Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(12):2052-2067. doi:10.7150/thno.16180 This issue Cite
Research Paper
Blocking the 4-1BB Pathway Ameliorates Crystalline Silica-induced Lung Inflammation and Fibrosis in Mice
Chao Li1, Sitong Du1, Yiping Lu1, Xiaowei Lu1, Fangwei Liu1, Ying Chen1, Dong Weng2,1, ![]() , Jie Chen1,
, Jie Chen1, ![]()
1. Division of Pneumoconiosis, School of Public Health, China Medical University, Shenyang, PR China.
2. Department of Respiratory Medicine, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, PR China.
Received 2016-5-16; Accepted 2016-7-30; Published 2016-9-9
Abstract

Long term pulmonary exposure to crystalline silica leads to silicosis that manifests progressive interstitial fibrosis, eventually leading to respiratory failure and death. Despite efforts to eliminate silicosis, clinical cases continue to occur in both developing and developed countries. The exact mechanisms of crystalline silica-induced pulmonary fibrosis remain elusive. Herein, we find that 4-1BB is induced in response to crystalline silica injury in lungs and that it is highly expressed during development of experimental silicosis. Therefore, we explore the role of 4-1BB pathway during crystalline silica-induced lung injury and find that a specific inhibitor blocking the pathway could effectively alleviate crystalline silica-induced lung inflammation and subsequent pulmonary fibrosis in vivo. Compared to controls, the treated mice exhibited reduced Th1 and Th17 responses. The concentrations of pro-inflammatory cytokines in bronchoalveolar lavage fluid (BALF), including tumor necrosis factor (TNF)-α, interferon (IFN)-γ and interleukin (IL)-17A following crystalline silica challenge were also reduced in inhibitor-treated mice. Although there was no significant alteration in Th2 cytokines of IL-4 and IL-13, another type of pro-fibrogenic cell, regulatory T cell (Treg) was significantly affected. In addition, one of the major participants in fibrogenesis, fibrocyte recruited less due to the blockade. Furthermore, we demonstrated the decreased fibrocyte recruitment was associated with chemokine reductions in lung. Our study discovers the 4-1BB pathway signaling enhances inflammatory response and promotes pulmonary fibrosis induced by crystalline silica. The findings here provide novel insights into the molecular events that control crystalline silica-induced lung inflammation and fibrosis through regulating Th responses and the recruitment of fibrocytes in crystalline silica-exposed lung.
Keywords: Crystalline silica, Silicosis, 4-1BB, Pulmonary fibrosis, Th responses, Fibrocyte, Molecular therapy.
Introduction
The industrialization process increases the occupational exposure of workers to respirable particles. Among these particles, crystalline silica accounts for the most part. Inhalation of crystalline silica causes silicosis, which is a potentially fatal lung disease [1]. It is characterized by chronic lung inflammation and irreversible fibrosis. Despite efforts to eliminate silicosis, clinical cases continue to occur in both developing and developed countries [2]. A recent survey highlighted deaths due to silicosis in young adults (aged 15-44 years) as being particularly noteworthy [3]. Chronic silicosis can develop or progress even after occupational exposure has ceased. As no therapy halts or reverses the disease progression, the available management is focused on controlling associated symptoms and providing supportive care to patients [3]. Although an effective therapeutic target is needed to postpone the progress of fibrogenesis, the complex molecular mechanisms involved are not well understood. Using protein array screening, we found 4-1BB is highly expressed during development of silicosis. However, the regulatory mechanism has not been identified. Revealing the functions of 4-1BB or its signaling pathway may uncover the mechanism(s) of crystalline silica-induced fibrosis.
4-1BB (CD137, TNF receptor superfamily 9) is a potent costimulatory molecule in T cell activation. T lymphocytes, particularly CD4+ T cells, play crucial roles in the development of crystalline silica-induced lung inflammation and fibrosis [4]. A previous study proposed that the interaction between 4-1BB and its ligand induces a Th1 response, suppresses the Th2 response and maximizes the function of effector T cells [5]. 4-1BB may also modulate the balance between Th17 and regulatory T cells in experimental autoimmune encephalomyelitis [6]. The mechanisms through which the 4-1BB pathway influence effector T cells in an experimental model of silicosis remain unknown.
4-1BB was originally described as being present on the surface of activated T cells, but its expression pattern includes other cell types. It functions by enhancing recruitment of circulating leukocytes [7]. Fibrocytes are circulating cells that share leukocyte (CD45, CD34) and mesenchymal cell markers (type I collagen and fibronectin) [8]. Fibrocytes appear in experimental fibrosis, such as renal fibrosis, skin wounds, bronchial asthma and pulmonary fibrosis [9-12]. During chronic COPD exacerbation in patients, fibrocytes were recruited and shown to be related to low lung function [13]. The number, activation, and differentiation of circulating fibrocytes have been correlated with asthma severity [14]. Fibrocytes express chemokine receptors such as CCR7, CXCR4, and CCR2 [8]. Several studies have asserted that key chemokines are involved in fibrocyte recruitment to sites of fibrosis [9, 15, 16]. Recruitment and differentiation of fibrocytes are believed to be a source of the extracellular matrix (ECM). However, effects of the 4-1BB pathway on fibrocyte accumulation in the pathogenesis of silicosis have not been investigated.
In this study, we attempted to dissect the mechanism(s) by which 4-1BB regulate crystalline silica-induced lung inflammation and fibrosis. To address this issue, we evaluated lung inflammation and fibrosis induced by crystalline silica, using an established experimental mouse model of silicosis [17-19]. Mice were treated with NQDI 1, an ASK1 inhibitor, as a method to block the downstream signaling of 4-1BB. The findings reveal that blocking the 4-1BB pathway could alleviate inflammatory reaction caused by crystalline silica and influence the lung cytokine milieu in this mouse model. Fibrocyte recruitment was also affected by this blockade. Our research reveals that blocking 4-1BB signaling represents a beneficial approach to ease crystalline silica-induced excessive lung inflammation and lessen pulmonary fibrosis. The 4-1BB pathway may be a novel therapeutic target for silicosis.
Materials and Methods
Animals. Female C57BL/6 mice were purchased from SLAC Laboratory Animal Co. Ltd. (Shanghai, China) at 6-8 weeks of age. All animals were housed in a specific pathogen-free environment and maintained on standard mouse chow at a temperature of 24±1ºC and 12/12 h light/dark cycles, with water ad libitum. All animal experiments were approved by the Animal Care and Use Committee at China Medical University under permit number CMU62043028, which complies with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Crystalline silica. Crystalline silica particulates were brought form the U.S. Silica Company (Frederick, MD, USA). Particulates size distribution: 97% <5 μm diameter and 80% <3 μm diameter; median diameter 1.4 μm. Equivalent spherical diameter was shown in Fig. S1. Some typical physical properties: Bulk density-compacted: 41 lbs/ft3, Bulk density-uncompacted 36 lbs/ft3, Mohs hardness 7, Hegman 7, Yellowness index 2.0, Specific gravity 2.65. Silicon dioxide accounts for 99.3% of its chemical composition. Particulates were ground in saline for 3 hours, boiled in 1 N HCl, washed, dried, and suspended in sterile saline. Suspensions were sonicated for 10 min before use.
Silicosis model. The silicosis model was induced according to previously published methods [17, 20]. Briefly, C57BL/6 mice were anesthetized with an injection of 10% chloral hydrate (0.4 ml/100 g) and placed on a platform. A suspension of 2.5 mg crystalline crystalline silica crystals in 50 μl sterile saline was directly administered by intra-tracheal instillation. Control mice received 50 μl sterile saline.
4-1BB Fusion protein (4-1BBIg) treatment. The 4-1BBIg was brought from Sino Biological Inc (Beijing, China). The recombinant mouse 4-1BB/Fc is a disulfide-linked homodimer. A DNA sequence encoding the mouse 4-1BB (NP_001070976.1) (Met 1-Leu 211) was fused with the Fc region of human IgG1 at the C-terminus. It could Immobilize mouse 4-1BB ligand at 10 μg/ml. The EC50 of mouse 4-1BBIg is 12.0-29.0 ng/ml. Isotype-matched human IgG1 was also obtained from Sino Biological Inc (Beijing, China). The 4-1BBIg treatment mice (n=3) was given 50μg or 100μg 4-BBIg intraperitoneally on day 1 and day 4 after crystalline silica instillation (Fig. 3B). As a control Ig group (n=3), isotype-matched human IgG1 (100μg) was given as the same schedule as 4-1BBIg treatment. Saline or crystalline silica treated mice were prepared as negative or positive controls (n=4). All mice were killed on day 7 to examine the phosphorylation of 4-1BB downstream kinases.
