Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(5):752-761. doi:10.7150/thno.14527 This issue Cite
Research Paper
Folate-modified Chitosan Nanoparticles Containing the IP-10 Gene Enhance Melanoma-specific Cytotoxic CD8+CD28+ T Lymphocyte Responses
Jian He1,*, Siliang Duan1,*, Xia Yu1,*, Zhiyong Qian2, Sufang Zhou1, Zhiyong Zhang3, Xianing Huang1, Yong Huang1, Jing Su1, Chunhui Lai1, Jinyu Meng4, Nuo Zhou1 ![]() , Xiaoling Lu1,5
, Xiaoling Lu1,5 ![]() , Yongxiang Zhao1
, Yongxiang Zhao1 ![]()
1. National Center for International Research of Biological Targeting Diagnosis and Therapy, Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research, Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China;
2. State Key Laboratory for Biotherapy and Cancer Center, West China Hospital, Sichuan University, Collaborative Innovation Center for Biotherapy, Chengdu, Sichuan 610041, China.
3. Department of Surgery, Robert-Wood-Johnson Medical School University Hospital, Rutgers University, The State University of New Jersey, New Brunswick, New Jersey 08901, USA.
4. Biomedical Polymers Laboratory, Soochow University, Suzhou, Jiangsu 215123, China;
5. The Department of Immunology, Guangxi Medical University, Nanning, Guangxi 530021, China.
* These authors contributed equally to this work.
Received 2015-11-25; Accepted 2016-2-1; Published 2016-3-21
Citation:
He J, Duan S, Yu X, Qian Z, Zhou S, Zhang Z, Huang X, Huang Y, Su J, Lai C, Meng J, Zhou N, Lu X, Zhao Y. Folate-modified Chitosan Nanoparticles Containing the IP-10 Gene Enhance Melanoma-specific Cytotoxic CD8+CD28+ T Lymphocyte Responses. Theranostics 2016; 6(5):752-761. doi:10.7150/thno.14527. https://www.thno.org/v06p0752.htm
Other stylesAbstract

Background: Adoptive immunotherapy with cytotoxic T lymphocytes (CTLs) has great potential for the treatment of some malignant cancers. Therefore, augmenting the responses of tumor-specific CTLs is significant for the adoptive immunotherapy of melanoma. This study aimed to investigate the anti-tumor response of a combination therapy employing folate-modified chitosan nanoparticles containing IP-10 (interferon-γ-inducible protein-10) plus melanoma TRP2-specific CD8+CD28+ T cells. Methods: We prepared folate-modified chitosan nanoparticles containing the mouse IP-10 gene (FA-CS-mIP-10), and induced melanoma TRP2-specific CD8+CD28+ T cells by co-culturing them with artificial antigen-presenting cells. B16-bearing mice were treated with FA-CS-mIP-10, melanoma TRP2-specific CD8+CD28+ T cells, a combination of both, and the saline control. Tumor volumes and the survival time of mice were recorded. The proportion of myeloid-derived suppressor cells (MDSCs) infiltrating the tumor microenvironment and regulatory T cells (Tregs) in the spleen was analyzed by flow cytometry. We also detected the proliferation and angiogenesis of tumors by immunohistochemistry and apoptosis by TUNEL. Results: The combination therapy inhibited the progression of melanoma in vivo. Compared with other treatments, it more efficiently inhibited tumor growth and increased the survival time of mice. After treatment with combination therapy, the proportion of MDSCs and Tregs decreased, while the percentage of CXCR3+CD8+ T cells increased. Furthermore, combination therapy inhibited proliferation and promoted apoptosis of tumor cells and significantly inhibited tumor angiogenesis in vivo. Conclusion: We describe a novel strategy for improving the anti-tumor response of CD8+CD28+ CTLs by combining them with FA-CS-mIP-10 nanoparticles.
Keywords: cytotoxic T lymphocytes, melanoma, folate, chitosan, interferon-γ-inducible protein-10.
Introduction
Malignant melanoma is a highly aggressive malignant neoplasm of melanocytes, the melanin-producing cells that originate from the skin, mucous membranes, uvea and leptomeninges of the central nervous system [1]. The incidence of melanoma has increased over the past decades, with 132,000 new melanoma cases being diagnosed globally each year. Surgical removal is the main treatment for early-stage melanoma patients while chemotherapy and radiotherapy are employed in patients with advanced melanoma. However, the prognosis of patients with melanoma is extremely poor due to its high metastatic potential [2, 3]. Previous studies have reported that immunotherapies based on adoptive transfer of tumor-specific cytotoxic T lymphocytes (CTLs) could efficiently repress tumor metastasis [4-6]. Although adoptive antigen-specific CTLs transfer is considered one of the most promising immunotherapies, the biological activity and distribution of these CTLs in vivo significantly limits their efficacy and application. Therefore, an effective approach for tumor eradication may be directed toward enhancing increase CTLs anti-tumor response.
In adoptive immunotherapy, the CTL anti-tumor immune response is highly dependent on antigen-presenting cells (APCs), especially dendritic cells (DCs), which efficiently present tumor-specific antigens and provide co-stimulatory signals for the activation and expansion of CTLs [7, 8]. Although the use of natural APCs was once considered the best choice for the treatment of cancer patients, this practice is time-consuming and expensive; it is also difficult to control for the quality of natural APCs. Therefore, several studies have instead used artificial APCs (aAPCs) for expanding tumor antigen-specific CTLs, demonstrating their high efficiency and availability [9, 10]. We previously developed a novel approach to promote efficient anti-tumor response via the induction of melanoma-specific CD8+CD28+ CTLs by co-culturing them with APCs in the presence of IL-21 and IL-15 [11]. However, in vivo, signals or factors from the tumor microenvironment limit the efficacy of CTLs by preventing them from reaching the tumor site and accelerating their exhaustion [12, 13]. Thus, to improve the therapeutic efficacy of adoptive tumor-specific CTLs, we need to increase survival of CTLs and facilitate their access to neoplastic areas.
