Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(12):2235-2249. doi:10.7150/thno.16425 This issue Cite
Research Paper
Multivalent Targeting Based Delivery of Therapeutic Peptide using AP1-ELP Carrier for Effective Cancer Therapy
Vijaya Sarangthem1*, Yunjae Kim1*, Thoudam Debraj Singh2*, Bo-Yeon Seo1, Sun-Ha Cheon1, Young-Jin Lee1, Byung-Heon Lee1, Rang-Woon Park1 ![]()
1. Department of Biochemistry and Cell Biology, School of Medicine, and Cell & Matrix Research Institute, Kyungpook National University, Daegu 41944, Republic of Korea.
2. Department of Nuclear Medicine, School of Medicine, Kyungpook National University, Daegu 41944, Republic of Korea.
* These authors contributed equally to the study.
Received 2016-6-8; Accepted 2016-8-28; Published 2016-9-25
Abstract

Elastin-like polypeptide (ELP)-based drug delivery has been utilized for various applications including cancer therapies for many years. Genetic incorporation of internalization ligands and cell-targeting peptides along with ELP polymer enhanced tumor accumulation and retention time as well as stability and activities of the drug conjugates. Herein, we described a unique delivery system comprised of genetically engineered ELP incorporated with multiple copies of IL-4 receptor targeting peptide (AP1) periodically and proapoptotic peptide (KLAKLAK)2 referred to as AP1-ELP-KLAK. It triggered thermal-responsive self-assembly into a nanoparticle-like structure at physiological body temperature and stabilized its helical conformation, which is critical for its membrane-disrupting activities. Increased IL-4 receptor specific cellular internalization was associated with the enhanced cytotoxic effect of (KLAKLAK)2 peptide. Additionally, multivalent presentation of targeting ligands by AP1-ELP-KLAK significantly enhanced intratumoral localization and prolonged the retention time compared to ELP-KLAK, non-targeted control. Systemic administration of AP1-ELP-KLAK significantly inhibited tumor growth by provoking cell apoptosis in various tumor xenograft models without any specific organ toxicity. Thus, our newly designed AP1-ELP-KLAK polymer nanoparticle is a promising candidate for effective cancer therapy and due to the simple preparative procedures of ELPs, this platform can be used as a good carrier for tumor-specific delivery of other therapeutics.
Keywords: IL-4 receptor, AP1, multivalent targeting, micelle, apoptosis, drug delivery.
Introduction
Recently, therapeutic peptides classified as biopharmaceuticals have gained attention as a new generation of cancer therapeutics with enhanced efficiency and diminished toxicity [1,2]. It can be easily synthesized and optimised relevelant to clinical application with lower immunogenicity [3]. Many studies have revealed that cationic peptides can be used as anti-tumor agents due to their ability to target anionic phospholipids and disrupt negatively charged membranes [4]. Despite their demonstrated activities at the molecular or cellular level, they often fail to have sufficient effects due to their unsatisfactory pharmacokinetic profiles, which include poor bioavailability, low stability, short half-life, and poor penetration across the biological membrane [5]. Thus, design of ideal delivery systems for these biologics is necessary, especially to achieve site-specific pharmacological actions in vivo. The antibacterial amphipathic KLAK peptide (KLAKLAK)2 is widely used as a membrane-disrupting agent in various tumor models. This peptide forms an α-helix with cationic amino acids on cell surfaces and preferentially disrupts anionic prokaryotic cytoplasmic and eukaryotic mitochondrial membrane integrity, triggering apoptotic program cell death [6-8]. As KLAK peptide is not efficiently internalized into tumor cells, it must be coupled with cell-penetrating moieties or receptor ligands in order to enter the target cell via receptor-independent and -dependent mechanisms. Recently, fusion of KLAK peptide with tumor-targeting motifs such as RGD [9], Interleukin-11 [10], and antibodies [11], have been exploited to selectively inhibit various tumor cells. Moreover, incorporation of therapeutic peptides into carriers such as liposomes, polyionic complexes, self-assembly nanogels, microspheres, and PEGylation has been shown to improve biological stability. Similarly, due to low bioavailability, high toxicity and prone to enzymatic degradation KLAK peptide are required to be conjugated with nanoparticles or chemically modified to form nano scaled self- assembles [12]. Despite their high efficacy, these models need to overcome several intracellular and extracellular biological barriers under in vivo conditions. This is sometimes less effective due to barriers in transforming drugs from inactive to active form as well as low drug loading efficiency, which makes it difficult to completely remove tumor cells [13-14]. Moreover, due to difficulties in formulation, site inflexibility, stoichoimetry, high costs, and purification, its wide application is very limited.
Besides thermal-sensitive biopolymer, elastin-like polypeptides (ELPs) offer many advantages as drug delivery systems since they can be designed and synthesized using genetic engineering techniques [15]. The sequence, molecular weights, numbers and locations of reactive sites for drug conjugation of ELPs can be precisely tuned to improve their pharmacokinetic properties [16], whereas the MW and polydispersity of synthetic polymers is hard to control. ELPs can be easily expressed with high yields (100-200 mg L-1) from E. coli and rapidly purified by exploiting their phase transition behavior [17-18]. The studies have shown that fusion of cell-targeting peptides or internalization ligands to the hydrophilic domain of diblock ELP stimulates self-assembly into a spherical micelle-like structure with multivalent display of functional peptides on the corona, thereby increasing binding activities, extravasation, and internalization rates [19-22]. Further, incorporation of the CD12 ligand NGR tripeptide and αVβ3 ligand RGD into the N-terminus of diblock ELP copolymers [V1A8G7]64/ELP[V]90 stimulates self-assembly into monodisperse spherical micelles presenting targeting ligands on theirs coronas and enhances binding activity as well as tumor vasculature-specific localization [23-24]. Unique features in cancer cells, such as overexpression of several receptors [25-26], up-regulation of certain enzymes, and lower pH [27-28], can be used in the development of several targeted drug carriers. Likewise, Interleukin-4 receptor (IL-4R) is highly expressed in a wide variety of solid tumors and confers resistance against drug-induced cancer cell death [29-30]. IL-4R-based immunotoxin and drug delivery has been utilized for the treatment of several tumor types [31-33]. Previously, our group reported an AP1 peptide that selectively binds to IL-4R in vivo using a phage display technique [34-35].
Herein, we postulate a new platform for peptide drug delivery systems for selective inhibition of tumors. In a previous study, we successfully generated an ELP-based multivalent targeting polymer (AP1-ELP) by incorporating a tandem repeat of AP1 along the ELP polymer backbone. We also observed that [AP1-V12]6 polymer consisting of six repeats of AP1 increased binding avidity and affinity towards IL-4R. Additionally, [AP1-V12]6 was highly accumulated in IL-4R-expressing tumor tissue after intravenous injection and was retained up to 24 h [36], demonstrating its efficacy as a potent drug carrier for cancer therapy. To enhance the proapoptotic activity of KLAK for inhibition of tumors based on IL-4R expression, we generated a novel chimeric polypeptide in which the carboxy terminus of ELP or AP1-ELP was genetically fused with the KLAK peptide sequence. The chimeric polypeptides ELP-KLAK and AP1-ELP-KLAK readily self-assembled into a spherical micelle forming a nanoparticle-like structure at physiological body temperature. AP1-ELP-KLAK was internalized efficiently by cancer cells and induced apoptosis in an IL-4R dependent manner. Further studies clearly confirmed strong tumor accumulation of AP1-ELP-KLAK with longer retention time as well as significant inhibition of tumor growth in various tumor models.
Materials and methods
Cell culture
MDA MB231 (human breast carcinoma), H460 (human lung carcinoma), and B16F10 (murine melanoma) cells were obtained from the American Type Culture Collection (ATCC). MDA MB231 cells were maintained in Dulbecco's modified Eagle's medium (DMEM), whereas H460 and B16F10 cells were grown in RPMI-1640 (Hyclone, Thermo Scientific, Logan, UT) medium supplemented with 10% fetal bovine serum (Hyclone), 100 U mL-1 of penicillin, and 100 µg mL-1 of streptomycin (Sigma Aldrich, St. Louis, MO, USA). Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.