NQDI 1 effectively inhibits phosphorylation of ASK1 and downstream MAPK cascades. A Bioinformatics analysis: protein-protein interaction network in STRING v.10. A screenshot from STRING shows a network associated with 4-1BB (Tnfrsf9). The upper right (red circle) shows cell signaling proteins in cells associated with 4-1BB. The lower left (blue circle) shows chemokines and chemokine receptors. Each node represents a protein and each line represents an interaction, color coded by evidence type. Gene and protein names are listed in the table. B Experimental protocol for the induction of silicosis and treatment scheme of 4-1BBIg. C Western blot analysis of lung from different treated mice (n=3-4) Figure shows the representative images of three independent experiments. D The levels of p-ASK1 were normalized to those of β-actin. Data are the mean of three independent experiments. Error bars indicate the mean ± S.D. # P<0.05 compared with the saline group; * P<0.05 compared with the crystalline silica group. E The chemical structure of NQDI 1. F-H Western blot analysis of ASK1 phosphorylation and downstream MAPK cascades (p38 and JNK/SAPK) at specific time points (n=3). The experiment was performed in triplicate.
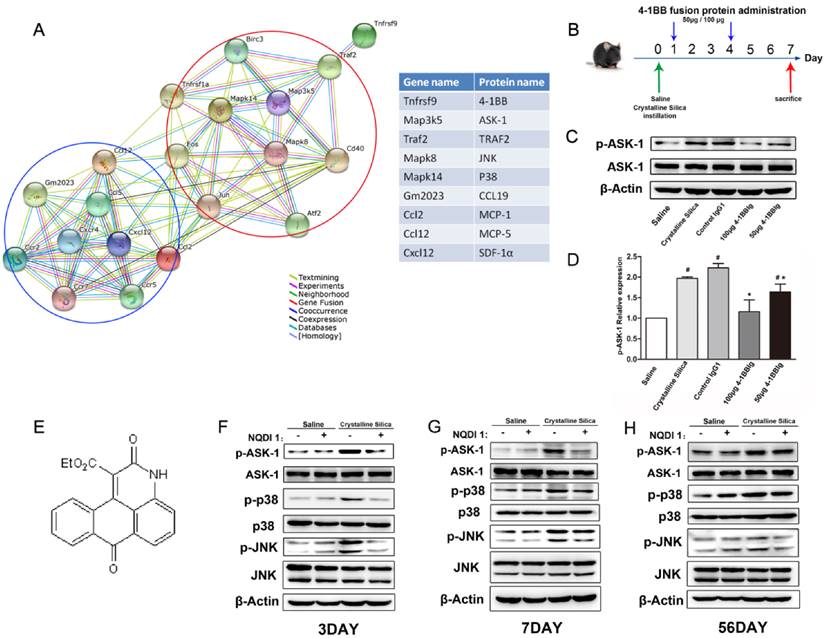
Experimental groups. Female C57BL/6 mice were randomly divided into four treatment groups (n=30 per group): (a) control, (b) control + NQDI 1, (c) crystalline silica, and (d) crystalline silica + NQDI 1. NQDI 1, a selective inhibitor of apoptosis signal-regulating kinase 1, was intraperitoneally (i.p.) administered as a single dose at 10 mg/kg after crystalline silica exposure. 3 and 7 days, mice were administered repeatedly every day for continued inhibition. After 7 days, mice were administered every other day (Fig. S4). NQDI 1 was dissolved at 10 mM in DMSO according to the manufacturer's instruction (Tocris, Bioscience, UK), and a stock solution was prepared and stored at -20ºC. Mice from groups (a) and (c) were treated with vehicle at the same molar concentration. The compound was also administered to the saline group as a control. NQDI 1 (Fig. 3E) is a chemical compound that belongs to 3H-naphtho[1,2,3-de]quinoline-2,7-diones and exhibits strong specific inhibitory activity toward ASK1 [21]. AST and ALT serum levels were measured to determine liver damage by NQDI 1. Test procedures were based on instructions provided by the manufacturer (Nanjing jian cheng Institute, China).
Cytokine microarray analysis: RayBiotech Mouse Cytokine Antibody Array (Cat# AAM-CYT-G2000) was used to profile cytokine proteins in the lungs in duplicate. Each experiment was performed in accordance with the manufacturer's instructions. Briefly, pre-coated antibody array membranes were incubated with 1 ml coating buffer for 30 min. Blocking buffer was then decanted and replaced with 100 μl sample dilutions. Membranes were incubated overnight at 4℃ with shaking. The next day, sample dilutions were decanted and membranes were washed with wash buffer and incubated with 100 μl biotin-conjugated antibody in the dark at room temperature for 2 hours. A mixture of biotin-conjugated antibodies was removed and 70 μl of 1X Streptavidin-Fluor was added to each sub-array. Incubation chambers were covered with adhesive film and incubated at room temperature for 2 hours with gentle shaking. After washing, signals were detected using a GenePix 4000B system (Axon Instruments, Foster City, CA, USA). GenePix Pro 6.0 software (Axon Instruments) was used for densitometric analysis. Values were normalized to the ratio of positive control values for each sample. Total normalized fluorescence values of replicate spots were averaged and expressed as the fold increase over control samples.
Cell preparation. Single-cell suspensions were prepared from the hilar lymph nodes (HLN), lungs and spleen of mice. Procedures were performed in accordance with previously published methods [20, 22]. Briefly, HLN were dissected with dissection needles and digested with 0.25% trypsin for 5 min at 37°C. PBS with 3% fetal bovine serum was used to quench the digestion. Samples were centrifuged at 1,500 rpm for 8 min at 4ºC. The HLN cell pellet was washed and resuspended in PBS. Lung tissue was minced into a slurry in digestion solution containing collagenase (15 mg), DNase I (250 KU units), and complete media (RPMI-10% FCS). The suspension was incubated on a rocker at 37°C for 30 min. Cells were dispersed by repetitive suction and passed through a 70-μm cell strainer. Red blood cell lysis buffer (RBCL) was used to lyse cells. After RBC lysis, lung cells were washed and resuspended in PBS. For splenocytes, the spleen was removed, ground, and mechanically dissociated in cold PBS. After lysis of RBCs, splenocytes were washed and resuspended in PBS.
Co-culture system. Primary lung cell and splenocytes were prepared as previously described. The cells were co-cultured with 50μg/cm2 crystalline silica [23]. 4-1BBIg was added to each plate at various concentration [24]. The cells were cultured in RPMI 1640 medium with 10% foetal bovine serum. The cultures were incubated at 37℃ in a humidified atmosphere with 5% CO2 for 3 days. Thereafter, the cells were sucked out and lysis for protein preparation and western blot analysis.
Identifying T cells and fibrocytes. To identify T cell types, HLN cells were stimulated with a leukocyte activation cocktail (BD Pharmingen, San Jose, CA, USA) for 5 h, followed by blocking with purified rat anti-mouse CD16/32 (BD Pharmingen) for 10 mins at 4°C. PerCP-Cy5.5 conjugated CD4 (BD Pharmingen) antibody was used for cell surface staining. For intracellular staining, Alexa Fluor 488-conjugated anti-IFN-γ (BD Pharmingen), PE-conjugated anti-IL-17A (BD Pharmingen), and Alexa Fluor 647-conjugated anti-Foxp3 (BD Pharmingen) were used to determine Th1, Th17 and regulatory T cells, respectively. Isotype control antibodies were used to determine positive cells. For lung fibrocytes, the cell surface was stained by CD45-PerCP-Cy5.5 antibody (BD Pharmingen) after Fc blocking. Cells were then washed and fixed/permeabilized using the Cytofix/Cytoperm kit (BD Pharmingen) and stained intracellularly for collagen 1 (rabbit anti-mouse biotin conjugated; Rockland Immunochemicals), followed by a streptavidin-PE secondary antibody (BD Pharmingen). Each sample was also stained by rabbit IgG (phycoerythrin) isotype control (Abcam). Analysis of cell markers was performed using a BD FACS CantoTMII flow cytometry system (BD Bioscience, San Jose, CA, USA). Dead cells and crystalline silica particles were gated out depending on forward scattering (FSC) and side scattering (SSC). Cells were analyzed with BD FACSDivaTM software v6.1.2 (BD Bioscience).
Cytokine and chemokine analysis. Mice were sacrificed at 3, 7, or 56 days after crystalline silica exposure. Bronchoalveolar lavage fluid (BALF) was obtained by cannulating the trachea, removing the lungs, and then injecting and retrieving 1 ml aliquots of sterile saline 2 times. BALF was centrifuged at 1,500 rpm for 8 min at 4°C. Cytometric bead arrays (mouse Th1/Th2/Th17 cytokine kit, BD Pharmingen) were used to determine secreted cytokine levels in BALF following the manufacturer's instructions. Generally, multiple capture beads were mixed together, including TNF-α, IL-6, IFN-γ, IL-2, IL-4, IL-17, and IL-10. The mixed capture beads were co-cultured with 50 μl BALF supernatant and detection reagent for 2 h. Beads were washed carefully and resuspended. Samples were analyzed using a FACS Canto II system (BD). Data were analyzed with FCAP Array software. To test other cytokine and chemokine levels, a mouse IL-13 enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Inc., Minneapolis, MN, USA), a mouse IL-1β ELISA kit (R&D), a mouse MCP-1 (CCL-2) ELISA kit (R&D), a mouse MCP-5 (CCL-12) ELISA kit (R&D), a mouse 6Ckine (CCL21) ELISA kit (R&D), and a mouse SDF-1α (CXCL-12) ELISA kit (R&D) were used in this study. A mouse 4-1BB (TNFSF9) ELISA kit was also used to confirm the results of cytokine array analysis. All experimental procedures were in accordance with the manufacturer's instructions. Cytokine levels in BALF and chemokine levels in lung homogenates from 5 or 6 different samples were tested three times in independent experiments. Before testing, lung homogenates were normalized to the same concentration of 1 μg/ml.
