Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(12):1456-1472. doi:10.7150/thno.11692 This issue Cite
Research Paper
EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model
Tao Wang1,2, Michael P. Gantier3,4, Dongxi Xiang2, Andrew G Bean5, Matthew Bruce5, Shu-Feng Zhou6, Mustafa Khasraw2, Alister Ward2, Li Wang7, Ming Q. Wei8, Hadi AlShamaileh2, Lijue Chen9, Xiaodong She9, Jia Lin10, Lingxue Kong9, Sarah Shigdar2 ![]() , Wei Duan2
, Wei Duan2 ![]()
1. School of Nursing, Zhengzhou University, Zhengzhou, P. R. China, 450001.
2. School of Medicine, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria, 3217, Australia.
3. Centre for Cancer Research, Hudson Institute of Medical Research, Clayton, Victoria 3168, Australia
4. Department of Molecular and Translational Science, Monash University, Clayton, Victoria 3168, Australia
5. CSIRO Australian Animal Health Laboratory, Private Bag 24, Geelong, Victoria 3220, Australia
6. Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL 33612, USA.
7. Department of Gynecologic Oncology, Henan Cancer Hospital, The Affiliated Cancer Hospital of Zhengzhou University, Zhengzhou 450008, China.
8. School of Medical Science & Griffith Health Institute, Gold Coast Campus, Griffith University, Southport, QLD 4222, Australia.
9. Institute for Frontier Materials, Deakin University, Waurn Ponds, Victoria, 3216, Australia
10. Department of Biochemistry and Molecular Biology, West China School of Preclinical and Forensic Medicine, Sichuan University, Chengdu, P. R. China, 610041.
Received 2015-1-25; Accepted 2015-7-30; Published 2015-10-20
Citation:
Wang T, Gantier MP, Xiang D, Bean AG, Bruce M, Zhou SF, Khasraw M, Ward A, Wang L, Wei MQ, AlShamaileh H, Chen L, She X, Lin J, Kong L, Shigdar S, Duan W. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics 2015; 5(12):1456-1472. doi:10.7150/thno.11692. https://www.thno.org/v05p1456.htm
Other stylesAbstract

Understanding the molecular basis of drug resistance and utilising this information to overcome chemoresistance remains a key challenge in oncology. Here we report that survivin, a key protein implicated in drug resistance, is overexpressed in cancer stem cell pool of doxorubicin-resistant breast cancer cells. Moreover, by utilising an active targeting system consisting of an RNA aptamer targeted against the epithelial cell adhesion molecule and a Dicer substrate survivin siRNA, we could deliver a high dose of the siRNA to cancer stem cells in xenograft tumours. Importantly, silencing of survivin with this aptamer-siRNA chimera in cancer stem cell population led to the reversal of chemoresistance, such that combined treatment with low dose of doxorubicin inhibited stemness, eliminated cancer stem cells via apoptosis, suppressed tumour growth, and prolonged survival in mice bearing chemoresistant tumours. This strategy for in vivo cancer stem cell targeting has wide application for future effective silencing of anti-death genes and in fact any dysregulated genes involved in chemoresistance and tumour relapse.
Keywords: Breast cancer, RNA interference, Cancer stem cell, Aptamer, Survivin
Introduction
Resistance to chemotherapy is a major mediator of treatment failure for solid malignancies [1]. Intensive efforts in developing chemosensitizing agents and either reversing or modulating chemoresistance have yielded only limited success to date [2]. Recent advances in tumour biology have revealed the extensive intratumoural genetic heterogeneity within human cancers [3], with the acquisition of resistance often mediated by outgrowth of resistant clones present at low frequency [4].
Emerging evidence suggests that cancer stem cells (CSCs) represent clones within a tumour that are responsible for treatment failure [5]. These CSCs possess unique properties, including resistance to treatment and evasion of cell death, as well as being capable of initiating and sustaining tumour growth in serial transplantation assays in immunocompromized animals [5]. The identification and targeting key molecules responsible for CSC survival will facilitate the development of effective CSC therapies.
Among known pro-survival proteins, survivin has been shown to exert a number of effects in cancer, where it is involved in inhibition of apoptosis, regulation of the mitotic spindle checkpoint and the promotion of angiogenesis and chemoresistance [6]. Largely undetectable in normal adult tissues, survivin becomes overexpressed in most tumours following chemotherapy and radiotherapy, and its overexpression correlates with poor prognosis in patients with multiple cancer types [7]. Furthermore, the overexpression of survivin has been found in CSCs in both solid and liquid cancers [8, 9]. Thus, targeting survivin overexpression represents a promising strategy to combat drug resistance [10]. However, the role of survivin in CSCs remains elusive, with the consequences of targeting survivin in CSCs as a means of chemosensitization yet to be elucidated.
As a revolutionary technology to control aberrant gene expression, RNA interference (RNAi) has great potential in combating tumour cells that evade chemotherapy through elevated expression of key genes, such as survivin. However, the translation of RNAi into the clinic has been hindered by the inability of effectively delivering a sufficient dose to specific cell populations [11]. An emerging strategy for targeted delivery of siRNA is the aptamer-guided RNAi [12]. Aptamers are synthetic, single-stranded nucleic acids that fold into complex 3-D structures capable of recognizing targets with high affinity and specificity [13]. Fusion of aptamer sequences to synthetic siRNAs has been shown to promote cell-specific siRNA internalization and gene knockdown following systemic administration [11, 14, 15]. Such a chimera approach possesses several advantages including simplified large-scale production with greater purity and consistency, and allowing unlimited gene targeting to virtually any cell type [16]. However, the use of an aptamer-siRNA chimera to target CSCs in vivo has not been reported.
One of the potential CSC markers for RNAi-medicated therapy is Epithelial cell adhesion molecule (EpCAM), which is present at low levels in normal epithelial cells but is highly overexpressed (up to 800-fold) in many solid cancers [17]. For example, enhanced EpCAM expression has been observed in ~98% primary breast cancers and is associated with poor prognosis [17]. A trifunctional antibody, catumaxomab, with specificity for EpCAM has shown clear clinical benefits in the treatment of malignant ascites associated with EpCAM-positive carcinomas [18]. Importantly, EpCAM is also a marker for CSCs in cancers of the prostate, colon, pancreas, breast, ovary, lung, and stomach/intestine [19-22], an attribute utilized to target survivin in CSCs with siRNA in this work.
Here, we describe a critical role of survivin in acquired chemoresistance in breast CSCs. By fusion with an EpCAM-specific aptamer, survivin siRNA could be delivered specifically to breast CSCs and successfully silence this anti-apoptosis gene in vivo. The resulting reversal of chemoresistance was accompanied by inhibited self-renewal of CSCs and extended survival of tumour-bearing mice. Therefore, aptamer-guided and CSC-targeted in vivo RNAi represents a promising novel avenue to overcome chemoresistance and achieve lasting remission.
Materials and methods
In vitro gene silencing
In the case of siRNA transfection, cells were trypsinized 24 h before transfection, resuspended at a concentration of 60,000 cells/mL in DMEM medium. Cells were transfected with 10 nM survivin or 50 nM MDR1 siRNAs using Lipofectamine 2000 (Invitrogen, 11668-030) according to the manufacturer's instruction (the guide strands of survivin siRNA and MDR1 siRNA are 5'-UGUAGAGAUGCGGUGGUCCTT-3' and 5'-GAAGCACTGGGATGTCCGGT-3', respectively). Twenty-four or 48 hours after the treatment, total RNA or protein were prepared for qRT-PCR or immunoblotting, respectively. In the case of chimera treatment, cells were cultured at 30% confluence and chimera was added directly. Twenty-four or 48 hours after the treatment, total RNA and protein were prepared, respectively.
Tumour implantation and treatment
To establish xenograft tumours, 4 × 106 MCF-7/Adr or 2 × 106 U118MG cells were resuspended in 150 µL of 50% Matrigel (BD, 354234), and injected into the 4th mammary fat pad of NOD-SCID female mice (6-8 week old). Once tumours arose, mice were randomized into treatment groups of 5 - 8 mice per group. A 7-day treatment protocol was initiated when the tumour volume reached 60 mm3. Specifically, Dox group was treated with PBS on day 1 and 5 mg/kg Dox on day 3, 5, 7. Chimera and negative control chimera groups were treated with 2 nmol/mouse chimera or control chimera on day 1, 3, 5 and PBS on day 7, while chimera + Dox and control chimera + Dox groups were treated with chimera or negative control chimera on day 1, followed by the combined chimera (or control chimera) and Dox on day 3 and 5 with single Dox injected on day 7. Mice were euthanized two days after the last treatments. Tumour fragments were processed for mRNA and protein assays or archived in neutral buffered formalin for apoptosis and proliferation assays.
In vivo limiting dilution assay
MCF-7/Adr tumour bearing mice were randomly assigned to groups (8 mice per group) and underwent various treatments. Two days after the last treatment, tumours from the same treatment groups were pooled and single cells suspension was prepared and secondary xeno-transplantation was carried out. Specifically, viable cells taken from each group were inoculated into three sub-groups of mice using a cell doses of 1 × 106, 2 × 106 and 4 × 106 /mouse, 8 mice per sub-group. The growth of tumour was evaluated daily during a 6-month period. The endpoint was defined as when the maximum diameter of a tumour reached 17 mm or consistent or rapid body weight loss of 20% or other health deteriorations. The estimated CSC frequency was calculated using the limiting dilution software package on the website of Walter and Eliza Hall Institute of Medical Research (http://bioinf.wehi.edu.au/software/elda/index.html).
