Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Material and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Material and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(1):132-143. doi:10.7150/thno.17032 This issue Cite
Research Paper
Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3β-E2F1 Axis Linking Reversely with Survival in Gliomas
Desheng Xiao 1,2, Jun Huang 3, Yu Pan 1,2, Hao Li 3, Chunyan Fu 1,2, Chao Mao4,5,6, Yan Cheng7, Ying Shi4,5,6, Ling Chen4,5,6, Yiqun Jiang4,5,6, Rui Yang4,5,6, Yating Liu4,5,6, Jianhua Zhou1,2, Ya Cao4,5,6, Shuang Liu8, Yongguang Tao4,5,6,8 ![]()
1. Department of Pathology, Xiangya Hospital, Central South University, Changsha, Hunan 410078 China;
2. Department of Pathology, School of Basic Medicine, Central South University, 172 TongZiPo Road, Changsha, Hunan, 410013 China;
3. Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, Hunan 410078 China;
4. Cancer Research Institute, Central South University, 110 Xiangya Road, Changsha, Hunan, 410078 China;
5. Key Laboratory of Carcinogenesis and Cancer Invasion (Central South University), Ministry of Education, Xiangya Hospital, Central South University, Hunan, 410078 China;
6. Key Laboratory of Carcinogenesis (Central South University), Ministry of Health, Hunan, 410078 China;
7. Department of Pharmacology, School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410078 China;
8. Center for Medicine Research, Xiangya Hospital, Central South University, Changsha, Hunan, 410008 China.
Received 2016-7-29; Accepted 2016-8-25; Published 2017-1-1
Citation:
Xiao D, Huang J, Pan Y, Li H, Fu C, Mao C, Cheng Y, Shi Y, Chen L, Jiang Y, Yang R, Liu Y, Zhou J, Cao Y, Liu S, Tao Y. Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3β-E2F1 Axis Linking Reversely with Survival in Gliomas. Theranostics 2017; 7(1):132-143. doi:10.7150/thno.17032. https://www.thno.org/v07p0132.htm
Other stylesAbstract

The signaling pathway-based stratification in chromatin modification could predict clinical outcome more reliably than morphology-alone-based classification schemes in gliomas. Here we reported a role of the chromatin-remodeling factor lymphoid-specific helicase (LSH) in gliomas. Among astrocytomas of grade I to III and glioblastoma of grade IV, LSH were almost completely expressed in all cases, and strongly correlated with astrocytomas progression and poor prognosis of patients with astrocytomas and glioblastoma. Ectopic expression of LSH promoted tumor formation. Up-regulation of transcription factor E2F1 in astrocytomas and glioblastoma was associated with the progression of gliomas and correlated with LSH expression. Chromatin immunoprecipitation (ChIP) analysis showed transcription factor E2F1 were recruited to the promoter region of LSH, and depletion of E2F1 decreased LSH expression and cell growth. Moreover, glycogen synthase kinase-3β (GSK-3β), an intact complex of E2F1, were also highly expressed in astrocytomas and linked with astrocytomas progression and poor prognosis of patients with astrocytomas and glioblastoma. Inhibition of GSK3β increased the enrichment of E2F1 to the LSH promoter, in turn, increased LSH expression. Lipoprotein receptor-related protein 6 (LRP6), an upstream regulator of GSK3β signaling pathway, was highly expressed in gliomas. Knockdown of LRP6 decreased LSH expression through decrease of recruitment of E2F1 to the LSH promoter leading to inhibition of cell growth. Taken together, this study reveals evidence demonstrating a mechanism by which upregulated promoted gliomas. A mechanistic link between LSH expression and activation of the LPR6/ GSK3β/E2F1 axis in gliomas illustrates a novel role of LSH in malignant astrocytomas and glioblastoma.
Keywords: LSH, LRP6, E2F1, GSK3β, Astrocytomas, Gioblastoma.
Introduction
Gliomas is the most frequently occurring and devastating human brain malignancy, which represent approximately 30% of all central nervous system tumors and 80% of malignant brain tumors [1, 2]. Despite technological advances in neuroimaging, surgery and adjuvant treatments including phototherapy, radiation and concomitant temozolomide, the prognosis for patients with gliomas remains dismal [2, 3]. Individuals receiving the current standard treatment for high grade gliomas, consisting of surgery, fractionated radiotherapy and concomitant chemotherapy with the DNA alkylating drug temozolomide, show a median survival of only 14.6 months with just 26.5% of individuals surviving for 2 years [4-6]. Gliomas are primary brain tumors that are thought to derive from neuroglial stem or progenitor cells. On the basis of their histological appearance, they have been traditionally classified as astrocytic, oligodendroglial or ependymal tumors and assigned WHO grades I-IV, which indicate different degrees of malignancy. Tremendous progress in genomic, transcriptomic and epigenetic profiling has resulted in new concepts of classifying and treating gliomas [7]. Recent reports indicate that molecular-based stratification in chromatin modification could predict clinical outcome more reliably than morphology-alone-based classification schemes in gliomas [7-11]. For this reason, it is vital to identify the molecular chromatin mediators and signaling pathways that drive gliomas progression for the development of targeted therapeutics to improve the overall survival of gliomas patients.
The SWI/SNF (mating type switching/sucrose non-fermenting) complex is an evolutionarily conserved multiunit complex of factors that utilize the energy of ATP hydrolysis to remodel nucleosomes and thereby affect gene expression [12]. LSH (lymphoid-specific helicase), also called HELLS (helicase, lymphoid specific) or PASG (proliferation-associated SNF2-like), a protein belonging to the SNF2 family of chromatin-remodeling ATPases, is critical for normal development of plants and mammals by establishing correct DNA methylation levels and patterns [13-16]. LSH maintains genome stability in mammalian somatic cells [17, 18]. LSH serves as a target for DeltaNp63alpha driving skin tumorigenesis in vivo and co-operates with the oncogenic function of E2F3 [19, 20]. Interestingly, LSH-mediated CG methylation can modulate gene activity and influence neuron lineage commitment [21]. Reports show that LSH contributes to the malignant progression of prostate cancer, melanoma, and nasopharyngeal carcinoma [20, 22, 23]. However, the clinical significance of LSH in gliomas and which mechanism involved in the regulation of LSH remain poorly known.
