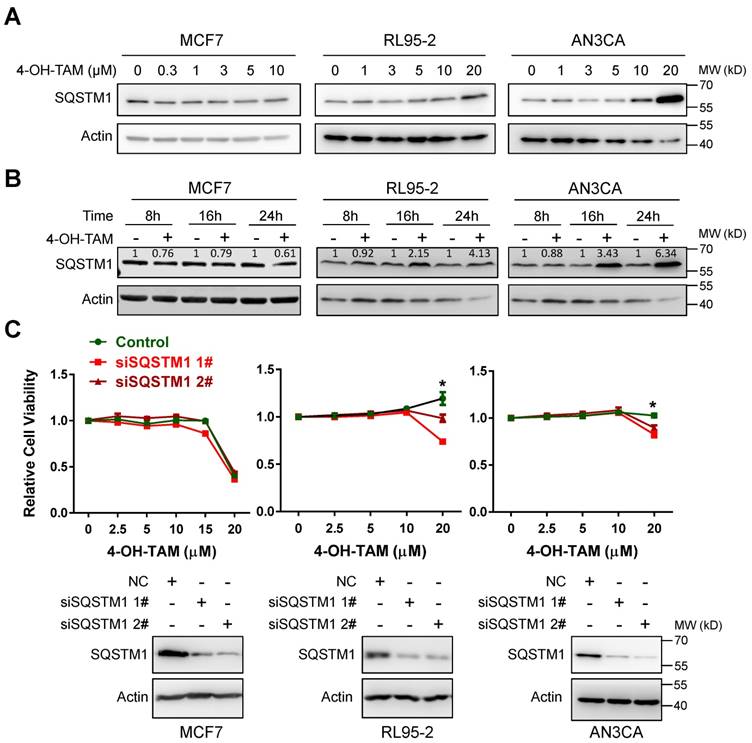
TAM promotes endometrial cell growth via upregulating SQSTM1 expression. A, MCF7, RL95-2 or AN3CA cells were incubated with various concentrations of 4-OH-TAM for 24 hours. SQSTM1 expression was detected by western blotting. Actin was used as the loading control. B, MCF7, RL95-2 or AN3CA cells were incubated with 4-OH-TAM (15 μM) for 8, 16 or 24 hours. SQSTM1 expression was detected by western blotting. Actin was used as the loading control. Relative SQSTM1 expression (SQSTM1/actin) was quantified and normalized to the relative SQSTM1 expression in the control. C, Various concentrations of 4-OH-TAM were added to MCF7, RL95-2 or AN3CA cells 24 hours after transfection with SQSTM1 siRNAs. MTS assay was performed 72 hours after transfection. The experiments were performed in triplicate and repeated three times. Representative results from one triplicated experiment were shown as Mean±SD. Student's T test was used for the statistical analysis. Asterisks indicate statistical significance. SQSTM1 knockdown was confirmed by Western blotting as in A and B.
TAM activated SQSTM1 transcription in a Nrf2-dependent manner
Next, we wonder how TAM increased SQSTM1 expression in endometrial cancer cells. Interestingly, both 4-OH-TAM and TAM increased SQSTM1 mRNA level in endometrial cancer cells but not breast cancer cells (Figure 2A, Supplemental Figure 3A, B and C). There is a canonical antioxidant response element (ARE) in SQSTM1 promoter region and Nrf2 could bind this ARE to activate SQSTM1 transcription under oxidative stress [33]. Therefore, we assumed that Nrf2 might be relevant to TAM-induced increase of SQSTM1 mRNA. Indeed, the interaction of Nrf2 with SQSTM1 promoter was enhanced upon TAM treatment (Figure 2B). Moreover, Nrf2 significantly increased the activity of luciferase driven by SQSTM1 promoter (Figure 2C), indicating that TAM might activate SQSTM1 transcription by Nrf2. In support of this, genetic knock-down of Nrf2 expression impaired TAM-promoted SQSTM1 expression and endometrial cell growth (Figure 2D, E and F). Consistently, chemical inhibitor of Nrf2, Brusatol, notably attenuated SQSTM1 expression and endometrial cell growth promoted by TAM (Figure 2G, H and I, Supplemental Figure 4A and B). Taken together, these data suggested that Nrf2 was implicated in TAM-induced SQSTM1 expression and endometrial cell growth.