ELP protein purification
For protein expression, modified pET 25b+ vector containing ELP-KLAK and AP1-ELP-KLAK genes were transformed with BL21 (DE3) competent E. coli cells (Invitrogen, Carlsbad, CA, USA). Starter cultures were prepared by inoculating transformed E. coli cells from glycerol stocks stored at -80 °C in 15 mL of Circle grow media (MP Biomedicals, CA, USA) containing 100 μg mL-1 of ampicillin (AMRESCO LLC, OH, USA). Cells were grown overnight at 37 °C in a shaking incubator. Starter cultures were inoculated in 800 mL of Circle grow media containing ampicillin and incubated for 37 °C until the optical density was between 0.8-1. After 3 h, 1 mM IPTG (Carbosynth Limited, Berkshire, UK) was added to induce protein synthesis for 4 h. Cells were then harvested by centrifugation at 4,000 rpm for 20 min at 4 °C and suspended in 10 mL of phosphate-buffered saline (PBS). ELP protein was purified using the inverse transition cycling (ITC) method [17]. After four rounds of ITC, protein purity and molecular weight were checked by SDS-PAGE (Bio-Rad Inc., Hercules, CA) followed by copper chloride staining (Sigma Aldrich, St. Louis, MO, USA). ELP concentration was measured using a Cary UV-visible spectrophotometer (Agilent Technologies, CA, USA) using an extinction coefficient of 5690 M-1 cm-1 for ELP-KLAK and AP1-ELP-KLAK. Purified proteins were further filtered with a 0.2 μM Whatmann syringe filter (GE Healthcare UK limited, Buckinghamshire, UK) to remove other cell contaminants.
Thermal characterization
Thermal characteristics (Tt) of ELP-KLAK and AP1-ELP-KLAK were determined by measuring the optical densities of protein solutions (10 μM) at different temperatures using a Cary UV-visible spectrophotometer equipped with a temperature controller (Agilent Technologies) at 350 nm. The absorbance was measured at temperatures ranging from 20 °C to 50 °C in 1°C min-1 increments.
MALDI TOF/TOF MS analysis
Molecular weights of ELP-KLAK and AP1-ELP-KLAK were examined using an UltrafleXtreme (Bruker). Protein samples were dissolved with 0.1% trifluoroacetic acid and mixed with an equal volume of matrix solution (1:1). Mixture (1 µl) was then applied to a standard steel target for drying at room temperature. The MS spectra were obtained after calibration with standards.
Dynamic light scattering (DLS)
Particle sizes of proteins were measured at temperatures ranging from 20 °C to 50 °C in 1 °C min-1 increments using a Wyatt dynaPro NanoStar (Wyatt technology, Santa Barbara, CA, USA). The size distribution of the hydrodynamic radius was measured by a DLS detector at 90 degrees.
Cryo-TEM imaging
For further validation of particle size, cryo-TEM images were taken using an FEI Tecnai transmission electron microscope (Oregon, USA). Filtered protein samples (25 μM) were incubated at 37 °C for 10 min to induce micelle formation, and a small drop of protein was air dried on carbon coated copper grids for 5 min. Further negative stained with uranyl acetate and examined by TEM.
Circular dichroism
Secondary structures of proteins were determined using a Jasco-1500 circular dichroism spectrometer [JASCO, Maryland]. Proteins samples (25 μM) were used for analysis, and CD spectra were measured at 37 °C in a 0.1 cm path length cell. The spectra were recorded from 190 to 250 nm at a scan speed of 50 nm min-1.
IL-4R expression analysis
MDA MB231 and H460 (5× 105) cells were incubated with APC anti-human CD124 (IL-4Rα) and respective IgG isotype antibody (Biolegend, San Diego) as a control for 1 h at room temperature. B16F10 cells were also incubated with APC anti-mouse CD124 (IL-4Rα) and respective IgG isotype antibody (Biolegend). The level of IL-4R expression was measured by flow cytometry (BD Bioscience, San Jose, CA, USA). Ten thousand events were collected for each sample, and the percentage of APC anti-human CD124 bound to cells was calculated by comparison with isotype control.
Confocal microscopy
MDA MB231 and H460 (8 × 104) cells were seeded on a four-chambered slide and incubated for 24 h. Cells were then treated with 10 μM Alexa 488 labeled ELP-KLAK and AP1-ELP-KLAK for 1 h at 4 °C and 37 °C. Cell nuclei and cell membranes were stained with Hoechst 33342 and Wheat germ agglutinin Alexa Fluor 594 conjugate (Molecular Probes, Inc., Eugene) after fixation with 4% paraformaldehyde. Images were captured and analyzed using a Zeiss LSM-510 Meta confocal microscope.
Cell viability assay
Cell cytotoxicity was measured using a Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan). This colorimetric assay monitors the number of viable cells in vitro. MDA MB231, B16F10, and H460 (5× 103) cells were seeded in 96-well plates and incubated for 16 h, followed by treatment with different concentrations of ELP-KLAK and AP1-ELP-KLAK proteins (0.625, 1.25, 2.5, 5, and 10 μM) for 48 h. Cell viability was determined by measuring the absorbance at 450 nm.
Apoptosis assay
MDA MB231 and B16F10 (5×105/well) cells were plated in 6-well plates overnight and then treated with different concentrations of ELP-KLAK and AP1-ELP-KLAK (5, 10, 20 μM) at 37 ºC for 4 h. Both floating and adherent cells were harvested, washed with PBS, and mixed with annexin V binding buffer. Further, 2 μg mL-1 of propidium iodide (PI) and 5 μL of FITC annexin V reagent were added to stain necrotic and apoptotic cells (BD Biosciences, San Diego, CA, USA). Annexin V and PI-positive cells were measured using flow cytometry.
Animal model
All studies were conducted in strict accordance with the rules recommended by the National Institutes of Health (NIH) for the Care and Use of Laboratory Animals. The protocol used for animal experiments were approved by the Committee on the Ethics of Animal Experiments of Kyungpook National University (Permit Number: KNU 2015-0043). Female nude mice (BALB/c nude; body weight, 20 ± 3 g) and C57BL/6 black female mice were kept in a specific pathogen-free environment with relative humidity, controlled light, and adequate access to food and water.
Serum stability test
Stability of conjugated dye was checked before beginning the in vivo experiments. FPR 675-labeled ELP-KLAK and AP1-ELP-KLAK were incubated in fresh mouse plasma for different time intervals (1, 3, 6, 12, and 24 h), and the protein was precipitated by using ITC. Two rounds of ITC were performed, and fluorescence intensities of the dye in the supernatant and pellet were measured at excitation and emission wavelengths of 675 nm and 698 nm, respectively, using a SpectraMax M plate reader (Molecular Devices, CA, USA. Percentage of released dye was determined in accordance with standard curve obtained from known quantities of labeled proteins.
Pharmacokinetics analysis
MDA MB231 tumor-bearing mice (100 mm3) were injected with FPR 675-labeled ELP-KLAK and AP1-ELP-KLAK (150 mg kg-1) intravenously (n = 8 for each group). At various time intervals (30 min, 1 h, 3 h, 6 h, and 24 h), 20 μl of blood was sampled from the tail vein, collected in heparinized tubes, and further centrifuged at 13,000 ×g for 5 min to collect plasma. Separated plasma (10 μL) was further diluted with 150 μL of PBS, and fluorescence intensity was measured using a SpectraMax M plate reader. Plasma fluorescence intensities were calculated from the standard curve generated using known concentrations of labeled proteins (0.625-20 μM). Pharmacokinetic parameters were determined by non-compartmental analysis.
In vivo biodistribution imaging
The IL-4R specific tumor targeting activities of ELP-KLAK and AP1-ELP-KLAK were examined using the eXplore Optix system (ART Advanced research technologies Inc., Montreal, Canada). Tumor xenograft mice were generated by implanting a MDA MB231 cell suspension (5×106 cells) with Matrigel (1:1 in PBS) (s.c) subcutaneously into the right flank of 5-week-old BALB/c nude female mice. When tumor size reached a volume of approximately 100-200 mm3, mice were anesthetized and injected with FPR 675-labeled ELP-KLAK (n = 10) control or 150 mg kg-1 of AP1-ELP-KLAK (n = 10) intraperitoneally or intraveneously. In vivo fluorescence images were taken before or after injection at various time points (1, 3, 6, 12, and 24 h, respectively).