Interferon-γ-inducible protein-10 (IP-10), also known as CXC motif chemokine 10 (CXCL10), is a member of the CXC chemokine family, which binds to the CXCR3 receptor to mediate chemotaxis, cell growth, apoptosis, and angiogenesis [14]. In the tumor microenvironment, IP-10 plays a key role by chemoattracting CXCR3+ cells, especially CD8+ T cells, toward the tumor site [15, 16]. Moreover, IP-10 was shown to significantly induce apoptosis of cancer cells and inhibit the formation of microvessels [17]. Thus, up-regulation of IP-10 in the tumor microenvironment may increase the efficacy of adoptive tumor-specific CTLs.
Chitosan (CS), derived from the N-deacetylation of chitin, is a promising non-viral nucleic acid delivery vector with high biocompatibility and biodegradability, low toxicity, and low cost [18, 19]. Recently, chitosan nanoparticles have been extensively investigated and widely used in vaccine delivery, parenteral or ocular drug delivery, and non-viral gene delivery [20]. In our previous study, we developed folate-modified chitosan nanoparticles containing the mouse IP-10 gene (FA-CS-mIP-10). These particles bound to the folate receptors of cancer cells and promoted IP-10 gene expression in the tumor microenvironment thereby preventing tumor growth [21]. In the present study, we investigated the in vivo anti-tumor response, of melanoma-specific CD8+CD28+ CTLs, together with the underlying mechanisms, when used in combination with folate-modified chitosan nanoparticles containing the mouse IP-10 gene (FA-CS-mIP-10).
Materials and Methods
Materials
Chitosan (Mw 7 kDa, deacetylation degree > 95%) was purchased from Aoxing Bio-Technology Co. (Zhejiang, China). Folate was purchased from Bio Basic Inc. (Markham, ON, Canada). Monoclonal antibodies against mouse CD11b-FITC and Ly6G-PE were obtained from Beckman Coulter (Brea, CA, USA). Monoclonal antibodies against mouse CD4-FITC, CD8-PerCP-Cy5.5, CD25-PE-Cy5, CXCR3-FITC, Foxp3-PE and Foxp3 staining buffer were purchased from eBiosciences (San Diego, CA, USA). The anti-CD31 monoclonal antibody was obtained from Abcam (Cambridge, MA, USA). Purified hamster anti-mouse CD28 antibody was purchased from BD Biosciences (San Jose, CA, USA). The anti-mouse PCNA monoclonal antibody was supplied by Boster Bio-Technology Co. (Wuhan, China). TdT-mediated dUTP-biotin nick end labeling (TUNEL) cell-death fluorescein detection kit was obtained from Roche Diagnostics (Hoffmann-La Roche, Basel, Switzerland). Dynal mouse CD8+ T isolation kit was purchased from Invitrogen (Carlsbad, CA, USA). Collagenase I was acquired from Solarbio Co. (Beijing, China). The H-2Kb-restricted, melanoma-specific antigenic peptide TRP2 (amino acids 180 - 188, N-SVYDFFVWL-COOH) was chemically synthesized by Nanjing GenScript Corporation (Nanjing, China) with a purity > 95%.
Cell lines and animals
The mouse melanoma cell line B16 was cultured in RPMI-1640 medium at 37°C in 5% CO2. The medium was supplemented with 10% fetal bovine serum (GIBCO Life Technologies, Grand Island, NY, USA), 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. C57BL/6 (H-2Kb) female mice (4 - 6 weeks old) were purchased from Weitonglihua Company (Beijing, China) and housed in laminar flow cabinets under pathogen-free conditions. All animal experiments were performed in accordance with the Federation of European Laboratory Animal Science Association guidelines and all protocols were approved by the Animal Ethics Committee of Guangxi Medical University, Nanning, Guangxi, China.
Preparation of FA-CS-mIP-10 nanoparticles
Chitosan nanoparticles were prepared as described previously [21]. Briefly, chitosan was first dissolved in 1% acetic acid (5 mg/ml) and, subsequently, sodium tripolyphosphate (2 mg/ml) and glutaraldehyde (5%) were added into the chitosan solution with continuously stirring. The resultant product was collected by dialysis against distilled water to which folate solution (5 mg/ml) was added and the solution was incubated for 1 hour at room temperature to obtain FA-CS. Subsequently, FA-CS (5 mg/ml in NaAc/HAc buffer, pH 5.5) and IP-10 plasmid (5 μg/μl in 25 mM Na2SO4) were pre-warmed at 55°C and mixed briefly by vortexing. The resultant nanoparticles were obtained by centrifugation at 100,000 g for 30 min at 4°C.
Mouse model of melanoma and animal treatments
B16 cells were harvested in logarithmic phase and digested by Trypsin (Sigma, Aldrich, St. Louis, MO, USA). A total of 1 × 106 cells in 200 μl phosphate buffered saline (PBS) were injected subcutaneously in the right abdominals of 20 C57BL/6 mice which were randomly divided into 4 groups (n = 5/group). On days 5, 10 and 15, two groups were injected via tail vein with (a) PBS, (b) 50 μg FA-CS-mIP-10, or into the peritoneal cavity (c) 1 × 107 TRP2-specific CD8+CD28+ T cells, and (d) a combination of FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells.
Tumors were measured using a vernier caliper every third day after inoculation and tumor volumes were calculated using the following formula: d1 × (d2)2 × 0.5, where d1 is the largest diameter and d2 is the perpendicular diameter. The survival time was recorded every day after inoculation.