Glycogen synthase kinase-3β (GSK-3β) is a serine/threonine protein kinase that plays key roles in multiple pathways, and its dysregulation is implicated in many disorders, such as neurodegenerative diseases and cancers [24]. Clearly, the function of GSK-3β as tumor suppressor or oncogene in cancer can differ depending on tumor type [25, 26]. GSK-3β is regulated by a number of different mechanisms, the best characterized being its autoinhibition following phosphorylation of its N-terminal serine-9 residue [27]. GSK-3β activity can also be regulated via interactions with other cellular proteins through the formation of protein-protein complexes. This includes proteins such as E2F1. GSK3β directly regulates E2F activity by physical interaction with the transactivation domain of E2F1 [28]. The E2F1 transcription factor plays a central role in cell cycle control, apoptosis and differentiation [29]. Recent studies concerning the functions of E2F1 gene seem to be contradictory as this gene was described both as an oncogene as well as a tumor suppressor gene [30, 31]. The nature of this duality is likely to depend on the cellular context.
Wnt signaling (hereafter referred to as Wnt/β-catenin-dependent signaling) plays pivotal roles in embryogenesis and adult tissue homeostasis through its regulation of diverse biologic functions, including cell fate specification, proliferation, and cell migration [32]. Wnt ligands are secreted glycoproteins that signal through Frizzled (FZD) receptor and low-density lipoprotein receptor-related protein 5 or 6 (LRP5 or LRP6) co-receptor, leading to downregulation of glycogen synthase kinase-3 (GSK-3) activity and the initiation of the canonical Wnt/β-catenin signaling cascade [33, 34]. Activation of canonical Wnt signaling culminates in the cytoplasmic accumulation of β-catenin and its translocation to the nucleus where it complexes with transcription factors and coactivators to drive context-dependent target gene expression [35, 36]. Deregulation of Wnt/β-catenin signaling occurs in many types of cancer either through key driver mutations or increased Wnt ligand-mediated receptor activation [37].
Little is currently known regarding the significance of LSH to gliomas survival. We therefore sought to evaluate the role of LSH in gliomas cell survival, proliferation and tumorigenicity both in vitro and in vivo and on the mechanistic consequences of inhibiting the GSK3β/E2F1 axis. Here, we demonstrate that E2F1 directly regulates LSH expression. Surprisingly, our data reveal that the kinase activity of GSK3β is not required to exert this control. Much more, binding of E2F1 to LSH appears to be the mode of action how E2F1 regulates LSH transcriptional activity. Our data point to LSH as a potentially attractive new target for human gliomas therapy.
Material and methods
Study Participants
The Research Ethics Committee of the Xiangya hospital approved the study. Written informed consent was obtained from all participants in all studies. In this study, we included patients who had gliomas of histologic grade (I, II, III, or IV). A total of 128 cases and controls from the Xiangya Hospital were used as the discovery set in this study and the characteristics of these patients were shown in Table 1.
Table 1
Patient characteristics.
| Groups | N=128 |
|---|---|
| Age (years) | |
| Median | 42 |
| Range | 7-72 |
| Gender | |
| Male | 67(52.3%) |
| Female | 61(47.7%) |
| Survival time (Months) | |
| Median | 36 |
| Range | 2-131 |
| 60 or more | 15(11.7%) |
| Under 60 | 113(88.3%) |
| Histological diagnosis | |
| Pilocytic astrocytoma (A I) | 4(3.1%) |
| Diffuse astrocytoma (A II) | 53(41.4%) |
| Anaplastic astrocytoma (AA III) | 39(30.5%) |
| Glioblastoma (GB IV) | 32(25%) |
Immunohistochemistry (IHC) analysis
Gliomas and related diseases biopsies were validated and obtained from Department of Pathology in Xiangya Hospital. IHC analysis of paraffin sections from gliomas tissues was described previously [38]. The sections were incubated with antibodies as indicated. The images were surveyed and captured using a CX41 microscope (OLYMPUS, Tokyo, Japan) with the Microscope Digital Camera System DP-72 (OLYMPUS, Tokyo, Japan) and differentially quantified by two pathologists who were from the Xiangya Hospital, Changsha, China.
GSK3β and LRP6 staining were considered positively by ascertaining cytoplasmic staining, whereas LSH and E2F1 were considered positively by nuclear expression. The determination results were obtained from semi-quantitative classification according to 10 or more visual fields (×200). The slides were first scored as 0 (negative), 1 (buff), 2 (pale brown), and 3 (tan). Positive expression of LSH, E2F1, GSK3β or LRP6 was scored as 0 (negative), 1+ (<10% of positively-staining tumor cells), 2+ (11-50% of positively-staining tumor cells), 3+ (50-75% of positively-staining tumor cells), and 4+ (>75% of positively-staining tumor cells). Both the scores by multiply were regarded as the determination result.
Antibodies, plasmids, siRNAs, chemicals and cell cultures
Primary antibodies LRP6, GSK3β, c-Myc, E2F1, p-GSK3β for IHC analysis and Western Blot were purchased from Cell Signaling (Danvers, MA). Primary antibody for LSH and β-actin were purchased from Sigma-Aldrich (St. Louis, MO). The TRC-shE2F1, and TRC-shLRP6 lentivirus plasmids were purchased from Genechem (www.genechem.com.cn). Gliomas cells including HS683, U373, SKMG-4, SF126, SHG-44, SF295, and U251 cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA), and were cultured in DMEM (GIBCO, Life Technologies, Basel, Switzerland) medium with fetal bovine serum (FBS) to a final concentration of 10%. All cell lines were maintained at 37°C with 5% CO2.
Western Blot, MTS assay, colony formation assay
Western Blot and MTS assay were essentially performed as previously described [38, 39]. Cell viability was measured using a CellTiter-Glo Luminescent Cell Viability Assay Kit (MTS) purchased from Promega Corp (Madison, WI) and used according to the manufacturer's protocol. Measuring the absorbance at 450 nm using a microplate reader assessed the number of viable cells.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were essentially performed as previously described [38, 39]. ChIP DNA was analyzed by qPCR with SYBR Green (Bio-Rad) in ABI-7500 (Applied Biosystems) using the primers specified in the following, site 1: LSH forward primer: 5'- CTGACATCCTCGTGCAGATAC-3', reverse primer: 5'- AGCCTGTAACTATCCTGTGAGA-3'; site 2: LSH forward primer: 5'- TACGGTTGCCACAGGTCAG-3', reverse primer: 5'- GCCCAGCCTTCTAGCCAAA-3'. The E2F1 antibodies used were as followed: E2F1 and normal mouse IgG (Millipore).