Ex vivo imaging and immunohistochemistry
To analyze ex vivo organ distribution, animals were euthanized at 6 h post-injection with CO2. All major organs (liver, kidney, spleen, heart, and lung) along with tumor tissues were isolated and washed in PBS and ex vivo fluorescence images were taken (n = 10) using the eXplore Optix system with FPR-675 fluorophore excitation at 675nm and emission at 698 nm. Fluorescence intensity within the region of interest (ROI) of each organ was analyzed. Tumor tissues were further fixed with 4% paraformaldehyde overnight and rapidly frozen. Tissue slices (8-μm thick) were prepared using a cryo-microtome and stained with anti-IL-4R antibody (R&D Systems; 1:100), followed by Alexa 488-labeled goat anti-mouse IgG secondary antibody (1:200). ELP-KLAK and AP1-ELP-KLAK tumor accumulation was observed under a confocal microscope after nuclei were stained with DAPI.
Tumor inhibition study
Female BALB/c nude mice were implanted with MDA MB231 cells as described above. When tumor size reached a volume of ~50 - 100 mm3 after 14 days of implantation, mice were divided into four groups (n = 20 per group). Treatments were initiated by daily IP injection of respective proteins (150 mg kg-1) or PBS as a control for 14 days. Tumor volume and body weight were monitored every other day. Tumor volume was measured using the formula: Volume (mm3 = (width2× length)/2. Mice were sacrificed when tumor volume reached 1,000 mm3 or ulceration occurred. Growth inhibition data were analyzed using Graph Pad Prism 5.
Tumor inhibition was further studied in another xenograft mice model. B16F10 (5×105) cells stably expressing the RFP gene were injected into the right flank of C57BL/6 black female mice subcutaneously. Proteins (AP1-ELP, ELP-KLAK, and AP1-ELP-KLAK) were administered intraperitoneally when tumor volume reached ~50 - 100 mm3 after 7 days (n = 10), continuously for 14 days. Tumor size was monitored using a caliper every other day. Mice were sacrificed when tumor volume reached 1,000 mm3.
Systemic toxicity in vivo
The hematological and biochemicals parameters were checked at end of the therapy in order to assess off-target toxicity result from frequent administration of polypeptides. For hematological parameters the complete blood was collected, numbers of white blood cells (WBC), red blood cell (RBC), platelets (PLT) and concentration of hemoglobin (HGB) were measured (Chemon Inc. (Suwon, Korea). Whereas for biochemical analysis serum was collected from total blood from each mice group and measured the contents including aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine (CRE), blood urea nitrogen (BUN), lactate dehydrogenase (LDH), creatine phosphokinase (CPK).
TUNEL staining
Tumor tissues from each group of mice were isolated and fixed with 4% paraformaldehyde. Cryo sections with a thickness of 8 μm were prepared from frozen tumor tissues and subjected to TUNEL staining to assess apoptotic cell death in tissue sections. Apoptotic cells were detected by deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay conjugated with FITC (TUNEL Apo-Green Detection Kit, Biotool.com, Seoul, Korea).
H and E staining
All major organs (liver, kidney, spleen, heart, and lung) along with tumor tissues were isolated after therapy from each treatment group and fixed with 4% paraformaldehyde. Paraffin-embedded blocks were prepared for each isolated tissue and sliced to a thickness of 3 μm. Hematoxylin and eosin (H&E) staining was conducted to assess morphology.
Statistical analysis
Statistical significance was determined using Student's t-test and one-way ANOVA for comparison of two or multiple groups. ***P<0.001, **P<0.01, and *P<0.05 were considered as statistically significant and indicated by asterisks in the Figures.
Results and discussion
Design and synthesis of KLAK containing polypeptides
Proapoptotic KLAK peptide was genetically incorporated along with [(V3G3A1)3-V7]3 or [AP1-V12]6 at the C-terminus separated by a short linker to avoid steric hindrance, resulting in ELP-KLAK or AP1-ELP-KLAK, respectively. Presence of multiples copies of AP1 along with ELP pentapeptide repeat (Val-Pro-Gly-Val-Gly), increased hydrophilicity thereby elevated transition temperature in comparison with related ELP protein [36]. For non-targeted ELP control, length and composition of fourth residue in pentapeptide repeat was arranged in specific ratio in order to have same thermal characteristic. Fig. 1a and (Fig. S1, Supplementary data) show the corresponding amino acid sequences of the chimeric polypeptides ELP-KLAK and AP1-ELP-KLAK. We hypothesized that fusion of KLAK peptides to these polypeptides triggered formation of a micelle-like structure with clinical applicability at physiological body temperature. Recombinant ELP-KLAK and AP1-ELP-KLAK proteins were successfully expressed and purified with a yield of ~50 mg L-1 using the inverse transition cycling (ITC) method. The purity and size of these polypeptides were confirmed by SDS-PAGE, followed by copper chloride staining (Fig. S2a, Supplementary data). Both polypeptides were observed to migrate more than theoretical MW estimated from ELP gene. The faint extra band with twice the size of expected proteins in SDS-PAGE was seen which might be a result of dimer formation caused by the presence of the Cys residues at the C-terminus. Accurate determination of molecular weight by MALDI-TOF/TOF MS confirmed the protein sizes of ELP-KLAK (35474.8 Da) and AP1-ELP-KLAK (43619.8 Da) (Fig. S2b, Supplementary data).
Characterization of ELP-KLAK and AP1-ELP-KLAK polypeptides. (a) Schematic representation of KLAK peptide containing ELP and AP1-ELP polymers with corresponding amino acid sequences. Blue and Red represent ELP and AP1 sequences; Green, KLAK peptide. Hydrodynamic radius (nm) and turbidity profiles of ELP-KLAK (b) and AP1-ELP-KLAK (c) were monitored at different temperatures. (d) TEM images of spherical nanoparticle like structures formed by ELP-KLAK and AP1-ELP-KLAK at 37 °C, scale bars indicate 100 nm. (e) Circular dichroism spectra of polypeptides were taken at 37 °C.
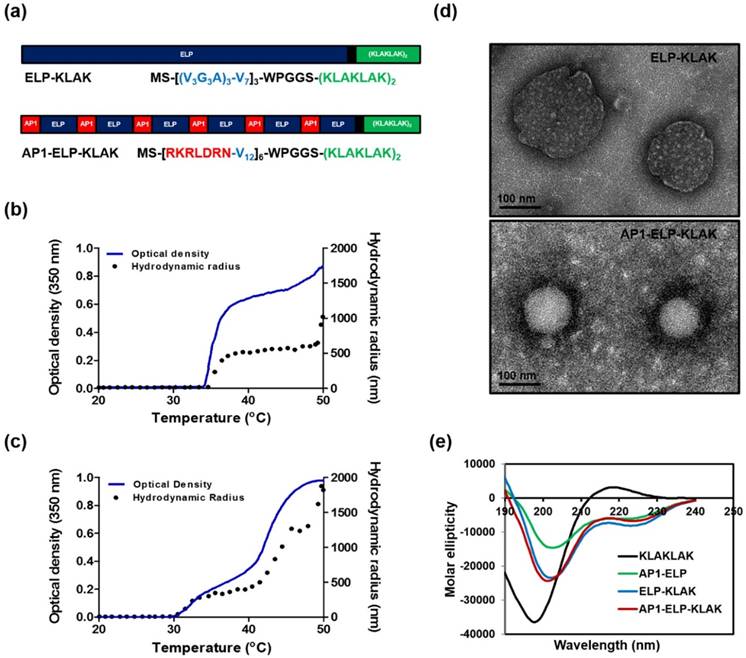
Thermal characterization and size determination
Their turbidity profiles were then measured to characterize thermal behavior as a function of temperature at a rate of 1 °C/min at OD350. Most of the monomeric ELPs underwent a single transition from highly soluble unimer to insoluble coacervation, whereas both polypeptides displayed two-step transition behavior from unimer to micelle to large aggregates in response to heat. The first transition consistent with self-assembly from unimer into micelle was observed in the range of 34 °C to 45 °C for ELP-KLAK (Fig. 1b) and around 34 °C to 42 °C for AP1-ELP-KLAK (Fig. 1c). The second transition was appeared when the temperature increased to higher than 45 °C, indicated by large micron-sized aggregate formation due to further assembly of micelles. Particle size determination by dynamic light scattering (DLS) demonstrated two-step transition behavior consistent with turbidity profiles. AP1-ELP-KLAK self-assembled to form nanoparticles with a size of ~300± 61 nm (Fig. 1c) while ELP-KLAK nanoparticles had a size of around ~500± 19 nm (Fig. 1b) at relevant physiological temperature. Elevation of temperature led to micron-sized aggregate formation in both cases. Importantly as micelle formation retained up to extended range of temperatures, indicated that both the polypeptides will remained in nanoparticle formulation upon systemic administration which is crucial of clinical application since aggregation into large micron size may hinder biodistribution by blocking the blood vessels. It was anticipated that the micelle formation pattern of the resultant polypeptides may be due to the presence of KLAK peptide, which might have increased the hydrophilic fraction. The stability of nanostructure like structure formed by polypeptides at 37 °C were further evaluated by incubating in mouse plasma. The measurement of turbidity as a function of temperature reveal the reduction in the range of micelle forming temperature but mere difference was observed with further incubation in plasma for various time intervals (Fig. S3a,b, Supplementary data). Additionally, a slight difference in the size of the nanostructure was observed upon proteins incubation with plasma when compared to proteins suspend in PBS (Fig. S3c-f, Supplementary data). Furthermore, the stability of the polypeptides in plasma upto 24 h was confirmed by SDS-PAGE (Fig. S4a,b, Supplementary data). These results validated that self-assembly of polypeptides into micelle like structure are stable in plasma at physiological temperature, thereby assuring the retention of nanoparticle like structure during systemic administration. To directly visualize the spherical nanoparticle structure, size, and shape, Cryo-TEM images were taken after incubating proteins at 37 °C. The TEM images clearly showed that ELP-KLAK and AP1-ELP-KLAK retained their spherical micelle-like structures with particle diameter sizes of 180~210 nm and 110~130 nm (Fig. 1d), respectively.