Real-time PCR. Total RNA was isolated from lung homogenates using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcribed into cDNA with PrimeScript RT kit (DRR047A, Takara, Japan) as previously described [17-20]. Primers were designed with Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/), and the sequences (supplementary material) were checked using a BLAST search (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The sequences of specific primer pairs are described below: 4-1BB,5'-CGTGCAGAACTCCTGTGATAAC-3' and 5'-GTCCACCTATGCTGGAGAAGG-3'; GAPDH,5'-AGGTCGGTGTGAACGGATTTG-3' and 5'-TGTAGACCATGTAGTTGAGGTCA-3'; T-bet, 5'-AGCAAGGACGGCGAATGTT-3' and 5'-GGGTGGACATATAAGCGGTTC-3'; GATA-3, 5'-CTCGGCCATTCGTACATGGAA-3' and 5'-GGATACCTCTGCACCGTAGC-3'; ROR-γt, 5'-GACCCACACCTCACAAATTGA-3' and 5'-AGTAGGCCACATTACACTGCT-3'; TGF-β1, 5'-CTCCCGTGGCTTCTAGTGC-3' and 5'-GCCTTAGTTTGGACAGGATCTG-3'; ICAM-1, 5'-GTGATGCTCAGGTATCCATCCA-3' and 5'-CACAGTTCTCAAAGCACAGCG-3'; VCAM-1, 5'-AGTTGGGGATTCGGTTGTTCT-3' and 5'-CCCCTCATTCCTTACCACCC-3'; COL-1A1, 5'-GCTCCTCTTAGGGGCCACT-3' and 5'-CCACGTCTCACCATTGGGG-3'. GAPDH was set as internal control for determining ΔCT values. Fold increases in expression were normalized to the saline control group by determining 2-ΔΔCT values.
Western blot analysis. For tissue lysis, the right lung of each mouse was homogenized in 200-300 μl of RIPA buffer isotonic cocktail containing protease and phosphatase inhibitors. For cell lysis, 100 μl RIPA buffer was added. The sample was sonicated and incubated on ice for 30 min and then centrifuged at 10,000 g for 10 min at 4°C. The resulting supernatant was re-centrifuged and saved. Total protein was estimated using the Pierce BCA Protein Assay Kit (Thermo Scientific, USA). Samples were stored at -70°C until analysis. Lung lysates of each mouse sample were diluted to a protein concentration of 3 μg/μl (1μg/μl for cell lysis) and boiled for 5 min. Then, 10 μl aliquots (i.e. 30 μg of protein/10 μg protein for cells) of each sample were loaded onto 8% SDS-acrylamide gels. Proteins were separated by application of a constant voltage of 100 V for 1.5 h and then transferred onto PVDF membranes at a constant voltage of 100 V for 1 h. After blocking nonspecific sites with TBS containing 0.1% Tween20 (TBST) and 5% defatted dried milk, membranes were washed and incubated with primary antibodies: rat anti-4-1BB (Abcam, 1:500), rat anti-4-1BBL (Santa, 1:200), rabbit anti-ASK1 (Abcam, 1:1000), rabbit anti-phospho-ASK1 (Cell Signaling Technology, 1:1000), rabbit anti-p38 (Cell Signaling Technology, 1:1000), rabbit anti-phospho-p38 (Cell Signaling Technology, 1:1000), rabbit anti-JNK (Cell Signaling Technology, 1:1000), rabbit anti-phospho-JNK (Cell Signaling Technology, 1:1000) and rabbit anti-β-Actin (Cell Signaling Technology, 1:1000) overnight at 4°C. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, goat anti-rabbit, 1:2000). Blots were developed with a high-performance luminol substrate solution (PexBio, Beijing, China). β-Actin was used as a loading control.
Hydroxyproline assay. Lung collagen deposition was estimated by measuring the hydroxyproline content of left lung homogenates following the manufacturer's instruction (Nanjing jian cheng Institute, China). Results are expressed as micrograms HYP per gram of wet lung weight using hydroxyproline standards.
Histological analysis. Lung samples were harvested at 3, 7 and 56 days. Lungs were removed, fixed in 4% paraformaldehyde and embedded in paraffin. Parrafin sections (6-μm thick) were used for H&E to visualize inflammatory infiltrates. Masson's trichome staining was used to visualize the extent of fibrosis. For IHC staining, an anti 4-1BB antibody (Abcam) and horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (H+L) secondary antibody (Abcam) were used. Rabbit IgG was used as an isotype control.
Statistics
All analyses were performed using SPSS software, version 19.0 (SPSS Inc., Chicago, IL, USA), and the analyzers were blind to the treatment of each group. All quantitative experiments were repeated at least once with consistent results. All sample data from independent experiments were calculated to obtain the mean ± S.D. One-way ANOVA followed by Student-Newman-Keuls test was used to compare treatment groups with the control. P<0.05 was considered statistically significant.
Results
Altered 4-1BB expression in the lungs of crystalline silica-injured mice
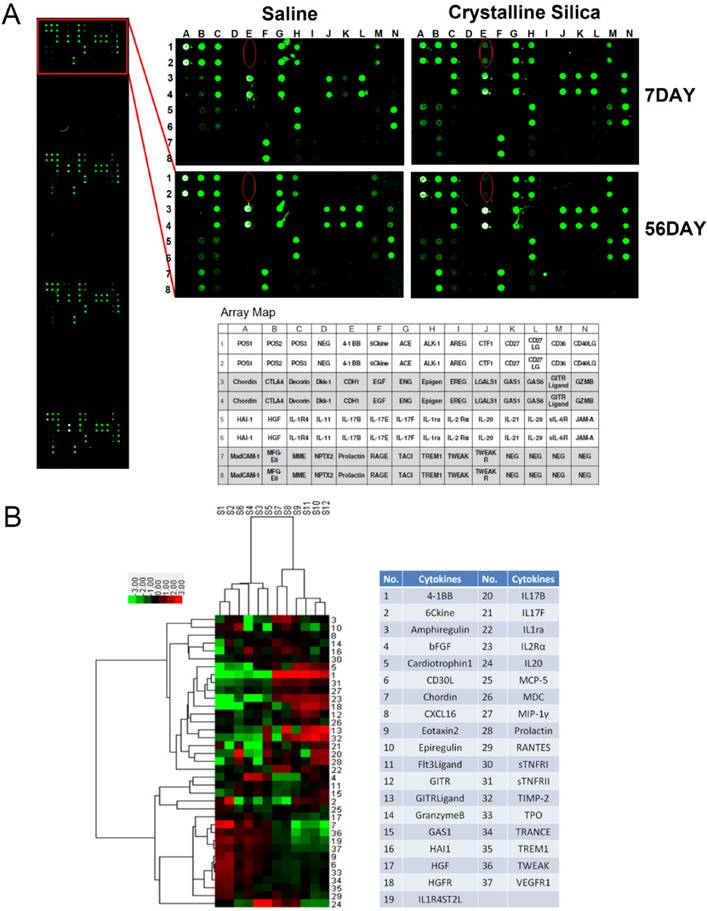
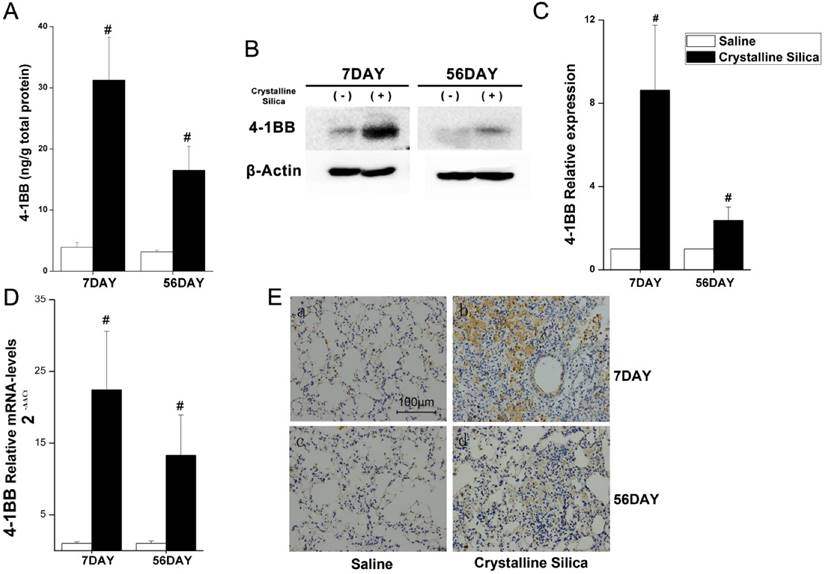
Silicosis is a disease characterized as progressive fibrosis. Fibrosis is a sequela of the inflammatory processes. During the inflammatory phase, both innate and adaptive immune mechanisms are operative [25]. To better understand the mechanism of progressive fibrosis, we performed a blind screening using crystalline silica-injured lung homogenates at specific time points to explore variations in protein profiles compared to control samples (Fig. 1A). Of 144 cytokines, 37 were significantly altered (Fig. 1B). We used hierarchical cluster analysis and confirmed that the significant alterations could distinguish whether lung tissues were damaged and duration of the injury (Fig. 1B). Using protein array analysis, a 770.86-fold increase in 4-1BB protein expression was observed in crystalline silica-injured lungs compared to control subjects after 7 days of exposure. An 81.65-fold increase was noted after 56 days. Increased 4-1BB protein levels were confirmed using ELISA and western blot analysis in the lung tissues from an independent experimental mouse model (Fig. 2A, B and C). We examined 4-1BB mRNA expression in the lungs using real-time PCR analysis. As shown in Fig. 2D, crystalline silica-injury stimulated 4-1BB mRNA expression. We observed a considerable increase in 4-1BB expression by immunohistochemical staining in sections of crystalline silica-injured lungs (Fig. 2E). These combined data verify the increased expression of 4-1BB in crystalline silica-injured lungs. 4-1BB may be critical in the pathogenesis of silicosis.