Nude mice and study approval
A xenograft tumor formation was essentially performed as previously described [38]. Mice were injected with C6 (2 × 106 cells/mice) and their corresponding stable clones with knockdown of LSH expression via mammary fat pad. Data were analyzed using Student's t test; a P value less than 0.05 was considered significant.
All procedures for animal study were approved by the Institutional Animal Care and Use Committee of the Central South University of Xiangya School of Medicine and conform to the legal mandates and federal guidelines for the care and maintenance of laboratory animals.
Statistics
Progression-free survival (PFS) was calculated from the day of first surgery until tumor progression, death, or end of follow-up. Overall survival (OS) was calculated from the day of first surgery until death or end of follow-up. Log- rank test was used to analyze survival data. When more than two groups were compared, we tested for the equality of groups regarding PFS or OS and present global p values indicating that at least two groups were different. Cox regression models were built to assess the association of clinical parameters and molecular groups and expression pro ling with OS. The statistic deviance (minus twice the logarithm of the maximized likelihood) was used to measure the model t. The experiments were repeated at least three times. Results are expressed as mean ± SD or SEM as indicated. A 2-tailed Student's t test was used for intergroup comparisons. A p value less than 0.05 was considered statistically significant (*p<0.05, ** p <0.01, *** p <0.001).
Results
LSH is highly expressed in glioblastomas and correlated with gliomas progression and poor prognosis of patients with gliomas
We first determined whether LSH expression was associated with gliomas of varying World Health Organization grades. We first identified these tumors with H&E staining (Figure 1A), a total of 128 archived paraffin-embedded gliomas specimens with extensive clinical follow-up, including 4 cases of pilocytic astrocytoma (World Health Organization grade I), 53 cases of diffuse astrocytoma (grade II), 39 cases of anaplastic astorcytoma (grade III), and 32 cases of glioblastomas (grade IV) were analyzed by IHC staining with antibody against human LSH. We found that LSH was up-regulated in all four grades of gliomas when compared with that in normal brain tissues. Overall, expression of LSH protein was detected in 127 of 128 glioma specimens (99.2%), whereas 0.8% of cases (1/128) were LSH-. Figure 1B showed examples of IHC stained tumor specimens of each of the four World Health Organization grade gliomas. Specifically, moderate to strong nuclear staining of LSH protein was evident in tumor cells in these primary gliomas tissues (Figure 1B). In contrast, minimal immunoreactivity of LSH was detected in control normal brain tissues (Figure 1B). In all, 18 cases were classified as weakly positive (immunoreactivity scores of 1-3), 94 were identified as medium positive (scores of 4-9), and the remaining 16 cases (12.4%) as strongly positive (scores of 10-12). Next, we addressed the question whether LSH is associated with the progression of gliomas. Quantitative IHC analysis revealed that density of LSH stain in newly diagnosed gliomas of each grade of I to IV was higher than that in normal brain tissues (p<0.05) and increased along with the tumor grades (Figure 1C).
Multivariate analysis showed that the expression level of LSH was independent of clinical risk factor such as gender, sex, and tumor size, but were correlated with shorter survival time, suggesting that LSH could be an independent prognostic factor. Of note, Kaplan-Meier analysis and the log-rank test revealed that levels of LSH expression in primary gliomas specimens were reversely correlated with patients' survival time with a high degree of statistical significance (p<0.01, Figure 1D).
To uncover the physiological role of LSH in gliomas, we established the stable expressed LSH in C6 cells, a mouse derived from glioblastoma cells (Figure 1E). We injected 2×106 of C6 cells (with or without LSH ectopic stable expression), and observed that stable ectopic expression of LSH significantly promoted tumor formation and tumor weight, while the body weight did not change significantly in either group. Taken together, up-regulation of LSH in newly diagnosed gliomas was associated with the progression of gliomas and correlated with the poor prognosis of the disease.
E2F1 expression level is linked with LSH expression in gliomas
Using bioinformatics, we found that a potential binding site of transcription factor E2F1 existed in the regulatory region of LSH gene. Therefore, we linked the potential connection between E2F1 and LSH. We detected E2F1 expression among these tumor tissues. Specifically, moderate to strong nuclear staining of E2F1 protein was evident in tumor cells in these primary gliomas tissues (Figure 2A). In contrast, minimal immunoreactivity of LSH was detected in control normal brain tissues (Figure 2A). In all, 0.8 % of cases (1/128) were E2F1-. 11 cases were classified as weakly positive (immunoreactivity scores of 1-3), 76 were identified as medium positive (score of 4-9), and the remaining 40 cases (31%) as strongly positive (scores of 10-12). Next, we addressed the question whether E2F1 is associated with the progression of gliomas. Quantitative IHC analysis revealed that density of E2F1 stain in gliomas of each grade of I to IV was higher than that in normal brain tissues (p<0.05) and increased along with the tumor grades (Figure 2B).
Multivariate analysis also showed that the expression level of E2F1 was independent of clinical risk factor such as gender, sex, and tumor size, but were correlated with shorter survival time. Of note, Kaplan-Meier analysis and the log-rank test revealed that levels of E2F1expression in primary gliomas specimens were reversely correlated with patients' survival time with a high degree of statistical significance (p <0.01, Figure 2C).
To confirm the potential correlation between LSH and E2F1, we screened LSH and E2F1 expression in a panel of glioblastoma cells using Western Blot analysis (supplementary Figure S1), data indicated a potential correlation between E2F1 and LSH existed, and then we selected two glioblastoma cell lines, U251 and HS683 for subsequent studies.
To further validate the physiological signaling pathway between LSH and E2F1 in glioblastoma, we generated stable E2F1 knockdown in U251 cells from two separated target sequence. The knockdown approach successfully reduced E2F1 protein to less than 10%. The knockdown of E2F1 resulted in significantly reduced LSH expression (Figure 2D). Furthermore, depletion of E2F1 reduced the growth of U251 cells in culture (Figure 2E). Taken together, up-regulation E2F1 in primary glioma tumors was associated with the progression of glioma and correlated with LSH expression.
