Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(1):67-80. doi:10.7150/thno.16752 This issue Cite
Research Paper
CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer
Chong Li1,2,7*, Shengwu Liu3,5*, Ruping Yan4*, Ning Han6, Kwok-Kin Wong3,5, Lei Li1, ![]()
1. Department of Urology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China.
2. Core Facility for Protein Research, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
3. Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
4. Department of Urology, The Second Affiliated Hospital of Kunming Medical College, Kunming 650101, China.
5. Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA.
6. Department of Life Science and Technology, China Pharmaceutical University, Nanjing 211198, China.
7. Beijing Jianlan Institute of Medicine, Beijing 100190, China.
* contributed equally to this work.
Received 2016-7-6; Accepted 2016-9-10; Published 2017-1-1
Citation:
Li C, Liu S, Yan R, Han N, Wong KK, Li L. CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics 2017; 7(1):67-80. doi:10.7150/thno.16752. https://www.thno.org/v07p0067.htm
Other stylesAbstract

Cancer stem cells (CSCs) are considered one of the key contributors to chemoresistance and tumor recurrence. Therefore, the precise identification of reliable CSC markers and clarification of the intracellular signaling involved in CSCs remains a great challenge in fields relating to cancer biology. Here, we implemented a novel chemoresistant prostate cancer patient-derived xenograft (PDX) model in NOD/SCID mice and identified CD54 as a candidate gene among the most highly enriched gene expression profiles in prostate tumors exposed to chronic cisplatin administration. Additional in vitro and in vivo assays showed that CD54 played a critical role in the self-renewal and tumorigenesis of prostate CSCs. Moreover, silencing CD54 greatly reduced the tumorigenesis of prostate cancers both in vitro and in vivo and significantly extended the survival time of tumor-bearing mice in a prostate cancer xenograft model. Dissection of the molecular mechanism revealed that the p38-Notch1 axis was the main downstream signaling pathway in CD54-mediated regulation of CSCs in prostate cancers. Together, these results established that CD54 could be a novel reliable prostate CSC marker and provided a new potential therapeutic target in prostate cancer via CD54-Notch1 signaling.
Keywords: prostate cancer, CD54, cancer stem cells, NOTCH1.
Introduction
Prostate cancer (PCa) has the highest incidence of cancer-related illness and is the second leading cause of cancer mortality in the U.S. [1]. An estimated 15% of newly diagnosed prostate cancer patients have an advanced disease at the time of diagnosis [2]. Androgen deprivation therapy (ADT) has been the standard treatment strategy for patients with advanced prostate cancer [3]. However, almost all patients develop castration resistance and relapse within 18-24 months following ADT [3]. Chemotherapy is regarded as a milestone treatment approach for castration-resistant PCa patients, with average survival of 3-6 months [4].
Increasing evidence revealed the critical role of cancer stem cells (CSCs) or cancer-initiating cells in tumorigenesis of many cancers [5, 6], including prostate cancer [7-9]. CSCs are a small population of cells that have the potential to differentiate into various tumor cells within a specific tumor environment [5]. CSCs are involved in chemotherapy resistance and become enriched after ADT treatment, which leads to poor outcomes and frequent recurrence in cancer patients [10-12].
Over the past 20 years, targeted cancer therapies have been extensively studied and developed based on the accumulated understanding of cancer biology [13]. Currently, precision medicine and targeted therapy are promising cancer treatments. Therefore, it is a priority to identify new CSC targets both in basic research and in clinical trials. The CD133+/α2β1 integrin/CD44+ phenotype could be a potential target for the PCa CSC population [14]. However, the lack of a unique marker has limited the use of these combination targeting therapies for PCa CSCs. Thus, a more specific and unique target for PCa CSCs still requires identification.
These ideal and novel unique markers for PCa CSCs could be used not only as CSC isolation diagnostic markers but also as potential therapeutic targets via antibodies, inhibitors, short interfering RNAs (siRNAs), etc. Here, we identified the PCa CSCs marker CD54 via generation of a novel human prostate cancer xenograft mouse model. CD54+ PCa cells had more tumorigenicity and were resistant to the administration of the chemotherapeutic drug cisplatin compared to CD54- PCa cells via activation of p38-Notch1 signaling. Targeting CD54 with the specific siRNAs suppressed CD54-induced p38-Notch1 signals, which resulted in the suppression of sphere formation by PCa cells and might represent a new potential therapeutic approach to suppress PCa CSCs.
Materials and Methods
Cell Lines and Primary Prostate Cancer Tissues. The cell lines LNCaP, RWPE-1, 22Rv1, DU145, and PC3 were obtained from the American Type Culture Collection (Rockville, MD, US). The prostate cancer tissues were obtained from the Second Affiliated Hospital of Kunming Medical College (Kunming, China) with informed consent. All human studies were reviewed and approved by the Institutional Review Board (IRB) of Institute of Biophysics, Chinese Academy of Sciences and conducted on the basis of the World Medical Association Declaration of Helsinki. Cell lines were used for less than 6 months after restoration and were routinely screened for Mycoplasma every 4 weeks. No genotypic authentication was conducted. Each cell line was used during the early passages.
Patient-derived xenograft mouse model. Tumor tissues were obtained from prostate cancer patients following either biopsy or radical prostatectomy and then were washed 3 times on ice with RPMI 1640 medium containing ampicillin (100 U/ml) and streptomycin (100 ug/ml). The tissue blocks were then morselized with scissors and dissociated in proteolytic (Accumax, Innovative Cell Technologies, Inc.), collagenolytic (200 U Type I and 20 U Type IV collagenase, Sigma-Aldrich) and DNase enzymes at 37 °C for 2 to 6 hours. The cells were washed 3 times and then centrifuged 1000 rpm for 5 min with RPMI 1640 at 4°C. The suspensions were then filtered with 100, 200, and 300 nylon mesh, and cells were stained with PE-conjugated anti-CD54 (eBioscience) antibody and a lineage mixture containing Cy7-PE-conjugated anti-CD45 (BD PharMingen), biotin-conjugated anti-CD31 (eBioscience), CD68 (eBioscience) and H2Kd (BD PharMingen 553564) antibodies. Flow cytometry analysis and cell sorting were performed on a BD FACS Aria (Becton Dickinson). For the PDX model, cancer cells were resuspended in testis extracts to establish the PDX mouse model.
For the preparation of testis extractions, the testis tissues were stripped into RMPI 1640 medium without serum, cut and emulsified with a homogenizer. The supernatant extracts were aliquoted into 100 µl and stored at -80°C before centrifugation at 12000 rpm for 20 minutes at 4 °C.
To establish the PDX mouse model, 8- to 10-week-old male BALB/c mice were anesthetized with an intraperitoneal injection of 1% sodium pentobarbital. The sorted prostate cancer cell suspension (cancer cell dissolved in testis extractions) was mixed on ice with extracellular matrix at a 1:1 ratio. Approximately 200 μl of the prostate tumor cell/extracellular matrix suspension was subcutaneously injected into the flanks of male NOD/SCID mouse using a 31 G needle. Tumor growth in the mice was monitored by palpation of the subcutaneous nodules every 3 days.
Knockdown of CD54 and overexpression of Notch-1 in prostate cancer cells. To construct a CD54 knockdown lentivirus, the target sequences were designed as follows: shCD54-1: 5'- GCTGACGTGTGCAGTAATA-3', shCD54-2: 5'- GGACATACAACTGGGAAAT-3'; and shCD54-3: 5'- CAGCGGAAGATCAAGAAAT-3'. In brief, both the shCD54 and control hairpins were cloned into the pSICO-R vector. Production of lentiviral particles and transduction of CSCs was performed according to protocols from the RNAi consortium at MIT. Prostate cancer cells were transfected with lentiviral constructs expressing either shRNA against CD54 or shCtrl for 24 hr. Positive cells were selected with puromycin for 2 weeks. The CD54-expressing cell lines were identified using real-time PCR.