Protein expression profile from crystalline silica-injured lungs of C57BL/6 mice. A Images show representative sample results (n=3) revealing a marked increase in 4-1BB, HAI-1 and Granzyme. The template shows the location of cytokines spotted onto the RayBiotech Mouse Cytokine Antibody Array G5. Red ellipses indicate 4-1BB. Dots are in duplicate. B Hierarchical cluster analysis and heatmap were utilized to verify the statistical results. Every row represents a protein and every column a mouse lung sample. S1-S3 are control samples at 7 days, S4-S6 are control samples at 56 days, S7-S9 are crystalline silica samples at 7 days, and S10-S12 are crystalline silica samples at 56 days. Red represents up-regulated protein expression and green represents down-regulated expression (crystalline silica vs saline, n=3). Cytokines are numbered in the column.
Increased 4-1BB expression in the lungs of crystalline silica-injured mice. A ELISA analysis was used to confirm increased expression in the lungs of crystalline silica-injured mice at indicated time points (n=5-8). B Western blot analysis of 4-1BB protein in the lung tissues of C57BL/6 mice at indicated time points (n=3). C Mean expression in the saline and crystalline silica groups are shown as fold change compared to mean expression in the saline group, which was ascribed a value of 1. Each bar represents the mean ± S.D. Within each time point, # indicates a significant difference from the saline group, # P<0.05 (n=3). D Real-time PCR analysis of 4-1BB mRNA expression in the lungs at indicated time points after saline or crystalline silica treatment (n=5). For statistical analysis, sample data from three independent experiments were calculated to obtain the mean ± S.D. E Immunohistochemistry of 4-1BB in lung sections after saline or crystalline silica treatment for 7 and 56 days. Representative staining images are shown (n=3). a-b, 7 days, c-d, 56 days. a and c are the saline group, b and d are the crystalline silica group. IgG was used as a control. All images are presented at higher magnification. Bars, 100 μm. A, C and D Error bars indicate the mean ± S.D.
The 4-1BB pathway could be effectively blocked by NQDI 1 in a mouse model of silicosis
After protein array screening, we performed bioinformatic analysis utilizing STRING, which is a database of known and predicted protein interactions. As Fig. 3A shows, several protein factors were involved, including ASK1, JNK, and p38. We used pathway analysis to explore how these protein factors are involved in 4-1BB signaling. Previous research reported that 4-1BB signaling results in TRAF2 recruitment, which can interact with and activate ASK1. ASK1 then activates downstream signaling cascades, including JNK/SAPK and p38 MAPKs [26]. As shown in Fig. 3B, we gave mice 4-1BB fusion protein (4-1BBIg) to block the interaction of 4-1BB and its ligand. We found that the phosphorylation of ASK1 was suppressed in response to treatment of 4-1BBIg in crystalline silica-treated mice (Fig. 3C and D). The expression of 4-1BB and 4-1BBL in lung were not significantly altered by this treatment (Fig. S2A-C). We also measured the body weight of treated mice. The body weight of 100μg 4-1BBIg treated mice was regained faster than crystalline silica treated mice (Fig. S2D). Besides, we performed in vitro study with primary lung cells and splenocytes to validate the causal relationship between 4-1BB and ASK1. Western blot analysis showed the phosphorylations of ASK1 were significantly decreased in accordance with 4-1BBIg concentration (Fig. S3D and E). Based on these evidence, we chose NQDI 1, a selective ASK1 inhibitor, to block the pathway (Fig. 3E). An optimal concentration range was determined using 5 mg/kg and 10 mg/kg on crystalline silica-injured C57BL/6 mice. At 5 mg/kg, ASK1 phosphorylation was reduced by 50% in the silicosis model mice at day 7. At 10 mg/kg, NQDI 1 prevented nearly 80% of ASK1 phosphorylation (Fig. S5), and was the concentration selected for use. To determine the influence of drug administration, we measured AST and ALT in the serum as indicators of liver injury. Animal body weights were recorded during the experiment. The data showed that NQDI 1 injection had no deleterious effects on the experimental animals (Fig. S6). We used western blot analysis to verify ASK1 phosphorylation and downstream MAPK cascades (p38 and JNK) in lung tissues at specific time points. As shown in Fig. 3F-H, ASK1 phosphorylation levels were effectively inhibited at all time points, while total ASK1 was not affected. Downstream p38 and JNK levels were affected to varying degrees. Given these results, we assert that NQDI 1 blocks the 4-1BB signaling pathway without adversely affecting experimental animals.
Blocking the 4-1BB signaling pathway alleviates crystalline silica-induced lung inflammation
Inhalation of crystalline silica induces lung inflammation. To investigate the pathogenesis of inflammation, we used lung slices with H&E staining to monitor pathological changes. As Fig. 4A shows, no obvious abnormalities were observed in the saline-treated group and the group treated with saline+NQDI 1, indicating that NQDI 1 did not induce obvious pathological changes. However, in the crystalline silica-treated group, particles damaged the lung architecture and led to massive inflammatory cell infiltration at days 3 and 7. Fibrotic cellular nodules were observed at day 56. In contrast, NQDI 1 treatment in crystalline silica-injured mice reduced the number of infiltrating inflammatory cells and the degree of alveolar thickness. The size of fibrotic cellular nodules was obviously reduced.
Inflammatory response due to crystalline silica alleviated by blocking the 4-1BB pathway. A Hematoxylin and eosin staining was used to observe histopathological changes in mice lungs after crystalline silica instillation. a1-d1, 3 days; a2-d2, 7 days; and a3-d3, 56 days. a1-a3, saline group; b1-b3, saline plus NQDI 1 group; c1-c3, crystalline silica group; d1-d3, crystalline silica+ NQDI 1 group. Scale bar, 100 μm. Magnification at 200× B-D ELISA and cytometric bead array analysis of TNF-α, IL-6, IL-1β secretions in BALF (n = 5-6; # P<0.05 compared with the saline group; * P<0.05 compared with the crystalline silica group). The experiment was performed in triplicate. Error bars indicate the mean±S.D.
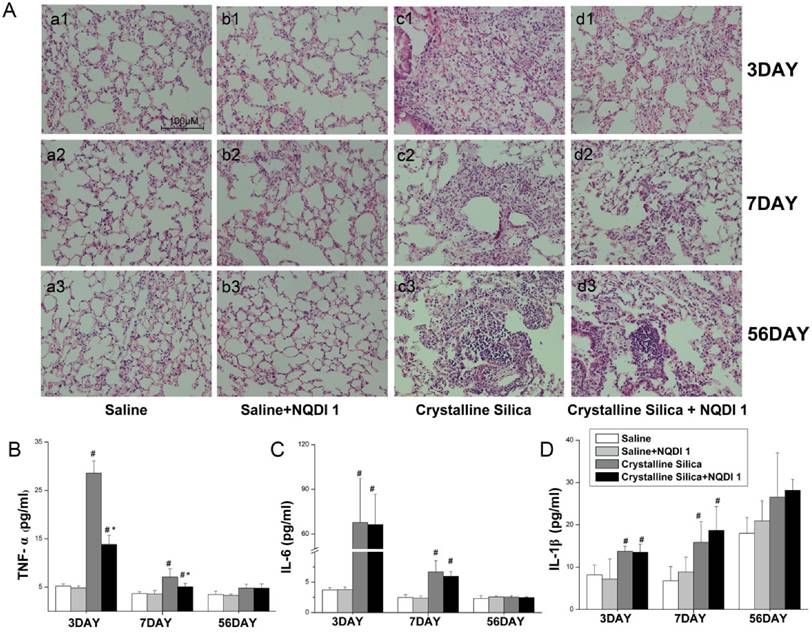
To further investigate inflammatory changes in the lungs, we examined three representative pro-inflammatory cytokines, TNF-α, IL-6 and IL-1β, in BALF by cytometric bead array (CBA) and ELISA analysis. TNF-α secretion increased after crystalline silica instillation at days 3 and 7 (Fig. 4B). TNF-α levels were obviously decreased by the inhibitor, particularly at day 3. Crystalline silica instillation also elevated IL-6 and IL-1β secretion at days 3 and 7. However, blocking the 4-1BB pathway did not alter secretion of the two cytokines (Fig. 4C, D). These combined data suggest that blocking the 4-1BB signaling pathway attenuates crystalline silica-induced lung inflammation.