Figure 1
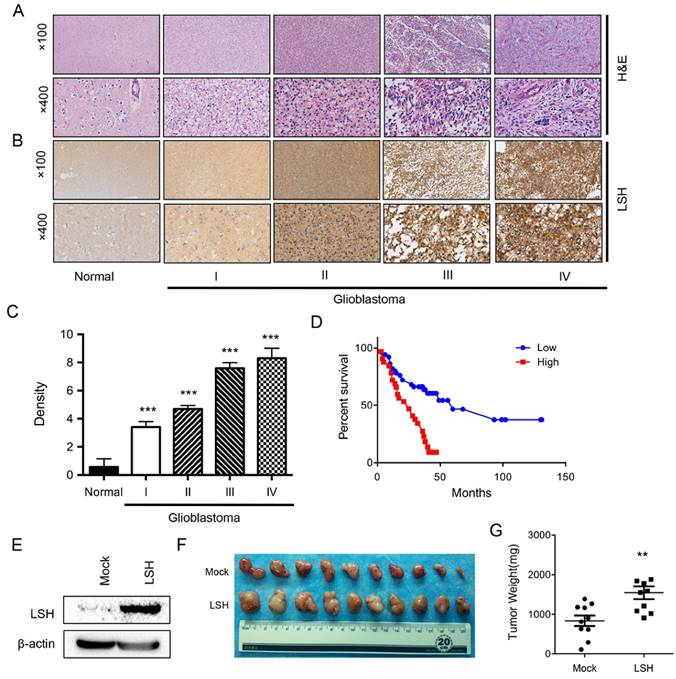
LSH is highly expressed in gliomas and correlated with gliomas progression and poor prognosis of patients with gliomas. (A) H & E staining was used to show different stages of gliomas. (B) A series of tissues samples was subjected to IHC with LSH-specific antibody. The panels shown here were representative of LSH staining in different stages gliomas including normal brain. (C) Anti-LSH staining intensity was quantified in three microscopic fields for each tissues section and expression level of LSH in different stages of gliomas as indicated. (D) Kaplan-Meier curves for overall survival of the related samples of LSH expression level measured in gliomas. (E) Ectopic stable expression of LSH in C6 cells. A xenograft model of tumor growth was established in nude mice to evaluate tumor formation (F) and tumor weight (G) of C6 cells with a stable expression of LSH to form tumors with. ** p <0.01, *** p <0.001.
Figure 2
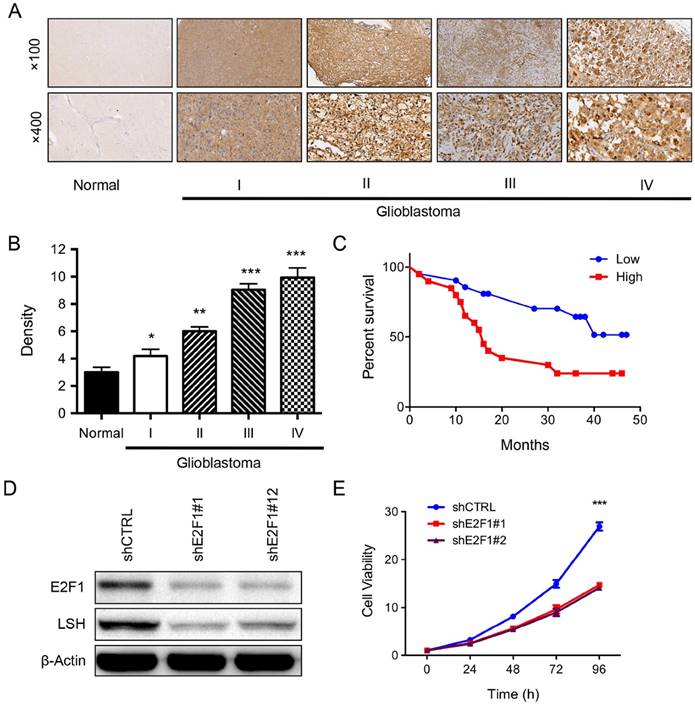
E2F1 expression level is linked with LSH expression in gliomas. (A) A series of tissues samples was subjected to IHC with E2F1-specific antibody. The panels shown here were representative of E2F1 staining in different stages gliomas including normal brain. (B). Anti-E2F1 staining intensity was quantified in three microscopic fields for each tissues section and expression level of LSH in different stages of gliomas as indicated. (C) Kaplan-Meier curves for overall survival of the related samples of E2F1 expression level measured in gliomas. (E). LSH expression was analyzed in the depletion of E2F1 in U251 cells by Western Blot analysis. (E) The MTT assay was performed to assess cell viability in U251 cells that were stably knockdown of E2F1 in U251 cells. *p<0.05, ** p <0.01, *** p <0.001.
GSK3β is highly expressed in gliomas and linked with E2F1 binding to the lsh promoter
On the basis that GSK3β binds to E2F1 and regulates its transcriptional activity [28], we first addressed whether GSK3β expression was associated with gliomas. In all, 23.4% of cases (30/128) were GSK3β-. 23 cases were classified as weakly positive (immunoreactivity scores of 1-3), 58 were identified as medium positive (score of 4-9), and the remaining 17 cases (13.2%) as strongly positive (scores of 10-12). Of the GSK3β+ cases, showed diffuse strong immunoreactivity in the cytoplasm of tumor cells (Figure 3A). Next, we demonstrated that the expression of GSK3β was significantly increased in advanced clinical stage IV compared to earlier stages of (p<0.05) (Figure 3B). Kaplan-Meier plotter performed on a cohort of these glioblastoma showed that lower expression of GSK3β linked with overall survival in all glioblastoma (Figure 3C).
SB216763 is a potent and selective, ATP-competitive GSK3 inhibitor that is equally effective at inhibiting human GSK3α and GSK3β. Then, we first treated H683 cells used SB216763, we performed a ChIP assay to examine E2F1 binding to the lsh promoter. E2F1 was recruited to the lsh promoter region, and its binding was increased significantly after the treatment of SB216763 (Figure 3). Similar findings were shown in U251 cells. However, both pGSK3βSer9 and β-catenin remained the same level in the depletion of E2F1 in U251 cells (supplementary Figure S2). Taken together, GSK3β in primary glioma tumors was associated with the progression of glioma and correlated with E2F1 binding to the lsh promoter region.
