Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(12):2015-2027. doi:10.7150/thno.15993 This issue Cite
Research Paper
Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1
Jenq-Lin Yang1, Wei-Yu Chen1, Yin-Ping Chen 1, Chao-Ying Kuo 1, Shang-Der Chen1,2,3, ![]()
1. Institute for Translation Research in Biomedicine;
2. Department of Neurology, Kaohsiung Chang Gung Memorial Hospital, 123 Ta Pei Road, Kaohsiung City 83301, Taiwan;
3. College of Medicine, Chang Gung University, 259 Wenhua 1st Road, Taoyuan City 33302, Taiwan
Received 2016-4-28; Accepted 2016-7-13; Published 2016-9-2
Abstract

Glucagon-like peptide-1 (GLP-1) is an intestinal-secreted incretin that increases cellular glucose up-take to decrease blood sugar. Recent studies, however, suggest that the function of GLP-1 is not only to decrease blood sugar, but also acts as a neurotrophic factor that plays a role in neuronal survival, neurite outgrowth, and protects synaptic plasticity and memory formation from effects of β-amyloid. Oxidative DNA damage occurs during normal neuron-activity and in many neurological diseases. Our study describes how GLP-1 affected the ability of neurons to ameliorate oxidative DNA damage. We show that activation of GLP-1 receptor (GLP-1R) protect cortical neurons from menadione induced oxidative DNA damage via a signaling pathway involving enhanced DNA repair. GLP-1 stimulates DNA repair by activating the cyclic AMP response element binding protein (CREB) which, consequently, induces the expression of apurinic/apyrimidinic endonuclease 1 (APE1), a key enzyme in the base excision DNA repair (BER) pathway. In this study, APE1 expression was down-regulated as a consequence phosphatidylinositol-3 kinase (PI3K) suppression by the inhibitor LY294002, but not by the suppression of MEK activity. Ischemic stroke is typically caused by overwhelming oxidative-stress in brain cells. Administration of exentin-4, an analogue of GLP-1, efficiently enhanced DNA repair in brain cells of ischemic stroke rats. Our study suggests that a new function of GLP-1 is to elevate DNA repair by inducing the expression of the DNA repair protein APE1.
Keywords: Glucagon-like peptide-1(GLP-1), Exendin-4, Oxidative DNA damage, Base excision repair (BER), Apurinic/apyrimidinic endonuclease 1 (APE1)
Introduction
Oxidative DNA Damage and Base Excision Repair (BER) in Neurons
Neurons are highly metabolic, non-dividing differentiated cells that produce abundant reactive oxygen species (ROS). On account of their high metabolic rate, they are subjected to elevated levels of oxidative stress and generate DNA base modifications and DNA strand breaks [1,2]. Oxidative DNA lesions are substantially elevated in acute brain trauma and neurodegenerative diseases [3-6]. Accumulation of DNA damage in neurons beyond repair capability may trigger aging processes, carcinogenesis, or neuronal death [7,8]. Enhancing DNA repair efficiency, therefore, is a crucial factor in maintaining DNA integrity and increasing neuronal survival against pathologically induced oxidative stress.
Base excision repair (BER) is the major process that repairs oxidative lesions, alkylated DNA bases, deaminated bases, single-strand breaks, and abasic sites in nuclear and mitochondrial DNA [9,10]. BER involves sequential steps of multiple enzymes starting with the recognition and removal of the damaged base (glycosylase), incising the sugar phosphate backbone (endonuclease), filling in the appropriate base (polymerase), and ligating the DNA nick (ligase). Apurinic/apyrimidinic endonuclease 1 (APE1), the most abundant apurinic endonuclease in human cells [11], incises the sugar phosphate backbone at the abasic site generating a one-nucleotide nick break. Class I and class II AP endonucleases incise DNA at a baseless site at the phosphate 3´ and 5´ ends leaving 3´-OH and 5´-phosphate termini [12]. Previous studies by the Bohr's and Mattson's groups demonstrated that APE1 is the only protein of the BER pathway up-regulated after glutamate-induced oxidative stress [13] and BDNF (Brain-derived neurotrophic factor) treatments [14]. Both studies reported that DNA repair efficiency of BER is enhanced by up-regulated APE1. In the present study, we found that stimulation of GLP-1 receptor (GLP-1R) enhanced APE1 expression and incision activity; also administration of Exendin-4 (EX-4) substantially reduced ischemia-induced nuclear DNA damage in brain cells. Our findings suggest that the activation of GLP-1R plays a neuroprotective function via enhancing APE1-mediated DNA repair efficiency.
Glucagon-like Peptide 1 (GLP-1) and Exendin-4 (EX-4)
GLP-1 is a 30-amino acid peptide, reported in 1985 by the Creutzfeldt group [15], that plays an important role in promoting blood sugar intake by cells. The proglucagon gene encodes GLP-1 [16], which is post-translationally processed into various active peptides in a tissue-specific manner [17,18]. GLP-1 is an incretin that stimulates cellular glucose up-take after meals and its analogue, EX-4, is used clinically to treat adult type II diabetes mellitus (T2DM) [17,19]. Fortunately, unlike the very short half-life of GLP-1 in circulation (less than two minutes), EX-4 resists dipeptidyl peptidase IV (DPP IV) thereby remaining longer in an active form in organisms. Expressions of GLP-1R are detected in pancreatic islet, central nervous system, muscle, kidney, heart, lung, gastrointestinal track, and adipose cells (see reviews [20,21]). The GLP1R gene, on human chromosome 6p21, encodes a 462-amino acid peptide, which is characterized as a G-protein coupled receptor. Activation of GLP-1R triggers two major signaling pathways: the phosphatidylinositol 3 kinase (PI3K), AKT pathway; and adenosine cyclase (AC), protein kinase A (PKA), mitogen-activated protein kinase kinase (MEK), and extracellular-signal-regulated kinase (ERK/MAPK) pathway [22]. Both downstream signaling pathways lead to phosphorylation of the cyclic AMP response element binding protein (CREB). Current data indicate that GLP-1 not only regulates blood-sugar, but also has a role in neuroprotection, including protecting cells from excitotoxic neuronal damage [23], alleviating tau-hyperphosphorylation [24] and Aβ deposition and plaque formation [25], and reducing ischemia-induced brain damage [26-28]. Nevertheless, what GLP-1 initiated mechanisms involve in the various neuroprotective functions is still under investigation. Our study demonstrated that activation of GLP-1R increased neuronal viability and DNA repair efficiency via PI3K-AKT- axis-mediated APE1 expression. GLP-1R, therefore, are a potential target to develop innovative treatments for neurodegenerative diseases and ischemic stroke.
Materials and Methods
Preparation of Primary Cortical Neurons and Treatments
Cultures of primary cortical neurons were prepared from 17-day-old embryos of Sprague-Dawley rats. Preparation procedures were described in a previous study [13]. All experimental procedures with animals were approved by Chang Gung Memorial Hospital Animal Care and Use Committee and complied with NIH guidelines. Experiments were performed in 8- to 10-day-old cultures in which greater than 95% of the cells were neurons. Cultured neurons were pretreated with 100 nM GLP-1 or 100 nM EX-4 overnight (~14 h) before menadione treatment. Menadione (2-methyl-1,4-naphthoquinone, Sigma-Aldrich) was freshly prepared in NeuroBasal medium. Neurons were treated with 40 µM menadione for 10 minutes at 37oC. Either GLP-1 or EX-4 was then added to 50% conditioned medium with a 100 nM final concentration. To examine downstream signal transduction by GLP-1R, a 10 μM MEK inhibitor (U0126, Cell Signaling) or a 20 μM PI3K inhibitor (LY294002, Cell Signaling) was added into cultured neurons two-hour prior to adding GLP-1 or EX-4. Cultures were incubated for 0.5, 1, 3, 6, 12 or 24 hrs, at which times the neurons were analyzed.