Induction of TRP2-specific CD8+CD28+ T cells
TRP2-specific CD8+CD28+ T cells were induced as described previously [11]. Murine lymphocytes from spleen or lymph nodes (3 × 106 - 4 × 106 cells/ml) were seeded into 24-well plates and co-cultured with H-2Kb:Ig-TRP2-aAPCs (3 × 105 - 4 × 105 cells/ml) in the presence of IL-21 (25 ng/ml) and IL-15 (10 ng/ml). The medium was changed every 3 days. After 2-3 weeks, CD8+ T cells were isolated by magnetic bead cell sorting using a Dynabeads cell separating system (Invitrogen, CA, USA).
Isolation of melanoma cells and tumor-infiltrating lymphocytes
Tumors were excised from mice and incubated in a solution with collagenase I (200 U/ml) and DNase I (200 μg/ml) for 1 hour at 37°C. Then, the suspensions were passed through a cell strainer and washed with PBS. Tumor-infiltrating lymphocytes (TILs) and tumor cells were enriched by gradient centrifugation. Subsequently, cell density was adjusted to 1 × 106 cells/100 μl for fluorescence-activated cell sorting (FACS) analysis.
Determination of the proportion of MDSCs and Tregs by flow cytometry
To analyze the proportion of myeloid-derived suppressor cells (MDSCs) in bone marrow cells, splenocytes, and TILs, a total of 1 × 106 cells/100 μL were stained with CD11b-FITC and Ly6G-PE-antibodies and incubated for 30 minutes at 4°C. The percentage of CXCR3+CD8+ T cells was determined by staining TILs (1 × 106 cells/100 μL) with CXCR3-FITC and CD8-PerCP-Cy5.5 antibodies and incubating for 30 minutes at 4°C.
For regulatory T cells (Tregs) analysis, after staining with CD4-FITC and CD25-PE-Cy5 antibodies, splenocytes (1 × 106 cells/100 μL) were fixed, permeabilized and subsequently stained with Foxp3-PE antibody. After the above staining protocols, cells were washed twice with PBS and then analyzed with a FACS Calibur flow cytometer (Beckman Coulter, Brea, CA, USA). Data were analyzed using Expo32 ADC software (Beckman Coulter, Brea, CA, USA).
Immunohistochemistry and Analyses of apoptosis
Tumors were excised from mice and fixed in 10% formalin, dehydrated in ethanol, and embedded in paraffin. Tumors were then cut into 4-μm sections, mounted on slides, and dried at 60°C. After deparaffinization and rehydration, sections were incubated with primary antibodies at 4°C overnight. The slides were then washed with PBS and incubated with the corresponding secondary antibodies for 30 min. Subsequently, slides were immersed in diaminobenzidine solution, cover slipped and mounted with permanent mounting media. Apoptosis was evaluated by TUNEL using an in situ cell-death fluorescein detection kit, according to the manufacturer's instructions. Images were captured with an Olympus microscope. Five visual fields were randomly captured for each section.
Statistical analyses
Data are presented as mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism 5 software (San Diego, CA, USA). Differences among groups were compared by two-tailed Student's t-test and one-way analysis of variance (ANOVA). Differences among tumor volumes were analyzed using ANOVA and Tukey's post-test. Survival rates were calculated by the log-rank (Mantel-Cox) test. P values < 0.05 are considered statistically significant. In the figures * denotes P < 0.05 and ** denotes P < 0.01.
Results
Characterization of folate-modified chitosan nanoparticles containing mouse IP-10 (FA-CS-mIP-10)
The morphological structure and size of FA-CS-Mip-10 were determined by scanning electron microscopy (SEM). As shown in Figure 1A, FA-CS-mIP-10 particles displayed a spherical shape of small volume with a nearly smooth surface. The size distribution of FA-CS-mIP-10 particles was determined by photon correlation spectroscopy (Figure 1B). The average particle diameter was 233.1 nm, which was in accordance with the criterion for nanometer materials.
Inhibitory effect of the combined treatment with FA-CS-mIP-10 and TRP2-specific CD8+CD28+T cells on C57BL/6 mice melanoma progression
We performed experiments to evaluate the effect of the combination treatment with FA-CS-mIP-10 and melanoma peptide TRP2-specific CD8+CD28+ T cells on melanoma progression in C57BL/6 mice. As shown in Figure 2, treatment with FA-CS-mIP-10 or TRP2-specific CD8+CD28+ T cells, alone or with a combination of both, significantly inhibited tumor growth and improved mice survival time. As expected, tumor volume was more noticeably inhibited by the combination treatment with FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells than with either FA-CS-mIP-10 or TRP2-specific CD8+CD28+ T cells alone (P < 0.05; Figure 2A). The survival rate for the group receiving the combination therapy was significantly higher than that of the control groups (P < 0.01; Figure 2B). These results indicated that the treatment of mice with FA-CS-mIP-10 in combination with TRP2-specific CD8+CD28+ T cells exerted a greater inhibitory effect on tumor growth.
Figure 1
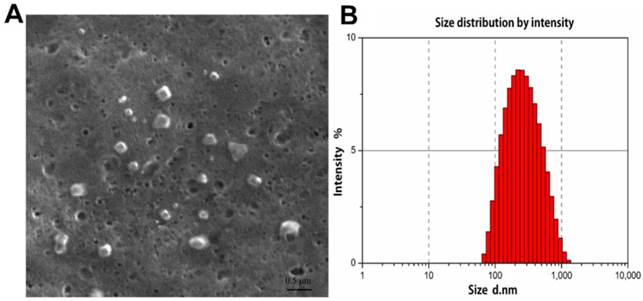
Characterization of FA-CS-mIP-10 nanoparticles: (A) SEM of FA-CS-mIP-10 nanoparticles (40000X). (B) Size distribution of FA-CS-mIP-10 nanoparticles was measured by photon correlation spectroscopy..