As tumor vessels are predicted to be more leaky due to irregular development of vasculature and uncontrolled angiogenesis with pores ranging in size from 200 nm to 2 µm, thus allow the diffusion of liposomes up to diameter size of 400 nm [37]. As a consequence, size of the self-assemble micelle formed by both the polypeptides can be well penetrable in tumor tissue. The difference in the size of particles observed by DLS and TEM may be due to the hydrophilic block of polypeptides used for DLS, including a large fraction of water in solution that increased hydrodynamic radii compared to TEM, which solely visualized particle size after quick drying [38-40]. Further, circular dichroism (CD) spectra confirmed a helical conformation, indicating that fusion of KLAK into these polypeptides further stabilized intact secondary structures (Fig. 1e). AP1-ELP polymer itself appeared to form α-helix structure, and fusion of KLAK peptide further increased its helix content while KLAK peptide mainly occurred in random coil conformation (Table S1, Supplementary data). On the other hand, ELP exhibited a highly disordered structure (data not shown).
Analysis of cell binding activity
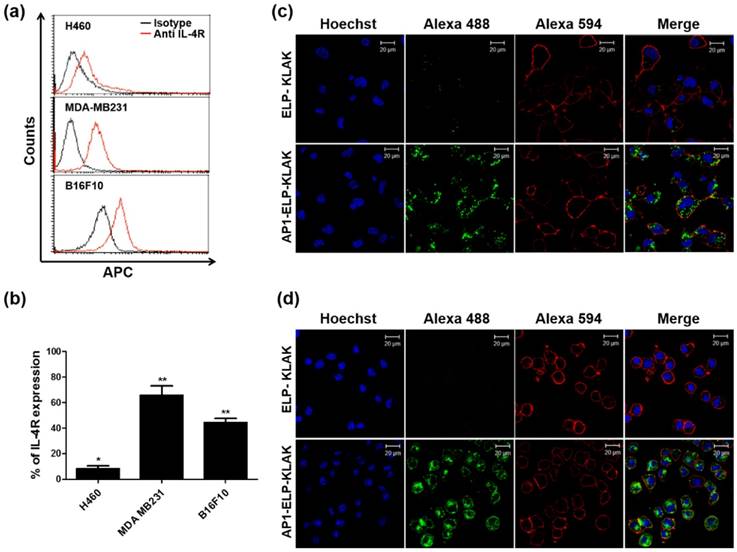
To investigate the binding activity of KLAK-containing polypeptides, the expression level of IL-4R was checked in three different cancer cell lines, H460, MDA MB231, and B16F10, by flow cytometry. In contrast to minimum IL-4R expression up to 9± 4% in H460 cells, IL-4R was highly expressed up to 67± 6% and 44 ± 5% in MDA MB231 and B16F10 cells, respectively (Fig. 2a,b). Cellular binding and internalization as determined by confocal microscopy clearly revealed greater accumulation of AP1-ELP-KLAK on the cell surface of IL-4R-overexpressing MDA MB231 (Fig. S5, Supplementary data) and B16F10 (Fig. S6, Supplementary data) cells at 4 °C. Further, AP1-ELP-KLAK was internalized upon incubation at 37 °C while no uptake was observed in the case of ELP-KLAK in MDA MB231 (Fig. 2c) and B16F10 cells (Fig. 2d), respectively. These solely validated that ELP does not bind or interact with cells either in unimer or micelle form but act as backbone for multivalent presentation of targeting ligands. Greater uptake obtained by AP1-ELP-KLAK may result from multivalent interaction with cell surface receptors followed by receptor mediated endocytosis. Both AP1-ELP-KLAK and ELP-KLAK displayed lower cell binding and uptake in H460 cells at 4 °C (data not shown) and 37 °C (Fig. S7, Supplementary data). Thus, these results verified that binding and uptake of polypeptides was highly dependent on the presence of targeting AP1 ligand and not due to the non-specific effect of hyperthermia.