Nuclear Fractionation
The nuclear fraction was collected to determine levels of DNA polymerase beta (Pol β), 8-oxoguanine DNA glycosylase (OGG1), nei endonuclease VIII-like 1 (NEIL1), and lamin A and C (lamin A/C). The neuronal cultures (around 5 millions neurons) were harvested by 1,000 rpm centrifugation and resuspended cell pellets were resuspended in 200 μl of pre-chilled buffer A (1.5 mM MgCl, 10 mM KCl, 0.5 mM DTT, and 10 mM HEPES pH 7.9). After a 30-minute incubation in ice, cells were homogenized and the cellular mixture was centrifuged at 228 g for 5 minutes at 4oC. Each pellet was resuspended in 200 μl of S1 buffer (0.245 M sucrose, 10 mM MgCl2) and layered over 200 μl of S3 buffer (0.865 M sucrose, 0.5 mM MgCl2). The two-layer mixture was centrifuged at 2,800 G for 10 minutes at 4 oC and the supernatant removed. The nuclear pellet was dissolved in 150 μl of RIPA buffer for Western blotting analysis.
Reverse-transcription PCR (RT-PCR)
GLP-1R mRNA from neuronal cells was detected by RT-PCR using the forward primer - “ATGAGAGACCTGCCCTTGGA” and the reverse primer - “CCCCAAGGCAAAGACAGGAT”. The Direct RT-PCR kit from Yeastern Biotech Co. was used to assay the GLP-1R mRNA. The cultured neurons, around fifty-thousand cells, were briefly rinsed with ice-cold PBS and pellets collected by centrifugation. Neuronal pellets were washed with Wash buffer and centrifuged to obtain a pellet. The clean neuronal pellet was added immediately to the cell lysis buffer to prevent RNA degradation. The RT and PCR procedures followed instructions in the user's manual of the kit.
Western Blotting Analysis
Neurons were extracted in RIPA buffer (150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1X protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail (Pierce), and 50 mM Tris; pH 8.0) and total protein within the lysate was determined according to the BCA™ protein assay kit (Pierce). The dilution factors for the primary antibodies were as following, APE1 (1:1,000; Santa Cruz), phosphor-AKT (1:1,000; Cell Signaling), phosphor-CREB (1:1,000; Cell Signaling), Ligase III (1:1,000; Santa Cruz), FEN1(1:1,000; Cell Signaling), Pol β (1:200; GeneTex), NEIL1 (1:1,000; Santa Cruz), OGG1 (1:1,000; Novus), UDG (1:1,000; Novus), lamin A/C (1:1,000; Cell Signaling), GLP-1R (1:1,000; Abcam/Alomone Lab) and actin (1:5,000; Sigma-Aldrich). The secondary antibodies (Vector Laboratories) were diluted 1:5,000. Immunolabeled proteins were visualized by using an enhanced chemiluminescence kit (Amersham).
Immunofluorescent labeling
Cultured neurons were briefly washed in cold PBS and fixed immediately with 4% paraformaldehyde in phosphate buffer saline (PBS) for 10 minutes. The formaldehyde fixed cells were incubated with permeablization buffer (0.5% Triton X-100, 100 mM glycine, 1% BSA, 0.7 mM EDTA) for 10 min on ice. The samples were then incubated with blocking buffer (10% bovine serum, 0.01% sodium azide in 1X PBS) at 37oC for 1 hr or 4oC overnight. Diluted primary antibodies (TUJ1, Sigma-Aldrich, 1:500; GLP-1R, Santa Cruz, 1:500) were incubated with samples for 1 hr at 37 oC. The samples were then rinsed with wash buffer (0.05% Tween-20 in 1xPBS) three times for 5 minutes at room temperature. A fluorophore conjugated secondary antibody (Alexa series, Invitrogen; 1:1,000 dilution), was added and the samples were incubated for 1 hr at 37 oC. Samples were rinsed briefly with wash buffer (3 x 5 minute) and mounted with Prolong Gold containing DAPI (nuclear marker, Invitrogen). Images of immunostained neurons were captured using a Olympus FV1000 confocal microscope system.
Measurement of BER protein enzymatic activities
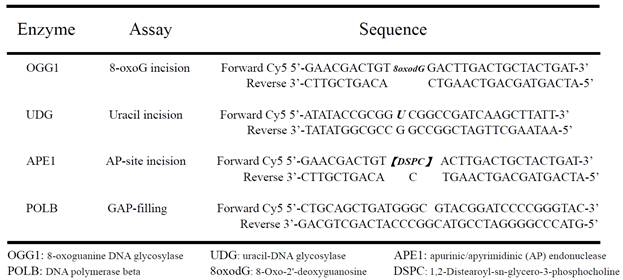
Cultured neurons were extracted by re-suspending and homogenizing in buffer I (10 mM Tris-HCl, 200 mM KCl, pH 7.8) and adding an equal volume of buffer II (10 mM Tris-HCl, 600 mM KCl, 2 mM EDTA, 40% (v/v) glycerol, 0.2% (v/v) Nonidet P-40, 2 mM dithiothreitol (DTT) 0.5 mM phenylmethylsulphonyl fluoride (PMSF) and 1X protease inhibitor cocktail (Roche; pH 7.8). The lysate was briefly sonicated to completely disrupt cell and nuclear membranes. A 16,000 g centrifugation at 4oC for 10 minutes was performed to remove cellular debris and DNA. The cell extract was dialyzed overnight with buffer III (25 mM HEPES-KOH, 100 mM KCl, 12 mM MgCl2, 1 mM EDTA 17% glycerol, 1 mM DTT, pH 8.0) at 4oC. A brief centrifugation was employed to remove the precipitate after dialysis. The protein concentration was determined by the BCA™ protein assay kit (Pierce). The enzymatic activity of OGG1, UDG, APE1, and Polβ was measured by specifically designed Cy5 fluorophore -conjugated lesion DNAs (Table 1). The procedures of incision and gap filling assays were preformed as in previous studies [29,30]. After electrophoresis, the gels were visualized by using a Molecular Dynamics image system (Bio-Rad Corp.)
The defect nucleotide sequences for assaying enzymic activities of OGG1, UDG, APE1 and POLB.
Measurement of Neuronal Viability
To measure cytotoxicity of oxidative insults, we directly counted neurons on the same spotted. Rat cortical neurons were plated and maintained in 4- or 6-well chamber slides as described above. One group of cells were pretreated with 100 nM GLP-1 overnight (~16 h) before adding freshly made menadione. Each sample was treated with 40 µM menadione for 10 min at 37oC. Cells were washed with warm Neurobasal medium after treatment and GLP-1 (100 nM) was included in the cultures that had been pretreated with GLP-1. Images of neurons in designated microscope fields (10X objective) were acquired at 0 hr, 6 hr, 12 hr, and 24 hr. Neuronal survival at post-menadione treatment time points were expressed as a percentage of the number of viable neurons present at the 0 h time point.