Figure 2
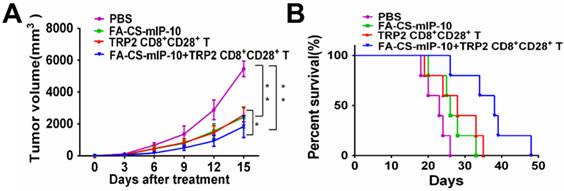
Effect of the treatment of B16-bearing mice with FA-CS-mIP-10 nanoparticles, TRP2-specific CD8+CD28+ T cells, or a combination of both on tumor growth and mice survival: (A) Mean tumor volume in mice from each treatment group measured every third day after treatment. (B) Percent survival in each treatment group was recorded daily after treatment. Each data point represents the mean of three independent experiments ± SD. * P <0.05; ** P < 0.01.
Combination therapy with FA-CS-mIP-10 and TRP2-specific CD8+CD28+T cells decreased the proportion of myeloid-derived suppressor cells
We further explored the effect of the combination therapy on myeloid-derived suppressor cells (MDSCs). Flow cytometry analysis showed that the proportion of MDSCs in TILs, spleen and bone marrow from mice receiving the combination therapy were 0.61%, 2.75%, and 15%, respectively (Figure 3A). The inhibitory effect on infiltrating MDSCs was significantly higher in mice receiving the combination therapy than that in the groups receiving other treatments (Figure 3B).
Combination therapy with FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells decreased the proportion of Tregs and increased the proportion of CXCR3+CD8+ T cells
The number of Tregs (CD4+CD25+FoxP3+ T cells) in spleen and of migrating CD8+ T cells in the tumor environment was determined by flow cytometry. The percentages of Tregs were 3.17% ± 0.218% in the PBS group, 2.06% ± 0.229% in the FA-CS-mIP-10 group, 2.16% ± 0.121% in the TRP2-specific CD8+CD28+ T cells group, and 1.78% ± 0.099% in the combination therapy group (Figure 4A and B). Thus, mice treated with a combination of FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells showed a significantly lower proportion of Tregs, compared with the other treatment groups (P < 0.05). As shown in Figure 4C and D, the percentages of migrating CD8+ T cells (CXCR3+CD8+ T cells) in the tumor environment were 0.50% ± 0.526% in the PBS group, 10.80% ± 1.716% in the FA-CS-mIP-10 group, 1.00% ± 1.317% in the TRP2-specific CD8+CD28+ T cells group, and 19.20% ± 1.727% in the combination therapy group. Interestingly, the number of CXCR3+CD8+ T cells in the FA-CS-mIP-10 group was considerably larger than that in the TRP2-specific CD8+CD28+ T cells group indicating that IP-10 could attract CD8+ T cells expressing CXCR3. Moreover, the combination therapy led to a significantly higher increase in the number of CXCR3+CD8+ T cells than did either treatment alone (P < 0.01). These data suggest that the combination of FA-CS-mIP-10 with TRP2-specific CD8+CD28+ T cells significantly decreased the number of Tregs in the spleen and increased the number of CXCR3+CD8+ T cells in the tumor.
Combination therapy with FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells inhibited proliferation and induced apoptosis in melanoma cells in vivo
We investigated the effect of the combination therapy on melanoma proliferation by immunohistochemical staining of tumor tissue with proliferating cell nuclear antigen (PCNA) antibody. As shown in Figure 5A and B, the number of PCNA-positive cells was lower in tumor sections from mice in the combination therapy group than that in tumor sections from mice in the other treatment groups (P < 0.01). We also analyzed apoptosis in tumor tissue from the different experimental groups by TUNEL. As is evident from shown in Figure 5C and D, the green fluorescence intensity was significantly stronger in cells from mice in the combination therapy group than in cells from mice in the other treatment groups (P < 0.01). These results indicate that the combination of FA-CS-mIP-10 with TRP2-specific CD8+CD28+ T cells may suppress melanoma growth through inhibition of proliferation and promotion of apoptosis in tumor cells in vivo.
Combination therapy with FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells decreased tumor microvessel density in mice
It is widely believed that tumor growth and metastasis are mainly dependent on angiogenesis. Immunohistochemical analysis by CD31 staining showed that the number of CD31-positive vessels in mice treated with the combination therapy was significantly lower than that observed in mice from all other treatment groups (P < 0.05, Figure 6A and B).
Discussion
In this study, we explored a new strategy to enhance the efficacy of adoptive immunotherapy with TRP2-specific CTLs against melanoma. A combination therapy with FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells was used to treat B16-bearing mice. We demonstrated that this combination therapy noticeably inhibited tumor growth and significantly prolonged survival time in mice compared to either therapy alone. We further investigated the mechanisms underlying the inhibition of tumor growth. Our results demonstrated that the combination of FA-CS-mIP-10 and TRP2-specific CD8+CD28+ T cells not only inhibited the angiogenesis and proliferation of tumor but also induced apoptosis by effectively decreasing the number of regulatory MDSCs and Tregs as well as increasing the number of CD8+ T cells in the tumor environment. Thus, our study has yielded important insights for improving the efficacy of adoptive CTLs transfer immunotherapy (Figure 7).
