Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(4):594-609. doi:10.7150/thno.14306 This issue Cite
Research Paper
Novel Bioluminescent Activatable Reporter for Src Tyrosine Kinase Activity in Living Mice
Weibing Leng1,2, Dezhi Li1,2, Liang Chen1, Hongwei Xia2, Qiulin Tang2, Baoqin Chen1, Qiyong Gong3, Fabao Gao3, Feng Bi1,2 ![]()
1. Department of Medical Oncology, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China;
2. Laboratory of Signal Transduction & Molecular Targeted Therapy, State Key Laboratory of Biotherapy, Sichuan University, Chengdu 610041, Sichuan, China;
3. Department of Radiology, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China.
Received 2015-11-3; Accepted 2016-1-27; Published 2016-2-24
Abstract

Aberrant activation of the Src kinase is implicated in the development of a variety of human malignancies. However, it is almost impossible to monitor Src activity in an in vivo setting with current biochemical techniques. To facilitate the noninvasive investigation of the activity of Src kinase both in vitro and in vivo, we developed a genetically engineered, activatable bioluminescent reporter using split-luciferase complementation. The bioluminescence of this reporter can be used as a surrogate for Src activity in real time. This hybrid luciferase reporter was constructed by sandwiching a Src-dependent conformationally responsive unit (SH2 domain-Srcpep) between the split luciferase fragments. The complementation bioluminescence of this reporter was dependent on the Src activity status. In our study, Src kinase activity in cultured cells and tumor xenografts was monitored quantitatively and dynamically in response to clinical small-molecular kinase inhibitors, dasatinib and saracatinib. This system was also applied for high-throughput screening of Src inhibitors against a kinase inhibitor library in living cells. These results provide unique insights into drug development and pharmacokinetics/phoarmocodynamics of therapeutic drugs targeting Src signaling pathway enabling the optimization of drug administration schedules for maximum benefit. Using both Firefly and Renilla luciferase imaging, we have successfully monitored Src tyrosine kinase activity and Akt serine/threonine kinase activity concurrently in one tumor xenograft. This dual luciferase reporter imaging system will be helpful in exploring the complex signaling networks in vivo. The strategies reported here can also be extended to study and image other important kinases and the cross-talks among them.
Keywords: Src tyrosine kinase, noninvasive molecular imaging, bioluminescence imaging (BLI), kinase activity, drug development.
Introduction
Src plays a crucial role in the crosstalk and mediation of many signaling pathways, including cell cycle progression, apoptosis, angiogenesis, adhesion and migration [1-3]. It is not surprising that aberrant activation of Src kinase contributes to diverse aspects of carcinogenesis. Deregulation and increased activity of Src has been observed in a variety of human cancers and usually correlates with poor clinical prognosis [4-7]. Growing evidence shows that inhibition of Src kinase activity has preclinical anti-tumor effects resulting in the development of many Src activity inhibitors [8,9]. However, Src inhibitors have shown disappointing therapeutic efficacy in clinical trials [9-11]. It remains challenging to realize the potential of basic research into preclinical or clinical application [12].
To overcome these challenges in the case of Src tyrosine kinase, we must improve early drug discovery strategies by combining our knowledge of the basic biology of the enzyme with the use of appropriate preclinical modeling. This approach would better mimic and thereby predict clinical response in the target tissue environment, rather than in reductionist systems. Molecular imaging, especially optical imaging, provides a new platform for noninvasive visualization of biological processes at the molecular level in vivo. Optical imaging, including fluorescence imaging and bioluminescence imaging (BLI) [13], is helping to bridge the gap between our understanding of critical biologic events and their clinical applications. Fluorescence resonance energy transfer (FRET) analysis has been widely used in characterizing the spatiotemporal dynamics regulation of Src activity in a single living cell [14-17]. Using fluorescence lifetime imaging microscopy (FLIM), the FRET-Src biosensor has also been used as a preclinical tool to assess drug delivery and efficacy in tumors [18]. However, the FRET assay suffers from several weaknesses, including the need for an external excitation source, insufficient detection sensitivity and dynamic range, the challenge for stable expression, and autofluorescence [19]. Furthermore, FRET assay requires sophisticated and expensive instrumentation and comprehensive post-imaging data analysis [20,21]. These demerits potentially limit its usefulness for in vivo application and for high-throughput screening (HTS) in drug development. As an alternative to fluorescence assay, bioluminescence assay is capable of ironing out these flaws and provides complementary advantages for preclinical applications in vivo.