Cell viability and apoptosis assay
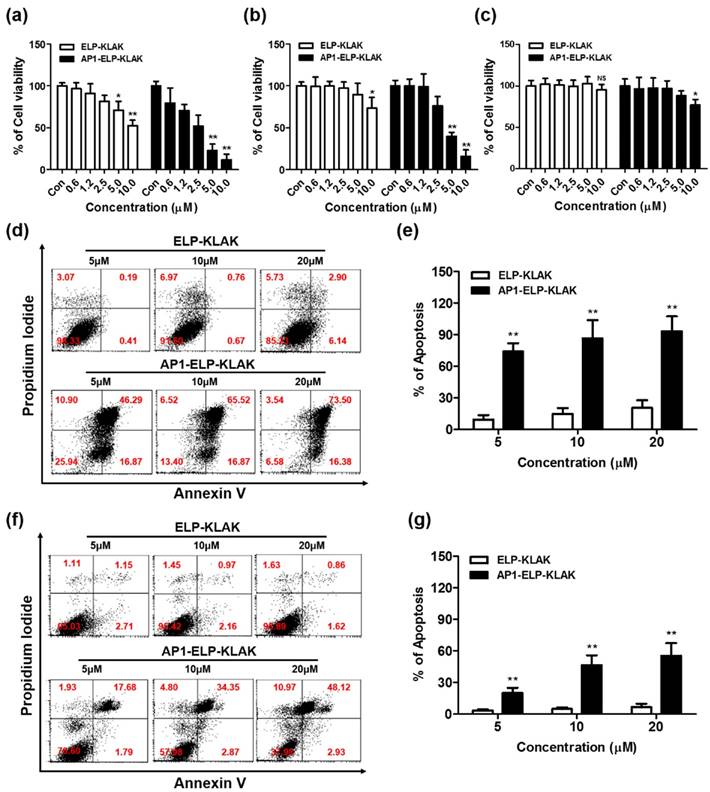
Next, the cytotoxicity effect induced by KLAK was examined in various cancer cell lines showing various expression levels of IL-4R. AP1-ELP-KLAK induced cell cytotoxicity in a concentration-dependent manner in MDA MB231 (Fig. 3a) and B16F10 cells (Fig. 3b), whereas no significant change was observed in H460 cells (Fig. 3c). The IC50 values for AP1-ELP-KLAK and ELP-KLAK were calculated to be 2.65 μM and 10.67 μM, respectively, in MDA MB231 cells. Likewise, IC50 values for AP1-ELP-KLAK and ELP-KLAK were 4.29 μM and 17.28 μM in B16F10 cells after 48 h, respectively. AP1-ELP-KLAK showed ~4 fold higher anti-proliferative activity than ELP-KLAK in both MDA MB231 and B16F10 cells. Neither polymer significantly reduced cell viability in H460 cells, suggesting that cytotoxic activity was highly dependent on the level of IL-4R expression. To determine the mechanism of AP1-ELP-KLAK-mediated cell death, MDA MB231 and B16F10 cells were stained with Annexin V after treatment with different concentrations of polypeptides for 4 h. A dose-dependent increase in the percentage of Annexin V-positive cells was observed in both AP1-ELP-KLAK treated cell lines. At a higher concentration of 20 μM, 90% of MDA MB231 cells underwent apoptosis while the same concentration of control ELP-KLAK did not induce apoptosis (Fig. 3d,e). In order to further confirmed that cancer cell death induced by AP1-ELP-KLAK polypeptide was solely depend on IL-4R mediated internalization, we performed additional experiments where cells were co-incubated with anti-IL-4R antibody or several endocytosis inhibitors in presence of AP1-ELP-KLAK, and level of apoptosis induction was determined by flow cytometry. Co-incubation of AP1-ELP-KLAK along with anti-IL-4R significantly reduced the percentage of apoptosis (Fig. S8a,b Supplementary data). Competitive inhibition of AP1-ELP-KLAK induced apoptosis by different concentration of anti-IL-4R antibody confirmed the specificity towards IL-4R. However co-incubation with endocytosis inhibitors such as dansylcadaverine and genistein (clathrin- or caveolae-dependent endocytosis) (Fig. S8c,d Supplementary data) had a little or not significant reduction in apoptosis [42]. Thus AP1-ELP-KLAK entered the cell through IL-4R mediated endocytosis neither by other mechanism of endocytosis. Simultaneously, AP1-ELP-KLAK induced 50% apoptosis in B16F10 cells (Fig. 3f,g), which clearly suggests that apoptosis level was dependent on IL-4R expression. This result is consistent with those of the cell viability assay and clearly indicate that AP1-ELP effectively delivered KLAK peptide into cancer cells and maintained a bioactive α-helical conformation to induce a higher level of apoptosis. Thus, periodic incorporation of AP1 peptide along the ELP polymer enhanced proapoptotic activity of KLAK by both active targeting of IL-4R and passive targeting in various cancer cells.
Determination of IL-4R expression and cellular binding. (a) Histogram showing IL-4R expression analyzed through flow cytometry after staining with anti-IL-4Rα APC. (b) Percentage of IL-4R expression in different cancer cells was analyzed compared to isotype (n = 5). *P<0.05, **P<0.01 (Student's t-test). Uptake of polypeptides by MDA MB231 (c) and B16F10 (d) cells after incubation with fluorescently labeled polypeptides at 37 °C for 1 h. Cell membrane and nuclei were stained with WGA Alexa 594 and Hoechst respectively. Scale bar, 20 μm.
Cytotoxicity and apoptosis assay. MDA MB231 (a), B16F10 (b), and H460 (c) cells were treated with different concentrations of ELP-KLAK and AP1-ELP-KLAK for 48 h. Cell viability was measured using CCK-8 kit. Both MDA MB231 (d, e) and B16F10 (f, g) cells were treated with KLAK containing polypeptides (5, 10, 20 μM) for 4 h, and level of apoptosis was measured by flow cytometry. Data are represented as mean ± s.d. (n = 5). *P<0.05, **P<0.01 (Student's t-test).
Pharmacokinetic and biodistribution of KLAK containing polypeptides
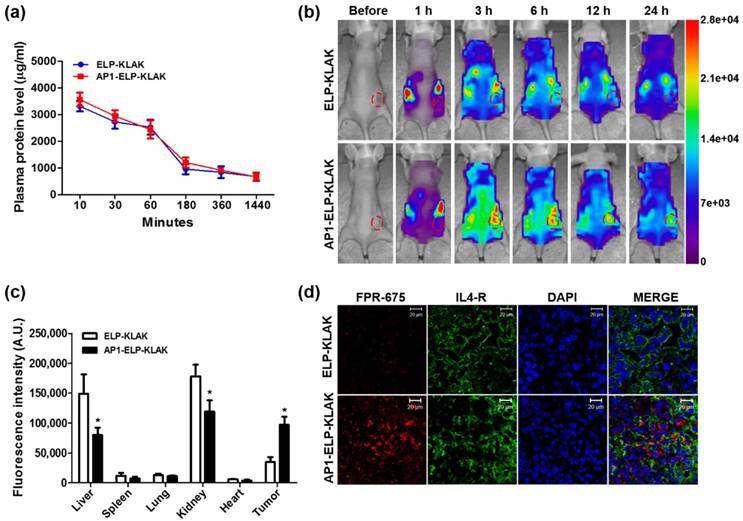
Due to higher intracellular internalization and induction of apoptosis in vitro, AP1-ELP-KLAK was further examined in order to determine pharmacokinetic properties and biodistribution in vivo. FPR-675 Flamma-labeled polypeptides were incubated in fresh plasma for different time intervals at 37 °C in order to insure stability of dye conjugation. Fluorescent intensity measured at 675 nm after two rounds ITC clearly showed that only 20% of dye was released over 24 h, suggesting that the labeled polymers were stable in plasma (Fig. S9, Supplementary data). Evaluation of pharmacokinetic properties in MDA MB231 xenograft mice after intravenous administration revealed that both polypeptides were readily cleared from the circulation with time. Both ELP-KLAK and AP1-ELP-KLAK polypeptides prolonged half-life by decreasing plasma clearance (Fig. 4a), with no significant difference in Cmax or AUC (Table S2, Supplementary data). Further, biodistribution of polymers was determined in order to analyze specific tissue localization after intraperitoneal and intravenous routes of administration. In vivo fluorescence images taken after IP injection using a near-infrared fluorescence (NIRF) live optical imaging system at different time intervals showed that AP1-ELP-KLAK polymer readily localized to tumor tissue in MDA MB231 xenograft mice and persisted longer than 24 h (Fig. 4b). On the other hand, ELP-KLAK showed minimal accumulation at tumor sites with high non-specific tissue localization. Thus, despite having nearly similar pharmacokinetic properties, AP1-ELP-KLAK displayed superior tumor accumulation compared to ELP-KLAK. This clearly indicates that improved pharmacokinetic properties were not a crucial factor in determining the tumor targeting activity of polypeptides. Ex vivo fluorescence images of excised tumor and organs collected at 6 h post-injection further showed 2.3-fold increased fluorescence intensity in target tumors of AP1-ELP-KLAK-injected mice compared to ELP-KLAK (Fig. 4c and Fig. S10, Supplementary data). Higher accumulation in the liver and kidney was observed in ELP-KLAK-injected mice compared to AP1-ELP-KLAK along with negligible levels in the lung, spleen, and heart. As protein molecular size significantly influences biodistribution, higher kidney accumulation shown by ELP-KLAK may be attributed to lower molecular weight [41]. Consistent with the in vivo and ex vivo results, immunohistological examination of tumor tissue revealed that AP1-ELP-KLAK was highly confined to IL-4R-overexpressing tumor tissue (Fig. 4d). The in vivo and ex vivo fluorescence images taken after IV injection of labelled polypeptides also showed no significant difference in biodistribution despite slight increased in liver and decreased in kidney accumulation in AP1-ELP-KLAK injected mice (Fig. S11, S12, Supplementary data) compared with that of IP injection. Later on for therapy experiments IP was preferred over IV injection due to easier and suitable alternative for frequent administration of polypeptides for a prolong period of time.
Pharmacokinetic and biodistribution of AP1-ELP-KLAK. (a) FPR 675-labeled ELP-KLAK and AP1-ELP-KLAK (150 mg kg-1) were injected intravenously into MDA MB231 tumor xenograft nude mice. Time course changes of fluorescence intensity in plasma were measured and fit into a non-compartmental pharmacokinetic model (n = 8). (b) To determine biodistribution in vivo, fluorescence images were collected at different time points after intraperitoneal injection (IP) of labeled polypeptides. Scale bars indicate normalized fluorescence intensity (NC). Representative optical images of ten experiments. (c) Fluorescence intensities of excised tumors and organs at 6 h post-injection (n = 10). *P<0.05 (Student's t-test). (d) Tumor tissue sections were stained and observed under confocal microscopy. Blue, nuclei stained with DAPI; Green, tumor cells stained with anti-IL-4R; Red, labeled polypeptide. Scale bar, 20 μm.