Figure 3
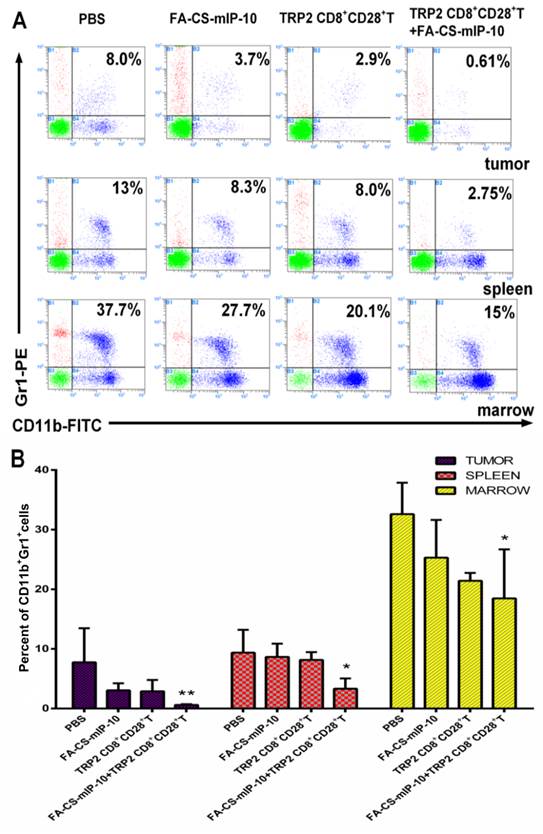
Effect of the treatment of B16-bearing mice with FA-CS-mIP-10 nanoparticles, TRP2-specific CD8+CD28+ T cells, or a combination of both on infiltrating MDSCs: MDSCs cells from the bone marrow, spleen, and tumor from treated mice were analyzed by flow cytometry. (A) Scatter plots of bone marrow, spleen, and tumor cells from mice in different treatment groups stained with Gr1-PE and CD11b-FITC antibodies. (B) Percent of CD11b+Gr1+ cells in bone marrow, spleen, and tumor from mice in different treatment groups. Each data point represents the mean of three independent experiments ± SD. * P < 0.05; ** P < 0.01.
Figure 4
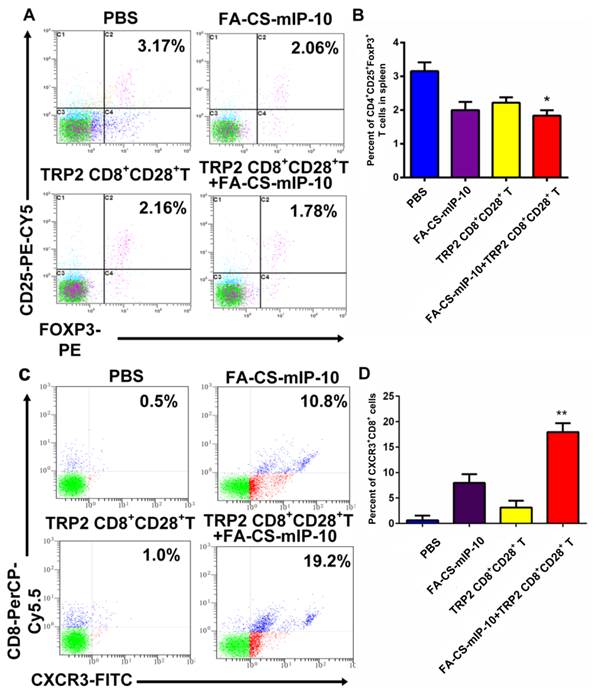
Effect of the treatment of B16-bearing mice with FA-CS-mIP-10 nanoparticles, TRP2-specific CD8+CD28+ T cells, or a combination of both on Tregs and infiltrating CXCR3+CD8+ T cells in tumor tissue: splenocytes and TILs from treated mice were analyzed by flow cytometry. (A) Scatter plots of CD4+ gated spleen cells from mice in different treatment groups stained with CD25-PE-Cy5 and Foxp3-PE. (B) Percent of CD4+CD25+Foxp3+ T cells cells in spleen from mice in different treatment groups. (C) Scatter plots of TILs from mice in different treatment groups stained with CD8-PerCP-Cy5.5 and CXCR3-FITC (D) Percent of CXCR3+CD8+ T cells in TILs from mice in different treatment groups. Each data point represents the mean of three independent experiments ± SD. * P < 0.05; ** P < 0.01.
CD28 is considered to be an important co-stimulatory molecular marker required as a secondary signal for the initiation of T cell activation [22]. Several clinical studies have recently shown that a subset of long-lived adoptively transferred CTLs existed in effectively treated patients. These CTLs expressed high levels of the CD28 surface marker, which was shown to be beneficial for CTLs proliferation and survival in vivo [6, 23]. Furthermore, in our previous study, we developed a novel approach to induce melanoma-specific CD8+CD28+ CTLs by co-culturing them with aAPCs in the presence of IL-21 and IL-15 [11].
To increase the number of melanoma-specific CD8+CD28+ CTLs in the tumor environment, we explored whether FA-CS-mIP-10 nanoparticles could increase the anti-tumor response of adoptively transferred CD8+CD28+ CTLs in vivo. IP-10 plays an important role in the recruitment of infiltrating T lymphocytes into the tumor and was also shown to inhibit tumor angiogenesis [17]. Liu et al. reported that transfection of dendritic cells with IP-10 plasmids increased the invasiveness of antigen-specific T cells [16].
In a previous study, we used folate modified chitosan as the gene delivery system [21]. Chitosan is a natural material derived from chitin that has great potential as a gene delivery biomaterial [18]. Folic acid receptors are expressed at a higher level on the cell surface of most tumors than that of normal tissues [24]. Therefore, we developed FA-CS nanoparticles containing IP-10 plasmids that were used to treat tumor-bearing mice. We showed that FA-CS-mIP-10 nanoparticles significantly induced IP-10 secretion and recruited CTLs into the tumor tissue during the anti-tumor response. We also demonstrated that more CXCR3+CD8+ T cells infiltrated the tumor tissue in mice treated with the combination therapy than in mice in the other treatment groups. Also, FA-CS-mIP-10 monotherapy recruited more CXCR3+CD8+ T cells than the TRP2-specific CD8+CD28+ CTLs monotherapy and the control PBS, indirectly implying that FA-CS-mIP-10 nanoparticles promoted migration of CXCR3+CD8+ T cells partly by up-regulating IP-10 expression.