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 3.03. An unpaired t test was used for comparisons between two experimental groups, and ANOVA was used for comparisons of more than two groups. Survival data were analyzed with the Mantel-Cox log-rank test. Unless otherwise indicated, all results were averaged from biological triplicates and values are reported as means ± SEM. A P value of less than 0.05 was considered statistically significant.
Results
Survivin is overexpressed in the CSC population of MCF-7/Adr cells
In order to investigate the role of survivin in chemoresistance in breast cancer, we generated a doxorubicin (Dox)-resistant sub-line of the human breast adenocarcinoma MCF-7 cells by chronic exposure of MCF-7 cells to a clinically relevant Dox concentration of 300 nM [23]. The resultant MCF-7/Adr cells displayed a 5-fold increase in resistance to Dox and mesenchymal-like morphology (Figure 1A and B), similar to that previously reported for drug resistant primary breast cancer cells derived from biopsies [24]. Furthermore, the MCF-7/Adr cells expressed 21-fold more survivin protein along with elevated levels of other proteins involved in drug efflux (ABCG2 and MDR1), anti-apoptosis (survivin and Bcl-2), and stemness (Notch1, Nanog, Nestin, SOX2 and OCT4) (Figure 1C and D).
To study the involvement of CSCs in chemoresistance, we utilized a combination of cell surface makers, EpCAM+/CD44+/CD24-, previously shown by others as a robust marker for the identification and isolation of breast CSCs [25]. Considering the heterogeneous property of tumours, the reliability of this marker combination in identifying CSCs in MCF-7/Adr cells was initially confirmed. As illustrated, in contrast to EpCAM-/CD44+/CD24- and EpCAM-/CD44-/CD24- MCF-7/Adr cells, the EpCAM+/CD44+/CD24- population showed prominent multi-potent differentiation (Figure 1E) and self-renew capacities (Figure 1G). Notably, MCF-7/Adr cells had an approximately 24-fold increase in the population of EpCAM+/CD44+/CD24- cells compared to the parental MCF-7 cells, accompanied by a 5-fold higher mammosphere forming efficiency (Figure 1F and G).
Next, we investigated whether silencing of survivin using a 21-mer siRNA [26] could reverse chemoresistance of MCF-7/Adr cells to Dox. As shown in Figure 2, transfection of MCF-7/Adr cells with this survivin siRNA resulted in more than 80% reduction of both survivin mRNA and protein. The specificity of survivin silencing was demonstrated by rescuing the siRNA-induced phenotype through the expression of an RNAi-resistant form of survivin cDNA, as well as the absence of siRNA-induced phenotype in cells treated with a scrambled siRNA control (Figures 2A and B). As demonstrated, the knockdown of survivin resulted in more than 4-fold increase in the sensitivity of MCF-7/Adr to Dox (Figure 2C). Interestingly, the increased chemosensitivity was associated with a significant reduction in the mammosphere formation efficacy of MCF-7/Adr cells (Figure 2D) and a 2-fold reduction in the abundance of EpCAM+/CD44+/CD24- cells (Figure 2E). Critically important was the fact that it was the EpCAM+/CD44+/CD24- population, but not the EpCAM+/CD44+/CD24- or EpCAM-/CD44-/CD24- population of MCF-7/Adr cells that expressed more survivin (>23-fold) than parental MCF-7 cells (Figure 2F). These data led to the hypothesis that the overexpression of survivin in CSCs was responsible for the acquired chemoresistance in MCF-7/Adr cells.
Figure 1
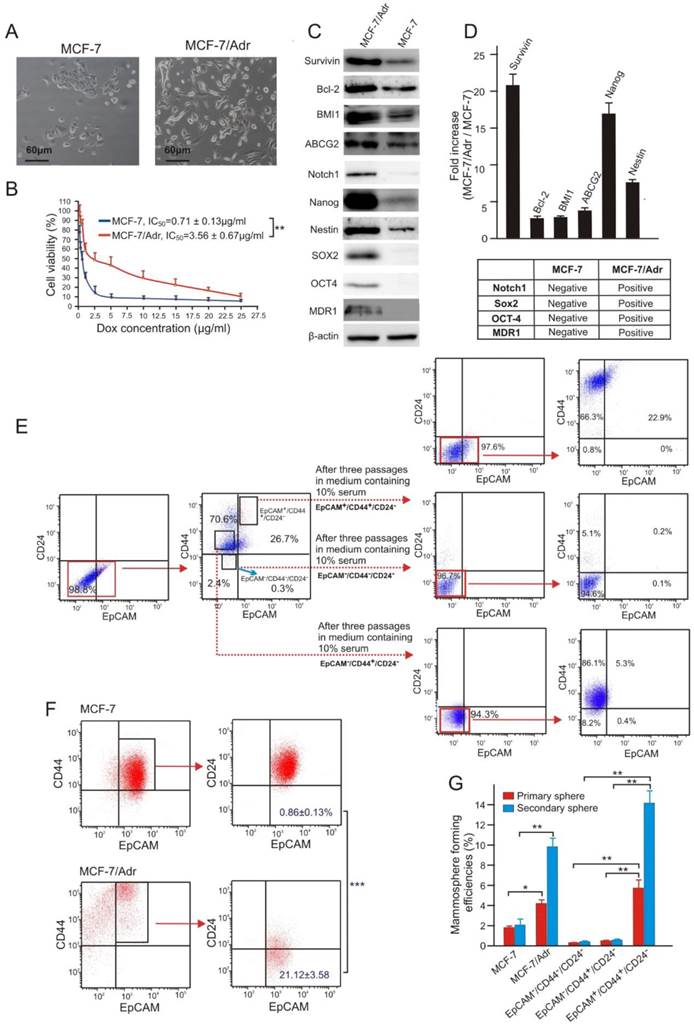
Doxorubicin-resistant MCF-7/Adr cells overexpress survivin and are enriched with CSCs. (A) Morphology of parental MCF-7 cells and its Dox-resistant derivative sub-line MCF-7/Adr. (B) Cell viability of MCF-7 and MCF-7/Adr cells treated with indicative concentration of Dox for three days. (C, D) Differential expression of proteins involved in drug-efflux, anti-apoptotic and stemness functions in MCF-7/Adr cells and parental MCF-7 cells. (C) Representative immunoblots. (D) Analysis of relative expression in MCF-7/Adr cells compared with that in MCF-7 cells as determined with immunoblotting. (E) Representative flow cytometric profiles illustrating multi-potent differentiation capacity of FACS-sorted EpCAM+/CD44+/CD24- MCF-7/Adr cells. (F) MCF-7/Adr cells have increased proportion of EpCAM+/CD44+/CD24- cells. (G) Increased mammosphere forming capacity in both primary and secondary mammospheres of MCF-7/Adr cells and FACS-sorted EpCAM+/CD44+/CD24- MCF-7/Adr cells (vs.MCF-7). Data are means ± SEM, n=3. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (as denoted).
Figure 2
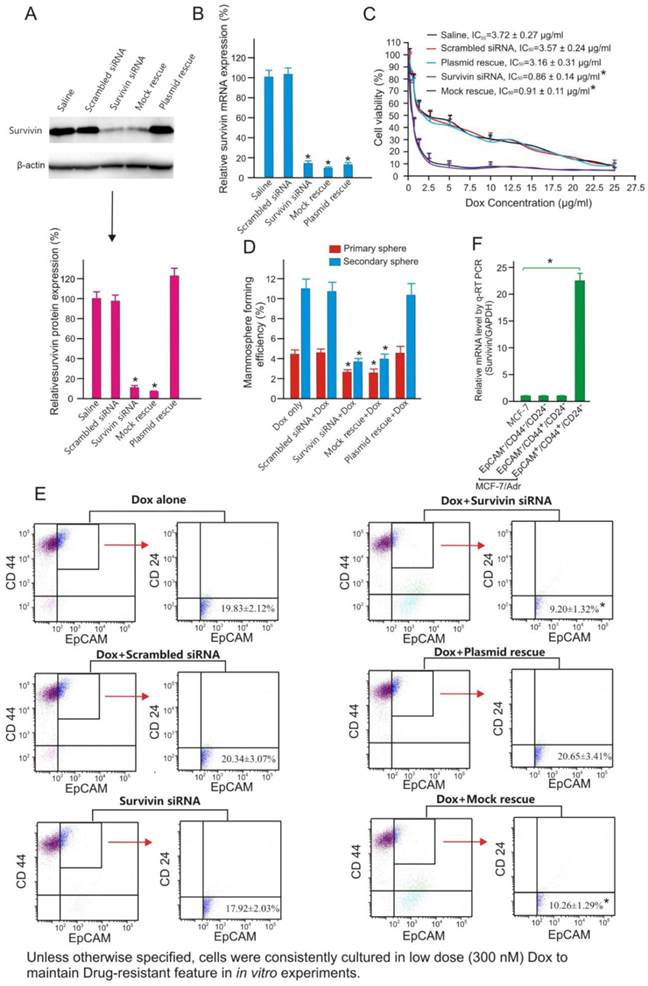
Survivin knockdown increases chemosensitivity and suppresses tumour-initiating capacity of MCF-7/Adr cells. (A) Examination of efficacy and specificity of survivin knockdown in MCF-7/Adr cells by immunoblotting 48 h after the treatment with saline or 10 nM of scrambled siRNA, survivin siRNA, survivin siRNA plus vector only (mock rescue) or survivin siRNA plus RNAi-resistant survivin replacement construct (plasmid rescue). Data are means ± SEM (n=3). *, P < 0.01 (vs. saline-treated). (B) Knockdown of surviving mRNA by chimera 24 h after treatment as in A. As the forward primer for survivin corresponds to the 5'-UTR region, and thus the transcripts from the rescue plasmid were not amplified in qRT-PCR (n=3). Data are means ± SEM (n=3). *, P < 0.01 (vs. saline-treated). (C) Cell viability of MCF-7/Adr cells treated as indicated and incubated with various concentration of Dox for 3 days. Data are means ± SEM (n=3). (D) Primary and secondary mammosphere forming efficiencies of MCF-7/Adr cells treated with 300 nM Dox or plus 10 nM survivin siRNA or controls as indicated for 72 hours (n=3). *, P < 0.01 (vs. Dox-treated). (E) Changes in the abundance of EpCAM+/CD44+/CD24- population in MCF-7/Adr cells treated as indicated and analyzed using flow cytometry (n=3). *, P < 0.01 (vs. Dox-treated). (F) Survivin mRNA levels as determined by qRT-PCR in parental MCF-7 cells as well as in EpCAM+/CD44+/CD24- , EpCAM-/CD44+/CD24- and EpCAM-/CD44-/CD24- populations of MCF-7/Adr cells purified by FACS (n=3). *, P < 0.01 (vs. MCF-7).