Figure 2
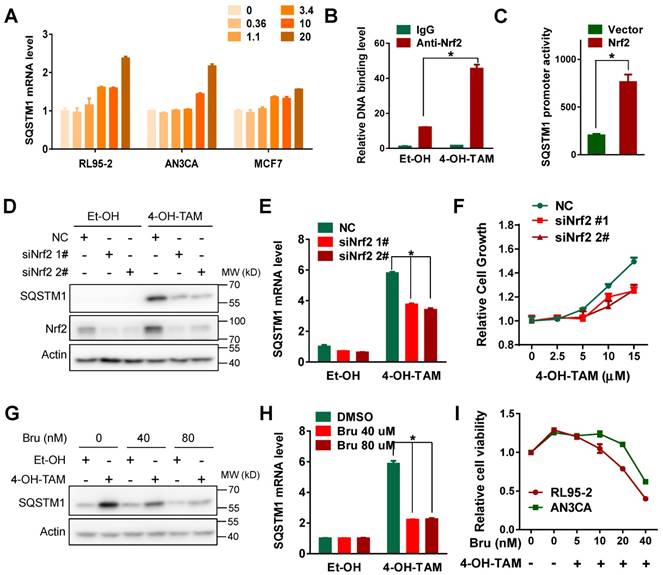
TAM activated SQSTM1 transcription in a Nrf2-dependent manner. A, SQSTM1 mRNA levels in RL95-2 (left), AN3CA (middle) or MCF7 (right) cells incubated with different concentrations of 4-OH-TAM for 24 hours were measured by qRT-PCR. B, ChIP-PCR experiment was performed to detect the binding of Nrf2 to SQSTM1 promoter in AN3CA cells before and after 4 hours of 4-OH-TAM (20μM) treatment. IgG was used as the negative control. The experiments were performed in triplicate and repeated three times. Representative results from one triplicated experiment were shown as Mean±SD. Student's T test was used for the statistical analysis. The Asterisk indicates the statistical significance. C, p62-pro/pGL2 or pGL2 vector were co-transfected with Nrf2 plasmid or vector in AN3CA cells. Forty-eight hours later, SQSTM1 promoter activity was measured by dual luciferase reporter assay. The experiments were repeated three times. Representative results from one triplicated experiment were shown as Mean±SD. Student's T test was used for the statistical analysis. The Asterisk indicates the statistical significance. D, Forty-eight hours after transfection of Nrf2 siRNAs, 4-OH-TAM (15 μM) was added for another 24 hours, and the effect of 4-OH-TAM on SQSTM1 and Nrf2 protein expression in AN3CA cells transfected with or without Nrf2 siRNAs were analyzed by Western blotting. E, Effects of 4-OH-TAM (as in D) on SQSTM1 mRNA expression in AN3CA cells transfected with or without Nrf2 siRNAs were analyzed by qRT-PCR. The experiments were repeated three times. Representative results from one triplicated experiment were shown as Mean±SD. Student's T test was used for the statistical analysis. The Asterisk indicates the statistical significance. F, Effects of 4-OH-TAM on growth of AN3CA cells transfected with or without Nrf2 knockdown were analyzed by MTS assay. Forty-eight hours after transfection of Nrf2 siRNAs, various concentrations of 4-OH-TAM were added for another 72 hours. Two way ANOVA test was used to compare the statistical difference (NC vs siNrf1 #1, p<0.01; NC vs siNrf1 #1, p<0.01). The experiments were repeated three time. Representative results from one triplicated experiment were shown as Mean±SD. G, Nrf2 inhibitor brusatol (Bru) (40 or 80 nM) was added together with 4-OH-TAM (15 μM) for 24 hours in AN3CA cells. SQSTM1 expression was determined by western blotting. H, SQSTM1 mRNA expression in AN3CA cells treated with brusatol (Bru) and 4-OH-TAM incubation (as in G) were analyzed by qRT-PCR. The results were shown as in E. I, The growth of AN3CA cells treated with various concentrations of brusatol (Bru) and 4-OH-TAM (15 μM) incubation were explored by MTS assay. The experiments were repeated three times. Representative results from one triplicated experiment were shown as Mean±SD.