Bioluminescence imaging has emerged as a sensitive technology to advance our understanding of disease mechanisms at the molecular level and accelerate drug discovery and development [22,23]. In particular, bioluminescence can circumvent cell and tissue autoluminescence resulting in a better signal to noise ratio [24]. This provides a complementary and alternative approach to traditional biochemical assays and the FRET assay, for preclinical evaluation of anticancer therapeutics in experimental cancer models. We reasoned that developing a method to noninvasively monitor and image Src activity in animals would be a valuable tool to further understand the biology of the Src kinase signaling pathway. To this end, we developed and characterized a novel bioluminescent activatable reporter based on the split-luciferase fragment complementation assay. This reporter system also allows monitoring of Src-targeted drug efficacy in intact cells and mice. The new imaging reporter would provide a better understanding of the role of the functional Src kinase in cancer biology and help accelerate the discovery and development of new anti-Src drugs.
Materials and Methods
Ethics statement
All experimental procedures with animals used in this study had been given prior approval by the Experimental Animal Manage Committee of Sichuan University under Contract 2015012A. Animal handling and all procedures on animals were carried out strictly according to the guidelines of the Animal Care and Use Committee of Sichuan University and the Animal Ethics Committee Guidelines of the Animal Facility of the West China Hospital. The nude mice were maintained under specific pathogen-free (SPF) conditions. Mice were gas anesthetized with isofluorane (2% isoflurane in 100% oxygen, 1 L/min) using the XGI-8 Gas Anesthesia Unit (Caliper Life Sciences) during all injection and imaging procedures.
Construction of plasmids
The N and C fragments of luciferase (Firefly or Renilla) were amplified by PCR from pGL3-Basic or pRL-tk (Promega), and the Myc-tag sequence was introduced by primer design. The fragment 370bp fragment, including nucleotide sequences of SH2 domain derived from Shc (aa 374-465), linker, and Src consensus substrate peptide, was synthesized by Genewiz Company (Su-zhou, China). This cassette and the luciferase fragments were seamlessly cloned into a HIV-1-based lentiviral expression vector, which was derived from pLVX-puro (Clontech). Gaussia luciferase, which was amplified from pGLuc-Basic (NEB), was cloned into the 5′ end of the puromycin resistance gene with a “self-cleaving” T2A sequence. For the Src reporter based on Renilla luciferase (rBSR), Firefly luciferase fragments were replaced by Renilla luciferase fragments, and the neomycin resistance gene was used for selection of stable transfectants. For the Akt reporter, the hybrid Firefly luciferase was cloned into pLVX-puro. Site-directed mutagenesis was performed using the QuickChangeTM site-directed mutagenesis kit (Stratagene). The GenBank accession numbers for Src reporters, Akt reporters, and Gluc-T2A-puro sequence are KT986061-KT986067.
Cell culture
HEK293T, Hela, HT29, MDA-MB-435S, Lovo, Colo320, SW480, SW48, SW1116, HCT116 and Caco-2 cells were purchased from ATCC. All cell lines were cultured in Dulbecco modified Eagle medium (DMEM, Gibco Laboratories, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco). Cell cultures were maintained in a 37°C incubator with 5% CO2.