Development of an aptamer-siRNA chimera for CSC-targeted RNAi
In order to effectively knock down survivin in CSCs, we adopted an aptamer-guided RNAi strategy by developing a CSC-targeted aptamer-siRNA chimera [27]. Specifically, an EpCAM targeted RNA aptamer [28] was fused with a 27-mer Dicer substrate survivin siRNA (designed based on the above 21-mer siRNA) to generate an EpCAM aptamer-survivin siRNA fusion (Figure 3A-C).
To engineer a chimera that has optimized chemical modifications, 10 different versions of the chimera (Figure S1A) were tested. Based on serum stability, Dicer processing efficiency and silencing efficacy, Chimera No. 10 that had an 8.3 h half-life in 50% human serum and produced a strong dose-dependent survivin gene silencing efficacy was selected (Supplementary Results and Figures S2A-C). This chimera consisted of a 25/27-nt siRNA Dicer substrate duplex for enhanced silencing potency through increased strand selectivity and polarized Dicer activity, with the guide strand placed on top favouring the loading of this 3' overhang-containing strand into RISC. A negative control of this chimera was also generated by introducing two point mutations in the guide strand with the same “miRNA-like seed region” and an identical passenger strand compared to the chimera (Figure 4A). This was designed to abolish silencing capacity of the siRNA portion while retain the potential of miRNA-mediated effects, thus facilitating the vigorous assessment of potential non-target effects of the chimera. Importantly, attachment of the survivin siRNA did not affect the EpCAM aptamer-dependent targeting, as the chimera bound to MCF-7/Adr cells with a dissociation equilibrium constant of approximately 39 nmol/L, and also bound specifically to various EpCAM-positive cells, including both primary breast cancer biopsies and cell lines (Figure 4B and Figure S2A-C). Of note, the relative mean fluorescence intensity with the chimera determined by flow cytometric studies corresponded well with the known level of EpCAM expression in the various cell lines tested (Figure S2B) [29].
Figure 3
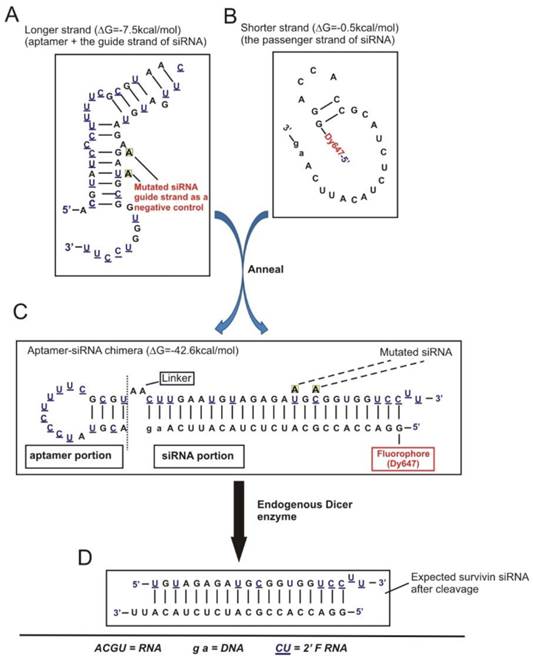
Schematic of design of EpCAM aptamer-survivin siRNA chimera. (A) Secondary structure for the longer strand of chimera in which an 18-nt RNA EpCAM aptamer was covalently linked with the guide strand of a 27-mer survivin siRNA through an AA linker. All pyrimidines were 2'-fluoropyrimidines. In the negative control version of the longer strand, two point mutations were introduced as indicated. (B) The structure of the shorter strand represents the passenger strand of the 27-mer survivin siRNA with 2-nt DNA at the 3'-end and a fluorophore at the 5'-end. (C) A stable aptamer-siRNA chimera with Gibbs' free energy of -42.6 kcal/mol was formed after annealing the longer and shorter stands. (D) The predicted 21-mer survivin siRNA after being processed by Dicer. The secondary structural predictions and Gibbs' free energy were generated or calculated using CLC Main Workbench software.
Figure 4
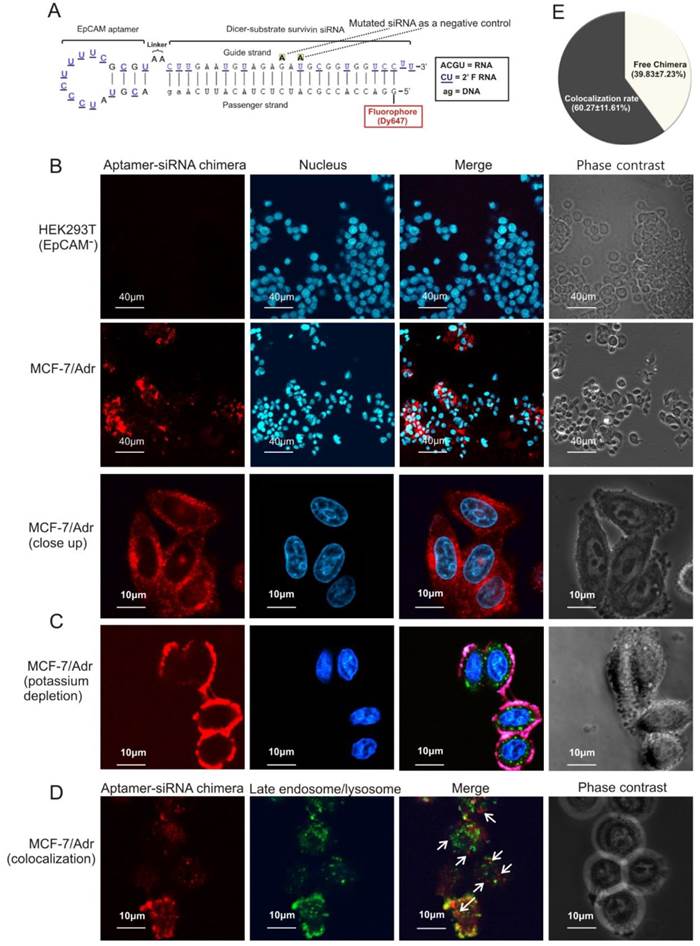
EpCAM aptamer-mediated survivin siRNA delivery in vitro. (A) Schematic diagram of EpCAM aptamer-survivin siRNA chimera. The optimized EpCAM aptamer-survivin siRNA chimera was constructed by linking an 18-nt RNA EpCAM aptamer via a 2-nt (AA) linker to a 27-mer Dicer substrate survivin siRNA, with the guide (antisense) strand on top, a 2-nt (UU)-overhang at the 3'-end of the guide strand and a Dy647 fluorophore conjugated at the 5'-end of the passenger strand. As a negative control, two point mutations in the guide strand (G→A and U→A) that abolish survivin silencing were introduced. (B) Cells were incubated with 100 nmol/L aptamer-survivin siRNA chimera at 37C for 30 min followed by washing and confocal microscopy. Red, Dy647 (chimera), and blue, Hoechst 33342 (nuclei). (C) Cells were subjected to potassium depletion followed by incubation of chimera and examined by confocal microscopy as in B. Green, LysoTracker® Green. (D) Following 1 h incubation of chimera as in B, cell surface fluorescence was quenched using trypan blue and further incubated for 2 h. Cells were treated with LysoTracker® Green in the last 30 min of the 2 h incubation followed by confocal microscopy analysis. Arrows depict cytoplasmic chimera. (E) Quantification of cytoplasmic chimera via confocal microscopy with the aid of ImagePro Premier 9.0 software.
Next, the ability of the chimera to internalize upon binding to cell surface EpCAM was studied. After incubation with the chimera for 30 min, a particular pattern of red fluorescence (for Dy467-labelled aptamer-siRNA chimera) was apparent (Figure 4B), indicating that the chimera was endocytozed. Internalization of the chimera was effectively arrested by potassium depletion, a treatment known to block receptor-mediated endocytosis [30], and the internalization of the chimera reassumed once the potassium depletion was removed (Figure 4C and Figure S2D). Thus, chimera internalization was mediated through a receptor-mediated endocytotic pathway.