TAM activated Nrf2 specifically in endometrial cells
However, SQSTM1 could stabilize Nrf2 protein level by promoting its dissociation from E3 ubiquitin ligase KEAP1 [13, 14]. Therefore, Nrf2 stabilization and subsequent nuclear translocation might be the consequence of increased SQSTM1 expression, thus forming a positive feedback loop to sustain Nrf2 activity. Indeed, both TAM and 4-OH-TAM significantly increased Nrf2 protein expression and nuclear translocation in endometrial cancer cells but not breast cancer cells (Figure 3A and B, Supplementary Figure 5A). The mRNA level of NQO1, a well-known Nrf2 target gene, was increased by 4-OH-TAM in endometrial cancer cells but not breast cancer cells (Figure 3C), confirming the specific activation of Nrf2 by 4-OH-TAM in endometrial cancer cells. However, knock-down of SQSTM1 expression had negligible effects on 4-OH-TAM- or TAM-induced elevation of Nrf2 expression and nuclear translocation (Figure 3D and Supplemental Figure 5B, C). Overexpression of SQSTM1 also failed to alter Nrf2 expression and nuclear translocation (Figure 3E and F). Altogether, these results indicated that TAM activated Nrf2 independent of SQSTM1.
Expression of Nrf2 and SQSTM1 in endometrial tissues
Next, we evaluated the expression of Nrf2 and SQSTM1 in primary endometrial tissues to further verify the clinical relevance of Nrf2-SQSTM1 axis. A total of 55 clinical samples including 27 endometrial hyperplasia and 28 normal endometrial tissues. Among 27 cases with endometrial hyperplasia, 18 cases were breast cancer patients received TAM treatment. Both Nrf2 and SQSTM1 were significantly upregulated in endometrial hyperplasia tissues when compared with their expressions in normal uterus (Figure 4A and B, chi-square test, p<0.01). Moreover, Nrf2 and SQSTM1 were significantly upregulated in endometrial tissues from patients with TAM treatment when compared with their expressions in endometrial tissues from patients without TAM treatment (Figure 4C, chi-square test, p<0.01). Importantly, the expression of Nrf2 was significantly correlated with SQSTM1 expression (Figure 4D, chi-square test, p<0.01), further implying the relevance of Nrf2-SQSTM1 axis to the pathogenesis of TAM-induced endometrial hyperplasia.
Figure 3
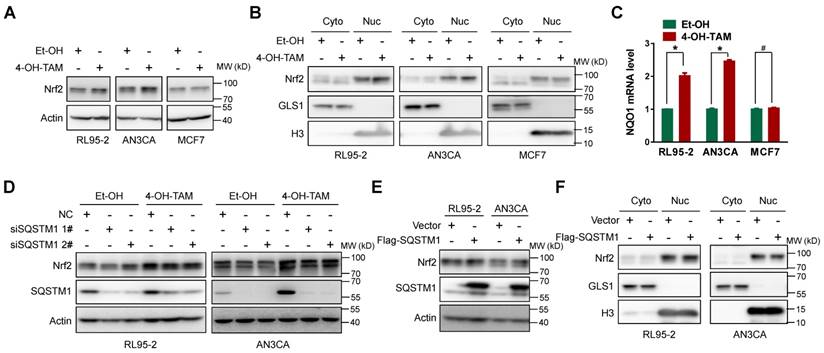
TAM activated Nrf2 specifically in endometrial cells. A, Nrf2 expression in RL95-2, AN3CA or MCF7 cells incubated with or without 4-OH-TAM (15 μM) for 24 hours were analyzed by western blotting. B, RL95-2, AN3CA or MCF7 cells were incubated with or without 4-OH-TAM (15 μM) for 24 hours. Nrf2 distributions in nuclear and cytoplasmic extracts were explored by western blotting. GLS1 was used as the cytoplasmic protein marker while H3 (Histone H3) was used as the nuclear protein marker. C, NQO1 mRNA level in RL95-2, AN3CA or MCF7 cells incubated with or without 4-OH-TAM (15 μM) for 24 hours were analyzed with qRT-PCR. The experiments were repeated three times. Representative results from one triplicated experiment were shown as Mean±SD. Student's T test was used for the statistical analysis. The asterisk indicates the statistical significance and the octothorpe stands for no significant difference. D, Forty-eight hours after transfection of SQSTM1 siRNAs, 4-OH-TAM (15 μM) was added for another 24 hours, the effect of 4-OH-TAM on Nrf2 expression in RL95-2 and AN3CA cells with or without SQSTM1 knockdown were explored by western blotting. E, Forty-eight hours after transfection of Flag-SQSTM1/Vector, Nrf2 expression in RL95-2 (left) or AN3CA (right) cells were detected by western blotting. F, Nrf2 distribution in nuclear and cytoplasmic extracts of RL95-2 (left) or AN3CA (right) cells with or without Flag-SQSTM1 over-expression were detected by western blotting.