We further explored the underlying mechanism of the tumor inhibitory effect of the combination therapy. The tumor microenvironment is viewed as a network of interacting cells, extracellular matrix, and signaling molecules that support tumor growth and help the tumor escape from the host immune response [25]. MDSCs and Tregs are major components of the immunosuppressive tumor microenvironment and play an important role by effectively interfering with host anti-tumor responses. Activated MDSCs secrete inducible nitric oxide synthase, arginase 1, NADPH oxidase, and immunosuppressive cytokines, which inhibit CTLs, DCs, and Tregs, leading to the amplification of the tumor microenvironment [26-28]. Tregs are key suppressive T cells that express CD4 and CD25 surface markers and Foxp3 transcriptional factors. Previous studies have reported that depletion of Tregs greatly inhibited tumor growth and promoted anti-tumor responses in tumor-bearing mice [29, 30]. We analyzed the proportion of MDSCs in the spleen, bone marrow, and tumor tissue in all experimental groups and demonstrated that the combination therapy significantly decreased the number of MDSCs. As expected, the number of Tregs in the spleen was also reduced after treatment with the combination therapy. Thus, our results suggested that the combination of FA-CS-mIP-10 and TRP2-specific CD8+CD28+ CTLs improved the suppressive tumor microenvironment by decreasing MDSCs and Tregs in vivo, resulting in delayed tumor growth.
Figure 5
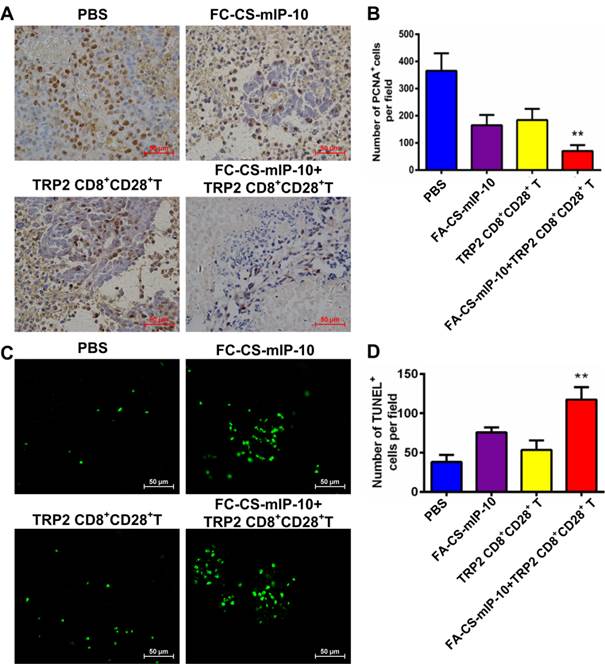
Effect of the treatment of B16-bearing mice with FA-CS-mIP-10 nanoparticles, TRP2-specific CD8+CD28+ T cells, or a combination of both on cell proliferation and apoptosis: (A) Visualization of melanoma cell proliferation in tumor tissue sections from mice in different treatment groups by immunohistochemical staining with anti-PCNA antibody (400X). (B) Average number of PCNA-positive cells per field in tumor tissue sections from mice in different treatment groups. (C) Visualization of melanoma cell apoptosis in tumor tissue sections from mice in different treatment groups by TUNEL. (D) Average number of TUNEL-positive cells per field in tumor tissue sections from mice in different treatment groups. Each data point represents the mean of three independent experiments ± SD. ** P < 0.01.
Figure 6
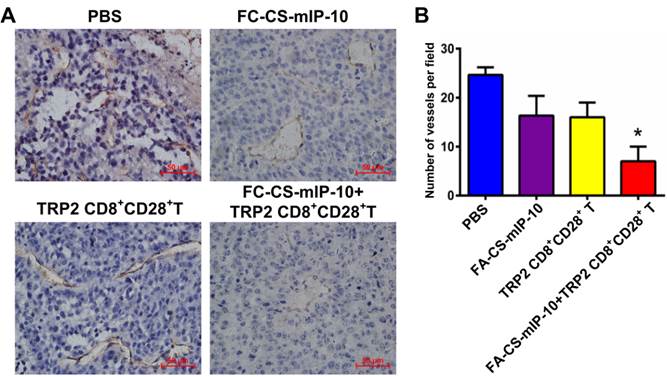
Effect of the treatment of B16-bearing mice with FA-CS-mIP-10 nanoparticles, TRP2-specific CD8+CD28+ T cells, or a combination of both on tumor angiogenesis: (A). Immunohistochemical staining of tumor tissue sections from mice in different treatment groups with anti-CD31 antibody (400X). (B) Average number of CD31-positive cells per field in tumor tissue sections from mice in different treatment groups. Each data point represents the mean of three independent experiments ± SD. * P < 0.05.