To confirm the role of GSK3β in the regulation of LSH, we treated U251 cells with SB216763 as indicated concentrations for 12 h, Western blot showed that the levels of LSH, pGSK3βSer9, and E2F1 increased after the treatment of SB216763, whereas total GSK3β and β-catenin remained the same level (Figure 4A). Similar findings were shown in HS683 after the treatment of SB216763 (Figure 4B). We further confirmed the inhibition of GSK3β with the treatment of SB216763 increased LSH expression (Figure 4C) as well as pGSK3βSer9, and E2F1 in both U251 and HS683 cells (Figure 4D). Taken together, we found that the inhibition of GSK3β signaling pathway increase LSH in glioblastoma cells.
LRP6 is highly expressed in gliomas
Wnt signaling is activated upon binding of Wnt ligands to Frizzled receptors and the low-density lipoprotein receptor-related protein (LRP) on the surface of cells and LRP6 is an upstream regulator of GSK3β signaling pathway [40], we then addressed whether LRP6 expression was associated with gliomas. In all, 14.0% of cases (18/128) were LRP6-. 19 cases were classified as weakly positive (immunoreactivity scores of 1-3), 89 were identified as medium positive (score of 4-9), and only the remaining 2 cases (1.50%) as strongly positive (scores of 10-12). Of the LRP6+ cases, showed diffuse strong immunoreactivity in the cytoplasm of tumor cells (Figure 5A).
Figure 3
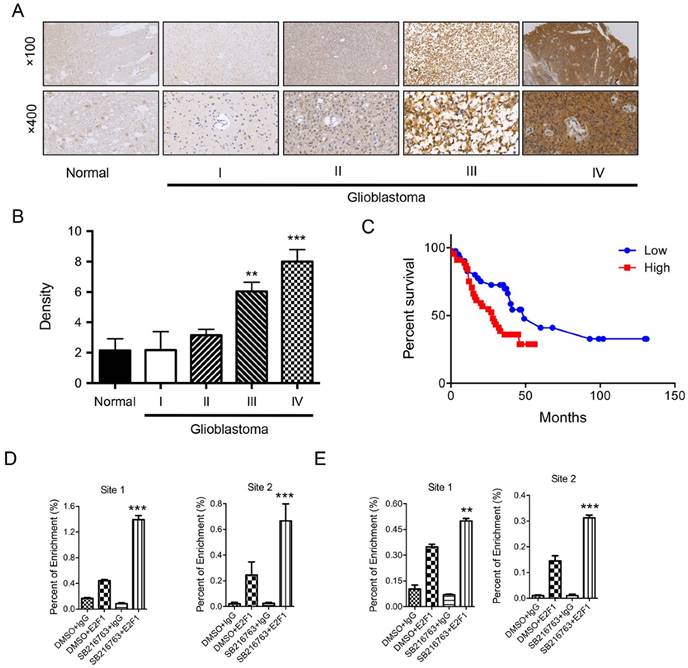
GSK3β is highly expressed in gliomas and linked with E2F1 binding to the lsh promoter. (A) A series of tissues samples was subjected to IHC with GSK3β -specific antibody. The panels shown here were representative of GSK3β staining in different stages gliomas including normal brain. (B). Anti- GSK3β staining intensity was quantified in three microscopic fields for each tissues section and expression level of LSH in different stages of gliomas as indicated. (C) Kaplan-Meier curves for overall survival of the related samples of GSK3β expression level measured in gliomas. ChIP analysis of HS683 (D) and U251 (E) cells was performed to detect E2F1 binding to the lsh promoter. ** p <0.01, *** p <0.001.
Figure 4
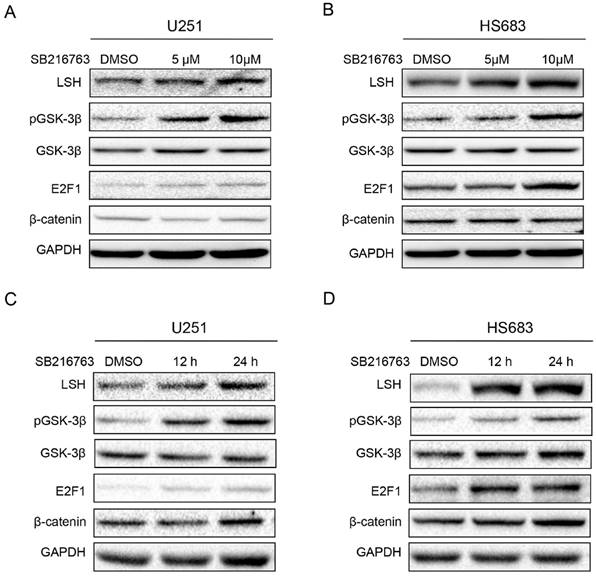
Inhibition of GSK3β increased LSH expression. U251 (A) and HS683 (B) with treatment of SB216763 with different concentration as indicated were examined for the expression of LSH, p-GSK3β, GSK3β, E2F1, β-catenin and GAPDH by Western analysis. U251 (C) and HS683 (D) with treatment of 10μM SB216763 with time as indicated were examined for the expression of LSH, p-GSK3β, GSK3β, E2F1, β-catenin and GAPDH by Western analysis.
Next, we addressed that the expression of LRP6 was significantly increased in advanced clinical stage IV compared to earlier stages of (p<0.05) (Figure 5B). Of note, Kaplan-Meier plotter performed on a cohort of these glioblastoma showed that lower expression of LRP6 linked with overall survival in all glioblastoma (Figure 5C). We analyzed the potential correlation between LRP6 and LSH, and we found that LSH expression level was positively related with LRP6 level (Figure 5D) and with E2F1 level (Figure 5E) respectively.
Furthermore, to address the connection of LRP6 and LSH, we generated stable LRP6 knockdown in U251 cancer cells. The knockdown approach successfully reduced LRP6 protein to less than 15%. The knockdown of LSH resulted in significantly reduced LSH expression (Figure 6A). Next, we performed a ChIP assay to examine E2F1 binding to the lsh promoter in the depletion of LRP6, knowndown of LRP6 significantly deceased the enrichment of E2F1 to the lsh promoter region (Figure 6B). Furthermore, depletion of LRP6 reduced the growth of U251 cells in culture (Figure 6C) and the colony number (Figure 6D). Taken together, activation of LRP6 was associated with the expression of LSH in gliomas, indicating that the LRP6-GSK3β-E2F1 axis was correlated with LSH expression in gliomas.