The Notch-1 plasmid was already established in our lab, and the pcDNA3.1-Notch-1 plasmid was transfected into prostate cancer cells using Lipofectamine 2000 (Invitrogen). The expression of Notch-1 was assayed by western blot.
Chemoresistant prostate cancer xenograft mouse model. Twelve non-castrated male NOD/SCID mice were obtained from the Animal Center of the Chinese Academy of Medical Science (Beijing, China). To generate the xenografts, 1×106 LNCaP cells were subcutaneously injected into the backs of NOD/SCID mice (n=24). When tumors reached a diameter of 5 mm after 3 d, the non-castrated mice were randomly divided into two groups (12 per group). Mice in the treatment group were intraperitoneally administered cisplatin, gemcitabine or docetaxel at doses of 5 mg/kg, 10 mg/kg, or 3 mg/kg, respectively, every 3 d for 30 d. Mice in the control group were treated with PBS vehicle. The xenograft volume was measured every 5 d. Mice were euthanized for tumor analysis after 30 d.
Tumor Luciferase Imaging System. In a subcutaneous mouse model, isolated primary LNCaP cells from xenograft mice were labeled with luciferase-GFP and transplanted into secondary NOD/SCID mice (6 mice per group). Tumors were monitored with an in vivo imaging system (IVIS) 21 d after secondary transplantation. Photon counts of the imaged mice are indicated with pseudo-color scales.
RNA Extraction and Microarray Analysis. Total RNA from freshly xenografted prostate tumor tissues from NOD/SCID mice treated with either cisplatin or vehicle was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, US) using a standard isopropanol/chloroform protocol. Gene expression patterns were analyzed with a human gene chip that contained clones of 35,000 human genes (GEO number: GSE78196) (CapitalBio Corp, Beijing, China). Microarray slides were scanned with a ScanArray 4000 Microarray Analysis System (Packard Bioscience, Meriden, CT, US), and data were analyzed with data analysis software (Dapple version 0.86 beta). Clones with normalized log ratios indicating 2-fold upregulation or downregulation were selected.
Real-time PCR. Total RNA from either cell lines or primary patient tumor samples was extracted with an RNA isolation kit (Tiangen Biotech, Beijing, China). RNA was subjected to cDNA synthesis with a PrimeScript RT reagent kit (Takara Bio, Shiga, Japan). cDNA was used as the template for real-time PCR analysis on an ABI 7200 analyzer (Applied Biosystems, Waltham, MA, US) with the fluorescent probe SYBR Green I (Tiangen Biotech). Relative expression levels of the genes were normalized to the housekeeping gene GAPDH. Each experiment was independently repeated at least 4 times.
FACS Analysis of Cell Proliferation and Apoptosis. LNCaP, PC3, and primary prostate tumor cells were stained with anti-human CD54-PE and isolated on a FACSAria-III (BD Biosciences, San Jose, CA, US). For the BrdU cell proliferation assays, primary cells from prostate cancer patients were treated with 30 μM BrdU for 4 h. Cells were fixed, permeabilized, DNase-treated, and stained with anti-BrdU antibody per the manufacturer's instructions (BD Biosciences). Cells were analyzed on a BD Biosciences LSR II flow cytometer.
For apoptosis assays, shCD54- or shCtrl-treated primary prostate cancer cells were stained with an Annexin V-FITC kit (Sigma-Aldrich) according to the manufacturer's instructions. Briefly, cells were washed twice with cold PBS, digested, collected, and resuspended in binding buffer. Cells were stained with annexin V-FITC and propidium iodide (BioVision, Milpitas, CA, USA) and incubated for 10 min at room temperature in the dark. After 200 μL of binding buffer was added, the annexin V-positive cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences). Each experiment was independently repeated at least 3 times.
Sphere-forming Assay. A single-cell suspension of prostate cancer cells was seeded at a density of 3×103 cells/well in 6-well plates with ultra-low attachment surfaces (Corning, Corning, NY, US). Cells were cultured in DMEM/F12 media (Gibco, Waltham, MA, US) supplemented with 20 ng/mL bFGF, 20 ng/mL EGF, 1% N2, 2% B27 (Invitrogen), and 100 mg/mL streptomycin (Gibco). The number of spheres was calculated 2 weeks after seeding. Each assay was repeated at least 3 times.
Colony Formation Assay. Prostate cancer cells were suspended in soft agar and culture media in 6-well plates at a density of 1,000 cells/well. After 2-3 weeks, the number of colonies (≥10 cells) within 5 microscope fields per well were counted and photographed. Each experiment was independently repeated at least 3 times.
Transwell Assay. shRNA-transfected cells from prostate cancer patients were harvested, suspended, and added to the upper compartments of transwell inserts (8 μm pore size; Corning). Lower chambers were filled with RPMI-1640 (Invitrogen) supplemented with 10% FBS (Invitrogen). After incubation for 16 h, cells in the upper chamber were scraped away with a swab. Cells that migrated to the lower layer of each membrane were stained with crystal violet, and 5 fields per well were counted under a light microscope. Each assay was repeated 3 times.
NOTCH1 Transcription Reporter System. LNCaP and PC3 cells were stably transduced with the GFP-NOTCH1 promoter to monitor transcriptional activation of NOTCH1. Cells were treated for 24 h with either vehicle or the p38 inhibitor BIRB 796 at a concentration of 10 μM. FACS analysis of GFP expression indicated NOTCH1 transcription. Each experiment was independently repeated at least 3 times.
Western Blots. Tumor cells collected from two prostate cancer patients were treated with either vehicle or BIRB 796 for 24 h. Cells were lysed, and proteins were separated using SDS-PAGE before transfer onto a nitrocellulose membrane. Membranes were blocked and then probed with primary antibodies and HRP-conjugated secondary antibodies (Sigma-Aldrich). Each experiment was independently repeated at least 4 times.
Primary Antibodies. The following primary antibodies were used: mouse anti-human CD54 (14-0549; eBioscience, San Diego, CA, US), mouse anti-human CD54-PE (12-0549; eBioscience), anti-activated Notch1 (Abcam, ab8925), rabbit anti-human HES1 (11988; Cell Signaling Technology), mouse anti-human phospho-p38 MAPK (Thr180/Tyr182) (9216; Cell Signaling Technology), rabbit anti-human p38 MAPK (ab197348; Abcam, Cambridge, UK), and mouse anti-β-actin (ab6276; Abcam).
Statistical Analysis. Kaplan-Meier analysis was used to estimate cumulative cause-specific survival rates and differences in mice survival between those receiving shCD54 xenografts and those receiving shCtrl xenografts. The influence of CD54 on sphere and colony formation capacities was analyzed using Student's t-test. In all statistical analyses, statistical significance in two-sided tests was indicated as P ≤ 0.05.
Results
Cisplatin treatment selects for tumorigenic cells and upregulates CD54 expression in prostate cancer cells from mouse xenografts. We used a xenograft prostate cancer model by subcutaneously transplanting LNCaP cells, a human prostate cancer cell line, into immunocompromised NOD/SCID mice. Mice (n=12 per group) that were intraperitoneally injected with the chemotherapeutic drug cisplatin showed significantly reduced tumor size and volume 30 d after treatment compared to vehicle-treated mice (Figure 1a-c).