Importantly, approximately 40% of the internalised chimera was found in the cytoplasm but did not colocalize with late endosomes or lysosomes (Figures 4D and E), indicating that the chimera was able to escape endosome to reach the cytoplasm, a necessary prerequisite to engage with the RNAi machinery.
Aptamer-guided survivin silencing sensitizes breast CSCs in vitro
Next, the ability of chimera to specifically silence survivin in vitro in the absence of transfection agents was determined. Indeed, the EpCAM aptamer- survivin siRNA chimera was able to knock down both survivin mRNA and protein by more than 80% in MCF-7/Adr cells and a range of different EpCAM-positive breast cancer cells, including cells dissociated from primary breast cancer biopsies (Figure S3A-C). The EpCAM-dependent targeting was demonstrated by the absence of survivin silencing in EpCAM-negative HEK293T cells. The specificity of the chimera-mediated gene silencing was also confirmed through the reversal of survivin knockdown by the introduction of a plasmid expressing an RNAi-resistant version of survivin cDNA. Of note, 300 nmol/L Dox was routinely included in the culture medium to maintain the Dox-resistance phenotype of MCF-7/Adr cells.
Importantly, as in the case of siRNA-induced survivin silencing, combined treatment using 20 nmol/L chimera and 300 nmol/L Dox, but not the chimera or Dox alone, successfully impaired self-renewal efficacy and reduced the CSC population in MCF-7/Adr cells (Figure S3D-F). Therefore, chimera-mediated survivin silencing restored the chemosensitivity of breast CSCs, at least in vitro.
PEGylation confers favourable pharmaceutical properties to aptamer-siRNA chimera
Although the chimera displayed specific targeting to EpCAM-positive xenograft MCF-7/Adr tumours in molecular imaging in nonobese diabetic-severe combined immunodeficiency (NOD/SCID) mice (Figure 5A), no survivin knockdown was observed after its intravenous administration (Figure S4). This was due to the short blood circulation time (4 hours) leading to insufficient tumour accumulation, as this version of the chimera had a size of ~4.4 nm which is significantly below the 10 nm renal filtration threshold (Figure 5B and D) [31].
In order to effectively silence survivin in CSCs in vivo, the chimera was further engineered by conjugating a terminal 20 kDa polyethylene glycol (PEG) to increase serum retention. Furthermore, a fluorescein isothiocyanate (FITC) was added at the 3'-end and a biotin or Dy647 fluorophore at the 5'-end of the passenger strand of the siRNA portion to allow accurate quantification and facilitate imaging of the chimera without compromizing Dicer processing efficiency (Figure 5C). Of note, the PEGylation of the chimera did not adversely affect the affinity and specificity to target EpCAM-positive cells (Figure S5). As expected, the conjugation of PEG increased the hydrodynamic diameter of the original chimera to approximately 15 nm, well above the 10 nm threshold for the glomerular filtration (Figure 5D) [31]. Accordingly, there was a significant increase in the accumulation of PEGylated chimera in the xenograft MCF-7/Adr tumour, evident from a 9.5-fold larger area under the fluorescence intensity curve (AUC) for the PEGylated chimera compared with that of the non-PEGylated counterpart (Figure 5E and F). The EpCAM-dependent in vivo delivery of this chimera to the EpCAM-positive xenograft tumour was confirmed by the lack of fluorescence in the control EpCAM-negative U118MG glioma during the first 4 h followed by a quick decline of the signal from 5 h onwards (Figure 5E and F).
Subsequently a biodistribution study was performed to assess the pharmaceutical properties of the chimera by injecting mice bearing MCF-7/Adr xenograft tumour of ~150 mm3 with 2 nmol/mouse of chimera followed by quantification of the aptamer-siRNA chimera using an ELISA-based approach (Figure S6). As shown in Figure 5G, the PEGylated chimera accumulated rapidly in highly perfused organs, such as the heart, liver, kidneys, spleen and lungs, and reached a peak concentration in these organs at 30 min, followed by a swift decline to a level between 0.76~4.8% of the respective peak concentrations from 6 h onwards. In contrast, the delivery of chimera to the xenograft tumour displayed a very different time course: while it only reached a third of its peak concentration at 30 min, the chimera reached the peak concentration in the tumour at 6 h time point and persisted for at least 9.5 h. Between 6 h and 10 h after administration, the xenograft tumour contained the highest chimera concentration compared to any other organs/tissues examined.
To ascertain the stability of the chimera following intravenous injection and optimal dose interval, time-related variations of the chimera in serum was studied using ELISA in healthy rats. Consistent with data from the biodistribution assay in mice, the pharmacokinetic analysis revealed that the PEGylated chimera exhibited a short t1/2 (α) for initial redistribution and was reasonably long-lasting, with a t1/2 (β) of nearly 5 h and a mean residence time of more than 6 h (Figure 5H and I). Taken together, the PEGylated chimera possessed favourable pharmaceutical attributes and was capable of robust delivery of siRNA to tumours in vivo.
Figure 5
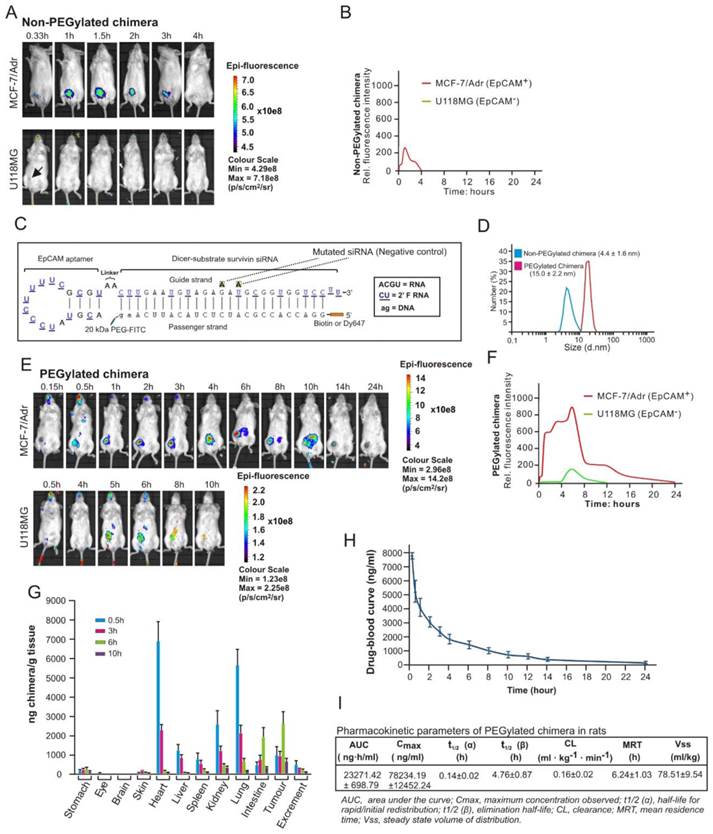
PEGylation confers chimera with desirable pharmaceutical features for in vivo RNAi. (A) A representative live animal image of non-PEGylated chimera. NOD-SCID mice bearing MCF-7/Adr or U118MG tumour implanted in the inguinal mammary fat pads or subcutaneously with a volume of approximately 60 mm3 received a single intravenous injection of 2 nmol non-PEGylated chimera labelled with Dy647 followed by serial fluorescence imaging at the indicated time points. Log-scale heat map of photon flux applies to all panels. p/s/cm2/sr: photons per second per cm2 per steradian. (B) The fluorescence-time curve of non-PEGylated chimera in tumours determined by Living Image software v2.50. (C) Schematic of PEGylated aptamer-siRNA chimera with a 20 kDa PEG-FITC conjugated to the 3'-end and a biotin or Dy647 to the 5'-end of the passenger strand of the siRNA portion. (D) Particle size of PEGylated and non-PEGylated versions of the chimera as determined by dynamic light scattering. (E) A representative live animal imaging data of tumour-bearing mice treated with PEGylated chimera labelled with Dy647 as in A. (F) The fluorescence-time curve of PEGylated chimera in tumour-bearing mice as determined in B. (G) Biodistribution of chimera in MCF-7/Adr tumour (150 mm3)-bearing mice after a single intravenous injection of 2 nmol of PEGylated chimera per mouse. (H) Drug-blood curve of PEGylated chimera in Sprague Dawley rats. A single dose of 100 nmol/kg of PEGylated chimera was injected intravenously and blood samples were collected at the indicated time points. (I) Pharmacokinetic parameters of PEGylated chimera in rats. The concentration of chimera in the blood and tissues were determined using ELISA.
Aptamer-guided RNAi efficiently silences survivin gene in vivo
After confirmation of the efficient delivery of PEGylated chimera to tumours (Figure 5), the ability of the chimera to knock down survivin gene in vivo was analyzed and found to be highly effective with a near 80% reduction in both survivin mRNA and protein levels (Figure 6A-C). The in vivo survivin silencing was EpCAM-dependent since the survivin levels in the EpCAM-negative U118MG tumours was not affected by chimera treatment. Finally, the siRNA mediated sequence specific cleavage of survivin miRNA after in vivo chimera treatment was verified by 5'-rapid amplification of cDNA ends (RACE)-PCR. As shown in Figure 6D, sequencing of the 5'RACE-PCR products demonstrated that an Argonaute2-mediated cleavage occurred between bases 10 and 11 relative to the 5'end of the guide survivin siRNA strand, suggesting that Dicer enters the chimera from the 3'-overhang of the longer strand and yields the expected 21-mer siRNA product (Figure 3D).