Discussion
Newly predicted gliomas drivers relative to the earlier TCGA analyses were genes associated with chromatin organization. Diffusely infiltrating gliomas in adults are now separated into three overarching tumor groups with distinct natural histories, responses to treatment and outcomes: isocitrate dehydrogenase (IDH)-mutant, 1p/19q co-deleted tumors with mostly oligodendroglial morphology that are associated with the best prognosis; IDH-mutant, 1p/19q non-co-deleted tumors with mostly astrocytic histology that are associated with intermediate outcome; and IDH wild-type, mostly higher WHO grade (III or IV) tumors that are associated with poor prognosis [8, 10, 11].
Figure 5
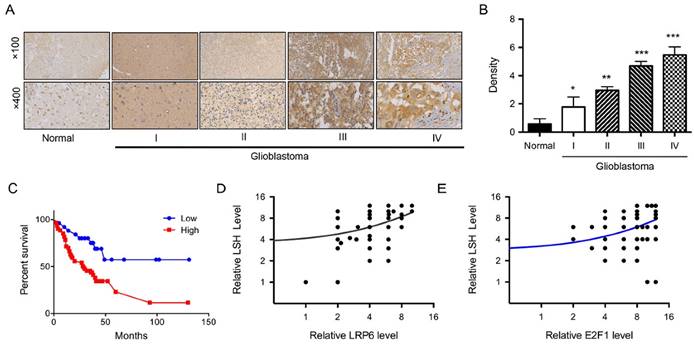
LRP6 is highly expressed in gliomas. (A) A series of tissues samples was subjected to IHC with LRP6-specific antibody. The panels shown here were representative of LRP6 staining in different stages gliomas including normal brain. (B) Anti-LRP6 staining intensity was quantified in three microscopic fields for each tissues section and expression level of LSH in different stages of gliomas as indicated. (C) Kaplan-Meier curves for overall survival of the related samples of LRP6 expression level measured in gliomas. ChIP analysis of HS683 (D) and U251 (E) cells was performed to detect E2F1 binding to the lsh promoter. The correlations of LSH with LRP6 (D) and E2F1(E) were analyzed. *p<0.05, ** p <0.01, *** p <0.001.
Figure 6
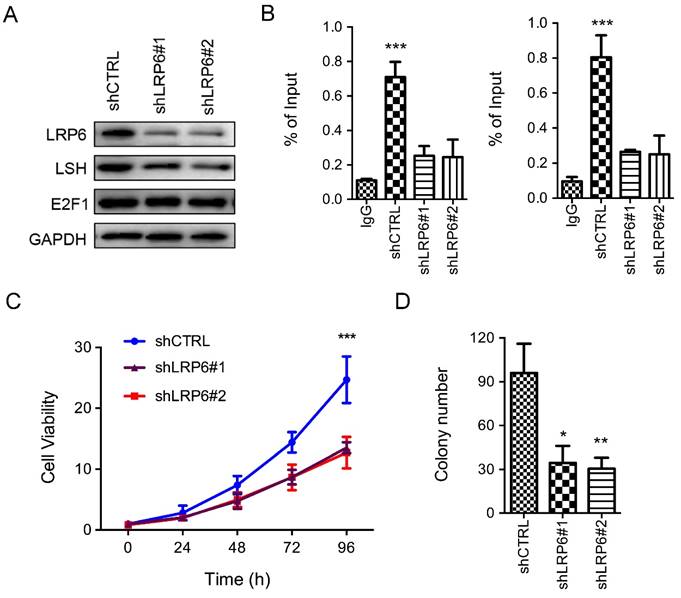
Depletion of LRP6 reduced LSH expression. (A) LSH expression was analyzed in the depletion of LRP6 in U251 cells by Western Blot analysis. (B) ChIP analysis of U251 cells was performed to detect E2F1 binding sites to the lsh promoter. (C) The MTT assay was performed to assess cell viability in U251 cells that were stably knockdown of LRP6. (D) Colony formation assay was performed to detect tumor cell formation in the depletion of LSH. *p<0.05, ** p <0.01, *** p <0.001.
However, about 80.6% of glioblastoma is characteristics of IDH wild-type [8]. We found about 22.9 % of glioblastoma was characteristics of IDH mutation, which is consistent with this finding (Data not shown). Here, for the first time we, addressed that LSH expression is closely linked with the progression of gliomas, LSH functions as a new chromatin modifier in gliomas.
Lymphoid-specific helicase (LSH) acts a chromatin modifier in DNA methylation [41], depletion of LSH increases DNA hypomethylation in 5-mC in repeats and somatic genes, leading to transcribed satellites repeat expression [42, 43]. Alterations in DNA methylation, including the hypomethylation of oncogenes and the hypermethylation of tumor suppressor genes, indicate that DNA methylation plays an important role in tumorigenesis including gliomas [7, 44, 45]. Here we first demonstrate that LSH might function as an oncogene in gliomas, the mechanism remains further identification. 5-hydroxymethylcytosine (5-hmC) is an epigenetic mark that can be converted from 5-methylcytosine (5-mC) by the tet-eleven translocation (TET) gene family, whereas IDH mutants could establishes CpG island methylator phenotype in glioma by remodelling the methylome [46-48]. Oncometabolite 2-Hydroxyglutarate (2-HG) is a competitive inhibitor of α-KG-dependent dioxygenases in gliomas that carry mutations of IDH1 and IDH2 [48, 49]. Loss of 5-hmC in solid tumors does not have driver properties in cancer initiation but rather reinforces the molecular networks functioning in cancer cells [50]. However, loss of 5-hmC is a frequent event in gliomas, independent of IDH1 mutation [51]. Interestingly, 5-hmC levels are significantly associated with long interspersed nucleotide element-1 (LINE-1) methylation that is regarded as a surrogate marker for global DNA methylation level, however, TETs mRNA expression is not associated with LINE-1 methylation, indicating other factor involves in the process [52]. We found that LSH promoted genome stability through silencing satellites expression by affecting 5-hmC levels in pericentromeric satellite repeats (Jia J, et al. Submitted). Recently, depletion of LSH maintained the propensity toward the neural lineage during in vivo differentiation [21], indicating that LSH directly control several neuron related genes that might be linked with gliomas progression in independent manner of DNA methylation.