Lentivirus production
The lentiviral plasmids of the reporters psPAX2 and pMD2.G were co-transfected into HEK293T cells in a 10cm dish using Lipofectamine 2000 (Invitrogen). Twelve hours after transfection, the medium was changed to 2% FBS-DMEM. Two days after transfection, the conditioned medium was collected, filtered through 0.4 μ filter, and used for infection.
Western blotting and coimmunoprecipitation
For Western blots, cells expressing the reporter were cultured in 6-well plates and were treated with stimulant, inhibitor or vehicle for the indicated times. Total protein lysate was prepared using lysis buffer containing protease inhibitors and phosphatase inhibitors. Protein was quantified using the BCA protein assay (Pierce Chemical Co.). Western blotting was performed as described previously [25]. Proteins were visualized using florescent-labeled secondary Abs and quantified by Odyssey infrared imaging system. The antibodies used in our study were as follows: EGFR, p-EGFR(Tyr1173), Src, p-Src(Tyr416), Akt, p-Akt(Ser473), Erk, p-Erk(Thr202/Tyr204), p-FAK(Tyr397) and p130Cas (Tyr410) antibodies were obtained from Cell Signaling Technology. β-actin and FAK antibodies were purchased from Santa Cruz Biotechnology Inc.
For coimmunoprecipitation, cells stably transfected with reporter were cultured on 60-mm culture dishes. After treatment, the cells were harvested in cell lysis buffer. One part of the whole-cell lysates was used for input. The proteins in the remaining lysates were coimmunoprecipitated with mouse anti-Myc antibody (clone 4A6; Millipore). The immune complexes were captured using protein G-coupled magnetic beads (Millipore) and then fractionated by SDS-PAGE. Phosphorylation of the Src reporter was detected with the anti-phospho-tyrosine antibody (Upstate).
Cells-based in vitro assay
Cells expressing the reporter were cultured in 24-, 48-, or 96-well plates and treated with stimulants, inhibitors or the vehicle. The stimulants used in our study were EGF (peprotech), PP1 (Cayman), dasatinib, and saracatinib (Selleck). All the bioluminescence was obtained in living cells. For the internal control bioluminescence, Gaussia luciferase activity was measured by adding coelenterazine (Regis, 1.5uM in D-PBS, 100ul/well) with the parameters: 1-min exposure; emission filter, 500nm; f-stop, 1; binning, 8; field of view, 15 cm. For the complemented Firefly activity, after administration of D-luciferin (Xenogen, 50ug/ml in Cell Culture Medium, 100ul/well), luminescence intensity (photons/second/square centimeter/steridian or p/s/cm2/sr) was measured by the charge-coupled device (CCD) camera of IVIS spectrum (Caliper Life Sciences, Hopkinton, MA) using the following parameters: 1-min exposure; emission filter, 600nm; f-stop, 1; binning, 8; field of view, 15 cm. The measure of Gluc activity was preferential to avoid mutual interference, because Gluc emission signal intensity is almost negligible at 600nm after minutes due to the rapid kinetics of coelenterazine. The value of each well is expressed in the normalized activity, which is calculated as the ratio of Firefly luciferase (Fluc) activity at 600nm to Gaussia luciferase (Gluc) activity at 500nm (Fluc/Gluc).