However, chemotherapy often results in drug resistance and increases the frequency of CSCs. To test whether cisplatin treatment enriched for CSCs, we harvested tumor cells from primary NOD/SCID mice that were either vehicle-treated or treated with cisplatin and injected these harvested cells into secondary NOD/SCID mice. Cells from cisplatin-treated tumors were more tumorigenic than cells from vehicle-treated tumors when transplanted into secondary mice recipients (Figure 1d, 6 mice per group, Figure S1), suggesting that cisplatin treatment resulted in enrichment for CSCs.
To identify the underlying molecular mechanism of drug resistance in prostate CSCs, we compared the expression profiles of LNCaP cells with or without cisplatin treatment by microarray analysis. The microarray results showed that the expression of many genes significantly changed after cisplatin treatment (Figure 1e and f, Supplementary Table 1). Among these genes, CD54 had one of the greatest differences in expression. CD54, also known as ICAM-1, is particularly interesting because it is a membrane protein and thus a potential marker for prostate CSCs.
To validate the microarray results, we performed real-time PCR and found that cisplatin treatment indeed induced significantly higher CD54 mRNA expression in LNCaP cells (Figure 1g). Furthermore, LNCaP cells treated with two other chemotherapy drugs, gemcitabine (GEM) and docetaxel (DOC), also had significantly higher CD54 expression than vehicle-treated cells (Figure 1h). Immunohistochemical staining of mouse tumors corroborated that CD54 expression was also higher after chemotherapeutic treatment (Figure 1h). Compared with the control prostate epithelia cell line RWPE-1, several other prostate cancer cell lines also had elevated CD54 mRNA (Figure 1i) and protein (Figure 1j) expression in vitro.
CD54+ Prostate Cancer Cells Resemble Cancer Stem Cells. After identifying elevated CD54 expression in cells from chemoresistant tumors, we next investigated the precise role of CD54 in the tumorigenicity of prostate CSCs. To analyze whether CD54 is a potential marker for prostate CSCs, we isolated CD54+ and CD54- cells from the two prostate cancer cell lines PC3 and LNCaP with a specific CD54 antibody (Figure S2) using flow cytometry (Figure 2a). Sphere-forming capacity is an important feature of CSCs. In sphere-forming assays with isolated cells, CD54+ cells had a significantly increased sphere-forming capacity than CD54- cells (Figure 2b). We also found that CD54+ prostate cancer cells such as PC3 had a greater colony-forming ability than CD54- cells (Figure 2c). In addition, compared to luminal marker expression, basal or progenitor cell marker expression was relatively high in CD54+ versus CD54- cells (supplementary Figure S3).
To further clarify the role of CD54 in vivo, we injected isolated CD54+ or CD54- cells from four prostate cancer lines (PC3, LNCaP, DU145, and 22RV1) into NOD/SCID mice. CD54+ cells from all cell lines developed significantly more tumors than CD54- cells (Supplementary Table 2). CSCs are resistant to many drugs, so we determined the IC50 of cisplatin on prostate cancer cell lines. CD54+ cancer cells had a significantly higher IC50, indicating that CD54+ cells were more drug resistant than CD54- cells (Figure 2d).
Figure 1
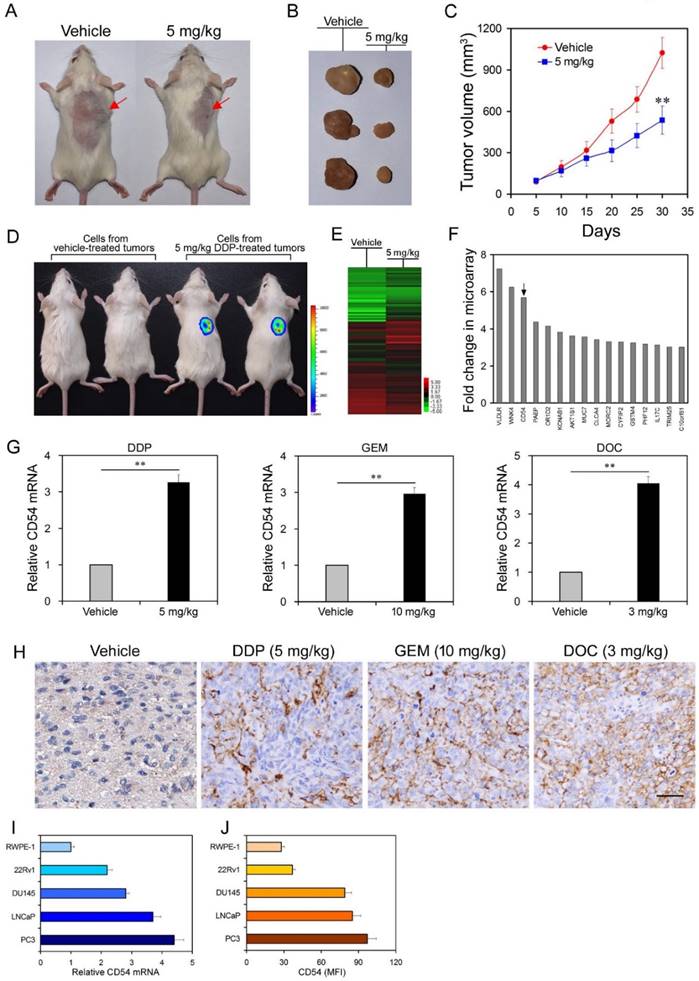
Differential Analysis of Gene Expression in Prostate Cancer Cells from Chemoresistant Xenografts in Mice. (a, b) NOD/SCID mice were injected with LNCaP prostate cancer cells. One group (n=12) was treated with 5 mg/kg of cisplatin, while the other group (n=12) was administered PBS as a control. Overall tumor sizes grossly differed between vehicle-treated mice and mice treated with cisplatin as determined via inspection of the whole animal and upon tumor excision after mice were euthanized 30 d following injection. (c) Tumor volumes from vehicle-treated mice and mice treated with cisplatin every 5 d after 5x106 cell injection during the 30 d interval. (d) Approximately 104 cells from DDP-treated tumors and vehicle-treated tumors were labeled with luciferase and transplanted into secondary NOD/SCID mice. Tumor formation was monitored with an in vivo imaging system. Pseudo-color scales indicate the photon counts of imaged mice. (e) Microarray analysis of gene expression from RNA extracted from combined vehicle-treated or cisplatin-treated tumors from each group. (f) Expression levels of selected genes after cisplatin treatment compared to expression levels in vehicle-treated cells. (g) Real-time PCR measurement of relative CD54 mRNA expression in vehicle-treated tumor cells and in tumor cells treated with cisplatin (DDP), gemcitabine (GEM), or docetaxel (DOC). (h) Immunohistochemical staining of CD54 expression in representative tumor tissues from vehicle-treated mice or mice treated with DDP, GEM, or DOC (scale bar=50 μm). (i, j) Relative CD54 mRNA and protein expression levels among the prostate cell line RWPE-1 and various prostate cancer cell lines. ** P < 0.01.