Blocking the 4-1BB pathway reduces the fibrotic phenotype after crystalline silica instillation
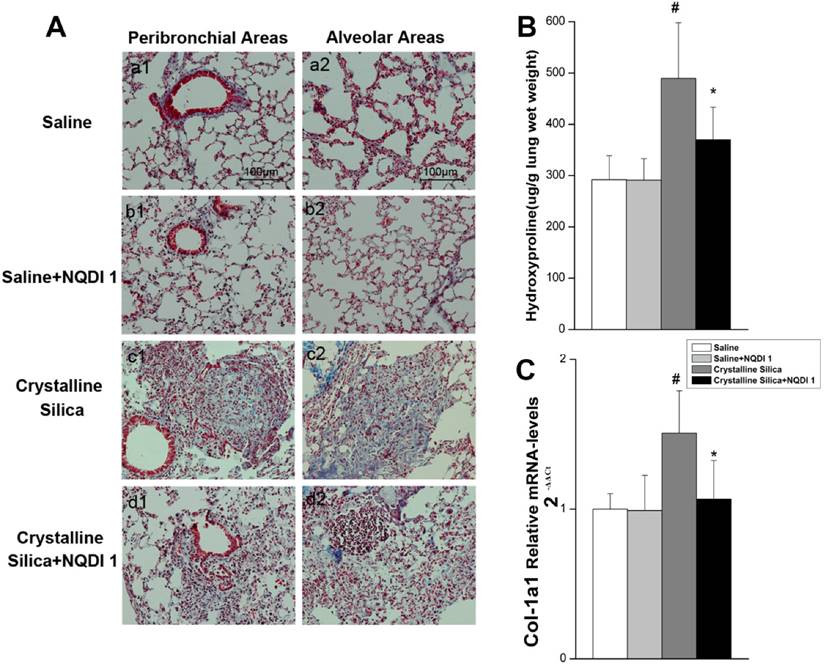
As observed by H&E staining, the size of cellular nodules was affected by NQDI 1 treatment at day 56. To further investigate the biological significance of the 4-1BB pathway during fibrogenesis, we examined lung slices using Masson's trichrome staining. As shown in Fig. 5A, crystalline silica-induced collagen deposition was apparent in the lungs of crystalline silica-treated mice, but not in mice treated with crystalline silica+NQDI 1. A reduction in collagen deposition was apparent in both the peribronchial and alveolar areas. Collagen in the lungs was determined by measuring hydroxyproline content. Reduced collagen in the lung tissue of NQDI 1-treated mice was confirmed (Fig. 5B). Attenuated fibrosis was further supported by decreased type I collagen mRNA levels (Fig. 5C). These data indicate that blocking the 4-1BB pathway could reduce collagen deposition in crystalline silica-injured mice.
Pulmonary fibrosis caused by crystalline silica attenuated by administrating NQDI 1. A Masson trichrome staining of collagen in lung sections of treated C57 mice at 56 days. Representative images are shown (n=5). a1-a2, saline group; b1-b2, saline+ NQDI 1 group; c1-c2, crystalline silica group; d1-d2, crystalline silica+ NQDI 1 group. a1-d1 are images of the peribronchial areas and a2-d2 are images of the alveolar areas. Scale bar, 100 μm. Magnification at 200×. B Hydroxyproline content in lung tissues from treated mice at 56 days were measured (n = 5). C Relative expression of Col-1A1 was assayed by real-time PCR (n=5 per group). All experiments were performed three times. # P<0.05 compared with the saline group; * P<0.05 compared with the crystalline silica group.
Th1 and Th17 responses could be effectively ameliorated through 4-1BB blockade
A previous study proposed that an interaction between 4-1BB and its ligand during inflammation may induce the Th1 response and maximize the function of effector T cells [27]. CD4+ T cells have been reported to play roles in fibrogenesis [28]. Our previous study using an experimental silicosis model reported that the Th balance shifts from Th1 dominant to Th2 dominant during fibrosis development. To examine whether attenuated inflammation and fibrosis are associated with changes in CD4+ T cells, we performed flow cytometry, real-time PCR, ELISA and CBA. As shown in Fig. 6A, the percentage of CD4+ T cells expressing IFN-γ, regarded as Th1 cells, was significantly decreased by NQDI 1 compared to cells in the crystalline silica group. The attenuated Th1 immune response was further supported by decreased Th1 transcription factor T-bet mRNA levels in the lungs (Fig. 6B). Levels of the Th1 cytokine INF-γ in BALF were also decreased by blocking the 4-1BB pathway (Fig. 6C). mRNA levels of the Th2 transcription factor GATA-3 in the lungs were increased in the late stages of fibrosis development (Fig. 6D), and levels decreased at day 56 by blocking the 4-1BB pathway. ELISA analysis showed that IL-13 in BALF slightly decreased without significance, and IL-4 levels were not altered by blocking the 4-1BB signaling pathway (Fig. 6E, F). These combined data suggest that the Th1 immune response is significantly attenuated in the inflammation stage, but that the Th2 response in the late stage of fibrosis is not affected.
Th1/Th2 and Th17 immune responses influenced by NQDI 1 in a mouse model of crystalline silica-induced fibrosis. A Percentage of CD4+ IFN-γ+ Th1 cells in hilar lymph nodes was assayed by flow cytometry. B Relative expression of Th1 nuclear transcription factor T-bet in the lungs was assayed by real-time PCR. C Secretion of the Th1 cytokine IFN-γ in BALF was assayed by CBA. D Relative expression of Th2 nuclear transcription factor GATA-3 in the lungs was assayed by real-time PCR. E, F Secretion of Th2 cytokines IL-4 and IL13 in BALF was assayed by CBA and ELISA. G Percentage of CD4+ IL-17A+ Th17 cells in HLN was assayed by flow cytometry. H Relative expression of Th17 nuclear transcription factor ROR-γt in the lungs was assayed by real-time PCR. i Secretion of the Th17 cytokine IL-17A in BALF was assayed by CBA. For all experiments, n=5-6 per group per time point. # P<0.05 compared with the saline group; * P<0.05 compared with the crystalline silica group.
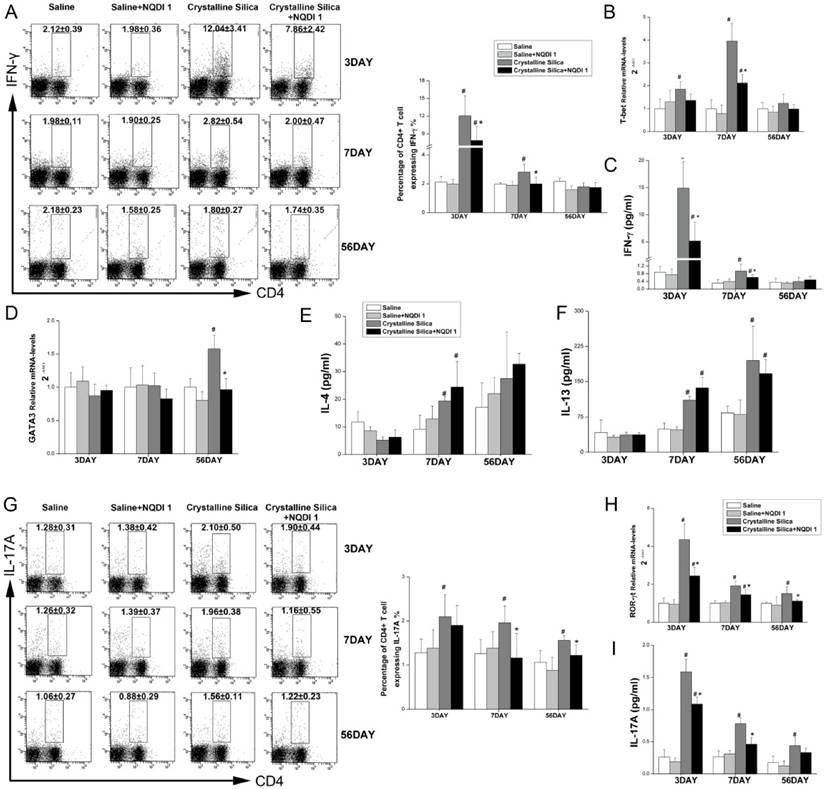
Th17 response was reported to be associated with Th1 response in inflammatory disease [29]. IL-17A also plays a role in crystalline silica-induced lung inflammation and fibrogenesis [17]. Our results show that blocking the 4-1BB signaling pathway significantly reduced the percentage of IL-17A-expressing CD4+ T cells (Th17 cells) (Fig. 6G) at days 7 and 56. Real-time PCR also showed reduced levels of the Th17 transcription factor ROR-γt in the lungs at all time points (Fig. 6H), supporting the notion that the Th17 immune response is also alleviated by blocking the 4-1BB pathway. We also measured the Th17 cytokine IL-17A in BALF to confirm the response (Fig. 6I). We demonstrated that attenuated inflammation by blocking the 4-1BB pathway is associated with a reduction in Th1 and Th17 cells and cytokines.