Middle Cerebral Artery Occlusion (MCAO)
The original procedures of MACO reperfusion were described in the study by Chen et al. [31]. 7-8 week old male SD rats, with a body weight 280-300 grams, were anesthetized by intraperitoneal injection of chloral hydrate (400 mg/kg body weight, Sigma-Aldrich). Incisions of 1.5-cm were made on anesthetized rat scalps at the midpoint between the right eye and right ear. The temporalis muscle was separated to expose the zygoma and sequamosal bone. An approximate 25-mm2 window was made with a dental drill 1 mm rostal to the anterior junction of the zygoma and the sequamosal bone. The dura mater was carefully pierced with a microsurgical needle. The exposed middle cerebral artery was carefully isolated and ligated for 60 minutes by 10·O surgical suture to induce ischemic stroke in the right hemisphere of the cortex. During the ligation period, one percent isoflurane was provided by ventilator to maintain sedation of animals. As soon as the middle cerebral artery (MCA) ligation was complete, both common carotid arteries (CCA) were ligated by aneurysm clips. Ligated CCA and MCA were released after 60 minutes. Twenty-four hours after the MCA reperfusion procedure, animals were anesthetized by isofluoran and perfused with 150 ml PBS. The focal ischemic brain was dissected and incubated with 2% triphenyl tetrazolium chloride (TTC) at 37oC for 60 minutes to determine the ischemic infarct area. The MCAO procedures of animals were approved by Chang Gung Memorial Hospital Animal Care and Use Committee and complied with NIH guidelines.
EX-4 was administered at the completion of the MCAO procedure and given daily until animals were sacrificed. The dose of EX-4 was 10 μg/kg body weight given by tail-vein injection.
Immunostochemistry for γH2AX labeling
Mouse/Rabbit HRP (DAB) Detection System (BIOTnA Biotech) were used to examine target-proteins in brains. The detecting procedures were followed the staining protocol of user's manual. The brief description is as following. The 25 μm thickness brain sections were covered thoroughly with hydrogen peroxide for 10 minutes. Brain sections were washed twice with 1x PBS (phosphate buffer saline) after incubation. The brain sections were briefly washed 3 times with PBS after a 30-minute incubation with penetration buffer (0.1 % Triton X-100 in PBS) at room temperature. The samples were incubated with immunoblocking buffer (buffer from kit) for 10 minutes at room temperature, then, wash samples were immediately washed twice with 1x PBS. The anti-γH2AX antibodies (1:250; Cell Signaling) and HRP-conjugated secondary antibodies (1:500; from the kit) were diluted with 1% BSA in 1x PBS and incubate with samples at 37oC for an hour respectively. Three 1x PBS washes were performed after primary antibody hybridization and four washes for secondary antibody hybridization. One milliliter of DAB substrate was mixed with 30 μl of DAB chromogen freshly. The tissue samples were incubated with DAB mixture for 3 minutes at room temperature. Then samples were washed 4 times. Hematoxylin solution was applied to stain nuclei of tissue samples by incubation 1 minute at room temperature. The sample slides were sealed with cover slips after dehydration by xylene. Sample slides were observed by Olympus confocal microscope (FV1000).
Terminal deoxynucleotide transferase-mediated dUTP Nick End Labeling (TUNEL) Assay
The In Situ Cell Death Detection Kit from Roche was used to detect apoptosis, double-strand, and single-strand DNA fragmentation. The staining procedures were adapted and optimized according to the product manual. The following is a brief description of procedures for frozen tissue sections. After washing out Tissue-Tek® (VWR) O.T.C. from embedded tissue slices by PBS, the tissue slides were fixed by 4% paraformaldehyde in PBS for an hour at room temperature. Slides were washed 5 times briefly in PBST (0.1% Triton X-100) and then immersed in 100% ethanol for 5 minutes. Next the slides were washed in decreasing concentrations of ethanol (100%, 95%, 85%, 70%, and 50%, 3 minutes each) for rehydration. Tissue slides were soaked in 0.85% NaCl and PBS each for 5 minutes, respectively. Fixation was performed by 4% formaldehyde in PBS for 15 minute. Following a twice brief PBS wash, permeabillization was obtained by placing samples in a Proteinase K solution (20 μg/ml proteinase K in PBS) for 10-20 minutes. Tissue slides were briefly washed once with PBS before and after a 5-minite re-fixation with 4% formaldehyde in PBS. Samples were equilibrated with 100 μl of Equilibration buffer (in kit) at room temperature for 5-10 minutes. For labeling, 50 μl of TdT reaction mix was added to the tissues and covered by cover slips. Slides then were incubated at 37oC for 60 minutes in the dark. After incubation, the cover slips were removed and reactions stopped by dipping the slides in 2x SSC for 15 minutes. After 3x5-minute PBS washes, 30 μl of mounting medium was added to the tissue sections and covered with cover slips. Sample slides were observed and images taken under a microscope (Olympus XM10).
Statistics
Statistical comparisons were made using Student's t-test. One-way ANOVA was used to evaluate significance of difference between groups, followed the Bonferroni t- test was used for multiple comparisons (* P<0.05, ** P<0.01, *** P<0.001). All values in graphs were the mean and standard error (SE) of determinations being made in at least 4-6 separate experiments.
Results
Cortical Neurons Express GLP-1 Receptor (GLP-1R) and Activation of GLP-1R Rescues Neurons against Menadione-induced Oxidative Insults
Reverse-transcription PCR (RT-PCR) and Western blotting techniques were used to detect the presence of GLP-1 receptor (GLP-1R) in rat primary cortical neurons; both mRNA and protein levels of GLP-1R were detected. Total RNA from 8-day old rat primary cortical neurons was isolated and one-step RT-PCR was used to probe for GLP-1R mRNA (Fig. 1A). The result of Western blotting demonstrated that GLP-1R proteins also were expressed in cortical neurons (Fig. 1B). Furthermore, we used immunocytochemistry to examine the presence and the distribution of GLP-1R in neurons. Images by confocal microscopy confirmed that GLP-1R expression was observed in cortical neurons and illustrated that GLP-1R are mostly located in neuronal cell bodies with some GLP-1R distributed in the axons (Fig. 1C). The GLP-1R were observed not only existing in cortical regions, but also were found expressing all over the rat brain including hippocampus, thalamus, hypothalamus, striatum, and amygdale regions (Fig. S1).
Activation of cortical neuron expressed GLP-1 receptor (GLP-1R) protected cortical neurons from oxidative insults. (A) mRNA and (B) protein expressions of GLP-1R were detected in the 8-day in vitro primary cortical neurons. (C) Immunofluorescence staining of GLP-1R (green), TUJ-1 (red), and DAPI (blue) in the 8-day in vitro primary cortical neurons. Merged bright field (100X) and immunostained (100X) images indicated that GLP-1R is mostly distributed in neuronal bodies but also exist in both axons and dendrites. (D) GLP-1or (E) EX-4 pretreated cortical neurons were continually treated with 100 nM GLP-1 or EX-4 after an oxidative insult and neuronal viability was measured at 0, 6, 12, 24 hr time points. Neuronal viability of ligand administrated neurons (GLP-1:~82%, EX-4:~67%) was approximately 20% higher than the menadione-treated group (GLP-1~65%, EX-4:~48%) 24-hour after menadione-induced oxidative insults. (M±SE; * p<0.05, ** p< 0.01, *** p< 0.001; compared to the 24-hr value of the menadione alone group, n=6).