Figure 2
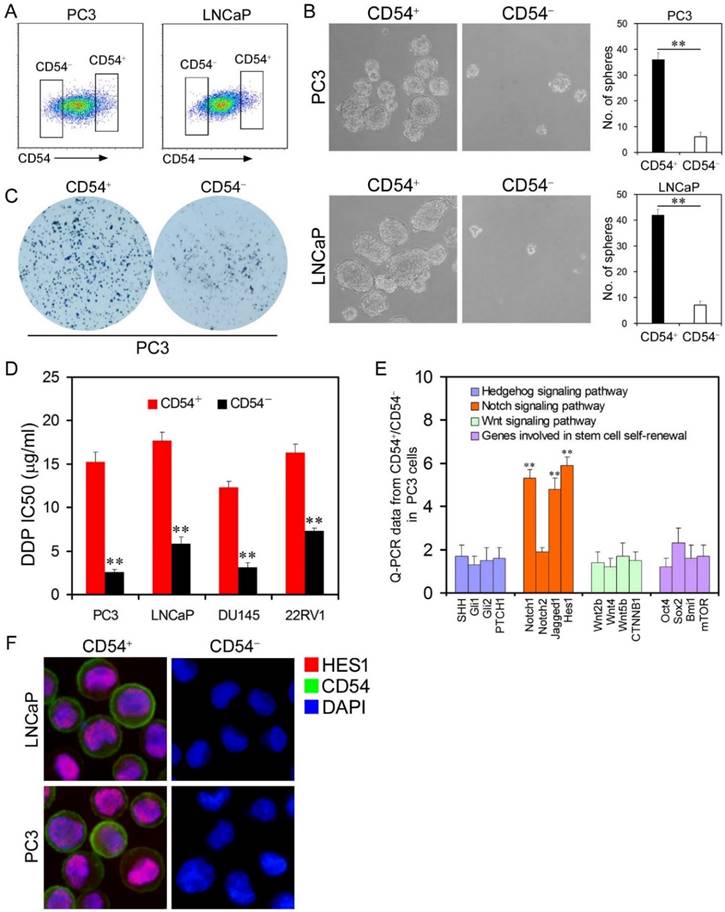
Cancer Stem Cell Traits of Differential Populations of CD54+ and CD54- Cells. (a) FACS histograms show separate gating for the isolation of CD54+ and CD54- PC3 and LNCaP cells. (b) Representative light micrograph fields and comparative quantification of the sphere-forming capacity of CD54+ and CD54- LNCaP and PC3 cells. (c) Representative microscope fields from colony formation assays for CD54+ and CD54- PC3 cells. (d) The cisplatin (DDP) IC50 of CD54+ and CD54- cells from the PC3, LNCaP, DU145, and 22RV1 prostate cancer cell lines. (e) q-PCR analysis of the expression of Hedgehog, Notch, and Wnt signaling pathway genes as well as genes involved in stem cell self-renewal in CD54+ and CD54- PC3 cells. (f) Representative micrographs of in situ protein expression of the key signaling pathway components HES1 and CD54 in DAPI-stained LNCaP and PC3 cells. ** P < 0.01.
Several signaling pathways are involved in regulation of the tumorigenesis of CSCs, including the Hedgehog [15, 16], Notch [17, 18], and Wnt signaling pathways [19, 20]. We compared the expression of some key molecules within these signaling pathways in CD54+ and CD54- PC3 cells and found that members of the NOTCH1 signaling pathway including NOTCH1, Jagged1 and HES1 were significantly elevated in CD54+ cells both at the mRNA (Figure 2e) and protein (Figure 2f) levels, indicating a close correlation with CD54 function.
CD54 Expression is Clinically Relevant. After identifying the critical role of CD54+ CSCs in prostate tumorigenesis in mice, we further explored whether CD54 expression was clinically relevant in prostate cancer patients. Detection of CD54 expression in a cohort of prostate patients showed that CD54 expression was significantly enhanced in cancer cells from patients with recurrent prostate cancer (Figure 3a). We also found that stronger CD54 expression was related to a higher 5-year mortality in prostate cancer patients (Figure 3b). Detailed clinical information regarding these findings is shown in supplementary Table 3.
To confirm CD54 expression differences during prostate cancer development, we compared CD54 expression in primary and recurrent tumors from two cancer patients. Data from fluorescent-activated cell sorting (FACS) assays (Figure 3c) and immunohistochemical staining (Figure 3d) showed increased CD54 expression after recurrence in both patients. Together, these data indicate CD54 expression was positively correlated to tumor recurrence.
Next we aimed to identify the function of CD54+ cells from prostate cancer patients. We collected tumor tissue from a prostate cancer patient and isolated CD54+ and CD54- tumor cells using FACS. CD54+ prostate cancer cells from both primary and secondary lesions had significantly stronger sphere-forming capacity (Figure 3e). NOD/SCID mice transplanted with primary or secondary CD54+ tumor cells from the same patient developed tumors, while an equal number of CD54- cells did not induce tumor development in these mice (Figure 3f, 6 mice per group). The detailed uptake rate and size of the primary or secondary xenografts in these mice are shown in Figure S4 and supplementary Table 4. In situ immunohistochemical analysis indicated CD54 expression heterogeneity in both primary and secondary xenografts of CD54+ tumors (Figure 3g).
CD54 Knockdown Inhibits Prostate Cancer Stem Cell Characteristics. Our data indicated that CD54+ cells showed properties of prostate CSCs. However, it was unknown whether CD54 was essential in maintaining CSC features. To explore the function of CD54 in prostate CSCs, we knocked down CD54 expression with three different shRNAs in LNCaP and PC3 cells. shCtrl (a non-targeting shRNA) was used as a negative control. Significant knockdown efficiency was confirmed by real-time PCR (Figure 4a) and FACS analysis (Figure 4b). Knockdown of CD54 significantly inhibited colony formation (Figure 4c) and decreased sphere-forming capacity (Figure 4d). Moreover, we confirmed that the addition of CD54 could increase the sphere-forming ability of PC3 cells. However, adding the CD54 inhibitor DAPT significantly abolished the sphere-forming capacity of PC3 cells (Figure S5).
After in vitro confirmation, we transplanted either control cells or cells with stable knockdown of CD54 into NOD/SCID mice. CD54 silencing significantly reduced tumor volume (Figure 4e) and tumor weight (Figure 4f). Exogenous expression of CD54 in the knockdown cells rescued these reductions. To assess the potential regulation roles of CD54 in CSCs, we inoculated single shRNA-treated cells into NOD/SCID mice via limiting dilution. The frequency of prostate CSCs was calculated by using ELDA software [21] based on the ratio between the number of tumors and the number of injection sites. Cells treated with shCD54-1 showed a sharp decrease in the frequency of prostate CSCs, while CD54 expression in these knockdown cells abrogated this decrease (Figure 4g). Because NOTCH1 signaling mediators were highly expressed in CD54+ cancer cells (Figure 2e and f), we examined whether NOTCH1 signaling was regulated downstream of CD54 in prostate CSCs. We found that NOTCH1 signaling, including Jagged1 and HES1, but not NOTCH2 signaling was significantly inhibited by CD54 knockdown in both LNCaP and PC3 cells. Exogenous CD54 expression rescued this inhibition (Figure 4h), indicating that CD54 plays a critical role upstream of NOTCH1 signaling in prostate CSCs.
Attenuated Proliferation and Increased Apoptosis upon CD54 Knockdown. Because CD54 silencing suppressed the characteristics of prostate CSCs, we evaluated the efficacy of CD54 silencing in bulk prostate cancer cells. We collected tumor tissue from a prostate cancer patient and evaluated the knockdown efficacy of CD54 shRNA on cancer cells. BrdU staining showed that shCD54 treatment reduced cell proliferation by keeping significantly fewer cells in S phase (Figure 5a). Ki67 staining revealed that shCD54 treatment also kept significantly more cells within G0 phase (Figure 5b). Similar proliferation data were found in 4 other patients (data not shown). shCD54 also significantly increased the apoptosis of cells collected from two patients (Figure 5c). Transwell assays showed that the migratory capacity of prostate cancer cells from both patients was also significantly decreased by shCD54 (Figure 5d).