Wnt morphogens play leading roles in embryonic development as well as in adult stem cell biology. Blocking of the interact complex between Wnt proteins and their coreptors including LRP6 in glioblastoma cells contributes to the anti-tumor effects [53]. Knockdown of LRP6 by miRNA and shRNA decrease cell growth in glioblastoma cells [54]. Here, we first show that the activation of LRP6/GSK3β/E2F1 in glioblastoma patients is linked with LSH overexpression. GSK3β inhibition in glioblastoma cells induces c-Myc-dependent recruitment of DNA methylation transferase 3A (DNMT3A), leading to O6-methylguanine DNA methyltransferase (MGMT) promoter methylation and consequent silencing of MGMT expression, a predictor for resistance to temozolomide, in tumor cells in patients with recurrent glioblastoma [55]. Here, we also found that GSK3β inhibition induces LSH expression, indicating that GSK3β involves in LSH expression in vivo. E2F1 is a potent and specific inhibitor of β-catenin/T-cell factor (TCF)-dependent transcription, and E2F1 deregulation suppresses β-catenin activity in an adenomatous polyposis coli (APC)/GSK3-independent manner, reducing the expression of key β-catenin targets [29].
In summary, this study reveals evidence demonstrating a mechanism by which upregulated promoted gliomas. A mechanistic link between LSH expression and activation of the LPR6/ GSK3β/E2F1 axis in gliomas illustrates a novel role of LSH in malignant gliomas. These results provide new insights into the mechanisms underlying the signaling pathway in LSH expression in the carcinogenesis of gliomas. Understanding the roles of LSH in glioma progression will not only advance our knowledge of gliomas, but also could establish LSH as a potential therapeutic target for the treatment of these deadly brain cancers.
Supplementary Material
Supplementary figures.
Acknowledgements
We would like to thank all laboratory members for their critical discussion of this manuscript.
Funding
This work was supported by the National Basic Research Program of China [2015CB553903(Y.T.)]; the National Natural Science Foundation of China [81372427 and 81672787(Y.Tao), 81271763 and 81672991(S.Liu), 81302354(Y.Shi), and 81472593 (Y.C.)]; and the Hunan Natural Science Foundation of China [12JJ1013(Y.T.)].
Contributions
Conception and design: Y.T. and S.L.
Development of methology: D.X., J.H., Y.P., H.L., C.F., C.M., Y.S., L.C., Y.J., R.Y., Y.L., J.Z.
Acquisition of data (provided animals, provided facilities, etc.): J D.X., J.H., Y.P., H.L., C.F., C.M.,
Analysis and interpretation of data (e.g. statistical analysis, biostatistitcs,computational analysis): D.X., J.H., Y.P., H.L., C.F., C.M., Y.Cheng, S.L.,
Writing, review, and/or revision of the manuscript: C.M., Y.Cheng, Y.S., L.C., Y.J., R.Y., Y.L., J.Z., Y.Cao, and S.L.
Administrative, technical, or material support (i.e. reporting or organizing data, constructing databases): D.X., J.H., Y.P., H.L., C.F., C.M., Y.Cheng, Y.S., L.C., Y.J., R.Y., Y.L., J.Z., Y.Cao, and S.L.
All authors have given final approval of the version to be published.
Conflict of interest
The authors declare no conflict of interest. This manuscript has been read and approved by all the authors, and not submitted or under consider for publication elsewhere.
References
1. Van Meir EG, Hadjipanayis CG, Norden AD. et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166-193
2. Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 2014;9:1-25
3. Stupp R, Hegi ME, Mason WP. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459-466
4. Messaoudi K, Clavreul A, Lagarce F. Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov Today. 2015;20:899-905
5. Sehedic D, Cikankowitz A, Hindre F. et al. Nanomedicine to overcome radioresistance in glioblastoma stem-like cells and surviving clones. Trends Pharmacol Sci. 2015;36:236-252
6. Stupp R, Mason WP, van den Bent MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996
7. Mazor T, Pankov A, Johnson BE. et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell. 2015;28:307-317
8. Ceccarelli M, Barthel FP, Malta TM. et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550-563
9. Erdem-Eraslan L, Gravendeel LA, de Rooi J. et al. Intrinsic molecular subtypes of glioma are prognostic and predict benefit from adjuvant procarbazine, lomustine, and vincristine chemotherapy in combination with other prognostic factors in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. J Clin Oncol. 2013;31:328-336
10. Cancer Genome Atlas Research N, Brat DJ, Verhaak RG. et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015;372:2481-2498
11. Eckel-Passow JE, Lachance DH, Molinaro AM. et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015;372:2499-2508
12. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481-492
13. Zemach A, Kim MY, Hsieh PH. et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193-205
14. Yu W, McIntosh C, Lister R. et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014;24:1613-1623
15. Tao Y, Xi S, Shan J. et al. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc Natl Acad Sci U S A. 2011;108:5626-5631
16. Myant K, Termanis A, Sundaram AY. et al. LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res. 2011;21:83-94
17. Fan T, Yan Q, Huang J. et al. Lsh-deficient murine embryonal fibroblasts show reduced proliferation with signs of abnormal mitosis. Cancer Res. 2003;63:4677-4683
18. Burrage J, Termanis A, Geissner A. et al. The SNF2 family ATPase LSH promotes phosphorylation of H2AX and efficient repair of DNA double-strand breaks in mammalian cells. J Cell Sci. 2012;125:5524-5534
19. Keyes WM, Pecoraro M, Aranda V. et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8:164-176
20. von Eyss B, Maaskola J, Memczak S. et al. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. EMBO J. 2012;31:972-985
21. Yu W, Briones V, Lister R. et al. CG hypomethylation in Lsh-/- mouse embryonic fibroblasts is associated with de novo H3K4me1 formation and altered cellular plasticity. Proc Natl Acad Sci U S A. 2014;111:5890-5895
22. Ryu B, Kim DS, Deluca AM. et al. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS One. 2007;2:e594
23. He X, Yan B, Liu S. et al. Chromatin remodeling factor LSH drives cancer progression by suppressing the activity of fumarate hydratase. Cancer Res. 2016
24. Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769-776
25. Atkins RJ, Stylli SS, Luwor RB. et al. Glycogen synthase kinase-3beta (GSK-3beta) and its dysregulation in glioblastoma multiforme. J Clin Neurosci. 2013;20:1185-1192
26. Tang QL, Xie XB, Wang J. et al. Glycogen synthase kinase-3beta, NF-kappaB signaling, and tumorigenesis of human osteosarcoma. J Natl Cancer Inst. 2012;104:749-763
27. Dajani R, Fraser E, Roe SM. et al. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721-732
28. Garcia-Alvarez G, Ventura V, Ros O. et al. Glycogen synthase kinase-3beta binds to E2F1 and regulates its transcriptional activity. Biochim Biophys Acta. 2007;1773:375-382
29. Morris EJ, Ji JY, Yang F. et al. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature. 2008;455:552-556
30. Bell LA, Ryan KM. Life and death decisions by E2F-1. Cell Death Differ. 2004;11:137-142
31. Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9:738-748
32. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469-480
33. Stamos JL, Chu ML, Enos MD. et al. Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6. Elife. 2014;3:e01998
34. Cheng PW, Chen YY, Cheng WH. et al. Wnt Signaling Regulates Blood Pressure by Downregulating a GSK-3beta-Mediated Pathway to Enhance Insulin Signaling in the Central Nervous System. Diabetes. 2015;64:3413-3424
35. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9-26
36. Chen S, Bubeck D, MacDonald BT. et al. Structural and functional studies of LRP6 ectodomain reveal a platform for Wnt signaling. Dev Cell. 2011;21:848-861
37. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11-26
38. Jiang Y, Yan B, Lai W. et al. Repression of Hox genes by LMP1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by HoxC8. Oncogene. 2015;34:6079-6091
39. Shi Y, Tao Y, Jiang Y. et al. Nuclear epidermal growth factor receptor interacts with transcriptional intermediary factor 2 to activate cyclin D1 gene expression triggered by the oncoprotein latent membrane protein 1. Carcinogenesis. 2012;33:1468-1478
40. Voskas D, Ling LS, Woodgett JR. Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol Rep. 2010;2:82
41. Ren J, Briones V, Barbour S. et al. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015;43:1444-1455
42. Huang J, Fan T, Yan Q. et al. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 2004;32:5019-5028
43. Yan Q, Cho E, Lockett S. et al. Association of Lsh, a regulator of DNA methylation, with pericentromeric heterochromatin is dependent on intact heterochromatin. Mol Cell Biol. 2003;23:8416-8428
44. Jiang Y, Liu S, Chen X. et al. Genome-wide distribution of DNA methylation and DNA demethylation and related chromatin regulators in cancer. Biochim Biophys Acta. 2013;1835:155-163
45. Liu S, Tao Y. Interplay between chromatin modifications and paused RNA polymerase II in dynamic transition between stalled and activated genes. Biol Rev Camb Philos Soc. 2013;88:40-48
46. Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. EMBO J. 2014;33:1198-1211
47. Shen L, Zhang Y. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr Opin Cell Biol. 2013;25:289-296
48. Turcan S, Rohle D, Goenka A. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479-483
49. Xu W, Yang H, Liu Y. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17-30
50. Ficz G, Gribben JG. Loss of 5-hydroxymethylcytosine in cancer: cause or consequence? Genomics. 2014;104:352-357
51. Muller T, Gessi M, Waha A. et al. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am J Pathol. 2012;181:675-683
52. Murata A, Baba Y, Ishimoto T. et al. TET family proteins and 5-hydroxymethylcytosine in esophageal squamous cell carcinoma. Oncotarget. 2015
53. Hara K, Kageji T, Mizobuchi Y. et al. Blocking of the interaction between Wnt proteins and their co-receptors contributes to the anti-tumor effects of adenovirus-mediated DKK3 in glioblastoma. Cancer Lett. 2015;356:496-505
54. Zhang W, Shen C, Li C. et al. miR-577 inhibits glioblastoma tumor growth via the Wnt signaling pathway. Mol Carcinog. 2015
55. Pyko IV, Nakada M, Sabit H. et al. Glycogen synthase kinase 3beta inhibition sensitizes human glioblastoma cells to temozolomide by affecting O6-methylguanine DNA methyltransferase promoter methylation via c-Myc signaling. Carcinogenesis. 2013;34:2206-2217
Author contact
![]() Corresponding author: Y.T. Key Laboratory of Carcinogenesis and Cancer Invasion of the Ministry of Education, Xiangya Hospital, Central South University. 87 Xiangya Road Changsha, Hunan, 410008 China Tel. +(86) 731-84805448; Fax. +(86) 731-84470589; Email: taoyongedu.cn.
Corresponding author: Y.T. Key Laboratory of Carcinogenesis and Cancer Invasion of the Ministry of Education, Xiangya Hospital, Central South University. 87 Xiangya Road Changsha, Hunan, 410008 China Tel. +(86) 731-84805448; Fax. +(86) 731-84470589; Email: taoyongedu.cn.
Citation styles
APA
Xiao, D., Huang, J., Pan, Y., Li, H., Fu, C., Mao, C., Cheng, Y., Shi, Y., Chen, L., Jiang, Y., Yang, R., Liu, Y., Zhou, J., Cao, Y., Liu, S., Tao, Y. (2017). Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3β-E2F1 Axis Linking Reversely with Survival in Gliomas. Theranostics, 7(1), 132-143. https://doi.org/10.7150/thno.17032.
ACS
Xiao, D.; Huang, J.; Pan, Y.; Li, H.; Fu, C.; Mao, C.; Cheng, Y.; Shi, Y.; Chen, L.; Jiang, Y.; Yang, R.; Liu, Y.; Zhou, J.; Cao, Y.; Liu, S.; Tao, Y. Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3β-E2F1 Axis Linking Reversely with Survival in Gliomas. Theranostics 2017, 7 (1), 132-143. DOI: 10.7150/thno.17032.
NLM
Xiao D, Huang J, Pan Y, Li H, Fu C, Mao C, Cheng Y, Shi Y, Chen L, Jiang Y, Yang R, Liu Y, Zhou J, Cao Y, Liu S, Tao Y. Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3β-E2F1 Axis Linking Reversely with Survival in Gliomas. Theranostics 2017; 7(1):132-143. doi:10.7150/thno.17032. https://www.thno.org/v07p0132.htm
CSE
Xiao D, Huang J, Pan Y, Li H, Fu C, Mao C, Cheng Y, Shi Y, Chen L, Jiang Y, Yang R, Liu Y, Zhou J, Cao Y, Liu S, Tao Y. 2017. Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3β-E2F1 Axis Linking Reversely with Survival in Gliomas. Theranostics. 7(1):132-143.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.