CSC-targeted survivin silencing results in markedly improved treatment outcome
Next, the efficacy of EpCAM-targeted survivin RNAi combined with Dox in extending overall survival of MCF-7/Adr tumour-bearing mice was assessed, since this represents a key indication of efficacy in elimination of CSCs in oncologic clinics [32]. Tumour-bearing (~30 mm3) NOD-SCID mice were randomized and treated with three cycles of alternating aptamer-siRNA chimera and Dox, with a 2-day interval between to allow the knockdown of survivin before the administration of Dox. Consistent with the in vitro results (Figure S3), the negative-control chimera alone had no effect on tumour growth or survival (Figure 7A and B). Knockdown of survivin by chimera alone elicited a moderate response. Treatment by Dox alone or combined with the negative control chimera merely retarded tumour growth initially, with the tumour-bearing mice eventually reached the designated animal ethics end points by Day 80 (Figure 7A). In contrast, in mice that received the combinatorial chimera and Dox treatment, the tumour growth rate was markedly inhibited, indicating a sustained reduction in tumour growth. Importantly, seven out of eight mice receiving the combinatorial treatment survived at Day 80, with an average tumour burden 2.7-fold lower than that reached in the group treated with Dox alone (Figure 7A and B). Therefore, targeting breast CSCs in vivo using combinatorial chimera and Dox treatment resulted in significantly extended overall survival of the tumour-bearing mice.
Figure 6
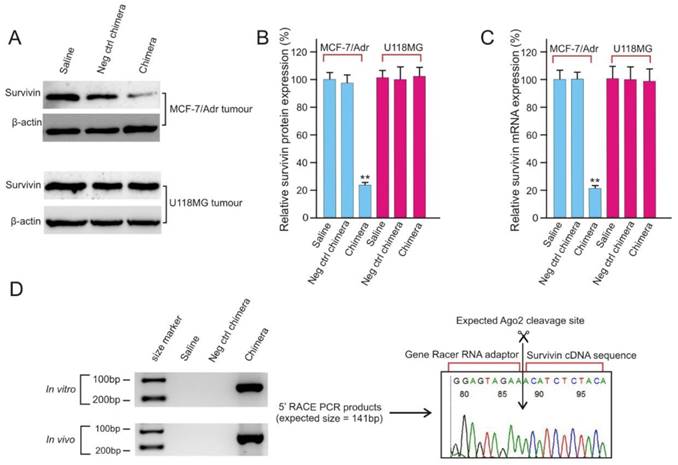
EpCAM aptamer-siRNA chimera is capable of silencing survivin in vivo. (A) Representative immunoblots for in vivo survivin silencing. NOD/SCID mice bearing orthotopic MCF-7/Adr tumour or U118MG glioma with a tumour volume of approximately 60 mm3 received a bolus intravenous injection of 2 nmol/mouse PEGylated chimera every other day over 5 days. Two days after the third injection, the tumours were removed and total protein was prepared. (B) Quantification of survivin expression in treated tumours using immunoblotting as in A. Data are means ± SEM (n=8). **, P < 0.01; compared with saline treated. (C) Quantification of survivin mRNA in treated tumours using qRT-PCR (n=8). (D) A 5'-RACE PCR assay to assess siRNA-mediated cleavage of survivin mRNA both in cell culture (top left panel) and in tumours in vivo (bottom left panel). The PCR product was cloned, sequenced and the cleavage sites were verified (right panel).
Figure 7
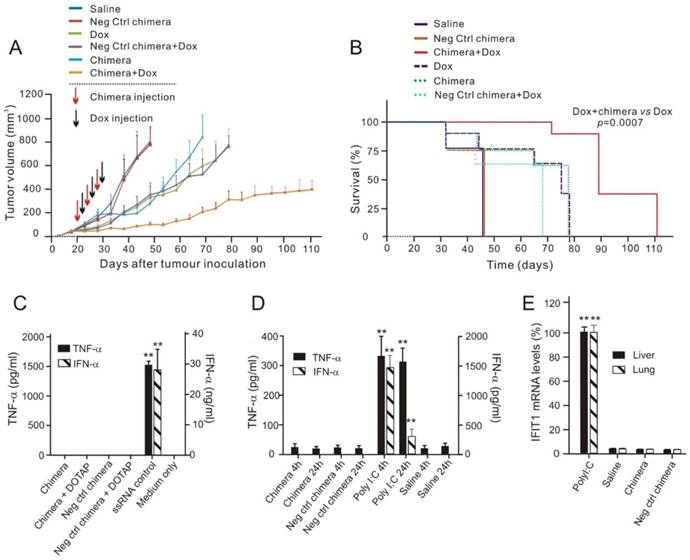
Chimera-mediated survivin silencing markedly improves tumour response to Dox without triggering interferon responses. (A) Tumour volume over time. Values represent the means ± SEM, (n=8). NOD/SCID mice bearing MCF-7/Adr orthotopic tumour with a tumour volume of ~30 mm3 were randomized into 6 groups and treated as described. (B) Kaplan-Meier survival curves of mice (n=8 per arm) bearing MCF-7/Adr tumour treated as indicated. Log rank (Mantel-Cox) p value is shown for aptamer-siRNA plus Dox treatment versus Dox alone. (C) Chimera oligonucleotides do not stimulate human peripheral blood mononuclear cells (PBMCs) in vitro. Human PBMCs were incubated overnight with 750 nmol/L chimera alone, DOTAP-complexed chimera or ssRNA control. IFN-α and TNF-α levels in the culture media was determined by ELISA. Data are representative of two independent experiments in biological triplicate. Data are representative of two independent experiments in biological triplicate. **, P < 0.001; compared with medium-treated group. (D) Chimera oligonucleotides do not cause a systemic innate immune response. Balb/c mice were treated with a single intravenous injection of 2 nmol/mouse chimera or 200 ng/mouse Poly I:C and blood was collected 4 h or 24 h post-treatment. Serum levels of IFN-α and TNF-α were determined using ELISA. Data are means ± SEM (n=6). **, P < 0.001; compared with saline-treated group. (E) Treatment of chimera in Balb/c mice for 24 h as indicated did not cause the elevation of IFIT1 mRNA in the liver and the lungs as determined by qRT-PCR (n=6). **, P < 0.001; compared with saline-treated group.
Given that activation of innate immune responses by synthetic oligonucleotides can lead to cellular toxicity and confound their in vivo therapeutic effects [33, 34], the potential immunostimulatory effects of the chimera were investigated. To study the possible activation of Toll-like receptor (TLR) 7 and 8 by synthetic RNAs, an in vitro assay using human peripheral blood mononuclear cells (PMBCs) and their ability to secrete interferon (IFN)-α and tumour necrosis factor (TNF)-α was employed [35]. High dose of chimeras (750 nmol/L), either naked or duplexed with the cationic liposome N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP) targeting endosomal TLR7/8, failed to induce significant levels of IFN-α and TNF-α in PBMCs, unlike the known TLR7/8 ligand, a single stranded RNA (Figure 7C) [36]. The in vivo innate immune response was investigated following i.v. injection of 2 nmol of the chimera in immunocompetent Balb/c mice [37]. Unlike the retinoic acid-inducible gene 1 (RIG-I) and TLR3 ligand polyinosinic:polycytidylic acid (poly I:C), which instigated a rapid and sustained induction of IFN-α and TNF-α, there was no detectable cytokine production after chimera treatment (Figure 7D).
Finally, to address the potential activation of intracellular RIG-I like pathways which may not necessarily lead to detectable circulating cytokines, the induction of the RIG-I target, interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) [38], in the liver and lung of Balb/c mice treated with 2 nmol chimera was also tested. While poly I:C led to a 100-fold increase in IFIT1 expression in both organs, there was no detectable induction following the administration of chimera (Figure 7E). Collectively, these data establish that the chimera does not activate the immune system, indicating that the suppression of tumour growth by the Dox/chimera co-treatment is not immune mediated.
Chimera-mediated RNAi reverses chemoresistance and impairs CSC self-renewal in vivo
To study how aptamer-siRNA chimera treatment enhances chemosensitivity, NOD/SCID mice bearing orthotopic MCF-7/Adr xenograft tumours were treated with three intravenous injections of the chimera or control chimera with or without 5 mg/kg Dox. The tumour weights in the mice treated with chimera combined with Dox were reduced by approximately 89%, being only one-third of those in the mice treated with Dox alone (Figure 8A). This was accompanied by a statistically significant reduction in Ki-67 index in tumours treated with combined chimera and Dox compared with those treated with Dox alone (Figure 8B and Figure S7A). Consistent with the changes in tumour weights, the combinatorial treatment of chimera and Dox resulted in approximately 10-fold more apoptotic cells than treatment with Dox alone, indicating an enhanced chemosensitivity (Figure 8C and Figure S7B).