Figure 4
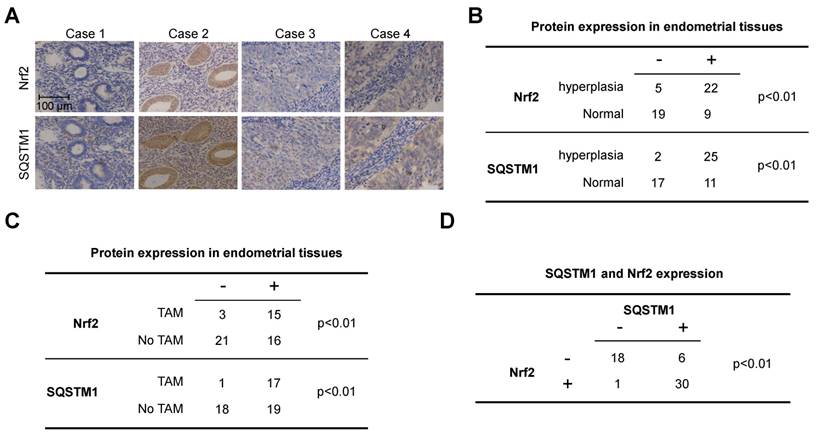
Expression of Nrf2 and SQSTM1 in clinical endometrial tissues. A, Represented images of Nrf2 or SQSTM1 expression in primary endometrial tissues. B, Summary of Nrf2 and SQSTM1 expression in normal and hyperplasia endometrial tissues. C, Summary of Nrf2 and SQSTM1 expression in endometrial tissues from cases received TAM treatment or not. D, Correlation of SQSTM1 and Nrf2 expression. Chi-Square test was used for the statistical analysis of staining results.
TAM activated Nrf2-SQSTM1 axis to promote endometrial hyperplasia in rats
To verify the relevance of Nrf2-SQSTM1 axis to TAM-induced endometrial hyperplasia, we established the endometrial hyperplasia animal model in rats treated with TAM (Figure 5A) [7]. After 7 weeks treatment, endometrial hyperplasia was successfully induced (Figure 5B and C, p<0.01). Meanwhile, both SQSTM1 mRNA and protein levels were increased after TAM treatment (Figure 5D and E). Interestingly, endometrial hyperplasia was greatly reversed upon the application of Nrf2 inhibitor brusatol (Figure 5B and C, p<0.01), accompanied by the significant reduction of SQSTM1 expression (Figure 5D and E).
TAM activated Nrf2 through phosphorylation at serine 40
Next, we tried to explore the mechanism underlying Nrf2 activation by TAM. 4-OH-TAM could promote Nrf2 nuclear translocation within 2 hours (Figure 6A). Meanwhile, there were little changes in the total expression level of Nrf2 at this time point (data not shown). Therefore, the increase of Nrf2 protein level was most likely the consequence rather than the cause of enhanced Nrf2 nuclear translocation. It has been reported that phosphorylation of Nrf2 at serine 40 (S40) could facilitate the translocation of Nrf2 to nucleus [31]. Indeed, S40 phosphorylation of Nrf2 was increased shortly after 1 hour of 4-OH-TAM incubation (Figure 6B). In addition, S40 phosphorylated Nrf2 in the nucleus was increased after 4-OH-TAM treatment in endometrial cancer cells (Figure 6C). Importantly, mutation of serine 40 into phosphorylation-defective alanine in Nrf2 (Nrf2-S40A) reduced its transcriptional activity and lost its ability to increase SQSTM1 expression (Figure 5D and E). In contrast, both wild type (Nrf2-WT) and phosphorylation-mimicking Nrf2 (Nrf2-S40E) retained their transcription activity to promote SQSTM1 expression (Figure 5D and E). Furthermore, TAM failed to increase the nuclear localization of Nrf2-S40A (Figure 5F), highlighting the importance of S40 phosphorylation to TAM-induced Nrf2 nuclear translocation. In conclusion, TAM activated Nrf2 by promoting its phosphorylation at serine 40.
PRKCD was involved in TAM-induced Nrf2 S40 phosphorylation
Previous reports demonstrated that oxidant stress could activate PRKCD (protein kinase C delta) to phosphorylate Nrf2 at serine 40 [31, 34, 35]. We, thus, explored the relevance of PRKCD to TAM-induced Nrf2 activation. As shown in Figure 7A, 4-OH-TAM indeed activated PRKCD in both RL95-2 and AN3CA cells. Moreover, 4-OH-TAM failed to stimulate Nrf2 phosphorylation (Figure 7B) as well as subsequent SQSTM1 mRNA and protein expression (Figure 7C and D), once PRKCD was knocked down. Taken together, TAM stimulated Nrf2 to promote SQSTM1 transcription through the activation of PRKCD.