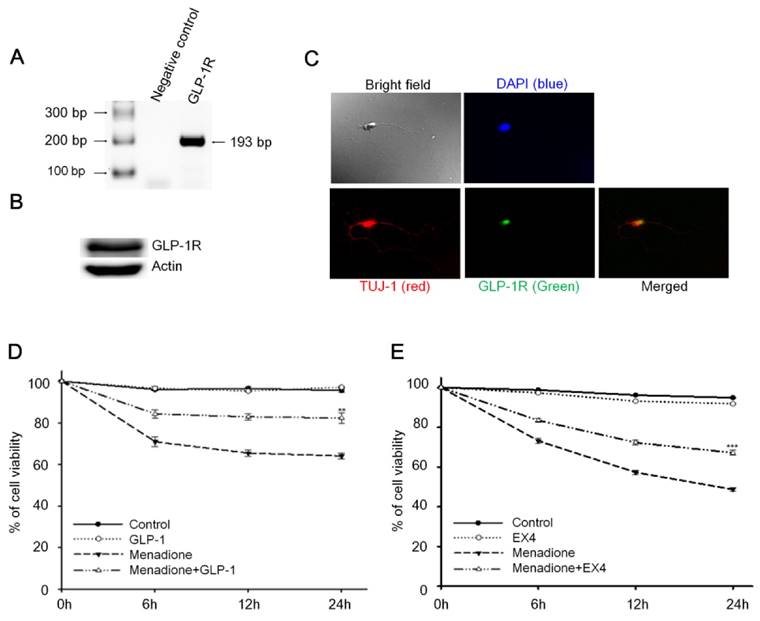
An earlier study by Li et al reported that activation of GLP-1R decreased hypoxia-induced neuronal death [32]. We postulated that GLP-1 protects neurons against oxidative stress since oxidative injury of neurons is the common form of pathogenesis in neurodegenerative diseases and brain traumas [33,34]. In this study, menadione was used as a ROS inducer to produce oxidative insults to GLP-1 treated primary cortical neurons. Menadione, which interrupts the mitochondrial electron transportation chain to induce reactive oxygen species (ROS), is used widely in cell culture assays [35]. Our neuronal viability assay showed that GLP-1(Fig. 1D) or EX-4 (Fig. 1E) treated cortical neurons were more tolerant of oxidative insults. These results indicated that activation of GLP-1R increases neuronal viability under oxidative stress.
Activation of GLP-1 Receptor Rescues Neurons by Selectively Up-regulating APE1 expression and Incision Activity
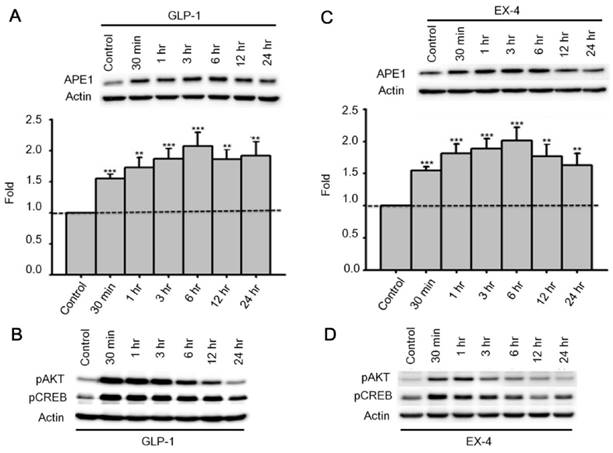
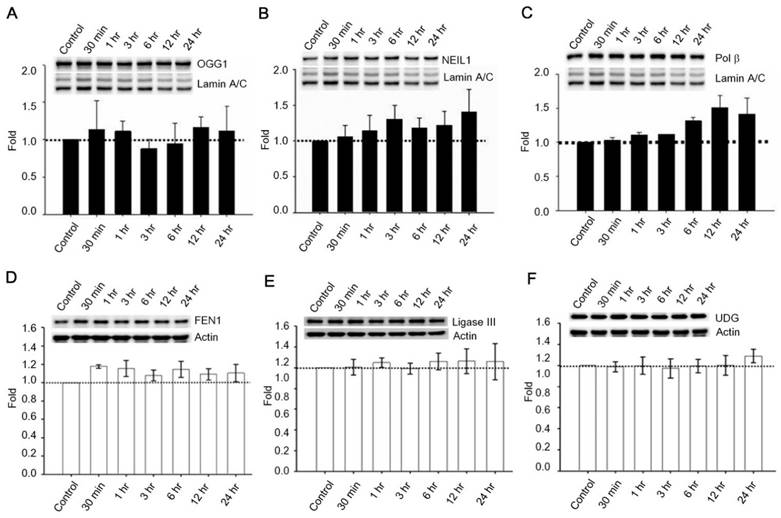
Past studies indicate that Menadione is able to induce oxidative DNA damage and single-strand DNA breaks as well as triggering apoptosis in different cells, including neurons [14, 35-37]. Thus we hypothesized that GLP-1 rescues neurons from menadione-induced cell death by elevating DNA repair efficiency and preserving DNA integrity. As described previously, BER is the predominant DNA repair pathway in neurons. The BER pathway involves the sequential activity of multiple enzymes, including various glycosylases, APE1, Polβ, and ligase III. We studied whether the BER proteins, 8-oxoguanine DNA glycosylase (OGG1), nei endonuclease VIII-like 1 (NEIL1), flap endonuclease 1 (FEN1), uracil DNA glycosylase (UDG), APE1, DNA polymerase beta (Polβ), and ligase III, were affected by GLP-1. Either GLP-1 or EX-4 (100 nM) was added to the primary neuronal cultures prior to harvesting cells at 30 min, 1, 3, 6, 12, and 24 hr for Western blotting. Western Blots of GLP-1 (Fig. 2 A) or EX-4 (Fig. 2C) treated cultures indicated that the APE1 protein was up regulated immediately starting at 30 min and reaching a peak at 6 hr. In addition, our preliminary results suggested that GLP-1 or EX-4 induced APE1 expression was dose-dependent (Fig. S2). A dose of GLP-1 or EX-4 (100 nM) increased APE1 expression by 1.5- to 2- fold. In addition, we observed that phosphorylation of AKT and CREB immediately occurred after treatment with GLP-1 or EX-4 (Fig. 2 B and D). According to this observation, the GLP-1 up-regulated APE1 expression possibly is mediated by the PI3K-AKT-CREB signaling pathway. The other BER proteins, OGG1, NEIL1, FEN1, UDG, Polβ, and Ligase III, were surveyed but showed no significant changes (Fig. 3).
APE1 expression was up-regulated by GLP-1 and it's analogue Exendin-4 (EX-4) in neurons. Rat primary cortical neurons were treated with 100 nM GLP-1 or EX-4 and were subsequently harvested at 30 min, 1 hr, 3 hr, 6 hr, 12 hr, and 24 hr time points. The APE1 protein levels were induced starting at 30 min continuing to 24 hr. These results clearly demonstrated that (A) GLP-1 or (C) EX-4 increased APE1 protein levels in cortical neurons. The immunoblots showed that phosphorylation of AKT and CREB were immediately increased 30 min after (B) GLP-1 or (D) EX-4 administration. The phosphorylation level of AKT returned to normal levels at 24 hr, and phophorylated CREB started decreasing after reaching a peak at 30 min. (M±SE; * p<0.05; ** p<0.01; ***p<0.001; compared to the value of control group, n=6).
Protein expressions of OGG1, NEIL1, Polβ, FEN1, UDG, and ligase III were not significantly influenced by GLP-1. Cortical neurons were treated with 100 nM GLP-1 and harvested in order to measure BER proteins at 30 min, 1 hr, 3 hr, 6 hr, 12 hr, and 24 hr time points. Nuclear fractions were collected for Western blotting; the blotting results suggested that protein levels of (A) OGG1, (B) NEIL1,and (C) Polβ were not significantly altered by GLP-1 administration. Whole cell lysates were used for FEN1, UDG, and ligase III Western blotting. Protein levels of (D) FEN1, (E) Ligase III, and (F) UDG were not affected by GLP-1. However, we observed that Polβ was slightly increased by GLP-1 treatment but not statistically significant. (M±SD; * p<0.05; ** p<0.01; ***p<0.001; compared to the value of control group, n=6).