Anti-tumor effect of AP1-ELP-KLAK. MDA MB231 tumor-bearing mice were intraperitoneally injected with PBS, AP1-ELP, ELP-KLAK, and AP1-ELP-KLAK (150 mg kg-1) daily for 14 days. Tumor volume (a) and body weight (b) were measured every other day during therapy (n = 20). (***P<0.001, significant difference for AP1-ELP-KLAK compared with PBS, AP1-ELP; **P<0.01, significant difference for AP1-ELP-KLAK compared with ELP-KLAK). (c) At the end of therapy, tumors were isolated and measured the tumor weight of each group. (d) Comparison of body weight during and after treatment. Data are represented as mean ± s.d. (n = 20). **P<0.01 (Student's t-test). (e) TUNEL staining of excised tumor tissues. Blue, nuclei stained with DAPI; Green, TUNEL positive nuclei. Scale bar, 50 μm.
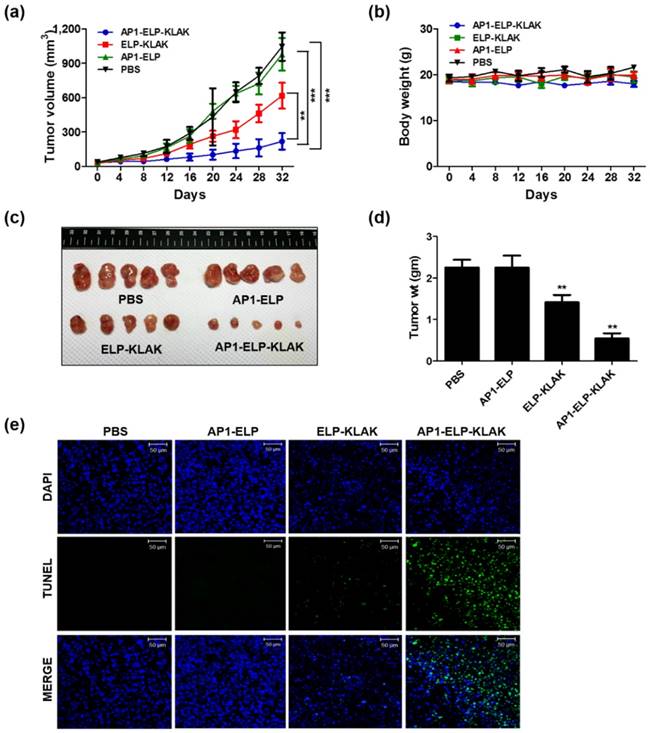
Antitumor effect of AP1-ELP-KLAK in various tumor models
Encouraged by the enhanced accumulation of self-assembled AP1-ELP-KLAK nanoparticles in tumor tissue, in vivo anti-tumor activity was further evaluated in MDA MB231 and B16F10 xenograft mice models. In relevant with the biodistribution and pharmacokinetic results, in vivo therapy was started by administrating 150 mg kg-1 of polypeptides daily via IP when the tumor volume reached 50-100 mm3. IP administration of AP1-ELP-KLAK inhibited tumor growth dramatically, whereas a slight but insignificant reduction in tumor growth was detected in the ELP-KLAK treated group in MDA MB231 bearing mice (Fig. 5a). On the other hand, tumors in the PBS and AP1-ELP treated groups grew more aggressively, reaching a size of 1,100 mm3 after 32 days. Slight reduction of tumor volume observed in ELP-KLAK treated mice may attributed to micelle like formation at body temperature which might result in tumor accumulation by enhanced permeability and retention (EPR) effect. But robust effect in tumor reduction shown by AP1-ELP-KLAK may anticipated as result of enhanced tumor accumulation by both passive EPR effect as well as active targeting based on the highly expressed IL-4R on surface of cancer cells. Excised tumor weight at the end of therapy was significantly reduced compared to the control (Fig. 5c,d). Furthermore, TUNEL assay detected more apoptotic cells in tumor tissue of the AP1-ELP-KLAK treated group but not in the control (Fig. 5e). These results further validated that AP1-ELP polymer can delivered KLAK specifically to the tumor site by penetrating various biological barriers as well as facilitated entry into cells and subsequently inducing massive apoptosis.
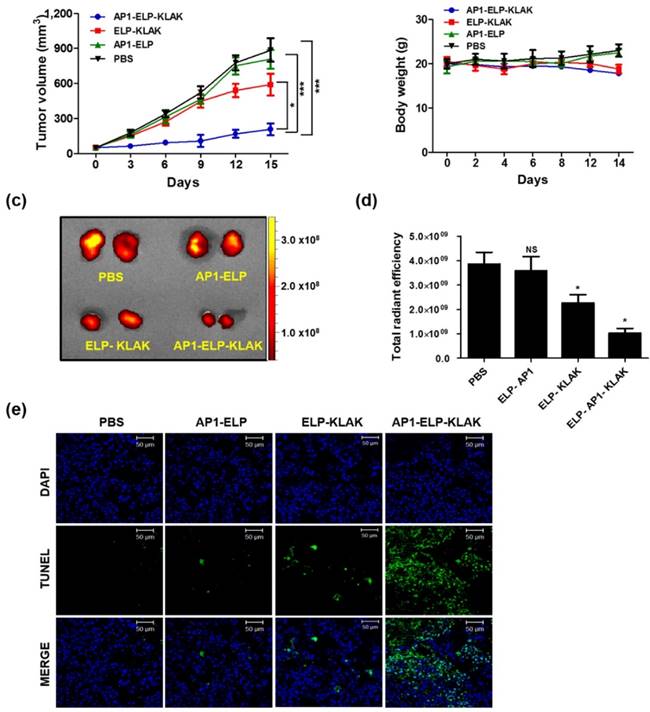
Similarly, AP1-ELP-KLAK significantly reduced tumor growth in B16F10-bearing mice (Fig. 6a) while control group tumor grew more aggressively, reached a volume of 1,000 mm3 in 14 days after implantation. Significant reduction of RFP fluorescent intensity in excised tumor tissue was detected in the AP1-ELP-KLAK-treated group compared to the control (Fig. 6c,d). Additionally, TUNEL assay revealed more apoptotic cells in the tumor section obtained from AP1-ELP-KLAK-treated mice (Fig. 6e). Daily injection of AP1-ELP-KLAK could reduce the growth of the B16F10 tumor significantly but fail to inhibit completely. Termination of polypeptide administration led to progression of tumor growth. It can be speculated that the cytotoxic effect of KLAK is highly dependent on the level of IL-4R expression. The minimal expression of IL-4R correlated with minimal internalization of cytotoxic peptide by B16F10 may lead to the recurrence of the tumor soon after the therapy. It was expected that combination of targeting peptide specific to B16F10 or adapting other therapeutic modalities such as radiotherapy, chemotherapy might allow total eradication of tumor cells.
Anti-tumor activity of AP1-ELP-KLAK. B16F10 tumor-bearing mice were IP-injected with PBS, AP1-ELP, ELP-KLAK, and AP1-ELP-KLAK (150 mg kg-1) daily for 14 days (n = 10). Comparison of relative tumor volume (a) and body weight (b) during therapy (n = 10). (***P<0.001, significant difference for AP1-ELP-KLAK compared with PBS, AP1-ELP; *P<0.05, significant difference for AP1-ELP-KLAK compared with ELP-KLAK). (c,d) Quantification of RFP fluorescence intensities in each group treated with PBS, AP1-ELP, ELP-KLAK, and AP1-ELP-KLAK. Data are represented as mean ± s.d. (n = 10). *P<0.05 (Student's t-test). (e) TUNEL staining of excised tumor tissues obtained from each treatment group. Blue, nuclei stained with DAPI; Green, TUNEL positive nuclei. Scale bar, 50 μm.