Figure 5
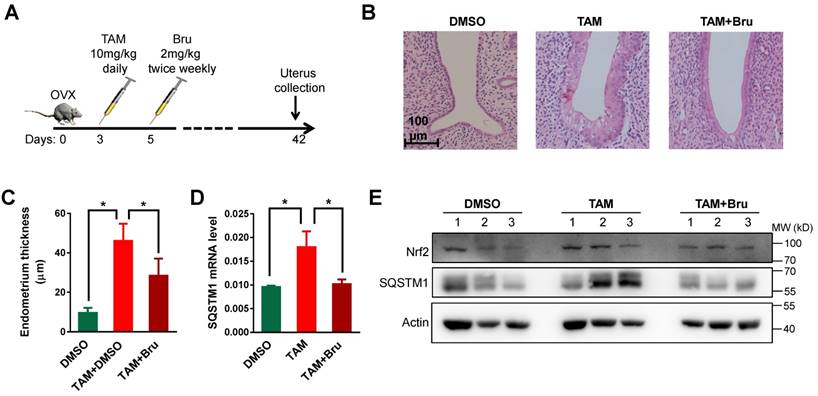
TAM activated Nrf2-SQSTM1 axis to promote endometrial hyperplasia in rats. A, TAM (10 mg/kg) was given daily to rats 3 days after ovariectomy to induce endometrial hyperplasia. Nrf2 inhibitor brusatol (Bru) (1 mg/kg) was given biweekly 5 days after ovariectomy. B, Representative HE staining of endometrium from rats treated with DMSO or TAM or TAM+Bru. C, The thicknesses of endometrium from 3 groups of rats treated with DMSO or TAM or TAM+Bru were summarized. The results of 5 mice in each group were shown as Mean±SD. Asterisks indicate statistical difference (Student's t test, p<0.05). D, Expressions of SQSTM1 mRNA in endometrium from rats were analyzed by qRT-PCR. Representative results from one triplicated experiment were shown as Mean±SD. Student's T test was used for the statistical analysis. The Asterisk indicates the statistical significance. E, Expressions of Nrf2 and SQSTM1 in endometrium from various rats treated as indicated were detected by Western blotting.
Figure 6
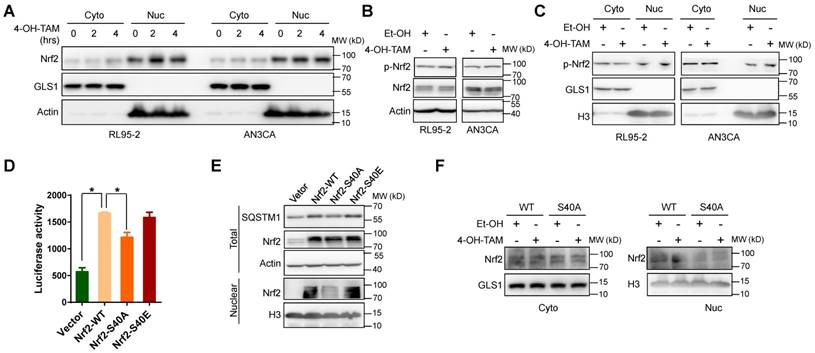
TAM activated Nrf2 through phosphorylation at ser40. A, Two or four hours after 4-OH-TAM (20 μM) incubation, nuclear and cytoplasmic distributions of Nrf2 in RL95-2 and AN3CA cells were explored by western blotting. B, Ser40 phosphorylation of Nrf2 (p-Nrf2) and total Nrf2 levels in RL95-2 and AN3CA cells after 1 hour 4-OH-TAM (20 μM) incubation were determined by western blotting. C, Nuclear and cytoplasmic distributions of p-Nrf2 in RL95-2 and AN3CA cells treated with or without 1-hour treatment of 4-OH-TAM (20 μM) were analyzed by western blotting. D, P62-pro/pGL2 or pGL2 vector were co-transfected with Nrf2 wild type (Nrf2-WT) or S40 phosphorylation deficient mutant (Nrf2-S40A) or S40 phosphorylation mimic mutant (Nrf2-S40E) or vector in AN3CA cells. Forty-eight hours later, the effects of wild type or mutated Nrf2 on SQSTM1 promoter activity were determined by luciferase activity assay. The experiments were performed in triplicate and repeated three times. Representative results were shown as Mean±SD. Asterisks indicate statistical significance (Student's T test, p<0.05). E, Forty-eight hours after Nrf2-WT or Nrf2-S40A or Nrf2-S40E or vector transfection in AN3CA cells, the expression and nuclear distribution of wild type or mutated Nrf2 and SQSTM1 protein expression were analyzed by western blotting. F, Nrf2-WT or Nrf2-S40A was transfected into AN3CA cells for 48 hours. Nuclear distributions of Nrf2-WT or Nrf2-S40A in AN3CA cells before and after 2h incubation of 4-OH-TAM (20 μM) were explored by western blotting.