Figure 7
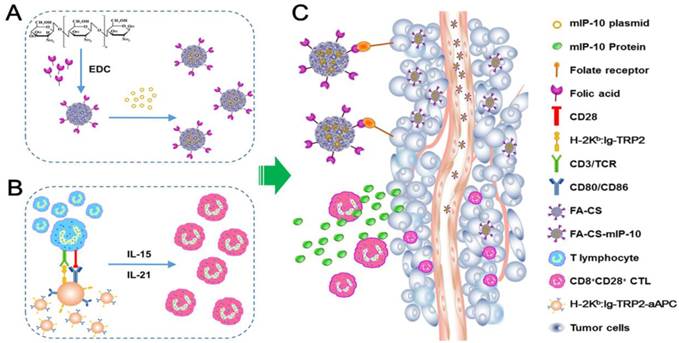
Schematic diagram for preparation and anti-tumor immunotherapy using FA-CS-mIP-10. (A) Preparation of FA-CS-mIP-10 nanoparticles. (B) Preparation of melanoma TRP2-specific CD8+CD28+ cytotoxic T lymphocytes (CTLs). (C) FA-CS-mIP-10 target to tumor cells, and FA-CS-mIP-10 nanoparticles increase the anti-tumor response of CD8+CD28+ CTLs.
IP-10 is thought to negatively regulate angiogenesis through directly inhibiting the expression of angiogenic factors as well as activating of monocytes, fibrocytes, epithelial cells, and keratinocytes [31, 32]. Moreover, IP-10 plays a role in cell proliferation and apoptosis [33, 34]. In this study, we observed that treatment of mice with FA-CS-mIP-10 alone or with the combination therapy caused less densely connected blood vessels in the tumor microenvironment, inhibiting tumor growth. Additionally, we showed that the combination therapy significantly induced apoptosis and delayed the proliferation of tumor cells.
In conclusion, we developed a novel combination therapy by using FA-CS-mIP-10 plus TRP2-specific CD8+CD28+ CTLs to treat B16-bearing mice. We demonstrated that this combination therapy induced proliferation of CXCR3+CD8+ T cells and inhibited infiltration of MDSCs into the tumor microenvironment, concomitantly decreasing Tregs in the spleen. Furthermore, the combination therapy significantly inhibited angiogenesis and proliferation of the tumor, while inducing apoptosis of tumor cells. Thus, we developed an effective strategy for improving the anti-tumor immune response of tumor-specific CD8+CD28+ CTLs in tumor-bearing mice.
Acknowledgements
This work was supported, in part, by grants from Programs for Changjiang Scholars and Innovative Research Team in University (No.IRT_15R13); National Natural Scientific Foundation of China (Nos. 81430055 and 81372452); International Cooperation Project of the Ministry of Science and Technology of China (No. 2015DFA31320); Project for Innovative Research Team in Guangxi Natural Science Foundation (2015GXNSFFA139001); Project of Science and Technology of Guangxi (Nos. 14125008-2-12, 1599005-2-10).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Erdei E, Torres SM. A new understanding in the epidemiology of melanoma. Expert Rev Anticancer Ther. 2010;10:1811-23
2. Bay C, Kejs AM, Storm HH. et al. Incidence and survival in patients with cutaneous melanoma by morphology, anatomical site and TNM stage: A Danish Population-based Register Study 1989-2011. Cancer Epidemiol. 2015;39:1-7
3. Jandus C, Speiser D, Romero P. Recent advances and hurdles in melanoma immunotherapy. Pigment Cell Melanoma Res. 2009;22:711-23
4. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237-51
5. Lu X, Jiang X, Liu R. et al. In vivo Anti-melanoma Efficiency of Allo-restricted CTLs Specific for Melanoma Expanded by Artificial Antigen-presenting cells. Cancer Immunol Immunother. 2009;58:629-38
6. Li Y, Liu S, Hernandez J. et al. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J Immunol. 2010;184:452-65
7. Azuma M, Ebihara T, Oshiumi H. et al. Cross-priming for antitumor CTL induced by soluble Ag+ polyI: C depends on the TICAM-1 pathway in mouse CD11c+/CD8alpha+ dendritic cells. Oncoimmunology. 2012;1:581-92
8. DeBenedette MA, Calderhead DM, Tcherepanova IY. et al. Potency of mature CD40L RNA electroporated dendritic cells correlates with IL-12 secretion by tracking multifunctional CD8+/CD28+ cytotoxic T-cell responses in vitro. J Immunother. 2011;34:45-57
9. Jiang X, Lu X, Liu R. et al. HLA Tetramer Based Artificial Antigen-Presenting Cells Efficiently Stimulate CTLs Specific for Malignant Glioma. Clin Cancer Res. 2007;13:7329-34
10. Lu X, Jiang X, Liu R. et al. Adoptive transfer of pTRP2-specific CTLs expanding by bead-based artificial antigen-presenting cells mediates anti-melanoma response. Cancer Lett. 2008;271:129-39
11. Yu X, He J, Mongkhoune S. et al. Artificial antigen-presenting cells plus IL-15 and IL-21 efficiently induce melanoma-specific cytotoxic CD8+ CD28+ T lymphocyte responses. Asian Pac J Trop Med. 2013;6:467-72
12. Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72-84
13. Laconi E. The evolving concept of tumor microenvironments. Bioessays. 2007;29:738-44
14. Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review). Oncol Lett. 2011;2:583-9
15. Lu XL, Jiang XB, Liu RE. et al. The enhanced anti-angiogenic and antitumor effects of combining flk1-based DNA vaccine and IP-10. Vaccine. 2008;26:5352-7
16. Liu M, Guo S, Hibbert JM. et al. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011;22:121-30
17. Lunardi S, Jamieson NB, Lim SY. et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget. 2014;5:11064-80
18. Yang CX, Nilsson L, Cheema MU. et al. Chitosan/siRNA Nanoparticles Targeting Cyclooxy-genase Type 2 Attenuate Unilateral Ureteral Obstruction-induced Kidney Injury in Mice. Theranostics. 2015;5:110-23