Because CSCs are essential in tumor chemoresistance, CD54 knockdown might overcome this resistance. To test this hypothesis, we knocked down CD54 in PC3 cells before adding cisplatin to the culture. shCD54 significantly reduced the resistance of PC3 cells to cisplatin (Figure 5e). We further measured the IC50 of cisplatin in cancer cells from 5 prostate cancer patients and found the shCD54 significantly reduced the IC50 values in all 5 patient samples (Figure 5f).
Figure 3
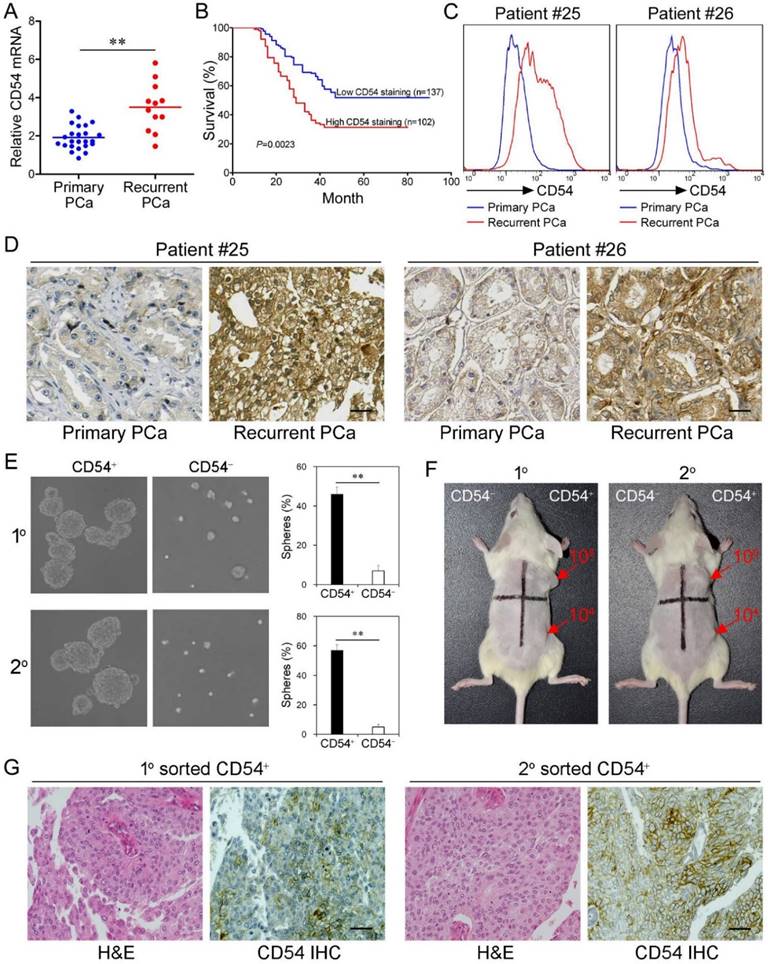
Clinical Relevance of CD54 Expression. (a) Relative CD54 mRNA expression in primary (n=24) and recurrent tumors (n=12) from prostate cancer patients. (b) Relative CD54 mRNA expression correlated with mortality of patients with prostate cancer by Kaplan-Meier survival analysis. (c) CD54 expression in primary and recurrent tumors from two prostate cancer patients. (d) Immunohistochemical analysis of in situ CD54 expression in primary and recurrent tumors from two prostate cancer patients. (e) Representative light micrograph of the sphere-forming capacity of CD54+ and CD54- sorted cells isolated from primary and secondary xenograft tumors of prostate cancer patients and quantification of the results. (f) Tumor-initiating capacity of differing numbers of different quadrants of CD54+ and CD54- tumor cells isolated from primary and secondary prostate cancer patient xenograft tumors of NOD/SCID mice 6 weeks after injection. Red numbers indicate number of cells injected, and red arrows indicate tumor formation. (g) Immunohistochemical analysis of in situ CD54 expression in both primary and secondary tumor samples collected from xenografted CD54+ tumors. H&E stained tissues are shown as a reference (scale bar=50 μm). ** P < 0.01.
Figure 4
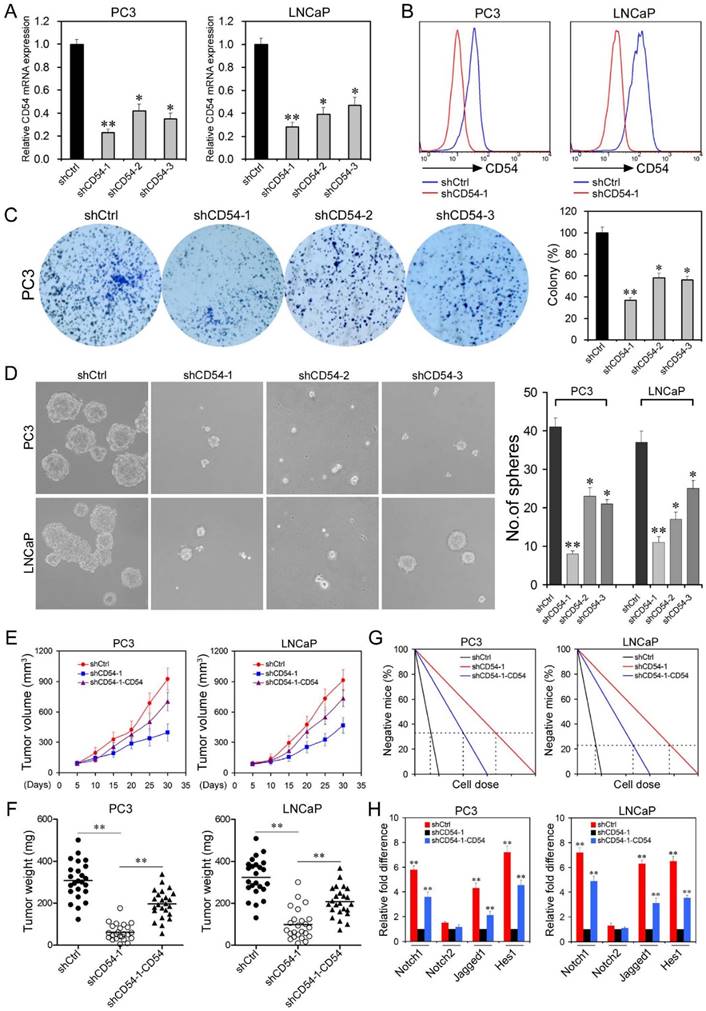
Effects of CD54 Knockdown on Prostate Cancer Stem Cells. (a) Real-time PCR analysis of relative CD54 mRNA expression in LNCaP and PC3 cells after transfection with control shRNA (shCtrl) or shRNAs targeting CD54 (shCD54-1, shCD54-2, and shCD54-3). (b) FACS analysis of CD54 expression in PC3 and LNCaP cells transfected with shCtrl or shCD54-1. (c) Representative light microscope fields and comparative quantification of the colony formation capacity of PC3 cells after transfection with shCtrl, shCD54-1, shCD54-2, or shCD54-3. (d) Representative light microscope fields and comparative quantification of the sphere-forming capacity of LNCaP and PC3 cells after transfection with shCtrl, shCD54-1, shCD54-2, or shCD54-3. (e) Tumor volume measurements, measured every 5 d, in NOD/SCID mice injected with LNCaP or PC3 cells transfected with shCtrl, shCD54-1, or shCD54-1 with reconstitution of endogenous CD54 expression. (f) Final weight of tumors from NOD/SCID mice injected with LNCaP or PC3 cells transfected with shCtrl, shCD54-1, or shCD54-1 with reconstitution of endogenous CD54 expression. (g) Frequency of CSCs in LNCaP or PC3 cells injected into NOD/SCID mice. (h) Relative fold difference of the expression of various signaling pathway components in LNCaP and PC3 cells transfected with shCtrl, shCD54-1, or shCD54-1 with reconstitution of endogenous CD54 expression. * P < 0.05; ** P < 0.01.