Figure 7
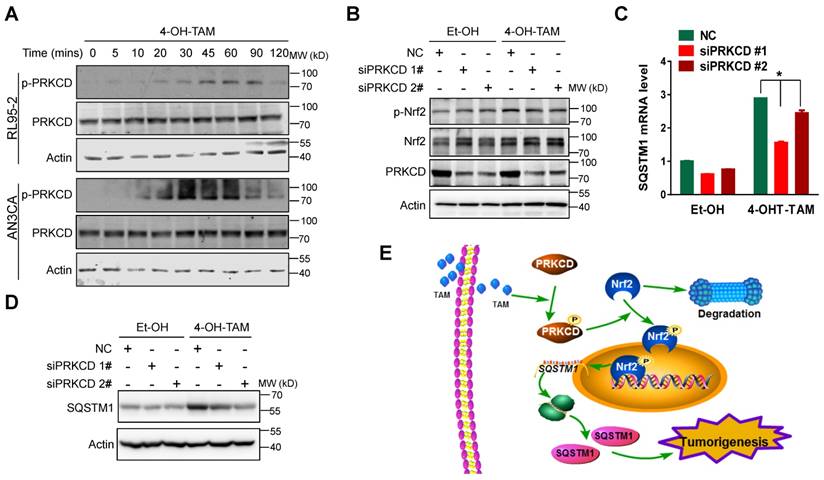
TAM activated PRKCD to activate Nrf2-SQSTM1 axis. A, Phosphorylation of PRKCD (p-PRKCD) and total PRKCD expression in RL95-2 and AN3CA cell upon 4-OH-TAM treatment at different times were analyzed by western blotting. B, Forty-eight hours after PRKCD siRNAs transfection, 4-OH-TAM (20 μM) was added for another 1 hour. The effect of PRKCD depletion on 4-OH-TAM-induced phosphorylation and expression of Nrf2 were explored by western blotting. The effects of PRKCD depletion on 4-OH-TAM induced SQSTM1 mRNA (C) and protein (D) expression were explored by qRT-PCR or western blotting, respectively. RT-PCR results were shown as Mean±SD. Asterisks indicate statistical significance (Student's T test, p<0.05, n=3). E, Working model: Tamoxifen activated PRKCD to promote Nrf2 S40 phosphorylation. Phosphorylated Nrf2 translocated to the nucleus and stimulate SQSTM1 transcription to promote endometrial hyperplasia, eventually leading to endometrial tumorigenesis.
Discussion
The increased incidence of endometrial hyperplasia even endometrial cancer restricted HR positive breast cancer patients to benefit from long-term TAM therapy. In the present study, we found TAM treatment promoted endometrial hyperplasia by activating PRKCD to stimulate Nrf2 activation and subsequent SQSTM1 transcription (Figure 7E).
In addition to function as substrate adaptor for autophagy, SQSTM1 plays an important role in the pathogeneses of various cancers. Its expression was increased in many tumors such as breast cancer, lung cancer and pancreatic cancer [22-24]. Meanwhile, SQSTM1 can be activated by post-translational modifications, like phosphorylation, to promote carcinogenesis [25]. Therefore, substantial efforts have been implemented to develop anti-cancer therapies based on SQSTM1-targeting. However, the inhibition of SQSTM1 function will inevitably result in the defect in autophagy and ubiquitin-proteasome system (UPS), leading to the stabilization of many oncoproteins such as Twist and MMPs [36]. Possibly due to such double-sword effects, the inhibition of SQSTM1 has not turned into promising outcomes. Instead, molecules upstream or downstream of SQSTM1 might be potential targets for anti-cancer therapy.