Figure 8
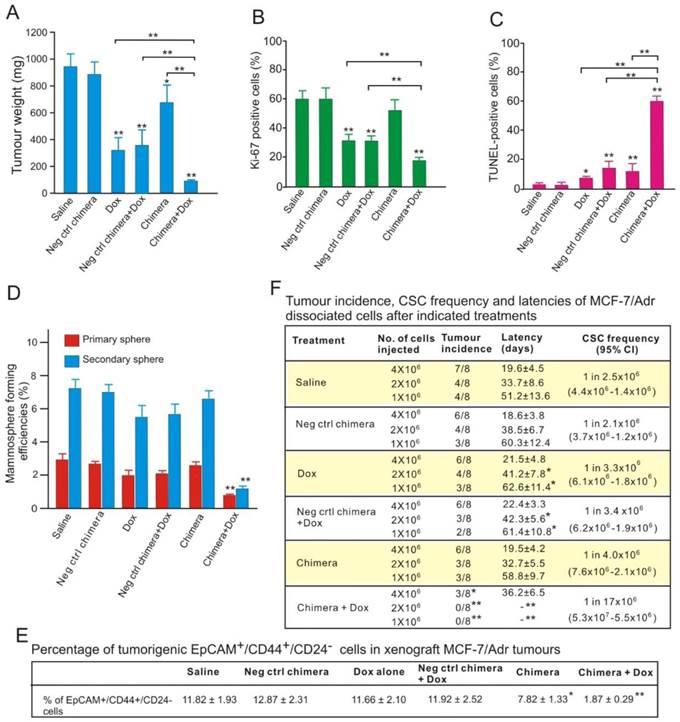
Aptamer-guided survivin RNAi sensitizes chemoresistant MCF-7-Adr to Dox. (A) Tumour weights after treatment. NOD/SCID mice bearing orthotopic MCF-7/Adr tumour with a volume of 60 mm3 were injected i.v. with 2 nmol/mouse PEGylated chimera or indicated controls on days 1, 3 and 5, with or without 5 mg/kg Dox on days 3, 5, and 7. Tumours were removed and analyzed on day 9. Data are means ± SEM (n=8). **, P < 0.001; compared with saline-treated group, unless otherwise denoted. (B) Ki-67 labelling index in treated tumours (n=8), **, P < 0.001; compared with saline-treated, unless otherwise denoted. (C) Percentage of apoptotic cells in treated tumours as determined by TUNEL assay (n=8). *, P < 0.01; **, P < 0.001; compared with saline-treated group, unless otherwise denoted. (D) Mammosphere formation efficiency of single cell suspension prepared from tumours treated as indicated (n=8), **, P < 0.001; compared with saline-treated. (E) Changes in the percentage of EpCAM+/CD44+/CD24- cells in the treated tumours (n=8). *, P < 0.01; **, P < 0.001; compared with saline-treated. (F) CSC frequency in tumours treated as determined by extreme limiting dilution assay (n=8). *, P < 0.01; **, P < 0.001; compared with saline-treated.
Interestingly, the combined chimera and Dox treatment, but not other treatments, led to a significant reduction of the proteins involved in stemness, such as Notch 1, Nanog, BIM-1 (Figure S7C). Conventional chemotherapy agents, such as Dox, are largely ineffective in eliminating CSCs [5]. The markedly increased apoptotic cell death together with the reduction in stemness proteins suggested that aptamer-guided in vivo survivin RNAi sensitized breast CSCs to Dox. To test this hypothesis, the in vitro self-renewal capacity of tumour cells prepared from treated tumours was examined. As shown in Figure 8D, the combinatorial treatment resulted in a 2.5-fold and 4.6-fold greater reduction in mammosphere formation efficacy in the primary and secondary sphere assays, respectively, compared with those treated with Dox alone. In accordance with the diminished self-renewal capacity, three doses of combinatorial chimera and Dox treatment of the tumour-bearing mice effectively eliminated approximately 84% of the breast CSCs (EpCAM+/CD44+/CD24- cells) in the treated xenograft tumours (Figure 8E).
To enumerate the frequency of CSCs directly, single cell suspensions prepared from treated MCF-7/Adr xenograft tumours were implanted into the secondary recipients of NOD/SCID mice at limiting cell doses. As shown in Figure 8F, different with those observed in other treatment groups, over a 6-month period, no tumour formation was observed in the 1 × 106 and 2 × 106 cell dose groups prepared from tumour-bearing mice treated with combined chimera and Dox. In the 4 × 106 cell dose group, tumour formed with a 5-fold lower frequency and a much longer latency in mice receiving combinatorial treatment than those treated with Dox alone. Thus, in the presence of Dox, survivin knockdown significantly decreased the frequency of CSCs in MCF-7/Adr xenograft tumours. Taken together, EpCAM aptamer-guided in vivo survivin gene silencing markedly sensitized the breast CSCs to Dox, lowered the frequency and impaired the self-renewal capacity of CSCs and thereby prohibited tumour initiation.
Aptamer-guided RNAi directly targets breast CSCs in vivo
To further confirm the direct targeting of CSC by aptamer-guided RNAi, the physical evidence of EpCAM aptamer-guided survivin siRNA to enter CSCs and the subsequent functional consequence of reversing chemoresistance of CSCs were studied. To this end, populations enriched for breast CSCs (EpCAM+/CD44+/CD24-) and non-CSCs (EpCAM-/CD44+/CD24- and EpCAM-/CD44-/CD24-) were sorted from cells treated in vitro as well as those dissociated from treated xenograft MCF-7/Adr tumours. Confocal microscopy confirmed that Dy647-labelled chimera was indeed internalized into breast CSCs in both cell culture and in vivo treated xenograft tumours (Figures 9A-C). Furthermore, this observed CSC targeting was EpCAM-dependent as no Dy647-labelled chimera was found in non-CSCs (Figure 9A and B). As shown in Figure 9B, the chimera was found in all in vitro cultured breast CSCs. Importantly, approximately 89% of breast CSCs dissociated from treated MCF-7/Adr tumours was found to uptake the chimera (Figure 9B), indicating excellent tumour penetration of the chimera in vivo. Consistent with the demonstrated cellular uptake, the chimera elicited robust knockdown of the survivin gene in breast CSCs but not in non-CSCs both in vivo and in vitro (Figure 9D).
Importantly, the silencing of survivin in breast CSCs led to a marked increase in the sensitivity of these cells to Dox-induced apoptosis in this Dox-resistant tumour model (Figure 9E). Consistent with the known inability of Dox to eliminate CSCs, treatment of mice bearing MCF-7/Adr tumours with Dox alone induced a very low level of apoptosis in breast CSCs but significantly more cell death in non-CSCs in the same tumour (Figure 9E). In contrast, when combined with the chimera, the same dose of Dox induced apoptosis in approximately 75% of the breast CSCs (Figure 9E). Furthermore, the combinatorial treatment with chimera and Dox resulted in a diminished mammosphere forming capacity of treated breast CSCs both in vitro and in vivo but not with that in non-CSC groups (Figure 9F). Taken together, these data demonstrate that aptamer-guided survivin silencing suppressed tumourigenicity and reversed the acquired chemoresistance in MCF-7/ADR xenograft tumours via direct targeting of breast CSCs in vivo.
Discussion
Previous work has shown that survivin is overexpressed in majority of breast cancers and such elevated expression is inversely correlated with the sensitivity of the breast cancer to chemotherapy and predicts adverse clinical outcome [39-42]. However, the consequence of survivin overexpression in CSCs remains to be explored.
Figure 9
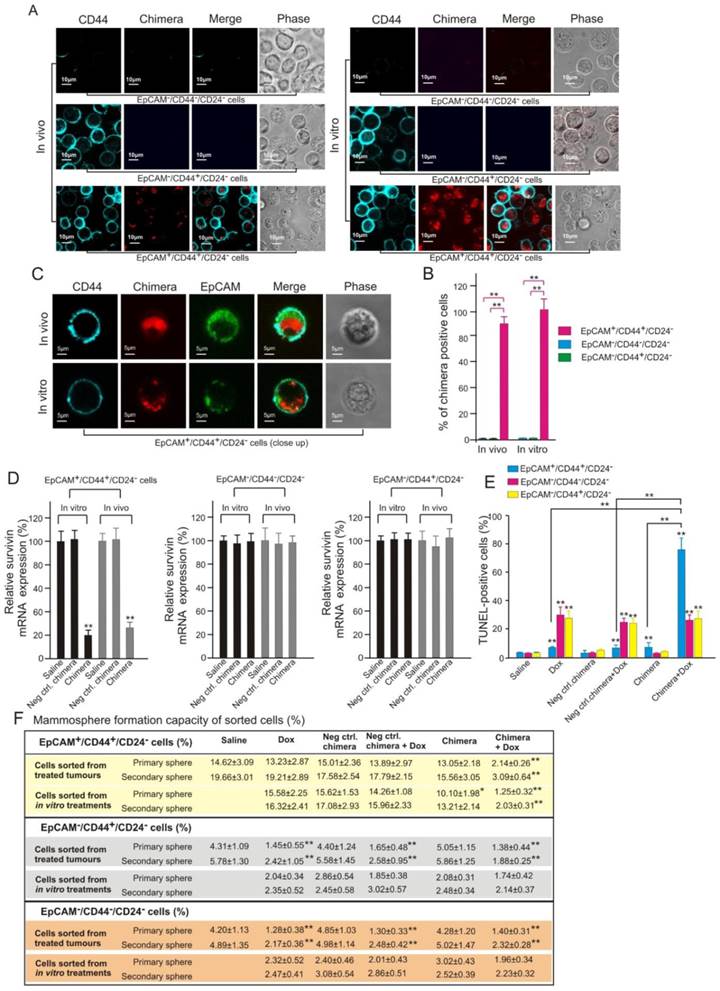
Effective targeting of CSC by aptamer-guided in vivo RNAi. (A) Confocal micrographs (low magnification) showing the presence or absence of chimera in FACS-sorted cells in those treated in vivo and in vitro, respectively. NOD/SCID mice bearing orthotopic MCF-7/Adr tumour were treated as described in Figure 8A. Tumours were removed on Day 9 and purified populations of EpCAM+/CD44+/CD24-, EpCAM-/CD44+/CD24- and EpCAM-/CD44-/CD24- cells were sorted by FACS. MCF-7/Adr cells treated with 20 nmol/L chimera and 300 nmol/L Dox for 3 days in vitro were similarly sorted. (B) Quantification of percentage of chimera-positive cells as in A with 100 cells counted in triplicate in each samples. Data are means ± SEM (n=6). **, P < 0.001; compared with saline-treated group. (C) Representative enlarged micrographs from A depicting internalization of chimera into EpCAM+/CD44+/CD24- cells. (D) Levels of survivin mRNA (via qRT-PCR) prepared from FACS-sorted tumour cells (in vivo) or cultured MCF-7/Adr cells (in vitro) treated as indicated, (n=6). **, P < 0.001; compared with saline-treated group. (E) Percentage of apoptotic cells in FACS-sorted cells dissociated from MCF-7/Adr tumours after indicated treatments, (n=6). **, P < 0.001; compared with saline-treated group, unless otherwise denoted. (F) Mammosphere forming efficiency of FACS-sorted cells dissociated from MCF-7/Adr tumours or cultured cells after indicated treatments (n=6). **, P < 0.001; compared with saline treatment (for in vivo treatment) or Dox-alone treatment (for in vitro treatment).