Blocking the 4-1BB signaling pathway may affect the number and function of regulatory T cells
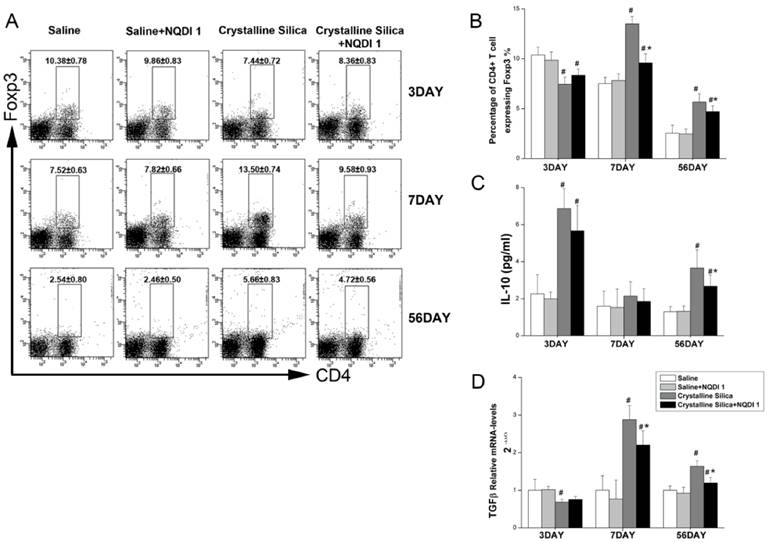
Regulatory T cells (Treg) are an important subset of regulatory immune cells and may suppress excessive inflammation [30]. Treg cells also participate in fibrogenesis. The Th2 response was not effectively altered by the blockade in the fibrosis stage. We explored whether Treg was affected by blocking the 4-1BB signaling pathway. As shown in Fig. 7A, the percentage of Foxp3+ Treg was increased in the crystalline silica group compared to the saline group at days 7 and 56. Percentages were both decreased by blocking the 4-1BB pathway. Interestingly, Foxp3+ Treg was significantly decreased in the crystalline silica group compared to the saline control group at day 3. NQDI 1 administration raised the percentage, but not significantly. We examined the levels of two cytokines, IL-10 and TGF-β, as a measurement of Treg function. Fig. 7C shows that the blockade did not alter secretion of IL-10 in BALF at days 3 and 7. At day 56, IL-10 was significantly decreased. Real-time PCR showed that TGF-β expression in the lungs was significantly decreased at days 7 and 56 (Fig. 7D). These results indicate that the number and function of Treg are affected by blocking the 4-1BB signaling pathway.
T regulatory cells influenced by blocking the 4-1BB pathway. A, B Percentage of CD4+ Foxp3+ T regulatory cells in HLN was assayed by flow cytometry. C, D Secretion of IL-10 in BALF was assayed by CBA. Relative expression of TGF-β in the lungs was assayed by real-time PCR. For all experiments, n=5-6 per group per time point. # P<0.05 compared with the saline group; * P<0.05 compared with the crystalline silica group.
Fibrocytes are involved in the less severe fibrotic phenotype by blocking the 4-1BB pathway
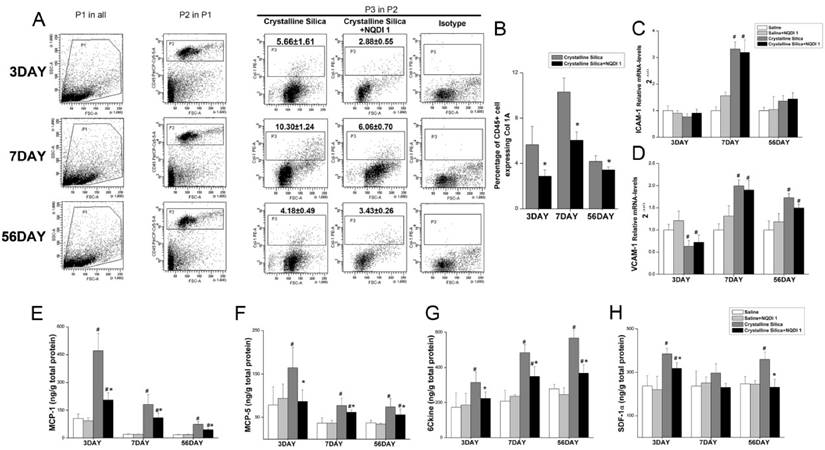
4-1BB signaling could enhance circulating leukocyte recruitment. Fibrocytes are circulating cells expressing leukocyte markers. Evidence shows that recruitment of fibrocytes from circulation play an important role in the development of fibrosis [16, 31, 32]. It has been reported that CD4+ T cells can control fibrocyte differentiation [33]. We investigated whether the decreased fibrotic phenotype is associated with fibrocytes. Single cell suspension of the lung tissue was used to determine fibrocyte recruitment. As shown in Fig. 8A, Gate P1 was used to gate out dead cells and crystalline silica particles, depending on forward scattering (FSC) and side scattering (SSC). Leukocytes were gated in depending on the cell surface marker CD45. The percentage of fibrocytes was calculated by P3, whose position is determined by isotype control staining. As shown in Fig. 8B, fibroctye recruitment was effectively decreased by the blockade at all time points. This result shows that, other than Th2 and Treg cells, reduced accumulation of fibrocytes is associated with a decreased phenotype of fibrosis.
Blocking the 4-1BB signaling pathway reduced fibrocyte recruitment into the lungs. A Identification of lung fibrocytes. Gate P1 was set to gate out dead cells and crystalline silica particles. Gate P2 was set to collect CD45-positive cells from P1. Gate P3 was placed to circle the Col-1 positive cells from P2. Isotype rabbit IgG was used to identify the appropriate position of gate P3. B Percentage of CD45+ Col-1+ fibrocytes in the lungs. C, D Relative expression of ICAM-1 and VCAM-1 in the lungs was assayed by real-time PCR (n=5). E-H ELISA analysis of MCP-1 (CCL-2), MCP-5 (CCL-12), 6Ckine (CCL-21), and SDF-1α (CXCL-12) expression in lung tissues (n = 5-6). # P<0.05 compared with the saline group; * P<0.05 compared with the crystalline silica group.
Chemokines are involved in decreased accumulation of fibrocytes
Leukocyte recruitment from circulation into tissues is a multistep process. Each step is orchestrated by different types of molecules, including adhesion molecules and chemokines [34]. To probe the cause of decreased fibrocyte recruitment, we examined two integrins, ICAM-1 and VCAM-1, in the lungs by performing real-time PCR. Relative VCAM-1 expression in crystalline silica-injured lungs was up-regulated to the saline controls at days 7 and 56, while ICAM-1 was only significantly increased at day 7 (Fig. 8C, D). Blocking the 4-1BB pathway did not alter mRNA levels of the two molecules, indicating that the adhesive molecules are not involved in decreased fibrocyte recruitment.
Previous studies have shown that fibrocytes express a set of chemokine receptors, such as CCR2, CCR7 and CXCR4. The ligands of these receptors (CCL-2, CCL-12, CCL-19, CCL-21 and CXCL-12) have been implicated in fibrocyte recruitment during fibrosis development [15, 16, 35]. We further examined expression of these chemokines in the lung tissue using ELISA. As shown in Fig. 8E-H, the chemokines MCP-1 (CCL-2), MCP-5 (CCL-12) and 6Ckine (CCL-21) were all increased in the lung tissue of the crystalline silica group compared to the saline controls at all time points. Blocking the pathway effectively reduced expression. SDF-1α (CXCL-12) expression was also decreased, but it was only statistically significant at days 3 and 56. These data indicate that reduced expression of related chemokines leads to a decrease in fibrocyte recruitment.
Discussion
Silicosis is a damaging occupational disease characterized by lung inflammation and progressive irreversible fibrosis. Progressive fibrosis leads to disability and mortality in patients, which results in a heavy burden to society. More effective treatments are needed to postpone progression of fibrosis, but the lack of a complete understanding of the molecular mechanisms involved is an obstacle. To better understand the mechanisms of progressive fibrosis, we used blind screening of lung homogenates from mice exposed to crystalline silica and found variations in protein expression profiles. 4-1BB showed the most significant changes at days 7 and 56 after exposure. We used a selective inhibitor of ASK1 to block 4-1BB downstream signaling pathway and found that this treatment could alleviate crystalline silica-induced lung inflammation and fibrosis. This finding adds new insight to understanding regulation of silicosis and may provide a molecular treatment target to silicosis.