In vivo mouse imaging experiments
To establish xenograft tumors, cells (1x106 cells/sample) stably transfected with the wild type or mutant reporter(s) were implanted subcutaneously in the bottom left or right flanks of 4-week old female nude mouse. BLI was performed pretreatment and after treatment with vehicle or inhibitors for indicated times when the xenografts reached a volume of 40 mm3. Mice were gas anesthetized with isofluorane (2% isoflurane in 100% oxygen, 1 L/min) using the XGI-8 Gas Anesthesia Unit (Caliper Life Sciences) during all injection and imaging procedures. The Renilla and Gaussia luciferases react with the same substrate, coelenterazine, to produce blue light with peak emission at approximately 480nm. The bioluminescence of luciferin-dependent and coelenterazine-dependent luciferases was obtained from the same mouse with different emission filters. For Firefly luciferase luminescence, the mice were imaged after i.p. injection of D-luciferin (150 mg/kg BW) using the following parameters: 2-min exposure; emission filter, 600nm; f-stop, 1; binning, 8; field of view, 15 cm. For Gaussia or Renilla luciferase activity, luminescence was measured after i.p. injection of coelenterazine (1mg/kg BW) with the parameters: 3-min exposure; emission filter, 500nm; f-stop, 1; binning, 8; field of view, 15 cm. The measure of Gluc or Rluc activity was preferential to avoid mutual interference, because Gluc or Rluc emission signal intensity is almost negligible at 600nm after minutes due to the rapid kinetics of coelenterazine. Although Firefly luciferase has glow kinetics, the bioluminescence intensity maintains only about 1 hour due to luciferin consumption. Little luminescence signal from last substrate injection was detected at emission filters 500nm and 600nm after 2 hours. New substrates needed for the bioluminescence at each time point.
High-throughput screening against a kinase inhibitor library with BSR
A compound library containing 84 small molecular weight kinase inhibitors from National Compound Resource Center in China was used to validate the efficacy of BSR for HTS. HT29-BSRwt treated with vehicle (DMSO) served as a normalized control. HT29-BSRwt cells cultured in 96-well plates were treated with 30uM of each compound for 60min in triplicates, according to the official recommendation for the preliminary screening concentration. BLI was performed as mentioned in vitro assay, and HT29-BSRmut as a positive control. The result of each compound sample was the average of normalized bioluminescence activity (Fluc/Gluc) in the triplicates.
Data analysis
For in vitro analyses, the value of each well is presented as normalized bioluminescence activity, which is calculated as the ratio of Firefly luciferase (Fluc) activity at 600nm to Gaussia luciferase (Gluc) activity at 500nm (Fluc/Gluc). The Gaussia luciferase activity was used as an internal control to normalize the cell number and the expression efficacy. For in vivo analyses, the value of the mutant on the right flank was used as a reference point for each bioluminescence imaging. The result was presented as normalized bioluminescence activity ratio of the wild-type value and the mutant value. Data were collected from at least 3 independent experiments with 3 or more replicates per experiment. Values are reported as mean±SD. All statistical analyses were performed with SPSS 14.0 for Windows software (SPSS Inc). In case only two groups were compared, a Student's t test was used. For multiple comparisons at different time points, analysis of variance (ANOVA) was performed. The EC50 values were fitted with sigmoidal dose-response curves using GraphPad Prism 5.0 software.
Results
Schematic of the bioluminescence-based activatable reporter for noninvasive observation of Src activity
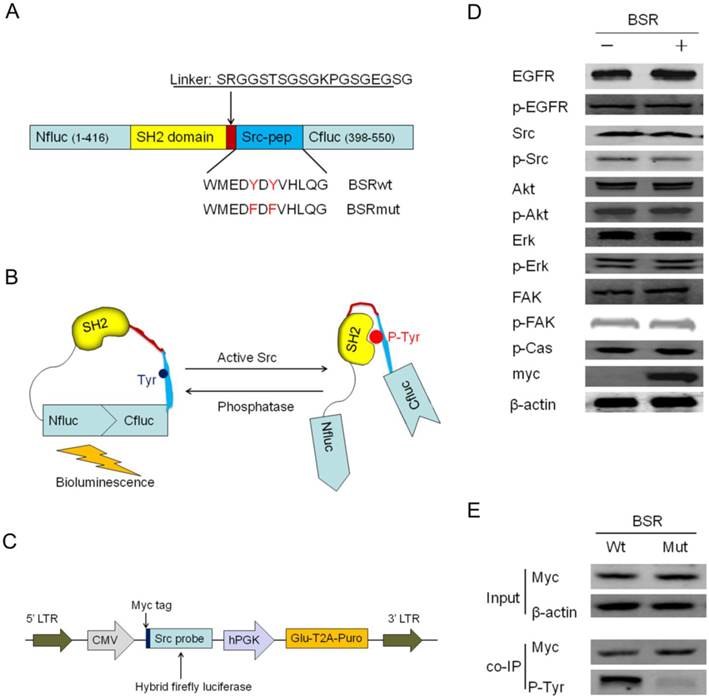
Our novel bioluminescent activatable reporter takes advantage of two facts. First, the crystal structure analysis of Firefly luciferase (PDB ID: 1LCI) has shown that the enzyme has a globular structure with a large N-terminal domain and a small C-terminal domain joined by a flexible hinge region [26]. Second, the active Src kinase can phosphorylate the potential Src substrate on tyrosine residues and then the phosphotyrosine (p-Tyr) -containing peptide can be recognized and bound by the SH2 domain [27].