Activation of mTOR, NF-kB and Nrf2 signaling has been considered to be the predominant mechanism underlying tumorigenic function of SQSTM1. By direct interaction with KEAP1, SQSTM1 can facilitate the disassociation of Nrf2 from KEAP1 to activate the anti-oxidant response [25, 37, 38]. Interestingly, Nrf2 could bind the anti-oxidant response element presented within the promoter of SQSTM1 and activate its transcription, thus forming a positive feedback loop for sustained expression of Nrf2 and SQSTM1 [33]. In this report, we found TAM activated this positive feedback loop to promote endometrial hyperplasia by stimulating Nrf2 phosphorylation. As the result, chemical inhibitor of Nrf2 successfully reversed TAM-induced endometrial hyperplasia in rats. Importantly, TAM-induced Nrf2 activation seems to be specific to endometrial cells but not breast cancer cells. TAM failed to induce Nrf2 nuclear translocation and subsequent SQSTM1 transcription in MCF7 cells, leading to the inhibition rather than promotion of cell growth. Therefore, inactivation of Nrf2 could be clinically valuable for the prevention of TAM-induced endometrial hyperplasia in breast cancer patients undertaking long-term TAM treatment.
Furthermore, we found TAM stimulated Nrf2 S40 phosphorylation and nuclear translocation through activating PRKCD. In fact, estrogen can activate PRKCD to prevent the ubiquitin-dependent proteasome degradation of oncogenic K-Ras protein [39]. Inhibition of PRKCD activation succeeded to block estrogen-induced cellular transformation in vitro and xenograft tumor growth in vivo. However, such a function seemed dependent on ERα-36 rather than the canonical ERα-66 isoform. Therefore, it would be interesting to know whether TAM activated PRKCD and subsequent Nrf2-SQSTM1 axis in endometrial cells through ERα-36. Moreover, elevated PRKCD expression or Nrf2 activity has been reported to be associated with resistance to TAM in breast cancer [40-42]. This was consistent with proliferative effect of TAM in uterus, as reported here. Therefore, inhibitors of PRKCD or Nrf2 might be also useful to overcome intrinsic or acquired TAM resistance in breast cancer.
In summary, TAM stimulated Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia by activating PRKCD. Nrf2 inhibitor brusatol effectively reversed TAM-induced endometrial hyperplasia. Therefore, blocking PRKCD-Nrf2-SQSTM1 signaling might be useful to overcome TAM resistance in breast cancer cells, in addition to prevent TAM-induced endometrial hyperplasia.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by Department of Healthcare of Ministry and Zhejiang (2015103070), National Natural Science Foundation of China (81372178; 81572715; 8170110497) and 151 talents program in Zhejiang Province.
Competing Interests
The authors have declared that no competing interest exists.
References
1. White IN. The tamoxifen dilemma. Carcinogenesis. 1999;20:1153-60
2. Smith LL, Brown K, Carthew P, Lim CK, Martin EA, Styles J. et al. Chemoprevention of breast cancer by tamoxifen: risks and opportunities. Crit Rev Toxicol. 2000;30:571-94
3. Park WC, Jordan VC. Selective estrogen receptor modulators (SERMS) and their roles in breast cancer prevention. Trends Mol Med. 2002;8:82-8
4. Conzen SD. Current status of selective estrogen receptor modulators (SERMs). Cancer J. 2003;9:4-14
5. Takahashi M, Shimomoto T, Miyajima K, Iizuka S, Watanabe T, Yoshida M. et al. Promotion, but not progression, effects of tamoxifen on uterine carcinogenesis in mice initiated with N-ethyl-N'-nitro-N-nitrosoguanidine. Carcinogenesis. 2002;23:1549-55
6. Brown K. Breast cancer chemoprevention: risk-benefit effects of the antioestrogen tamoxifen. Expert Opin Drug Saf. 2002;1:253-67
7. Wu H, Chen Y, Liang J, Shi B, Wu G, Zhang Y. et al. Hypomethylation-linked activation of PAX2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature. 2005;438:981-7
8. Lin BC, Suzawa M, Blind RD, Tobias SC, Bulun SE, Scanlan TS. et al. Stimulating the GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes endometrial cell proliferation. Cancer Res. 2009;69:5415-23
9. Strissel PL, Ellmann S, Loprich E, Thiel F, Fasching PA, Stiegler E. et al. Early aberrant insulin-like growth factor signaling in the progression to endometrial carcinoma is augmented by tamoxifen. Int J Cancer. 2008;123:2871-9
10. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45
11. Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836-41
12. Yu H, Su J, Xu Y, Kang J, Li H, Zhang L. et al. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur J Cancer. 2011;47:1585-94
13. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213-23
14. Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O. et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275-84
15. Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625-9
16. Kim JY, Ozato K. The sequestosome 1/p62 attenuates cytokine gene expression in activated macrophages by inhibiting IFN regulatory factor 8 and TNF receptor-associated factor 6/NF-kappaB activity. J Immunol. 2009;182:2131-40
17. Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A. et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134-46
18. Proud CG. A new link in the chain from amino acids to mTORC1 activation. Mol Cell. 2011;44:7-8
19. Wei H, Wang C, Croce CM, Guan JL. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014;28:1204-16
20. Cai-McRae X, Zhong H, Karantza V. Sequestosome 1/p62 facilitates HER2-induced mammary tumorigenesis through multiple signaling pathways. Oncogene. 2015;34:2968-77
21. Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K. et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun. 2016;7:12030
22. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT. et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343-54
23. Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z. et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105-20
24. Feng L, Ma Y, Sun J, Shen Q, Liu L, Lu H. et al. YY1-MIR372-SQSTM1 regulatory axis in autophagy. Autophagy. 2014;10:1442-53
25. Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R. et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618-31
26. Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88:93-100
27. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291-5
28. Lee JS, Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005;224:171-84
29. Niture SK, Khatri R, Jaiswal AK. Regulation of Nrf2-an update. Free Radic Biol Med. 2014;66:36-44
30. Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK. et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74:7430-41
31. Niture SK, Jain AK, Jaiswal AK. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci. 2009;122:4452-64
32. Jain AK, Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem. 2006;281:12132-42
33. Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A. et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576-91
34. Rushworth SA, Ogborne RM, Charalambos CA, O'Connell MA. Role of protein kinase C delta in curcumin-induced antioxidant response element-mediated gene expression in human monocytes. Biochem Biophys Res Commun. 2006;341:1007-16
35. Cho MK, Kim WD, Ki SH, Hwang JI, Choi S, Lee CH. et al. Role of Galpha12 and Galpha13 as novel switches for the activity of Nrf2, a key antioxidative transcription factor. Mol Cell Biol. 2007;27:6195-208
36. Hua F, Li K, Yu JJ, Lv XX, Yan J, Zhang XW. et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nature communications. 2015;6:7951
37. Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z. et al. TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol Cell. 2016;61:720-33
38. Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279-89
39. Koo KH, Jeong WJ, Cho YH, Park JC, Min do S, Choi KY. K-Ras stabilization by estrogen via PKCdelta is involved in endometrial tumorigenesis. Oncotarget. 2015;6:21328-40
40. Kim SK, Yang JW, Kim MR, Roh SH, Kim HG, Lee KY. et al. Increased expression of Nrf2/ARE-dependent anti-oxidant proteins in tamoxifen-resistant breast cancer cells. Free Radic Biol Med. 2008;45:537-46
41. Nabha SM, Glaros S, Hong M, Lykkesfeldt AE, Schiff R, Osborne K. et al. Upregulation of PKC-delta contributes to antiestrogen resistance in mammary tumor cells. Oncogene. 2005;24:3166-76
42. Li Z, Wang N, Fang J, Huang J, Tian F, Li C. et al. Role of PKC-ERK signaling in tamoxifen-induced apoptosis and tamoxifen resistance in human breast cancer cells. Oncol Rep. 2012;27:1879-86
Author contact
![]() Corresponding author: Dr. Hongchuan Jin, email: jinhcedu.cn
Corresponding author: Dr. Hongchuan Jin, email: jinhcedu.cn
Citation styles
APA
Feng, L., Li, J., Yang, L., Zhu, L., Huang, X., Zhang, S., Luo, L., Jiang, Z., Jiang, T., Xu, W., Wang, X., Jin, H. (2017). Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics, 7(7), 1890-1900. https://doi.org/10.7150/thno.19135.
ACS
Feng, L.; Li, J.; Yang, L.; Zhu, L.; Huang, X.; Zhang, S.; Luo, L.; Jiang, Z.; Jiang, T.; Xu, W.; Wang, X.; Jin, H. Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics 2017, 7 (7), 1890-1900. DOI: 10.7150/thno.19135.
NLM
Feng L, Li J, Yang L, Zhu L, Huang X, Zhang S, Luo L, Jiang Z, Jiang T, Xu W, Wang X, Jin H. Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics 2017; 7(7):1890-1900. doi:10.7150/thno.19135. https://www.thno.org/v07p1890.htm
CSE
Feng L, Li J, Yang L, Zhu L, Huang X, Zhang S, Luo L, Jiang Z, Jiang T, Xu W, Wang X, Jin H. 2017. Tamoxifen activates Nrf2-dependent SQSTM1 transcription to promote endometrial hyperplasia. Theranostics. 7(7):1890-1900.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.