4-1BB is a member of the TNF receptor superfamily and a potent T cell costimulatory molecule [36]. Aggregation with its ligand recruits TRAF2, which can activate ASK1, resulting in activation of the JNK/SAPK and p38 MAPK pathways [26]. 4-1BB receptors play a costimulatory role in T cell immunity and boost T cell responses. Although strong T cell immunity is elicited when animals are treated with agonist antibodies, it can simultaneously distort immune homeostasis [37] (Fig S7C). JNK and p38 MAPKs are involved in the pathway, and their activations are associated with inflammation and fibrosis. However, clinical trials of a p38 inhibitor in rheumatoid arthritis have failed [38], and a trial of a JNK inhibitor ended because the benefits/risk profile did not support continuation [39]. Neither of the two MAPKs are suitable to block the pathway. In addition, 4-1BB knock-out research showed that severe inflammation occurred in some diseases, including bleomycin-induced pulmonary fibrosis [40]. It is possible that lack of 4-1BB blocks its interaction with 4-1BBL and influences 4-1BB ligand signaling in cells [40] (Fig. S7B). In a small scale animal experiment (Fig. 3B), we administrated 4-1BB fusion protein (4-1BBIg) to crystalline silica-injured mice in the expectation to immobilize 4-1BBL and inhibit its interaction to 4-1BB (Fig. S7D). Western blot analysis showed mice given 4-1BBIg exhibits blocked phosphorylation of ASK1 in a dose-dependent manner (Fig. 3C and D). In vitro study also exhibited similar result (Fig. 3D and E). However, the results of in vitro study showed that the addition of 4-1BBIg to the co-culture system increased the expression of 4-1BBL in splenocytes (Fig S3A and C), while the level of 4-1BB was unchanged (Fig. S3A and B). It is possible that the 4-1BBIg could block 4-1BB signal pathway while enhance the 4-1BBL signal (Fig.S7D). Evidence showed that blocking 4-1BB pathway results in blocked ASK-1 activation. Therefore, in the large scale animal experiment, we set the kinase involved in the 4-1BB signaling pathway, ASK1, as the blocking target and used a small molecular chemical NQDI 1 at an optimal concentration (Fig. 9A and Fig. S7E). The administration of NQDI 1 would not influence the interaction of 4-1BB and its ligand, which does not affect the 4-1BBL signal. Moreover, a small molecular inhibitor has clinical and industrial advantages than fusion protein in consideration of economic benefits.
A The signaling pathway involved in 4-1BB. B Schematic diagram of effects of blocking the 4-1BB signaling pathway in crystalline silica-induced lung fibrosis.
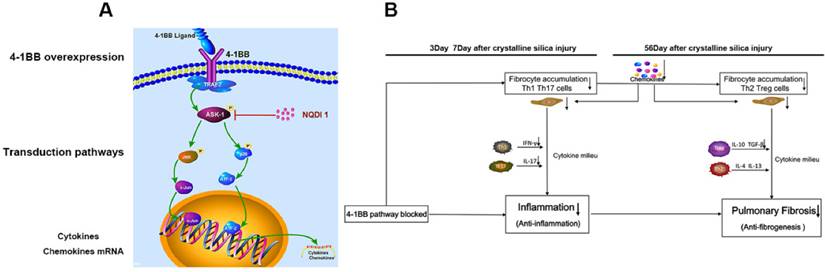
Several studies have shown evidence that injection of an anti-4-1BB monoclonal antibody (mAb) could ameliorate and prevent development of Th2-meditated allergic airway inflammation. Protection is associated with reduced Th2 cytokines and increased secretion of the Th1 cytokine IFN-γ [5, 27]. 4-1BB stimulating research has shown that although Th1 polarization of T cells was induced, IL-13 production could be enhanced in response to excessive IFN-γ secretion [41]. There is still controversy regarding Th2 cytokine release after 4-1BB stimulation. In this study, we blocked the pathway and found that the Th1 response was alleviated. As for the Th2 response, our data demonstrate that long-term injection of NQDI 1 in animals with silicosis effectively decreased expression of the Th2 transcription factor GATA-3 in the lungs. However, the Th2 cytokines IL-4 and IL-13 in BALF were not effectively reduced. IL-4 and IL-13 could be released by multiple cells [42] and there is a possibility that reduced cytokine secretion from Th2 cells could be compensated by other sources. Future studies will be needed to dissect the effect between 4-1BB signaling and Th2 response. Th17 and IL-17A are important inflammatory factors. IL-17A neutralization could decrease early inflammation and delay progression of crystalline silica-induced lung fibrosis. In the present study, we showed a reduction in Th17 cells and IL-17A after blocking the 4-1BB pathway. These results show that reduced lung inflammation by blocking the 4-1BB pathway is linked with alleviated Th1 and Th17 responses, while the relationship between an ameliorated fibrotic phenotype and Th2 response is not closely related.
Regulatory T cells (Foxp3 expressing CD4+ T cells) play important roles in lung inflammatory diseases [43]. Research on the relationship between Treg and 4-1BB showed that 4-1BB costimulation boosted proliferation of Treg [44]. However, several other studies have put forth different theories. Zhang et al. [45] showed that an agonistic 4-1BB antibody could promote Treg expansion while maintaining Foxp3 expression. However, Choi et al. reported that costimulation of 4-1BB did not contribute to Treg proliferation and that the 4-1BB signal had an adverse effect on the suppressive function of Treg cells [46]. In the present study, we showed that blocking the 4-1BB pathway effectively decreased the percentage of Foxp3 expressing CD4+ T cells in HLN. However, 3 days after crystalline silica injury, the percentage of Treg was lower in the crystalline silica group than in the controls and was slightly higher in the crystalline silica + NQDI 1 group than other crystalline silica counterparts. It is possible that in the acute stage of inflammation, naïve T cells (Th0) have a tendency to differentiate into pro-inflammatory Th1 and Th17 cells, leading to a lower percentage of Treg cells in the crystalline silica group at day 3. The decreased percentage of Treg by blocking 4-1BB signaling at days 7 and 56 may partly account for reduced collagen deposition.
Although 4-1BB was originally described as a surface molecule present on leukocytes, recent studies have revealed that it is expressed in inflamed blood vessels, hypoxic endothelial cells and even atherosclerotic lesions, leading to significantly enhanced monocyte infiltration [7, 47]. In protein array analysis, we found several chemokine variations, some of which were associated with fibrocyte recruitment. Fibrocytes are mesenchymal cells that arise from monocyte precursors and differentiate into fibroblasts and myofibroblasts. Fibrocytes are a potential source of lung fibroblasts [9, 48, 49] that produce collagen. Cytokines and additional protein factors released by fibrocytes during injury may lead to recruitment of diverse populations of effector cells [50, 51]. Thus, fibrocytes may be involved in the pathogenesis of fibrosis not only by synthesizing collagen but also by regulating the infiltration and activation of other cell types. We demonstrated that blocking the 4-1BB pathway could effectively reduce fibrocyte recruitment. Reduced recruitment was associated with decreased chemokines. Mitogen-activated protein kinases (MAPKs) have been implicated as playing key regulatory roles in the production of pro-inflammatory cytokines and chemokines. 4-1BB is a cell surface molecular triggering the two MAPKs, JNK and p38 activation. Teijeira et al. reported that CD137 in inflamed lymphatic endothelial cells could increase CCL21 production and enhance migration of dendritic cells [47]. Tsou et al. indicated that ASK-1 mediates CCL2 expression in human synovial fibroblasts [52], which may provide evidence for a relationship between 4-1BB signaling and chemokine release.
Niedermeier et al. reported that development of fibrocytes was under the control of CD4+ T cells [33]. Treating mice with Th1 cytokines resulted in a significant reduction in the number of fibrocytes. Bellini et al. also reported that fibrocytes may proliferate and express a predominant profibrotic or proinflammatory phenotype depending on the local cytokine milieu [53]. In addition, development of rheumatoid arthritis could be promoted by fibrocyte and T cell interaction [54]. T cells may have an influence on fibrocytes. CD4+ T cells may play a role in fibrogenesis by regulating fibrocyte development. Other research has also indicated that CD4+ T cells participate in fibrogenesis through releasing related cytokines (IL-4, IL-5, IL-13) rather than producing collagen directly [28]. Blocking the 4-1BB pathway could reduce cytokine release by effector T cells and decrease fibrocyte recruitment.
Acute inflammatory responses are central to elimination of foreign bodies, but often cause harm to tissues. Lung exposed to crystalline silica first leads to Th1 dominant response to remove foreign bodies. After that Th2 immune response would take place to constrain Th1-driven acute inflammation and repair damaged tissues. 4-1BB, a costimulatory molecular, works as an immune accelerator leading to excessive secretions of IFN-γ and TNF-α. In some disease like cancer or microbe infections, the immune response should be strengthened to eliminate the tumor cells or microbes. However, in the lung exposed to crystalline silica, Th1 immune response needs not to be that strengthened for crystalline silica dust is a kind of inorganic material that cannot be dissolved and cleared. Excessive inflammatory response is a kind of injury to lung cells, which leads to overzealous Th2 response resulting in progressive fibrosis. Taken together, these data provide strong evidence blocking the 4-1BB pathway attenuated crystalline silica-induced pulmonary inflammation and subsequent fibrosis (Figure 9b): i) inflammation stage: The acute Th1 and Th17 response was alleviated, accompanied by down regulated expression of IFN-γ IL-17A inflammatory mediators. ii) fibrosis stage: The number of Th2 and Treg cells into the lungs were reduced, accompanied by down regulated expression of IL-10 TGF-β fibrogenesis mediators. Besides, the recruitment of fibrocytes was dropped throughout development of silicosis. Decreased release of chemokines leads to the reduction of fibrocyte recruitment. All these factors are believed to be involved in the less severe fibrotic phenotype. We conclude that blocking the 4-1BB pathway alleviates crystalline silica-induced lung inflammation and fibrosis. All aspects of this study provide novel insights into an efficient target molecular for postponing the fibrogenesis of silicosis.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81573117) and Program for Liaoning Innovative Research Team in University (LT2015028).