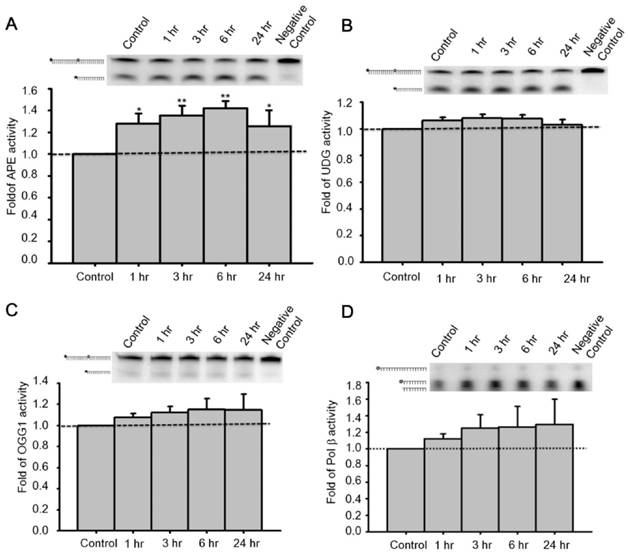
To determine enzymatic activity of pertinent proteins, primary neurons were treated with 100 nM GLP-1 and harvested at 1, 3, 6, and 24 hr for total protein extraction. Additionally, specifically designed defective oligonucleotides were used to measure the activities of DNA incision (APE1, OGG1, and UDG) and gap filling (Polβ) BER enzymes in GLP-1 treated neurons (Table 1). OGG1 and UDG are two common BER glucosylases incising oxidized purine/pyrimidine bases, APE1 is the key enzyme to incise the DNA sugar backbone, and Polβ is the BER polymerase that adds nucleotides. Enzyme activities in whole cell lysates were measured in response to GLP-1 treatment. The incision activity of APE1 was significantly up-regulated by GLP-1(Fig. 4A). The activities of UDG and OGG1 were unchanged after GLP-1 treatment (Fig. 4 B-C). Polβ seemed to respond slightly to GLP-1, however, it was not significant (Fig. 4D). The protein levels of UDG, OGG1, Polβ, and APE1were comparable to their activities. APE1 is the only protein of the BER pathway up-regulated in response to GLP-1 treatment. These results suggested that up-regulated APE1 has an important role in the GLP-1 mechanism which protects neurons against oxidative DNA damage.
Administration of GLP-1 enhanced the enzymatic activity of APE1, but not UDG, OGG1 or Polβ. Cortical neurons were treated with GLP-1 (100 nM) and harvested to measure protein activity at 1, 3, 6, 12, and 24 hr time points. The incision activities of (A) APE1, (B) UDG, and (C) OGG1 were examined by various Cy5-labeled defect-oligonucleotides which could be recognized by APE1, UDG, and OGG1 respectively. (D) The gap-filling activity of Polβ was assayed by Cy5-labeled duplex-oligonucleotides lacking deoxycytidine. The DNA incision activity of APE1 (panel A) was increased significantly by GLP-1 treatment, but not the activities of UDG (panel B), OGG1 (panel C), or Polβ (panel D). However, the incision activity of OGG1 and the gap-filling activity of Polβ were slightly increased by GLP-1 treatment but were not statistically significant. (M±SE; *p< 0.05; ** p< 0.01; ***p<0.001; compared to the value of control group, n=6).
The Downstream PI3K-AKT Signaling Axis of GLP-1R Regulates Expression of APE1 Agonists of GLP-1R
Although BER proteins and repair mechanisms are well known, regulation of expression and activity of repair proteins are not clearly understood. Two signaling pathways, AC-PKA-MEK-Erk and PI3K-AKT, are triggered immediately after stimulation of GLP-1R in neurons [21]. Previous results indicated that both GLP-1 and EX-4 immediately stimulated phosphorylation of AKT and downstream CREB (Fig. 2 B and D). We postulated that APE1 expression was mediated by the PI3K-AKT-CREB signaling pathway during activation of GLP-1R.
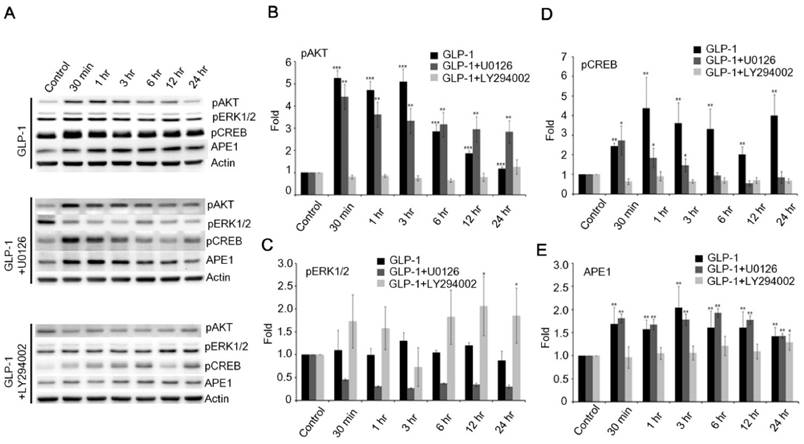
Therefore, the phosphorylation of ERK1/2 was examined following the stimulation of GLP-1R. The phosphorylation of ERK1/2 was slightly increased, but not as dramatically as AKT in response to GLP-1- (upper panel of Fig. 5A) or EX4- (upper panel of Fig. S3A) stimulated GLP-1R. To elucidate how GLP-1 regulates APE1 expression, the inhibitors of MEK (U0126) and PI3K (LY294002) were applied to suppress the MEK-ERK1/2 and PI3K-AKT axes respectively. U0126 robustly suppressed phosphorylation of ERK1/2 in response to the stimulation of GLP-1R by GLP-1 (middle panel of Fig. 5 A and C) or EX-4 (middle panel of Fig. S3 A and C), but did not affect phosphorylation of AKT and CREB or APE1 expression (Fig. 5 A, D, and E; Fig. S3 A, D, and E). On the contrary, administration of LY294002 significantly suppressed GLP-1- (lower panel of Fig. 5 A, B, and D) or EX-4- (lower panel of Fig. S3 A, B, and D) induced phosphorylation of AKT and CREB as well as APE1 expression (Fig. 5E and Fig. S3E), on the other phosphorylation of ERK1/2 seemed slightly increase but not significantly affected (Fig. 5C). These results indicated that the PI3K-AKT-CREB axis is the major signaling pathway in the regulation of APE1 expression in response to GLP-1R stimulation in cortical neurons.
The downstream PI3K-AKT signaling axis of GLP-1R is the major pathway that regulates expression of APE1. (A) Western blot analyses of pAKT, pERK1/2, pCREB, APE1, and actin levels in neuronal cultures treated with 100 nM of GLP-1(100nM), GLP-1 plus MEK inhibitors U0126, or GLP-1 plus PI3K inhibitor LY294002. (B) LY294002 specifically inhibited GLP-1-induced AKT phosphorylation. (C) U0126 specifically inhibited GLP-1-induced pERK1/2 phosphorylation. (D) pCREB levels and (E) APE1 were specifically inhibited by LY294002 but not U0126. The results suggested that PI3K-AKT-CREB is the predominant downstream signaling pathway of GLP-1R elevating APE1 expression. (M±SE; *p< 0.05; ** p< 0.01; ***p<0.001; compared to the value of control group, n=4).