Depending on the experimental systems used and approaches adopted, suppression of the survivin function alone could lead to decreased cell viability, increased apoptosis and inhibition of tumour growth [43, 44]; or no alteration of cell viability or stemness (our results and those from Hong et al [45]). Certainly, tumour heterogeneity plays a large role in this tumour- and/or cell type-specific discrepancy. Indeed, Guvenc and colleagues have elegantly demonstrated a significant difference in cell viability after disruption of the survivin-LLP-3 complex between the same types of glioma cells with different p53 status [44]. In their study, however, the high concentration (13.6-38.1 µM) of the pharmacological inhibitor used and extended incubation period (72 h) adds another layer of complexity to the specificity in targeting of survivin [44].
Nonetheless, the consensus emerged from our data and those of others [43, 44] suggests that targeting survivin in CSCs will improve treatment outcome. In this study, we observed overexpression of survivin in the more aggressive ER-/PR-/HER2- breast cancers biopsies while not in three of the four PR+ or HER2+ samples (Table S1). This is consistent with a recent report in which over expression of survivin was detected in 45.6% of the 136 triple negative clinical breast cancer samples and correlated with metastasis and poor prognosis [46]. More importantly, we confirmed that the critical role of survivin in chemoresistance of CSCs in human breast cancer, consistent with recent findings that by disrupting the interaction between survivin and Ran, one can sensitize glioma stem cells to temozolomide [44]. Importantly, compared with that in the non-CSCs, the level of survivin were elevated 23-fold in the CSC population of Dox-resistant MCF-7/Adr cells (Figure 2F). This is in sharp contrast to some other potential therapeutic targets of CSCs (e.g. BMI-1) which are expressed equally in CSCs and non-CSCs in at least certain cancers [47].
Interestingly, the expression of MDR1, one of the putative genes promoting chemoresistance in breast cancer [48, 49], was also elevated in MCF-7/Adr cells. However, silencing MDR1 had no effect on the self-renewal capacity or the abundance of the CSCs (Figure S8), suggesting MDR1 is not critically involved in the survival and maintenance of CSCs in MCF-7/Adr cells. Given the finding that survivin overexpression is restricted to the CSC population and plays a critical role in chemoresistance and survival of MCF-7/Adr cells, targeting survivin in CSCs constitutes a promising therapeutic strategy for eradicating CSCs and improving the therapeutic outcome of chemotherapeutic drugs in this model.
To effectively down-regulate survivin in breast CSCs, a fully synthetic aptamer-siRNA chimera, relying on our 18-nt EpCAM aptamer [28] was developed. With the longer strand consisting of only 47 nucleotides (nt), this chimera, to our knowledge, is the shortest effective aptamer-siRNA chimera ever developed. A chimera of such length is amenable to large-scale production via chemical synthesis, unlike that of previously reported chimeras that were produced via in vitro transcription using T7 RNA polymerase [14, 15].
Given the short circulation time observed with the original chimera, a 20 kDa PEG was introduced and the resultant PEG-conjugated chimera displayed prolonged circulation half-life and enhanced accumulation in the EpCAM-positive MCF-7/Adr tumours, indicating the engagement of the enhanced permeability and retention (EPR) effect. However, it only displayed a late onset, low concentration and short duration of accumulation in the EpCAM-negative U118MG tumour, suggesting that the EPR effect alone is necessary but not sufficient for delivering the chimera to tumours (Figure 5E and F). Only the combined effect of the EPR effect and active targeting via the EpCAM aptamer ensured a swift, high level and durable delivery of aptamer-siRNA to the EpCAM-positive tumour. In addition, time-dependent biodistribution studies revealed that, after a single injection, the PEGylated chimera reached a concentration of approximately 800-2800 ng/g tumour tissue between 0.5 h to 10 h post-injection (assuming the specific weight of tumour is 1.0), which is equivalent to 22-65 nM (Figure 5G). Such a concentration and duration of siRNA in the tumour are known to be sufficient to silence most target genes [50, 51].
As suggested, one key limiting factor of cancer treatment is the inability of many therapeutic agents to penetrate the core of a tumour mass [52]. Our studies indicate that the PEGylated chimera is able to penetrate the core of the MCF-7/Adr tumour, since the chimera was detected in ~90% of CSCs dissociated from treated xenograft tumours (Figure 9B). Furthermore, intracellular chimera could be detected in most of the CSCs (Figure 9A and C), essential for its engagement with the cellular RNAi machinery. This study therefore provides proof of principle that the challenge of CSC targeted siRNA in vivo delivery can be addressed by targeting a cell surface marker using aptamer-siRNA chimeras.
As reported, apart from overexpressed on CSCs, EpCAM also presents on bulk cancer cells [25, 53]. Therefore, this EpCAM aptamer mediated siRNA delivery system targets both CSCs and bulk cancer cells. In this paradigm, through targeted silencing of survivin in EpCAM-positive tumour cells, Dox was then capable of killing CSCs in addition to bulk tumour cells. Since CSCs and non-CSCs may interconvert [54, 55] and survivin has been implicated in the regulation of such interconvertion [56], the bimodal treatment strategy presented here to target CSCs and their progenies simultaneously appears advantageous, and indeed imperative.
One key implication of this work is that by targeting a CSC related cell surface marker using an aptamer, one can efficaciously deliver siRNA via receptor-mediated endocytosis into CSCs in vivo, thus opening a new front for translating RNAi to the oncologic clinic by silencing any dys-regulated gene expressed in CSCs. This work also sheds light on strategies to overcome chemoresistance, which despite intensive efforts remaining a key limitation clinically [57]. The CSC population represents a major cellular source of chemoresistance within a tumour. However, the targeting of CSCs is hampered by pathway redundancy and potential toxicities [58, 59]. Our study provides a new approach to target CSCs in vivo. By combining targeted delivery of siRNA into CSCs and treatment of Dox, a classical chemotherapy drug known to be ineffective in killing CSCs, can be transformed to an agent capable of eradicating CSCs, thereby reversing chemoresistance and inhibiting both self-renewal and tumourigenic potential (Figures 7 and Figure 8). These findings suggest a potential avenue for not only reversing chemoresistance but also achieving a lasting tumour remission.
In conclusion, we have demonstrated, for the first time, the feasibility and efficacy of effective delivery of siRNA to target CSCs in vivo. When combined with Dox treatment, CSC-targeted survivin silencing led to the reversal of chemoresistance, elimination of CSCs and prolonged survival. The aptamer-guided CSC gene silencing combined with rationally selected chemotherapeutic agent(s) may open up new and practical avenues to combat chemoresistance and treatment relapse.
Supplementary Material
Supplementary methods, Supplementary results, Supplementary tables and figures.
Acknowledgements
The authors thank Hamulati Hasimu for technical assistance with animal experimentations. T. Wang was supported by a Deakin University Postgraduate Research Scholarship. This work was supported by grants from Indo-Australia Science and Technology Fund. (Grant No. ST040007), Victorian Cancer Agency Platform Technology Capacity Building Grant (Grant No. PTCP-02) and CASS Foundation (Australia).