Figure 5
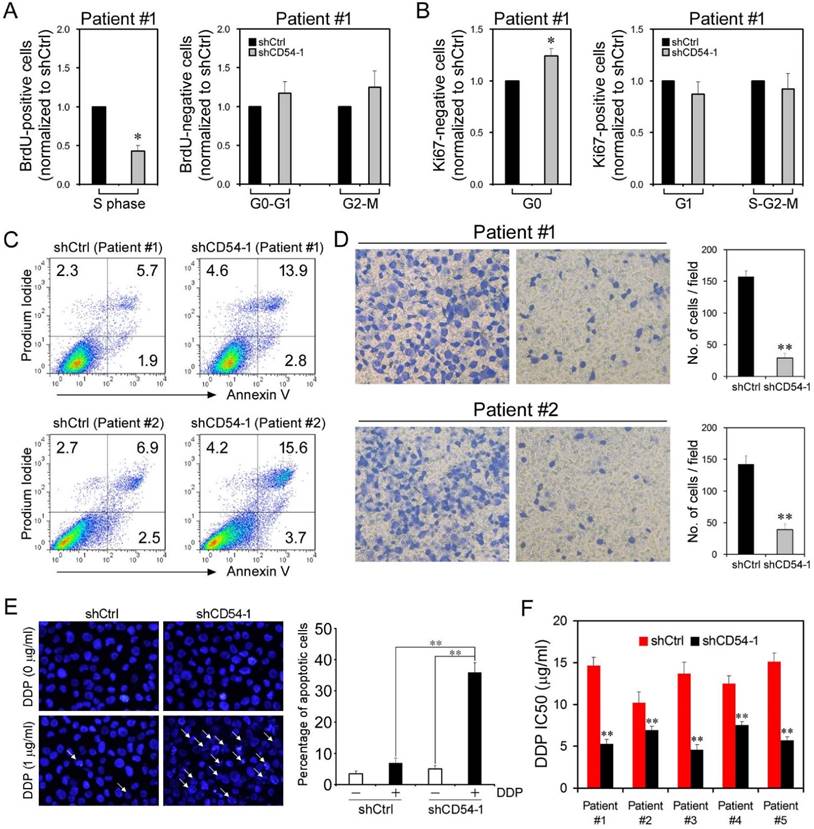
Effects of CD54 Knockdown on Cell Proliferation and Apoptosis. (a) BrdU analysis of cell cycle progression in prostate cancer patient cells transfected with shCtrl or shCD54-1. (b) Ki67 analysis of cell cycle progression in prostate cancer patient cells transfected with shCtrl or shCD54-1. (c) FACS analysis of propidium iodide- and annexin V-stained prostate cancer patient cells (from two different patients) transfected with shCtrl or shCD54-1. (d) Representative light micrograph fields and comparative quantification of the migratory capacity of prostate cancer patient cells (from two different patients) transfected with shCtrl or shCD54-1 via Transwell assay. (e) Representative microscope fields and comparative quantification of apoptosis in vehicle-treated or cisplatin (DDP)-treated PC3 cells transfected with shCtrl or shCD54-1. Arrows indicate apoptotic cells. (f) The cisplatin (DDP) IC50 values of 5 prostate cancer patient cell lines transfected with shCtrl or shCD54-1. * P < 0.05; ** P < 0.01.
CD54 Drives Tumor Formation via p38-Mediated NOTCH1 Regulation. Because of the relevance of NOTCH1 in CD54 signaling, we explored CD54-mediated signaling in further detail. Since we detected elevated NOTCH1 signaling in CD54+ compared to CD54- PC3 cells (Figure 2e and f), we measured NOTCH1 expression in prostate cancer cell lines. Compared with the prostate cell line RWPE-1, all prostate cancer cell lines tested expressed higher levels of NOTCH1 mRNA (Figure 6a). Moreover, NOTCH1 expression positively correlated with CD54 expression in these cell lines. NOTCH1 and CD54 expression were also correlated in 50 prostate cancer patients (Figure 6b), strongly suggesting that NOTCH1 is involved in CD54 signaling.
Knockdown of CD54 in LNCaP and PC3 cells significantly inhibited the expression of NOTCH1 and HES1, a NOTCH1-targeted transcriptional repressor [22, 23], and the phosphorylation of p38, a downstream kinase of CD54/ICAM-1 [24] (Figure 6c). To validate whether CD54 regulates CSCs through NOTCH1, we overexpressed NOTCH1 in CD54-silenced PC3 cells and found that NOTCH1 overexpression significantly rescued the sphere-forming capacity, which was inhibited by CD54 knockdown alone (Figure 6d).
As p38 phosphorylation was linked to CD54 signaling, we further aimed to clarify the relationship between p38 and NOTCH1 in prostate cancer cells. We transfected a GFP-tagged NOTCH1 promoter into LNCaP and PC3 cells and found that NOTCH1 transcription was significantly suppressed by BIRB 796, a p38 inhibitor (Figure 6e). Consistent with the cell line data, BIRB 796 inhibited p38 phosphorylation and NOTCH1 expression in cancer cells from two patients with prostate cancer (Figure 6f).
Next, we evaluated the therapeutic effect of shCD54 against prostate cancer patient tumor samples in mice xenografts. We treated tumor cells from two patients with prostate cancer with either shCD54 or shCtrl and transplanted the treated cells into 12 NOD/SCID mice. The PDX tissues were validated for CD54 expression by IHC before xenotransplantation into mice (Figure S6). CD54 silencing significantly reduced the tumor volume (Figure 6g) and enhanced the survival time (Figure 6h) of mice, indicating the potential for CD54 knockdown as a therapy against prostate cancer.
Discussion
CSCs play key roles in the tumorigenesis, metastasis and drug resistance of many cancer types [5, 6]. The CSC hypothesis posits that cancers are maintained in a hierarchical organization of rare ''cancer stem cells'' that divide rapidly into amplifying cells and differentiated tumor cells [25]. Due to this unique survival mechanism, this rare population of cells is resistant to the current chemotherapies because the treatments are largely targeted at eradicating the rapidly proliferating tumor bulk [26]. However, the stem cells, which express drug transporters, are more likely to survive. These cells repopulate the tumor, resulting in a heterogeneous tumor composed of stem cells and committed but variably differentiated offspring [27]. It has been reported that CSCs are resistant to chemotherapy and enriched after treatment in many cancer types, such as breast, lung, and liver cancer [28-31]. Although there are a few identified CSC markers [32], specific and reliable CSC markers are lacking for several individual cancer types. Furthermore, many of the known CSC markers are not expressed exclusively on CSCs; therefore, more specific CSC markers are needed for the precise clarification of the tumorigenic function of CSCs and for CSC-based targeted therapies. We used chemotherapy to enrich CSCs in a prostate cancer xenograft mouse model and performed microarray analyses to identify genes with elevated expression after chemotherapy treatment of prostate cancer cells. This analysis revealed CD54 as a potential marker for prostate CSCs.