Administration of Exendin-4 Enhances DNA Repair Capability via enhancing APE1expression in the Ischemic Brain of Rat
Many recent studies have reported that the stimulation of GLP-1R plays a protective role in neurons against neurodegenerative disorders, brain trauma, and ischemic stroke [25,26,28,32,38-40]; nevertheless, how subsequent mechanisms respond to the GLP-1R activation in neuroprotection remains under investigation. Results of current studies suggested to us that GLP-1 induced neuroprotection is mediated via enhanced DNA repair capacity and maintenance of DNA integrity. We utilized the rat cerebral ischemia model to test whether activation of GLP-1R increases the DNA repair capability of brain cells. The procedure of middle cerebral artery occlusion (MCAO) reperfusion was performed to induce cerebral ischemic stroke, and animals were sacrificed on day 1, 2, 3, 4, 5, 7, and 10 post- MCAO procedure. The images of TTC (triphenyl tetrazolium chloride) staining showed the infarct was located in the frontal cortex (Fig. 6A). The vehicle group and experimental group were given 1x phosphate buffer saline (100 μl) or EX-4 (100 μl,10 μg/kg body weight) respectively by tail vein injection immediately after completing the MCAO procedure and were given daily until sacrificed. The protein level of APE1 was examined form cortical regions of both vehicle and EX-4 administrated groups. The Western blotting results demonstrated that APE1 level of EX-4 administrated group was dramatically higher than vehicle treated animals in ischemic brains starting from the first day to 10th day after MCAO surgery (Fig. 6 B-C).
Administration of EX-4 significantly reduced ischemia-induced nuclear DNA damage. (A) Cerebral ischemic strokes in rat brains were induced by MCAO procedures and injuries developed in the frontal cortex region. (B-C) APE1 expression was robustly elevated in EX-4 administrated animals comparing to vehicle treated animals. (D) In this study, TUNEL assay and measurement of γH2AX (Phosphorylation of histone 2AX) were applied to assess DNA damage and repair after an ischemic stroke in rats. Five regions of the brains, two in the infarct (filled-line circles) and three in the penumbra (dot-line circles), were analyzed for injured brain cells. (E-F) The results of the TUNEL assay demonstrated that daily administration of EX-4 rapidly reduced nuclear DNA fragmentation compared with the vehicle group in ischemic brains. (G-H) γH2AX is a common biomarker of nuclear DNA damage, which is wildly used to determine DNA damage. Immunohistological results of γH2AX also showed that EX-4 administrated animals had less DNA damage than vehicle treated animals. These results suggested that EX-4 treated animals had less nuclear DNA damage and better DNA repair to protect brain cells against ischemic injury. (M±SE; *p< 0.05; ** p< 0.01; ***p<0.001; compared to the number of MCAO without EX-4, n=4).
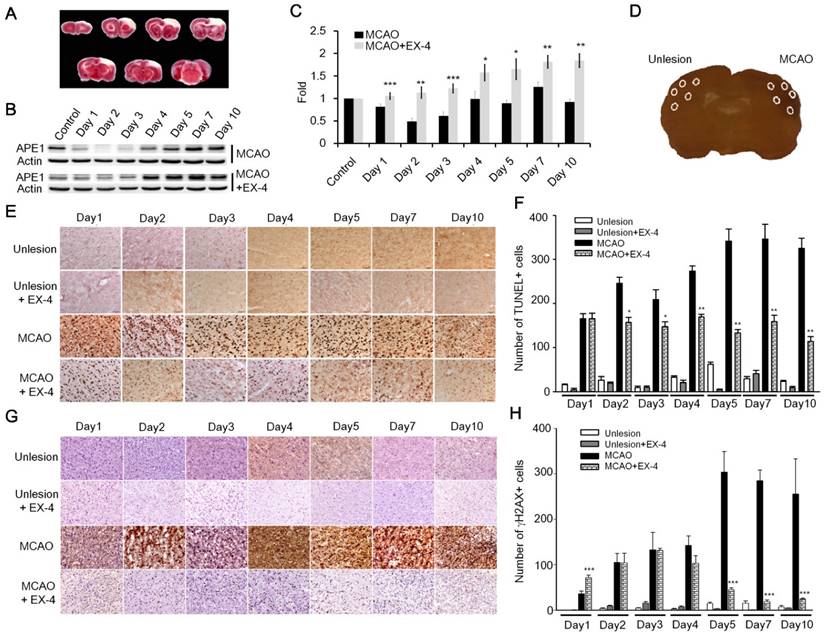
TUNEL assay and γH2AX (phosphorylated histone 2AX) labeling were performed on the fixed and sectioned brains to determine nuclear DNA damage. TUNEL assay relies on the enzymatic activity of terminal deoxynucleotide transferase to incorporate fluorophore -labeled dUTP into the 3'-hydroxyl termini of DNA fragments, especially at strand-breaks of DNA. TUNEL assay is originally employed to determine DNA damage. Nowadays, TUNEL assay is used widely for detecting apoptosis in situ, but this method is not limited to detection in cells undergoing apoptosis. The assay can be also employed to detect DNA damage (DNA nicks formed by lyase or APE1 as well as single-strand and double-strand breaks) and DNA repair efficiency [41]. In addition, γH2AX is an action in response to DNA double-strand breaks, blocked replication, and barricaded transcription, hence, γH2AX is a common and sensitive biomarker of nuclear DNA damage and is wildly used to assess DNA damage and repair [42]. Five areas (0.48 mm2 each) from each ipsilateral and contralateral hemispheres were chosen, two in the infarct and three in the penumbra areas, to count positive nuclear staining by TUNEL or γH2AX (Fig. 6D). In results of TUNEL assay, nuclear DNA damaged cells of the vehicle treated group increased starting the second day of the post MCAO procedure and remained at a high level until day 10; but the EX-4 treated MCAO group had fewer DNA damaged cells and a trend of decreasing DNA damage over time (Fig. 6E). Statistically, EX-4 treated ischemic brains had significantly fewer DNA damaged cells or better DNA repair efficiency than the vehicle treated group (Fig. 6F). The immunohitological results of γH2AX demonstrated that significantly lower DNA damage was observed in EX-4 administrated ischemic brains than in vehicle treated ischemic brains (Fig. 6 G-H). The results of TUNEL and γH2AX, directly and indirectly illustrated that stimulation of GLP-1R effectively reduced nuclear DNA damage, or in other words, efficiency enhanced DNA repair in brain cells. Taking together of these results, which suggests that higher DNA repair efficiency is the consequence of upregulated APE1 by activation of GLP-1R.