Study approval
All animal protocols used in this research have been approved by the Deakin University Animal Welfare Committee. Human breast cancer tissues were obtained from consenting patients through Barwon Health, Victoria, Australia, with relevant institutional review board approval.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714-26
2. Yu M, Ocana A, Tannock IF. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: why has it failed to provide clinical benefit? Cancer Metastasis Rev. 2013;32(1-2):211-27
3. Gerlinger M, Rowan AJ, Horswell S. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883-92
4. Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368(9):842-51
5. Maugeri-Sacca M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17(15):4942-7
6. Altieri DC. Targeting survivin in cancer. Cancer Lett. 2013;332(2):225-8
7. Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14(16):5000-5
8. Carter BZ, Qiu YH, Huang XL. et al. Survivin is highly expressed in CD34(+)38(-) leukemic stem/progenitor cells and predicts poor clinical outcomes in AML. Blood. 2012;120(1):173-80
9. Lu X, Deng Q, Li H, Suo Z. Altered characteristics of cancer stem/initiating cells in a breast cancer cell line treated with persistent 5-FU chemotherapy. Exp Ther Med. 2011;2(5):821-6
10. Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CHA. Treat cancers by targeting survivin: Just a dream or future reality? Cancer Treat Rev. 2013;39(7):802-11
11. Zhou J, Bobbin ML, Burnett JC, Rossi JJ. Current progress of RNA aptamer-based therapeutics. Front Genet. 2012;3:234
12. Xiang D, Shigdar S, Qiao G. et al. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine. Theranostics. 2015;5(1):23-42
13. Cho EJ, Lee JW, Ellington AD. Applications of Aptamers as Sensors. Annu Rev Anal Chem. 2009;2:241-64
14. Dassie JP, Liu XY, Thomas GS. et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27(9):839-U95
15. McNamara JO 2nd, Andrechek ER, Wang Y. et al. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24(8):1005-15
16. Shum KT, Zhou J, Rossi JJ. Aptamer-based therapeutics: new approaches to combat human viral diseases. Pharmaceuticals (Basel). 2013;6(12):1507-42
17. Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev. 2012;38(1):68-75
18. Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs. 2010;2(2):129-36
19. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313-9
20. Li C, Heidt DG, Dalerba P. et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030-7
21. Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8(10):806-23
22. Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs. 2009;1(1):12-25
23. McDermott M, Eustace AJ, Busschots S. et al. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front Oncol. 2014;4:40
24. Creighton CJ, Li X, Landis M. et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820-5
25. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983-8
26. Carvalho A, Carmena M, Sambade C, Earnshaw WC, Wheatley SP. Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J Cell Sci. 2003;116(14):2987-98
27. Zhou J, Rossi JJ. Aptamer-targeted cell-specific RNA interference. Silence. 2010;1(1):4
28. Shigdar S, Qian C, Lv L. et al. The Use of Sensitive Chemical Antibodies for Diagnosis: Detection of Low Levels of Epcam in Breast Cancer. PLoS One. 2013:8 (2)
29. Shigdar S, Lin J, Yu Y, Pastuovic M, Wei M, Duan W. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102(5):991-8
30. Vercauteren D, Vandenbroucke RE, Jones AT. et al. The Use of Inhibitors to Study Endocytic Pathways of Gene Carriers: Optimization and Pitfalls. Mol Ther. 2010;18(3):561-9
31. Choi HS, Liu W, Misra P. et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165-70
32. Huff CA, Matsui W, Smith BD, Jones RJ. The paradox of response and survival in cancer therapeutics. Blood. 2006;107(2):431-4
33. Kleinman ME, Yamada K, Takeda A. et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452(7187):591-U1
34. Robbins M, Judge A, Ambegia E. et al. Misinterpreting the Therapeutic Effects of Small Interfering RNA Caused by Immune Stimulation. Hum Gene Ther. 2008;19(10):991-9
35. Gantier MP, Williams BRG. Monitoring Innate Immune Recruitment by siRNAs in Mammalian Cells. Rna Interference: From Biology to Clinical Applications. Methods Mol Biol. 2010;623:21-33
36. Gantier MP, Tong S, Behlke MA. et al. Rational Design of Immunostimulatory siRNAs. Mol Ther. 2010;18(4):785-95
37. Gantier MP, Williams BRG. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 2007;18(5-6):363-71
38. Marques JT, Devosse T, Wang D. et al. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol. 2006;24(5):559-65
39. Jha K, Shukla M, Pandey M. Survivin expression and targeting in breast cancer. Surg Oncol. 2012;21(2):125-31
40. Ryan BM, Konecny GE, Kahlert S. et al. Survivin expression in breast cancer predicts clinical outcome and is associated with HER2, VEGF, urokinase plasminogen activator and PAI-1. Ann Oncol. 2006;17(4):597-604
41. Zhao YC, Wang Y, Ni XJ. et al. Clinical significance of Smac and survivin expression in breast cancer patients treated with anthracycline-based neoadjuvant chemotherapy. Mol Med Rep. 2014;9(2):614-20
42. Petrarca CR, Brunetto AT, Duval V, Brondani A, Carvalho GP, Garicochea B. Survivin As a Predictive Biomarker of Complete Pathologic Response to Neoadjuvant Chemotherapy in Patients With Stage II and Stage III Breast Cancer. Clin Breast Cancer. 2011;11(2):129-34
43. Aliabadi HM, Landry B, Mandipoor P, Uludag H. Induction of Apoptosis by Survivin Silencing through siRNA Delivery in a Human Breast Cancer Cell Line. Mol Pharmaceut. 2011;8(5):1821-30
44. Guvenc H, Pavlyukov MS, Joshi K. et al. Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res. 2013;19(3):631-42
45. Hong SE, Kim EK, Jin HO. et al. S6K1 inhibition enhances tamoxifen-induced cell death in MCF-7 cells through translational inhibition of Mcl-1 and survivin. Cell Biol Toxicol. 2013;29(4):273-82
46. Zhang M. et al. Prognostic value of survivin and EGFR protein expression in triple-negative breast cancer (TNBC) patients. Target Oncol. 2014;9(4):349-57
47. Kreso A, van Galen P, Pedley NM. et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20(1):29-36
48. Perez J, Bardin C, Rigal C, Anthony B, Rousseau R, Dutour A. Anti-MDR1 siRNA restores chemosensitivity in chemoresistant breast carcinoma and osteosarcoma cell lines. Anticancer Res. 2011;31(9):2813-20
49. Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human "MDR1" gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci U S A. 1987;84(9):3004-8
50. Kim SH, Jeong JH, Lee SH, Kim SW, Park TG. Local and systemic delivery of VEGF siRNA using polyelectrolyte complex micelles for effective treatment of cancer. J Control Release. 2008;129(2):107-16
51. Lee H, Lytton-Jean AK, Chen Y. et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012;7(6):389-93
52. Saggar JK, Yu M, Tan Q, Tannock IF. The tumor microenvironment and strategies to improve drug distribution. Front Oncol. 2013;3:154
53. Martowicz A, Rainer J, Lelong J, Spizzo G, Gastl G, Untergasser G. EpCAM overexpression prolongs proliferative capacity of primary human breast epithelial cells and supports hyperplastic growth. Mol Cancer. 2013;12:56
54. Chaffer CL, Brueckmann I, Scheel C. et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108(19):7950-5
55. Iliopoulos D, Hirsch HA, Wang GN, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108(4):1397-402
56. Dahan P, Gala JM, Delmas C. et al. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: possible involvement in radioresistance. Cell Death Dis. 2014:5
57. Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8(5):411-24
58. Ntziachristos P, Lim JS, Sage J, Aifantis I. From Fly Wings to Targeted Cancer Therapies: A Centennial for Notch Signaling. Cancer Cell. 2014;25(3):318-34
59. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497-512
Author contact
![]() Corresponding authors: Professor Wei Duan, School of Medicine, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria, 3217, Australia. Tel: +61 3 52271149; Fax: +61 3 52272945; E-mail: wduanedu.au (W. Duan) and sarah.shigdaredu.au (S. Shigdar)
Corresponding authors: Professor Wei Duan, School of Medicine, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria, 3217, Australia. Tel: +61 3 52271149; Fax: +61 3 52272945; E-mail: wduanedu.au (W. Duan) and sarah.shigdaredu.au (S. Shigdar)
Citation styles
APA
Wang, T., Gantier, M.P., Xiang, D., Bean, A.G., Bruce, M., Zhou, S.F., Khasraw, M., Ward, A., Wang, L., Wei, M.Q., AlShamaileh, H., Chen, L., She, X., Lin, J., Kong, L., Shigdar, S., Duan, W. (2015). EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics, 5(12), 1456-1472. https://doi.org/10.7150/thno.11692.
ACS
Wang, T.; Gantier, M.P.; Xiang, D.; Bean, A.G.; Bruce, M.; Zhou, S.F.; Khasraw, M.; Ward, A.; Wang, L.; Wei, M.Q.; AlShamaileh, H.; Chen, L.; She, X.; Lin, J.; Kong, L.; Shigdar, S.; Duan, W. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics 2015, 5 (12), 1456-1472. DOI: 10.7150/thno.11692.
NLM
Wang T, Gantier MP, Xiang D, Bean AG, Bruce M, Zhou SF, Khasraw M, Ward A, Wang L, Wei MQ, AlShamaileh H, Chen L, She X, Lin J, Kong L, Shigdar S, Duan W. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics 2015; 5(12):1456-1472. doi:10.7150/thno.11692. https://www.thno.org/v05p1456.htm
CSE
Wang T, Gantier MP, Xiang D, Bean AG, Bruce M, Zhou SF, Khasraw M, Ward A, Wang L, Wei MQ, AlShamaileh H, Chen L, She X, Lin J, Kong L, Shigdar S, Duan W. 2015. EpCAM Aptamer-mediated Survivin Silencing Sensitized Cancer Stem Cells to Doxorubicin in a Breast Cancer Model. Theranostics. 5(12):1456-1472.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.