CD54, also known as ICAM-1 (intercellular adhesion molecule 1), is an adhesion molecule belonging to the immunoglobulin superfamily [33]. It is constitutively expressed on endothelial cells and some immune cells at low levels [34-36]. CD54 expression is upregulated upon cytokine stimulation during the inflammatory response [37, 38]. CD54 is also expressed in many tumor types and is involved in the tumorigenesis of various cancers such as lung cancer, breast cancer, melanoma, and myeloma [39-43]. Our study showed that CD54 expression is also upregulated in prostate cancer cells compared to control cells. Furthermore, CD54 expression is enriched after chemotherapy and may contribute to chemoresistance. We also identified that CD54+ prostate cancer cells show characteristics of CSCs both in vitro and in vivo. Recent studies also showed that CD54 is a potential CSC marker of esophageal squamous cell carcinoma [44], glioblastoma [45], and prostate cancer [45]. Together, these data suggested that CD54 was a reliable marker for CSCs.
Considering the essential role of CD54 in prostate CSCs, we further aimed to clarify CD54 signaling within prostate CSCs. Many signaling pathways have been identified in CSCs, including the Hedgehog [15, 16], Notch [17, 18], and Wnt signaling pathways [19, 20]. In addition, the JAK/STAT pathway also interacts with CD54 and vascular cell adhesion molecule (VCAM) during the formation of tumor spheroids from prostate cancer stem cells [46]. A recent study demonstrated that the lncRNA ICR specifically regulates CSC properties in CD54+ hepatocellular carcinoma [47]. We compared the expression of some main signaling mediators in CD54+ and CD54- prostate cancer cells and identified NOTCH1 as a key mediator in prostate CSCs. Using inhibitor assays, we also found that p38 phosphorylation occurred upstream of NOTCH1 in CD54 signaling. It has been reported that notch-1 could be activated by their counterligands CD54 and jagged-1, which were involved in the plasticity of vascular niches [47]. The LFA-1/ICAM-1 interaction could upregulate notch signaling during T-cell migration [48]. Taken together, our data identified a critical role of the CD54-p38-NOTCH1 axis in prostate CSCs.
Figure 6
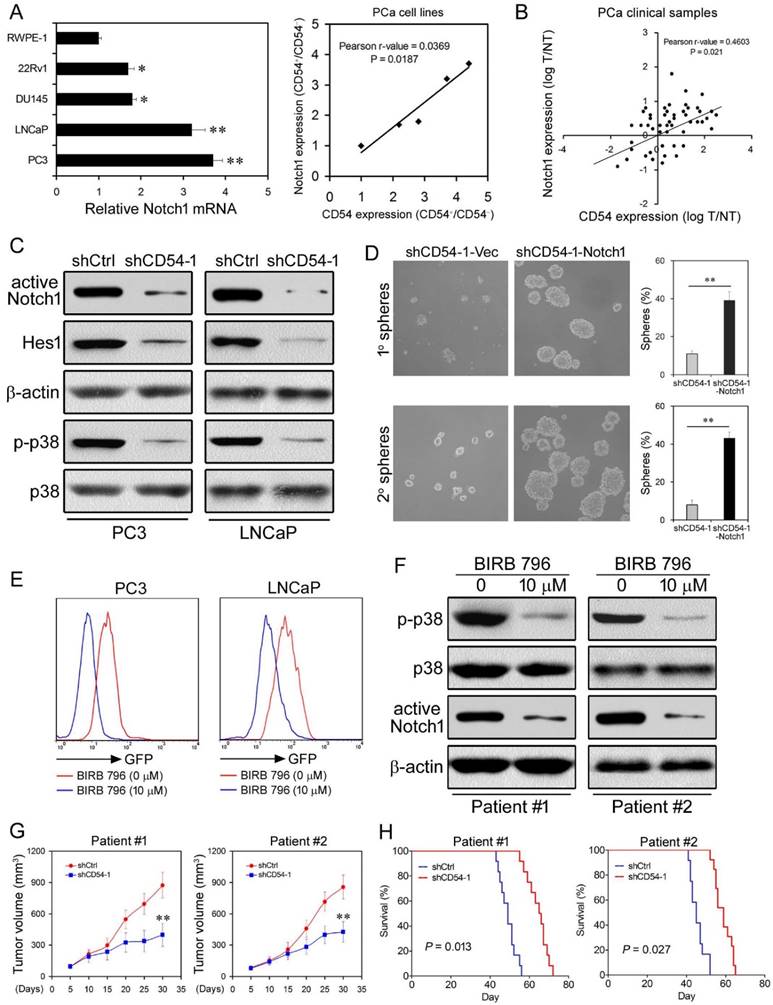
CD54 Drives Tumor Formation via p38-Mediated NOTCH1 Regulation. (a) Relative NOTCH1 mRNA expression and positive correlation between NOTCH1 and CD54 expression in a control prostate cell line (RWPE-1) and in prostate cancer cell lines (22Rv1, DU145, LNCaP, and PC3). (b) Positive correlation between NOTCH1 and CD54 expression in tumors from prostate cancer patients. (c) Western blots of active NOTCH1 and HES1 expression as well as p38 phosphorylation in LNCaP and PC3 cells transfected with shCtrl or shCD54-1. β-actin is shown as a loading control, and total p38 is a control for p38 phosphorylation. (d) Representative light micrograph fields and comparative quantification of the sphere-forming capacity of PC3 cells at primary and secondary levels after transfection with either shCD54-1 or shCD54-1 with subsequent exogenous expression of NOTCH1. € FACS analysis of GFP-NOTCH1 promoter expression in LNCaP and PC3 cells with or without treatment using the p38 inhibitor BIRB 796. (f) Representative Western blots of p38 phosphorylation and NOTCH1 expression in tumor cells from two prostate cancer patients with or without BIRB 796 treatment. (g) Tumor volume measurements of NOD/SCID mice injected with tumor cells (from two prostate cancer patients) transfected with shCtrl or shCD54-1. (h) Survival curves of NOD/SCID mice injected with tumor cells (from two prostate cancer patients) transfected with shCtrl or shCD54-1. * P < 0.05; ** P < 0.01.
Tumor samples from prostate cancer patients showed that CD54 expression was higher in recurrent and metastatic cancer cells than in primary tumors. CSCs play a key role in tumor metastasis [49, 50]. It has been proposed that CSCs can be stationary (to establish tumor growth) or mobile (to promote tumor metastasis) [51], and these two distinct populations of CSCs have been found in human cancer tissues [52]. CSCs could be induced into the epithelial mesenchymal transition (EMT), which allows for the cells to become mobile or migrating CSCs. Migrating CSCs cells divide asymmetrically. One daughter cell continues proliferating and differentiating, and the remaining CSC migrates a short distance before either undergoing a new asymmetric division or beginning to disseminate through blood or lymphatic vessels to produce a metastasis [51-53]. In addition to the correlation of CD54 expression with prostate cancer recurrence, CD54+ cells from cancer patients had higher tumor-initiating capacity in xenograft models than CD54- cells, confirming the role of CD54+ cells as CSCs.
Having identified the role of CD54+ CSCs in prostate tumorigenesis, we used shRNA to knockdown CD54 expression in prostate cancer cells. CD54 silencing inhibited the tumorigenic capacity and prolonged the survival time of xenografted mice. Our findings provide support that the development of therapeutic strategies to target CD54 has promising potential to translate into new clinical treatment modalities against prostate cancer. Moreover, downstream p38-NOTCH1 signaling pathways can also be considered as targets for developing prostate cancer treatments.