Discussion
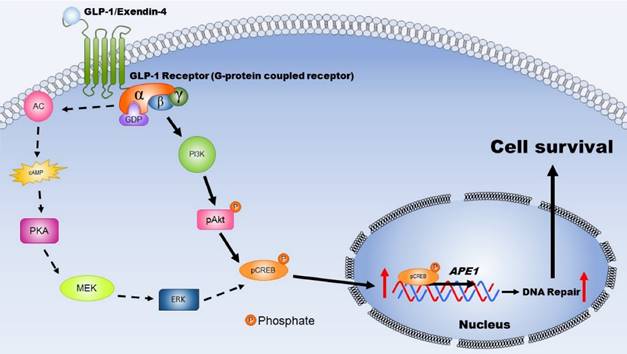
GLP-1, an incretin, is not only secreted from intestinal L cells, but also is expressed in neurons of the central nervous system [43]. Many studies have revealed that GLP-1[32,44], EX-4 [45-47], and other agonists of GLP-1R [40,48] have a neuroprotective role against neurodegenerative cell death, traumatic brain injury, and ischemic stroke. Previous reports indicate that the neuroprotective effects of GLP-1 and EX-4 involve reducing infarct size and stoke volume [28,32] as well as anti-apoptotic effects [28,39]. However, other mechanisms of GLP-1 involvement in neuronal protection are still under investigation. Our findings in the present study suggest a novel biological action of GLP-1R: enhanced DNA repair efficiency resulting in elevating resistance of neurons to ischemia-induced oxidative DNA damage (Fig. 7). The effects of GLP-1 and its analogue (EX-4) in protecting cortical neurons and enhancing DNA repair are due to up-regulating the protein level and incision activity of APE1 (Fig. 2 and Fig. 4). Overexpression of APE1 protects neurons against MPTP-induced neuronal death [49], in addition, up-regulation of APE1 reduces oxidative DNA damage and protects hippocampal neurons from ischemic injury [50]. Our study demonstrated that administration of EX-4 significantly enhanced APE1 expression and reduced MACO-induced DNA damage in rat brains (Fig. 6). Selectively suppressing APE1 production with shRNA produces a significant increase of oxidative DNA damage and attenuates the ability of BDNF to enhance DNA repair [14]. All evidence indicates that APE1 plays a pivotal role in neuronal DNA repair and viability.
This schematic diagram illustrates the mechanisms by which activated GLP-1R elevate repair of oxidative DNA damage and increase neuronal survival. The expression GLP-1 and its receptors occurs in the central nervous system and is involved in neuronal protection. Two major signaling pathways are triggered, including PKA-Erk (dotted-line arrow) and PI3K-AKT (filled-line arrow), when GLP-1R is activated. Results of our study suggested that PI3K-AKT is the key downstream signaling pathway regulating APE1 expression, elevation DNA repair efficiency, and increasing neuronal survival after oxidative insults. (AC: adenylyl cyclase; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; MEK: mitogen-activated protein kinase kinase; ERK: mitogen-activated protein kinase; PI3K: phosphatidylinositide 3-kinase; AKT: protein kinase B; CREB: cAMP response element binding protein).
The previous studies of Mattson's group demonstrated that signaling by activated glutamate receptors and TrkB (BDNF receptor) enhance nuclear DNA repair in neurons by a mechanism involving up-regulation of the BER protein APE1, but not other BER proteins [14,51]. Many other studies also have shown that APE1 is up-regulated by a CREB-mediated signaling pathway [52,53]. Although GLP-1R and TrkB are characterized as a G-protein-coupled receptor and a tyrosine kinase receptor respectively, our findings show that GLP-1R shares the same signaling pathway, PI3K-AKT-CREB axis, to up-regulate APE1 expression and to protect neurons. Accumulating evidence suggests GLP-1 is not only an incretin, but also has a neurotransmitter role [54-57]. GLP-1 and its analogues promote neurite outgrowth and neuronal survival under conditions of metabolic, oxidative and excitotoxic stress in cultured neuronal cells [58-60]. The role of GLP-1 in the CNS is not limited to energy metabolism and may have similar functions as BDNF, a neurotrophic factor.
GLP-1R are G-protein coupled receptors expressed in many tissues and organs, including the central nerve system [20,21]. Binding of ligands to GLP-1R engages multiple kinase signaling cascades including PI3K, AKT, AC, PKA, MEK, and ERK. Stimulated GLP-1R exert protective actions in various tissues or cells by one of two signaling pathways, such as GLP-1 was reported that has including a protective effect against cardiac ischemia by reducing infarct size via the PI3K-AKT signaling pathway [61]; EX-4 improves endothelial function and proliferation in type 2 diabetes patients with coronary heart disease [62]; GLP-1 prevents apoptosis of pancreatic beta cells through both PI3K-AKT and PKA-ERK signaling pathways [63]. In our study, GLP-1 and EX-4 promoted neuronal survival through up-regulated APE1, which is mediated only by the PI3K-AKT axis.
Oxidative DNA damage in neurons is common in neurodegenerative disorders, ischemic stroke, and brain trauma, but no therapeutic mechanism has been established to connect neuroprotective actions to neuronal DNA repair. Our findings of a new biological function for GLP-1 suggest that downstream signaling of stimulated GLP-1R enhanced DNA repair capability by up-regulating PI3K-AKT-CREB-mediated APE1 expression to ameliorate ischemia-induced DNA damage and cell death. Enhancing DNA repair capability in neurons by the administration of long-lasting agonists of the GLP-1R, such as EX-4, is a potential strategy for the prevention or therapeutic intervention in ischemic stroke, brain trauma, and neurodegenerative diseases.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by research grants from Chang Gung Medical Foundation CMRPG8B0571, CMRPG8B0572, CMRPG8B0573, and CMRPG8C1221, CMRPG8C1222, and CMRPG8C1223; and from Taiwan Ministry of Science and Technology 103-2320-B-182A-011-MY3 and 103-2314-B-182-026-MY2. We also would like to thank Dr. Kathryn Fischbach for her critically reading and editing written-English of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Barzilai A. The contribution of the DNA damage response to neuronal viability. Antioxid.Redox.Signal. 2007;9:211-18
2. De BR, van LN. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169-85
3. Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech.Ageing Dev. 2012;133:157-68
4. Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83:266-82
5. Slemmer JE, Shacka JJ, Sweeney MI, Weber JT. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr.Med.Chem. 2008;15:404-14
6. Smith JA, Park S, Krause JS, Banik NL. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem.Int. 2013;62:764-75
7. Best BP. Nuclear DNA damage as a direct cause of aging. Rejuvenation.Res. 2009;12:199-208
8. Martin LJ, Liu Z, Pipino J, Chestnut B, Landek MA. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb.Cortex. 2009;19:1273-93
9. Englander EW. Brain capacity for repair of oxidatively damaged DNA and preservation of neuronal function. Mech.Ageing Dev. 2008;129:475-82
10. Martin LJ. DNA damage and repair: relevance to mechanisms of neurodegeneration. J.Neuropathol.Exp.Neurol. 2008;67:377-87
11. Barzilay G, Hickson ID. Structure and function of apurinic/apyrimidinic endonucleases. Bioessays. 1995;17:713-19
12. Myles GM, Sancar A. DNA repair. Chem.Res.Toxicol. 1989;2:197-226
13. Yang JL, Tadokoro T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. J.Biol.Chem. 2010;285:28191-99
14. Yang JL, Lin YT, Chuang PC, Bohr VA, Mattson MP. BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease. Neuromolecular.Med. 2014;16:161-74
15. Schmidt WE, Siegel EG, Creutzfeldt W. Glucagon-like peptide-1 but not glucagon-like peptide-2 stimulates insulin release from isolated rat pancreatic islets. Diabetologia. 1985;28:704-07