19. Lai CH, Ye BB, Hou XQ. et al. Anti-Tumor Effect of Folic Acid-Conjugated Chitosan Nanoparticles Containing IL-33 Gene in Hepatocellular Carcinoma. Cell Communications. 2014;1:30-40
20. Hu L, Sun Y, Wu Y. Advances in chitosan-based drug delivery vehicles. Nanoscale. 2013;5:3103-11
21. Lai CH, Yu X, Zhuo HQ. et al. Anti-Tumor Immune Response of Folate-Conjugated Chitosan Nanoparticles Containing the IP-10 Gene in Mice with Hepatocellular Carcinoma. J Biomed Nanotechnol. 2014;10:3576-89
22. Sepulveda H, Cerwenka A, Morgan T. et al. CD28, IL-2-independent costimulatory pathways for CD8 T lymphocyte activation. J Immunol. 1999;163:1133-42
23. Powell DJ Jr, Dudley ME, Robbins PF. et al. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241-50
24. Yang T, Li B, Qi SB. et al. Co-delivery of Doxorubicin and Bmi1 siRNA by Folate Receptor Targeted Liposomes Exhibits Enhanced Anti-Tumor Effects in vitro and in vivo. Theranostics. 2014;4:1096-11
25. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267-96
26. Filipazzi P, Huber V, Rivoltini L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immun. 2012;61:255-63
27. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162-74
28. Fujimura T, Kambayashi Y, Aiba S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology. 2012;1:1433-4
29. Golovina TN, Vonderheide RH. Regulatory T Cells Overcoming Suppression of T-Cell Immunity. Cancer J. 2010;16:342-7
30. Poehlein CH, Haley DP, Walker EB. et al. Depletion of tumor-induced Treg prior to reconstitution rescues enhanced priming of tumor-specific, therapeutic effector T cells in lymphopenic hosts. Eur J Immunol. 2009;39:3121-33
31. Zhuo HQ, Peng Y, Yao Q. et al. Tumor Imaging and Interferon-γ-Inducible Protein-10 Gene Transfer Using a Highly Efficient Transferrin-Conjugated Liposome System in Mice. Clin Cancer Res. 2013;19:4206-17
32. Wang LL, Chen P, Luo S. et al. CXC-chemokine-ligand-10 gene therapy efficiently inhibits the growth of cervical carcinoma on the basis of its anti-angiogenic and antiviral activity. Biotechnol Appl Biochem. 2009;53:209-16
33. Sui Y, Stehno-Bittel L, Li S. et al. CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci. 2006;23:957-64
34. Feldman ED, Weinreich DM, Carroll NM. et al. Interferon gamma-inducible protein 10 selectively inhibits proliferation and induces apoptosis in endothelial cells. Ann Surg Oncol. 2006;13:125-33
Author contact
![]() Corresponding authors: Yongxiang Zhao, M.D., Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061 E-mail: yongxiang_zhaocom Other corresponding authors: Xiaoling Lu, Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061 Email: luwuliucom Nuo Zhou, M.D., Ph.D., Professor, Department of Oral and Maxillofacial Surgery, the Affiliated Hospital of Stomatology, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061 E-mail: nuozhoucom.
Corresponding authors: Yongxiang Zhao, M.D., Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061 E-mail: yongxiang_zhaocom Other corresponding authors: Xiaoling Lu, Ph.D., Professor, National Center for International Research of Biological Targeting Diagnosis and Therapy / Guangxi Key Laboratory of Biological Targeting Diagnosis and Therapy Research / Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061 Email: luwuliucom Nuo Zhou, M.D., Ph.D., Professor, Department of Oral and Maxillofacial Surgery, the Affiliated Hospital of Stomatology, Guangxi Medical University, Nanning, Guangxi 530021, China. Tel: (86) 771-5317 061; Fax: (86) 771-5317 061 E-mail: nuozhoucom.
Citation styles
APA
He, J., Duan, S., Yu, X., Qian, Z., Zhou, S., Zhang, Z., Huang, X., Huang, Y., Su, J., Lai, C., Meng, J., Zhou, N., Lu, X., Zhao, Y. (2016). Folate-modified Chitosan Nanoparticles Containing the IP-10 Gene Enhance Melanoma-specific Cytotoxic CD8+CD28+ T Lymphocyte Responses. Theranostics, 6(5), 752-761. https://doi.org/10.7150/thno.14527.
ACS
He, J.; Duan, S.; Yu, X.; Qian, Z.; Zhou, S.; Zhang, Z.; Huang, X.; Huang, Y.; Su, J.; Lai, C.; Meng, J.; Zhou, N.; Lu, X.; Zhao, Y. Folate-modified Chitosan Nanoparticles Containing the IP-10 Gene Enhance Melanoma-specific Cytotoxic CD8+CD28+ T Lymphocyte Responses. Theranostics 2016, 6 (5), 752-761. DOI: 10.7150/thno.14527.
NLM
He J, Duan S, Yu X, Qian Z, Zhou S, Zhang Z, Huang X, Huang Y, Su J, Lai C, Meng J, Zhou N, Lu X, Zhao Y. Folate-modified Chitosan Nanoparticles Containing the IP-10 Gene Enhance Melanoma-specific Cytotoxic CD8+CD28+ T Lymphocyte Responses. Theranostics 2016; 6(5):752-761. doi:10.7150/thno.14527. https://www.thno.org/v06p0752.htm
CSE
He J, Duan S, Yu X, Qian Z, Zhou S, Zhang Z, Huang X, Huang Y, Su J, Lai C, Meng J, Zhou N, Lu X, Zhao Y. 2016. Folate-modified Chitosan Nanoparticles Containing the IP-10 Gene Enhance Melanoma-specific Cytotoxic CD8+CD28+ T Lymphocyte Responses. Theranostics. 6(5):752-761.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.