Conclusion
Prostate cancer stem cells (CSCs) are considered one of the main reasons for tumor recurrence after chemotherapy. Here, we show that CD54 is a reliable new marker for prostate CSCs. CD54 plays a critical role in the self-renewal and tumorigenesis of prostate CSCs. Targeting CD54-Notch1 signaling could be a potential novel strategy for future clinical prostate cancer treatment.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81672956, 81472413, 81130041, 81472679), Shenzhen Science and Technology Development Funds Program (CXZZ20130516153248144); Shenzhen Strategic Emerging Industry Development Special Funds Program (JSGG20130411091246833); Major Program of Scientific Research Foundation from Yunnan Provincial Department of Education (2014Z052); Science and Technology Program of Yunnan Province (2014RA067).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
2. Chang AJ, Autio KA, Roach M 3rd, Scher HI. High-risk prostate cancer-classification and therapy. Nat Rev Clin Oncol. 2014;11:308-23
3. Wong YN, Ferraldeschi R, Attard G, de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat Rev Clin Oncol. 2014;11:365-76
4. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN. et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England journal of medicine. 2004;351:1502-12
5. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
6. Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012;13:e83-9
7. Stoyanova T, Cooper AR, Drake JM, Liu X, Armstrong AJ, Pienta KJ. et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc Natl Acad Sci U S A. 2013;110:20111-6
8. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211-5
9. Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR. et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell stem cell. 2012;10:556-69
10. Vidal SJ, Rodriguez-Bravo V, Galsky M, Cordon-Cardo C, Domingo-Domenech J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. 2014;33:4451-63
11. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R. et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22:373-88
12. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci U S A. 2014;111:E5429-38
13. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nature reviews Cancer. 2012;12:237-51
14. Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:2862-70
15. Merchant AA, Matsui W. Targeting Hedgehog-a cancer stem cell pathway. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:3130-40
16. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J. et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776-9
17. Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B. et al. Targeting Notch to target cancer stem cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:3141-52
18. McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS. et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci U S A. 2012;109:E2939-48
19. Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013;25:254-64
20. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-50
21. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of immunological methods. 2009;347:70-8
22. Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J. et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18:268-81
23. Moriyama M, Osawa M, Mak SS, Ohtsuka T, Yamamoto N, Han H. et al. Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J Cell Biol. 2006;173:333-9
24. Wang Q, Doerschuk CM. The p38 mitogen-activated protein kinase mediates cytoskeletal remodeling in pulmonary microvascular endothelial cells upon intracellular adhesion molecule-1 ligation. J Immunol. 2001;166:6877-84
25. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annual review of medicine. 2007;58:267-84
26. Al-Hajj M. Cancer stem cells and oncology therapeutics. Current opinion in oncology. 2007;19:61-4
27. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature reviews Cancer. 2005;5:275-84
28. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C. et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109-23
29. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749-58
30. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell stem cell. 2011;9:50-63
31. Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L. et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009;106:16281-6
32. Medema JP. Cancer stem cells: the challenges ahead. Nature cell biology. 2013;15:338-44
33. Yang Y, Jun CD, Liu JH, Zhang R, Joachimiak A, Springer TA. et al. Structural basis for dimerization of ICAM-1 on the cell surface. Mol Cell. 2004;14:269-76
34. Clark PR, Manes TD, Pober JS, Kluger MS. Increased ICAM-1 expression causes endothelial cell leakiness, cytoskeletal reorganization and junctional alterations. J Invest Dermatol. 2007;127:762-74
35. Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep. 2009;61:22-32
36. Long EO. ICAM-1: getting a grip on leukocyte adhesion. J Immunol. 2011;186:5021-3
37. Qureshi MH, Cook-Mills J, Doherty DE, Garvy BA. TNF-alpha-dependent ICAM-1- and VCAM-1-mediated inflammatory responses are delayed in neonatal mice infected with Pneumocystis carinii. J Immunol. 2003;171:4700-7
38. Ren G, Zhao X, Zhang L, Zhang J, L'Huillier A, Ling W. et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol. 2010;184:2321-8
39. Dowlati A, Gray R, Sandler AB, Schiller JH, Johnson DH. Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab-an Eastern Cooperative Oncology Group Study. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:1407-12
40. Hamai A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F. et al. ICAM-1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer research. 2008;68:9854-64
41. Guo P, Huang J, Wang L, Jia D, Yang J, Dillon DA. et al. ICAM-1 as a molecular target for triple negative breast cancer. Proc Natl Acad Sci U S A. 2014;111:14710-5
42. Lin YC, Shun CT, Wu MS, Chen CC. A novel anticancer effect of thalidomide: inhibition of intercellular adhesion molecule-1-mediated cell invasion and metastasis through suppression of nuclear factor-kappaB. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:7165-73
43. Veitonmaki N, Hansson M, Zhan F, Sundberg A, Lofstedt T, Ljungars A. et al. A human ICAM-1 antibody isolated by a function-first approach has potent macrophage-dependent antimyeloma activity in vivo. Cancer Cell. 2013;23:502-15
44. Tsai ST, Wang PJ, Liou NJ, Lin PS, Chen CH, Chang WC. ICAM1 Is a Potential Cancer Stem Cell Marker of Esophageal Squamous Cell Carcinoma. PloS one. 2015;10:e0142834
45. Guichet PO, Guelfi S, Teigell M, Hoppe L, Bakalara N, Bauchet L. et al. Notch1 stimulation induces a vascularization switch with pericyte-like cell differentiation of glioblastoma stem cells. Stem cells. 2015;33:21-34
46. Duzagac F, Inan S, Ela Simsek F, Acikgoz E, Guven U, Khan SA. et al. JAK/STAT pathway interacts with intercellular cell adhesion molecule (ICAM) and vascular cell adhesion molecule (VCAM) while prostate cancer stem cells form tumor spheroids. Journal of BUON: official journal of the Balkan Union of Oncology. 2015;20:1250-7
47. Guo W, Liu S, Cheng Y, Lu L, Shi J, Xu G. et al. ICAM-1-related non-coding RNA in cancer stem cells maintains ICAM-1 expression in Hepatocellular Carcinoma. Clinical Cancer Research. 2015 pii: clincanres.3106.2014
48. Verma NK, Dempsey E, Long A, Davies A, Barry SP, Fallon PG. et al. Leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction induces a novel genetic signature resulting in T-cells refractory to transforming growth factor-beta signaling. The Journal of biological chemistry. 2012;287:27204-16
49. Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell stem cell. 2014;14:306-21
50. Adorno-Cruz V, Kibria G, Liu X, Doherty M, Junk DJ, Guan D. et al. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer research. 2015;75:924-9
51. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nature reviews Cancer. 2005;5:744-9
52. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M. et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell stem cell. 2007;1:313-23
53. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C. et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820-7
Author contact
![]() Corresponding author: Lei LI, Department of Urology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China; Phone: 0086-29-85323661; E-mail: lilydrcom.
Corresponding author: Lei LI, Department of Urology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China; Phone: 0086-29-85323661; E-mail: lilydrcom.
Citation styles
APA
Li, C., Liu, S., Yan, R., Han, N., Wong, K.K., Li, L. (2017). CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics, 7(1), 67-80. https://doi.org/10.7150/thno.16752.
ACS
Li, C.; Liu, S.; Yan, R.; Han, N.; Wong, K.K.; Li, L. CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics 2017, 7 (1), 67-80. DOI: 10.7150/thno.16752.
NLM
Li C, Liu S, Yan R, Han N, Wong KK, Li L. CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics 2017; 7(1):67-80. doi:10.7150/thno.16752. https://www.thno.org/v07p0067.htm
CSE
Li C, Liu S, Yan R, Han N, Wong KK, Li L. 2017. CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics. 7(1):67-80.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.