16. Bell GI, Sanchez-Pescador R, Laybourn PJ, Najarian RC. Exon duplication and divergence in the human preproglucagon gene. Nature. 1983;304:368-71
17. Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J.Biol.Chem. 1986;261:11880-89
18. Orskov C, Holst JJ, Knuhtsen S, Baldissera FG, Poulsen SS, Nielsen OV. Glucagon-like peptides GLP-1 and GLP-2, predicted products of the glucagon gene, are secreted separately from pig small intestine but not pancreas. Endocrinology. 1986;119:1467-75
19. Gros L, Thorens B, Bataille D, Kervran A. Glucagon-like peptide-1-(7-36) amide, oxyntomodulin, and glucagon interact with a common receptor in a somatostatin-secreting cell line. Endocrinology. 1993;133:631-38
20. Christ E, Wild D, Reubi JC. Glucagonlike peptide-1 receptor: an example of translational research in insulinomas: a review. Endocrinol.Metab Clin.North Am. 2010;39:791-800
21. Harkavyi A, Whitton PS. Glucagon-like peptide 1 receptor stimulation as a means of neuroprotection. Br.J.Pharmacol. 2010;159:495-501
22. Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17:819-37
23. Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J.Pharmacol.Exp.Ther. 2002;302:881-88
24. Yang Y. et al. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J.Alzheimers.Dis. 2013;37:637-48
25. McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer's disease. Neuropharmacology. 2014;76(Pt A):57-67
26. Briyal S, Gulati K, Gulati A. Repeated administration of exendin-4 reduces focal cerebral ischemia-induced infarction in rats. Brain Res. 2012;1427:23-34
27. Darsalia V. et al. Glucagon-like peptide-1 receptor activation reduces ischaemic brain damage following stroke in Type 2 diabetic rats. Clin.Sci.(Lond). 2012;122:473-83
28. Teramoto S. et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, provides neuroprotection in mice transient focal cerebral ischemia. J.Cereb.Blood Flow Metab. 2011;31:1696-705
29. Weissman L. et al. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545-55
30. Weissman L, de Souza-Pinto NC, Mattson MP, Bohr VA. DNA base excision repair activities in mouse models of Alzheimer's disease. Neurobiol.Aging. 2009;30:2080-81
31. Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD. A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke. 1986;17:738-43
32. Li Y. et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc.Natl.Acad.Sci.U.S.A. 2009;106:1285-90
33. bdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol.Neurobiol. 2015;51:966-79
34. Kiselyov K, Muallem S. ROS and intracellular ion channels. Cell Calcium. 2016
35. Criddle DN. et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J.Biol.Chem. 2006;281:40485-92
36. Kulkarni A, McNeill DR, Gleichmann M, Mattson MP, Wilson DM III. XRCC1 protects against the lethality of induced oxidative DNA damage in nondividing neural cells. Nucleic Acids Res. 2008;36:5111-21
37. Woods JA, Young AJ, Gilmore IT, Morris A, Bilton RF. Measurement of menadione-mediated DNA damage in human lymphocytes using the comet assay. Free Radic.Res. 1997;26:113-24
38. Briyal S, Shah S, Gulati A. Neuroprotective and anti-apoptotic effects of liraglutide in the rat brain following focal cerebral ischemia. Neuroscience. 2014;281C:269-81
39. Holscher C. The role of GLP-1 in neuronal activity and neurodegeneration. Vitam.Horm. 2010;84:331-54
40. Ji C. et al. A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson's disease by increasing expression of BNDF. Brain Res. 2015
41. Kanoh M. et al. Significance of myocytes with positive DNA in situ nick end-labeling (TUNEL) in hearts with dilated cardiomyopathy: not apoptosis but DNA repair. Circulation. 1999;99:2757-64
42. Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679-86
43. Han VK, Hynes MA, Jin C, Towle AC, Lauder JM, Lund PK. Cellular localization of proglucagon/glucagon-like peptide I messenger RNAs in rat brain. J.Neurosci.Res. 1986;16:97-107
44. Zhang H. et al. Pro-GLP-1, a Pro-drug of GLP-1, is neuroprotective in cerebral ischemia. Eur.J.Pharm.Sci. 2015;70:82-91
45. Korol SV, Jin Z, Birnir B. The GLP-1 Receptor Agonist Exendin-4 and Diazepam Differentially Regulate GABAA Receptor-Mediated Tonic Currents in Rat Hippocampal CA3 Pyramidal Neurons. PLoS.One. 2015;10:e0124765
46. Lietzau G, Nystrom T, Ostenson CG, Darsalia V, Patrone C. Type 2 diabetes-induced neuronal pathology in the piriform cortex of the rat is reversed by the GLP-1 receptor agonist Exendin-4. Oncotarget. 2016
47. Tsukamoto M, Niimi N, Sango K, Takaku S, Kanazawa Y, Utsunomiya K. Neurotrophic and neuroprotective properties of exendin-4 in adult rat dorsal root ganglion neurons: involvement of insulin and RhoA. Histochem.Cell Biol. 2015;144:249-59
48. Zhang H. et al. An Orally Active Allosteric GLP-1 Receptor Agonist Is Neuroprotective in Cellular and Rodent Models of Stroke. PLoS.One. 2016;11:e0148827
49. Huang E. et al. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat.Cell Biol. 2010;12:563-71
50. Leak RK. et al. Apurinic/apyrimidinic endonuclease 1 upregulation reduces oxidative DNA damage and protects hippocampal neurons from ischemic injury. Antioxid.Redox.Signal. 2015;22:135-48
51. Yang JL, Takahashi T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated Up-regulation of APE1. J.Biol.Chem. 2010
52. Stetler RA. et al. Apurinic/apyrimidinic endonuclease APE1 is required for PACAP-induced neuroprotection against global cerebral ischemia. Proc.Natl.Acad.Sci.U.S.A. 2010;107:3204-09
53. Yang JL, Takahashi T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated Up-regulation of APE1. J.Biol.Chem. 2010
54. Larsen PJ, Tang-Christensen M, Jessop DS. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. Endocrinology. 1997;138:4445-55
55. Larsen PJ, Holst JJ. Glucagon-related peptide 1 (GLP-1): hormone and neurotransmitter. Regul.Pept. 2005;128:97-107
56. Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann.N.Y.Acad.Sci. 2008;1144:97-112
57. Trapp S, Hisadome K. Glucagon-like peptide 1 and the brain: central actions-central sources? Auton.Neurosci. 2011;161:14-19
58. Luciani P. et al. Differentiating effects of the glucagon-like peptide-1 analogue exendin-4 in a human neuronal cell model. Cell Mol.Life Sci. 2010;67:3711-23
59. Perry T. et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J.Pharmacol.Exp.Ther. 2002;300:958-66
60. Perry T. et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J.Neurosci.Res. 2003;72:603-12
61. Iwasa M. et al. Antidiabetic drug voglibose is protective against ischemia-reperfusion injury through glucagon-like peptide 1 receptors and the phosphoinositide 3-kinase-AKT-endothelial nitric oxide synthase pathway in rabbits. J.Cardiovasc.Pharmacol. 2010;55:625-34
62. Erdogdu O, Nathanson D, Sjoholm A, Nystrom T, Zhang Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/AKT-dependent pathways and requires GLP-1 receptor. Mol.Cell Endocrinol. 2010;325:26-35
63. Favaro E. et al. The ghrelin gene products and exendin-4 promote survival of human pancreatic islet endothelial cells in hyperglycaemic conditions, through phosphoinositide 3-kinase/AKT, extracellular signal-related kinase (ERK)1/2 and cAMP/protein kinase A (PKA) signalling pathways. Diabetologia. 2012;55:1058-70
Author contact
![]() Corresponding author: Shang-Der Chen Email: chensdcgmh.org.tw; Tel: +886-7-7317123; Fax: +886-7-7317123 ext. 2293
Corresponding author: Shang-Der Chen Email: chensdcgmh.org.tw; Tel: +886-7-7317123; Fax: +886-7-7317123 ext. 2293