Systemic side effect of AP1-ELP-KLAK in the liver, kidney, heart and myeloid function of MDA MB231 tumor-bearing mice. Blood was collected at the end of treatment and analyzed for (a) ALT, alanine aminotransferase; (b) AST, aspartate aminotransferase; levels to check liver function, (c) BUN, blood urea nitrogen; (d) CRE, creatinine; level for kidney, (e) CPK, creatine phosphokinase; (f) LDH lactate dehydrogenase; levels for heart function. Data are mean ± SD (n = 8). Blood was collected at the end of treatment and analyzed for hematological parameters (g) RBC, red blood cells; (h) WBC, white blood cells; (i) hemoglobin, HGB; (j) platelets, PLT. Data are mean ± SD (n= 8).
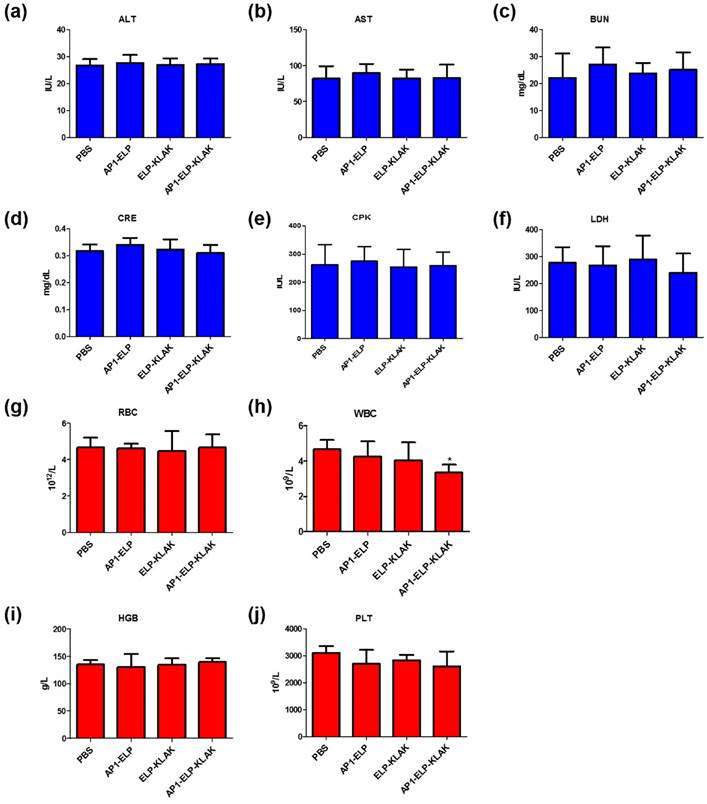
To evaluate the biological safety of AP1-ELP-KLAK, we monitored the changes in body weight during the treatment process, together measured various blood biochemistry and hematology parameters after therapy. Continuous injection of these polypeptides during therapy did not change body weight of mice (Fig. 5b and Fig. 6b). Various biochemistry parameters were tested to assure liver, heart, and kidney function. No significant fluctuation in any of these parameters was observed after treatment and was well within the normal reference range. These results confirm that no obvious liver, heart, or kidney toxicity was induced by ELP-KLAK or AP1-ELP-KLAK treatment (Fig. 7a-f). Hematological analysis showed that all of the important hematological markers, such as white blood cells, red blood cells, hemoglobin, and platelets, in the treated groups appeared to be normal as compared with the control groups treated with PBS (Fig. 7g-j). Taken together, no noticeable toxicity of ELP-KLAK or AP1-ELP-KLAK was detected from the blood biochemistry and hematological examination. Low systematic toxicity after polypeptide treatment was further confirmed by H & E staining, which showed no change in the physiological structures of major organs (Fig. S13, Supplementary data). However, dead tumor cells were detected only in the AP1-ELP-KLAK treated group. AP1-ELP-KLAK inhibited tumor growth in both MDA MB231 and B16F10 tumor models, optimization of a therapeutic protocol is important depending on the aggressiveness of tumor growth in a xenograft system.
Conclusion
In this study we reported a new multivalent based drug delivery system of a therapeutic peptide for effective cancer therapy in various tumor models. Thus, proapoptotic KLAK peptide can be incorporated along with ELP or a multivalent-targeted AP1-ELP carrier as a single polypeptide and purified with high yield from E.coli, in contrast to the complicated scale-up process of synthetic polymers with multiple ligand-binding sites. Interestingly, both ELP-KLAK and AP1-ELP-KLAK polypeptides self-assembled into a nanoparticle-like structure at physiological body temperature without any structural changes or effects on bioactivity. Thus, it was anticipated that they self-assembled into a micelle-like structure and circulated throughout the body upon systematic administration. IL-4R specific internalization by AP1-ELP-KLAK could be attributed to higher cytotoxicity and apoptosis in IL-4R highly expressing cancer cells in vitro. More interestingly, both polypeptides exhibited similar pharmacokinetic properties and plasma clearance profiles. However, only AP1-ELP-KLAK significantly accumulated in tumor tissue and almost inhibited tumor growth up to 80 ̴ 85% without any systemic toxicity, whereas ELP-KLAK inhibited tumor growth only up to 35 ̴ 40%. During therapy, our unique polypeptides, AP1 peptide along the ELP polymer enhanced proapoptotic activity of KLAK by both active targeting of IL-4R and passive targeting to tumor tissue. Moreover, incorporation with ELP and AP1-ELP reduced system toxicity and highly increased activities against tumor cells. Further optimization of molecular weight must be considered to avoid liver and kidney accumulation, thereby enhancing tumor targeting activity of AP1-ELP polymer. Almost all ELPs that have been addressed for drug delivery purposes have exploited local hyperthermia to trigger phase transition for optimal effect. But increase in temperature is not possible for all type of tumor thus triggering of nanoparticle formation at physiological temperature with better tumor targeting and accumulation effect might expand the application of ELP in drug delivery. Consequently, this strategy can be further optimized for delivery of other varieties of anti-cancer drugs. Collectively, we proposed a distinctive multivalent cancer targeting system that can deliver therapeutic peptides selectively to tumor sites to effectively inhibit tumor progression.
Supplementary Material
Supplementary tables and figures.
Abbreviations
ELP: elastin-like polypeptide; IL-4R: interleukin-4 receptor; ITC: inverse transition cycling; Tt : transition temperature; TEM: transmission electron microscope; CD: circular dichroism; DLS: dynamic light scattering; OD: optical density; Da: daltons; PBS: phosphate buffer saline; i.p: intra-peritoneal; PI: propidium iodide; s.c: sub cutaneous; H & E: hematoxylin and eosin; EPR: enhanced permeability and retention; NIRF: near-infrared fluorescence; ALT: alanine aminotransferase; AST: aspartate aminotransferase; LDH: lactate dehydrogenase; CPK: creatine phosphokinase isoenzymes; CRE: creatinine; BUN: blood urea nitrogen; WBC: white blood cells; RBC: red blood cells; PLT: platelets; HGB: hemoglobin.
Acknowledgements
This study was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea (0720550-2) and a National Research Foundation of Korea (NRF), (No. 2014R1A5A2009242). Together this research was also funded by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) and Ministry of Health & Welfare, Republic of Korea (HI15C3272 & HT15C0003).
Author contributions
Vijaya Sarangthem, Yunjae Kim and Thoudam Debraj Singh equally contributed to this work, performed, acquired and analyzed all the data, while Bo-Yeon Seo, Sun-Ha Cheon, and Young-Jin Lee help in performing some in vivo experiments and Byung-Heon Lee help in revising the manuscript. Design of experiments, concept and wrote the manuscript by Vijaya Sarangthem, Thoudam Debraj Singh and Rang-Woon Park. The whole study is supervised by Rang-Woon Park.
Competing Interests
The authors declare that they have no competing financial interests.