Conflict of interest
The authors declare no competing financial interests with this manuscript.
References
1. Lopes-Pacheco M, Bandeira E, Morales MM. Cell-Based Therapy for Silicosis. Stem Cells Int. 2016;2016:5091838
2. Leung CC, Yu ITS, Chen W. Silicosis. The Lancet. 2012;379:2008-18
3. Bang KM, Mazurek JM, Wood JM, White GE, Hendricks SA, Weston A. Silicosis mortality trends and new exposures to respirable crystalline silica—United States, 2001-2010. MMWR Morb Mortal Wkly Rep. 2015;64:117-20
4. Re SL, Lison D, Huaux F. CD4+ T lymphocytes in lung fibrosis: diverse subsets, diverse functions. J Leukoc Biol. 2013;93:499-510
5. Polte T, Foell J, Werner C, Hoymann HG, Braun A, Burdach S. et al. CD137-mediated immunotherapy for allergic asthma. J Clin Invest. 2006;116:1025-36
6. Kim YH, Choi BK, Shin SM, Kim CH, Oh HS, Park SH. et al. 4-1BB triggering ameliorates experimental autoimmune encephalomyelitis by modulating the balance between Th17 and regulatory T cells. J Immunol. 2011;187:1120-8
7. Drenkard D, Becke FM, Langstein J, Spruss T, Kunz-Schughart LA, Tan TE. et al. CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. FASEB J. 2007;21:456-63
8. Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol. 2011;11:427-35
9. Sakai N, Wada T, Yokoyama H, Lipp M, Ueha S, Matsushima K. et al. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci U S A. 2006;103:14098-103
10. Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556-62
11. Inomata M, Kamio K, Azuma A, Matsuda K, Kokuho N, Miura Y. et al. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-induced murine pulmonary fibrosis. Respir Res. 2014;15:16
12. Singh SR, Sutcliffe A, Kaur D, Gupta S, Desai D, Saunders R. et al. CCL2 release by airway smooth muscle is increased in asthma and promotes fibrocyte migration. Allergy. 2014;69:1189-97
13. Dupin I, Allard B, Ozier A, Maurat E, Ousova O, Delbrel E. et al. Blood fibrocytes are recruited during acute exacerbations of chronic obstructive pulmonary disease through a CXCR4-dependent pathway. J Allergy Clin Immunol. 2016;137:1036-42.e7
14. Shipe R, Burdick MD, Strieter BA, Liu L, Shim YM, Sung SS. et al. Number, activation, and differentiation of circulating fibrocytes correlate with asthma severity. J Allergy Clin Immunol. 2016;137:750-7.e3
15. Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:175-81
16. Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY. et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438-46
17. Chen Y, Li C, Weng D, Song L, Tang W, Dai W. et al. Neutralization of interleukin-17A delays progression of silica-induced lung inflammation and fibrosis in C57BL/6 mice. Toxicol Appl Pharmacol. 2014;275:62-72
18. Song L, Weng D, Dai W, Tang W, Chen S, Li C. et al. Th17 can regulate silica-induced lung inflammation through an IL-1β-dependent mechanism. J Cell Mol Med. 2014;18:1773-84
19. Liu T, Dai W, Li C, Liu F, Chen Y, Weng D. et al. Baicalin Alleviates Silica-Induced Lung Inflammation and Fibrosis by Inhibiting the Th17 Response in C57BL/6 Mice. J Nat Prod. 2015;78:3049-57
20. Liu F, Liu J, Weng D, Chen Y, Song L, He Q. et al. CD4+ CD25+ Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS One. 2010;5:e15404
21. Volynets GP, Chekanov MO, Synyugin AR, Golub AG, Kukharenko OP, Bdzhola VG. et al. Identification of 3 H-Naphtho [1, 2, 3-de] quinoline-2, 7-diones as Inhibitors of Apoptosis Signal-Regulating Kinase 1 (ASK1). J Med Chem. 2011;54:2680-6
22. Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB. et al. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L341-53
23. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA. et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035-40
24. Haga T, Suzuki J, Kosuge H, Ogawa M, Saiki H, Haraguchi G. et al. Attenuation of experimental autoimmune myocarditis by blocking T cell activation through 4-1BB pathway. J Mol Cell Cardiol. 2009;46:719-27
25. Wick G, Grundtman C, Mayerl C, Wimpissinger T-F, Feichtinger J, Zelger B. et al. The immunology of fibrosis. Annu Rev Immunol. 2013;31:107-35
26. Cannons JL, Choi Y, Watts TH. Role of TNF Receptor-Associated Factor 2 and p38 Mitogen-Activated Protein Kinase Activation During 4-1BB-Dependent Immune Response. J Immunol. 2000;165:6193-204
27. Sun Y, Blink SE, Liu W, Lee Y, Chen B, Solway J. et al. Inhibition of Th2-mediated allergic airway inflammatory disease by CD137 costimulation. J Immunol. 2006;177:814-21
28. Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210
29. Wan YY, Flavell RA. How diverse—CD4 effector T cells and their functions. J Mol Cell Biol. 2009;1:20-36
30. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636-45
31. Andersson-Sjöland A, de Alba CG, Nihlberg K, Becerril C, Ramírez R, Pardo A. et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129-40
32. Strieter RM, Keeley EC, Hughes MA, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J Leukoc Biol. 2009;86:1111-8
33. Niedermeier M, Reich B, Gomez MR, Denzel A, Schmidbauer K, Göbel N. et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A. 2009;106:17892-7
34. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678-89
35. Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C. et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675-84
36. Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23-68
37. Lee SW, Salek-Ardakani S, Mittler RS, Croft M. Hypercostimulation through 4-1BB Distorts Homeostasis of Immune Cells. J Immunol. 2009;182:6753-62
38. Hammaker D, Firestein G. “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis. 2010;69:i77-i82
39. Ma FY, Tesch GH, Nikolic-Paterson DJ. ASK1/p38 signaling in renal tubular epithelial cells promotes renal fibrosis in the mouse obstructed kidney. Am J Physiol Renal Physiol. 2014;307:F1263-73
40. Park SJ, Kim HJ, Lee JS, Cho HR, Kwon B. Reverse signaling through the co-stimulatory ligand, CD137L, as a critical mediator of sterile inflammation. Mol Cells. 2012;33:533-7
41. Shin SM, Kim YH, Choi BK, Kwon PM, Lee HW, Kwon BS. 4-1BB triggers IL-13 production from T cells to limit the polarized, Th1-mediated inflammation. J Leukoc Biol. 2007;81:1455-65
42. Dong J, Ma Q. In vivo activation of a T helper 2-driven innate immune response in lung fibrosis induced by multi-walled carbon nanotubes. Arch Toxicol. 2016 [Epub ahead of print]
43. Ryanna K, Stratigou V, Safinia N, Hawrylowicz C. Regulatory T cells in bronchial asthma. Allergy. 2009;64:335-47
44. Zheng G, Wang B, Chen A. The 4-1BB costimulation augments the proliferation of CD4+ CD25+ regulatory T cells. J Immunol. 2004;173:2428-34
45. Zhang P, Gao F, Wang Q, Wang X, Zhu F, Ma C. et al. Agonistic anti-4-1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand J Immunol. 2007;66:435-40
46. Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS. et al. 4-1BB-dependent inhibition of immunosuppression by activated CD4+ CD25+ T cells. J Leukoc Biol. 2004;75:785-91
47. Teijeira A, Palazon A, Garasa S, Marre D, Auba C, Rogel A. et al. CD137 on inflamed lymphatic endothelial cells enhances CCL21-guided migration of dendritic cells. FASEB J. 2012;26:3380-92
48. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C. et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047-53
49. Jimenez SA, Piera-Velazquez S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 2016;51:26-36
50. Kleaveland KR, Moore BB, Kim KK. Paracrine functions of fibrocytes to promote lung fibrosis. Expert Rev Respir Med. 2014;8:163-72
51. García de Alba C, Buendia-Roldán I, Salgado A, Becerril C, Ramírez R, González Y. et al. Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am J Respir Crit Care Med. 2015;191:427-36
52. Tsou HK, Chen HT, Chang CH, Yang WY, Tang CH. Apoptosis signal-regulating kinase 1 is mediated in TNF-alpha-induced CCL2 expression in human synovial fibroblasts. J Cell Biochem. 2012;113:3509-19
53. Bellini A, Marini MA, Bianchetti L, Barczyk M, Schmidt M, Mattoli S. Interleukin (IL)-4, IL-13, and IL-17A differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. 2012;5:140-9
54. Galligan CL, Keystone EC, Fish EN. Fibrocyte and T cell interactions promote disease pathogenesis in rheumatoid arthritis. J Autoimmun. 2016;69:38-50
Author contact
![]() Corresponding author: Jie Chen e-mail: Jchenedu.cn, Tel: 86 24 31939079 and Dong Weng e-mail wengdongedu.cn, Tel: 86 21 65115006 2206 Fax: 86 21 65111298.
Corresponding author: Jie Chen e-mail: Jchenedu.cn, Tel: 86 24 31939079 and Dong Weng e-mail wengdongedu.cn, Tel: 86 21 65115006 2206 Fax: 86 21 65111298.