To develop this intra-molecular activatable reporter, we constructed a hybrid luciferase by inserting the SH2 domain and the Src consensus substrate peptide (Src-pep) between the amino-(Nluc) and carboxyl-(Cluc) terminal domains of the Firefly luciferase molecule (Fig. 1A). Analogous reporters have been successfully developed to noninvasively image phosphorylation events of many kinases, such as AKT [28,29], FADD [30], EGFR [31], C-MET [32] and TGFβ [33]. The functional basis of this approach is that the proximity of the N and C terminals of the SH2 domain [31,32,34] and the flexible linkers enable the inactive luciferase fragments to reassemble the active luciferase molecule. In the presence of Src kinase activity, phosphorylation of the Src consensus substrate at tyrosine residues (Tyr 662 and 664) would result in its intra-molecular interaction with the docking pocket of the SH2 domain, thus sterically preventing reconstitution of a functional luciferase. Thus, the activatable reporter is designed to increase bioluminescent activity following inhibition of the Src substrate phosphorylation (Fig. 1B).
Apart from the hybrid luciferase, the bioluminescent Src reporter (BSR) also constitutes some other essential elements (Fig. 1C): (a) a lentiviral vector-based HIV for stable transformation, (b) a coelenterazine-dependent luciferase for internal control (considering the capacity of the lentiviral vector, Gaussia luciferase (Gluc), the smallest known coelenterazine-using luciferase, was the ideal candidate; Gaussia luciferase was co-expressed with the puromycin resistance gene via the “self-cleaving” T2A sequence), (c) flexible linker sequences for minimizing steric hindrance, and (d) a Myc-tag for Western blot analysis and immunoprecipitation. To validate that the reporter was indeed correctly expressed in living cells, we transfected BSR into HT29 cells containing high Src expression and activity [35]. The expressions of the recombinant protein and many signaling molecules were examined through Western blotting. The activities of several important molecules of Src signaling cascades, such as Erk, Akt, FAK, and p130Cas, were not affected, suggesting that the reporter did not perturb endogenous cellular signaling (Fig. 1D). We immunoprecipitated BSR using anti-Myc antibody to validate that the reporter is a substrate for the Src kinase. An increase in phosphorylation of the reporter was observed when the immunoprecipitated BSR was probed with phosphotyrosine antibody, indicating that the hybrid luciferase didn't disturb the phosphorylation of the Src consensus substrate peptide (Fig. 1E).