References
1. Antosova Z, Mackova M, Kral V & Macek T. Therapeutic application of peptides and proteins: parenteral forever? Trends Biotechnol. 2009;27:628-35
2. Mulder KC, Lima LA, Miranda VJ, Dias SC, Franco OL. Current scenario of peptide-based drugs: the key roles of cationic antitumor and antiviral peptides. Front. Microbiol. 2013;4:321
3. Sun L. Peptide-based drug development. Mod. Chem. Appl. 2013;1:e103
4. Schweizer F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009;625:190-4
5. Talmadge JE. Pharmacodynamic aspects of peptide administration biological response modifiers. Adv. Drug Delivery Rev. 1998;33:241-52
6. Javadpour MM, Juban MM, Lo WC, Bishop SM, Alberty JB, Cowell SM. et al. De novo antimicrobial peptides with low mammalian cell toxicity. J. Med. Chem. 1996;39:3107-13
7. Ellerby H M, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD. et al. Anti-Cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999;5:1032-8
8. Mai JC, Mi Z, Kim SH, Ng B, Robbins PD. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001;61:7709-12
9. Smolarczyk R, Cichon T, Graja K, Hucz J, Sochanik A, Szala S. Antitumor effect of RGD-4C-GG-D(KLAKLAK)2 peptide in mouse B16(F10) melanoma model. Acta Biochim Pol. 2006;53:801-5
10. Zurita AJ, Troncoso P, Cardo-Vila M, Logothetis CJ, Pasqualini R, Arap W. Combinatorial screenings in patients: the interleukin-11 receptor alpha as a candidate target in the progression of human prostate cancer. Cancer Res. 2004;64:435-9
11. Marks AJ, Cooper MS, Anderson RJ, Orchard KH, Hale G, North JM. et al. Selective apoptotic killing of malignant hemopoietic cells by antibody-targeted delivery of an amphipathic peptide. Cancer Res. 2005;65:2373-7
12. Toft DJ, Moyer TJ, Standley SM, Ruff Y, Ugolkov A, Stupp SI. et al. Coassembled cytotoxic and pegylated peptide amphiphiles form filamentous nanostructures with potent antitumor activity in models of breast cancer. ACS Nano. 2012;6:7956-65
13. He ZY, Chu BY, Wei XW, Li J, Edwards CK 3rd, Song XR. et al. Recent development of poly(ethylene glycol)-cholesterol conjugates as drug delivery systems. Int. J. Pharm. 2014;469:168-78
14. Pathak RK, Marrache S, Choi JH, Berding TB, Dhar S. The prodrug platin-A: simultaneous release of cisplatin and aspirin. Angew. Chem. 2014;53:1963-7
15. Meyer DE, Chilkoti A. Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: examples from the elastin-like polypeptide system. Biomacromolecules. 2002;3:357-67
16. Liu W, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR. et al. Tumor accumulation, degradation and pharmacokinetics of elastin-like polypeptides in nude mice. J. Controlled Release. 2006;116:170-8
17. Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat. Biotechnol. 1999;17:1112-5
18. Trabbic-Carlson K, Liu L, Kim B, Chilkoti A. Expression and purification of recombinant proteins from Escherichia coli: comparison of an elastin-like polypeptide fusion with an oligohistidine fusion. Protein Sci. 2004;13:3274-84
19. Meyer DE, Chilkoti A. Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules. 2004;5:846-51
20. Bidwell GL 3rd, Raucher D. Cell penetrating elastin-like polypeptides for therapeutic peptide delivery. Adv. Drug Delivery Rev. 2010;62:1486-96
21. Kim W, Chaikof EL. Recombinant elastin-mimetic biomaterials: emerging applications in medicine. Adv. Drug Delivery Rev. 2010;62:1468-78
22. Liu W, MacKay JA, Dreher MR, Chen M, McDaniel JR, Simnick AJ. et al. Injectable intratumoral depot of thermally responsive polypeptide-radionuclide conjugates delays tumor progression in a mouse model. J. Controlled Release. 2010;144:2-9
23. Simnick AJ, Amiram M, Liu W, Hanna G, Dewhirst MW, Kontos CD. et al. In vivo tumor targeting by a NGR-decorated micelle of a recombinant diblock copolypeptide. J. Controlled Release. 2011;155:144-51
24. Simnick AJ, Valencia CA, Liu R, Chilkoti A. Morphing low-affinity ligands into high-avidity nanoparticles by thermally triggered self-assembly of a genetically encoded polymer. ACS Nano. 2010;4:2217-27
25. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013;12:991-1003
26. Qin SY, Feng J, Rong L, Jia HZ, Chen S, Liu XJ. et al. Theranostic GO-based nanohybrid for tumor induced imaging and potential combinational tumor therapy. Small. 2014;10:599-608
27. Ko JY, Park S, Lee H, Koo H, Kim MS, Choi K. et al. pH-sensitive nanoflash for tumoral acidic pH imaging in live animals. Small. 2010;6:2539-44
28. Kruger HR, Schutz I, Justies A, Licha K, Welker P, Haucke V. et al. Imaging of doxorubicin release from theranostic macromolecular prodrugs via fluorescence resonance energy transfer. J. Controlled Release. 2014;194:189-96
29. Obiri NI, Hillman GG, Haas GP, Sud S, Puri RK. Expression of high affinity interleukin-4 receptors on human renal cell carcinoma cells and inhibition of tumor cell growth in vitro by interleukin-4. J. Clin. Invest. 1993;91:88-93
30. Obiri NI, Siegel JP, Varricchio F, Puri RK. Expression of high-affinity IL-4 receptors on human melanoma, ovarian and breast carcinoma cells. Clin. Exp. Immunol. 1994;95:148-55
31. Conticello C, Pedini F, Zeuner A, Patti M, Zerilli M, Stassi G. et al. IL-4 protects tumor cells from anti-CD95 and chemotherapeutic agents via up-regulation of antiapoptotic proteins. J. Immunol. 2004;172:5467-77
32. Yang L, Horibe T, Kohno M, Haramoto M, Ohara K, Puri RK. et al. Targeting interleukin-4 receptor alpha with hybrid peptide for effective cancer therapy. Mol. Cancer Ther. 2012;11:235-43
33. Wu XL, Kim JH, Koo H, Bae SM, Shin H, Kim MS. et al. Tumor-targeting peptide conjugated pH-responsive micelles as a potential drug carrier for cancer therapy. Bioconjug. Chem. 2010;21:208-13
34. Hong HY, Lee HY, Kwak W, Yoo J, Na MH, So IS. et al. Phage display selection of peptides that home to atherosclerotic plaques: IL-4 receptor as a candidate target in atherosclerosis. J. Cell. Mol. Med. 2008;12:2003-14
35. Park K, Hong HY, Moon HJ, Lee BH, Kim IS, Kwon IC. et al. A new atherosclerotic lesion probe based on hydrophobically modified chitosan nanoparticles functionalized by the atherosclerotic plaque targeted peptides. J. Controlled Release. 2008;128:217-23
36. Sarangthem V, Cho EA, Bae SM, Singh TD, Kim SJ, Kim S. et al. Construction and application of elastin like polypeptide containing IL-4 receptor targeting peptide. PLoS One. 2013;8:e81891
37. Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP. et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cut-off size. Cancer Res. 1995;55:3752-6
38. Janib SM, Liu S, Park R, Pastuszka MK, Shi P, Moses AS. et al. Kinetic quantification of protein polymer nanoparticles using non-invasive imaging. Integr. Biol. 2013;5:183-94
39. Olariu CI, Yiu HP, Bouffier L, Nedjadi T, Costello E, Williams SR. et al. Multifunctional Fe3O4 nanoparticles for targeted bi-modal imaging of pancreatic cancer. J. Mater. Chem. 2011;21:12650-9
40. Palma RD, Peeters S, Bael MJV, Rul HV, Bonroy K, Laureyn W. et al. Silane ligand exchange to make hydrophobic superparamagnetic nanoparticles water dispersible. Chem. Mat. 2007;19:1821-31
41. Ryu JS, Raucher D. Elastin-like polypeptides: the influence of its molecular weight on local hyperthermia-induced tumor accumulation. Eur. J. Pharm. Biopharm. 2014;88:382-9
42. Macewan SR, Chilkoti A. Digital switching of local arginine density in a genetically encoded self-assembled polypeptide nanoparticle controls cellular uptake. Nano Letter. 2012;12:3322-8
Author contact
![]() Corresponding author: Rang-Woon Park, MD, PhD. E-mail: nwparkac.kr, phone No.: +82 53 420 4822, Fax No.: +82 53 422 1466.
Corresponding author: Rang-Woon Park, MD, PhD. E-mail: nwparkac.kr, phone No.: +82 53 420 4822, Fax No.: +82 53 422 1466.