Schematic of the activatable reporter based on split-luciferase complementation assay for detecting Src activity. (A) The model shows a diagrammatic representation of the domain structure of the activatable reporter and the sequences of the linker and Src kinase substrate. Two versions of the reporter were developed: the BSRwt molecule, which contains the wild-type Src-pep sequences and the BSRmut molecule, which contains two tyrosine-to-phenylalanine substitutions at the putative phosphorylation sites. (B) BSR functions in Src-dependent phosphorylation of the Src-pep substrate. In the presence of Src kinase (Src on), the phosphorylation of Src-pep results in its intra-molecular interaction with the SH2 domain, sterically preventing the complementation of split luciferase fragments and generating minimal bioluminescence activity. In the absence of Src activation (Src off), the reconstitution of N- and C-terminal luciferase domains restores the bioluminescent activity. (C) The lentiviral vector map of BSR reporter. Via the “self-cleaving” T2A sequence, Gaussia luciferase and the puromycin resistance gene were co-expressed under the control of hPGK promoter to serve as an internal control and selection of stable transductants, respectively. (D) Cell lysates from HT29 and HT29-BSRwt cells were analyzed by Western blotting using antibodies specific for total EGFR, p-EGFR(Tyr1173), total Src, p-Src(Tyr416), total Akt, p-Akt(Ser473), total Erk, p-Erk(Thr202/Tyr204), total FAK, p-FAK(Tyr397) and p130Cas (Tyr410). β-actin was used as a loading control and myc as the BSR expression control. (E) Myc antibody was used to immunoprecipitate the BSR reporter molecule and Western blot analysis was performed by using phospho-tyrosine antibody.
Characterization of the Src-dependent, phosphorylation-sensitive bioluminescent reporter in living cells
According to the schemes presented above, construction of a specific Src reporter required a specific substrate peptide. The Src consensus substrate peptide WMEDYDYVHLQG, derived from p130cas, was considered to be the best candidate for two reasons: first, it is not a substrate for other kinases [14] and second, this peptide contains the YDYV motif, which serves as a Shc-SH2 binding site with moderate binding affinity when phosphorylated [36]. Therefore, after careful experimentation, this substrate peptide and Shc-SH2 domain (amino acids 374-465) [31,32] were selected as the best partners to construct the bioluminescent Src reporter (BSRwt). Two phenylalanine mutations of the corresponding phosphorylation sites were also generated by site directed mutagenesis as a control vector for imaging experiments (BSRmut) (Fig. 1A). Since Src is overexpressed and highly activated in the HT29 colorectal cell line, we generated stable HT29 cell lines expressing the BSRwt or BSRmut to validate the phosphorylation dependence of the BSR reporter. First, we generated stable HT29 cell line expressing the full-length Firefly luciferase for the control of direct drug effect on FL activity. The results showed no significant changes among the vehicle, dasatinib, saracatinib and EGF treatments (Fig. 2A).
Characterization of the BSR reporter in vitro (A) The changes of the full-length Firefly luciferase activity in HT29 cells with the vehicle, dasatinib, saracatinib and EGF treatments. (B) HT29-BSRwt or HT29-BSRmut cells cultured in 24-well plates were treated with the vehicle or 10uM PP1 for 60min. A pseudocolor data of Firefly luciferase imaging represents four separate experiments performed with quadruple culture wells. The changes in normalized bioluminescence activity (Fluc/Gluc) were plotted as fold induction over the value of BSRmut/pp1 (-) (line2). Data are presented as means ± S. E. Statistical analysis was done by one group t test for significance at the p< 0.01 level. (C) Western blotting analysis was performed using the antibody specific for phospho-tyrosine, phospho-Src, and total Src. For coimmunoprecipitation, Myc antibody was used to immunoprecipitate the BSR reporter molecule and Western blot analysis was performed by using phospho-tyrosine antibody. (D) Bioluminescence imaging of HEK293T-BSRwt co-expressed with the activated mutant of Src family kinase (Src Y527F, Yes Y535F or Fyn Y531F). (E and F) Bioluminescence imaging of the steady-state level of Src phosphorylation in a panel of colorectal cancer cells (SW480, Caco-2, HCT116, HT29, SW48, Colo320, SW1116 and Lovo) with the BSR reporter. Western blot analysis of the indicated cells was performed using antibodies specific for phospho-Src and total Src, β-actin and myc as the controls.
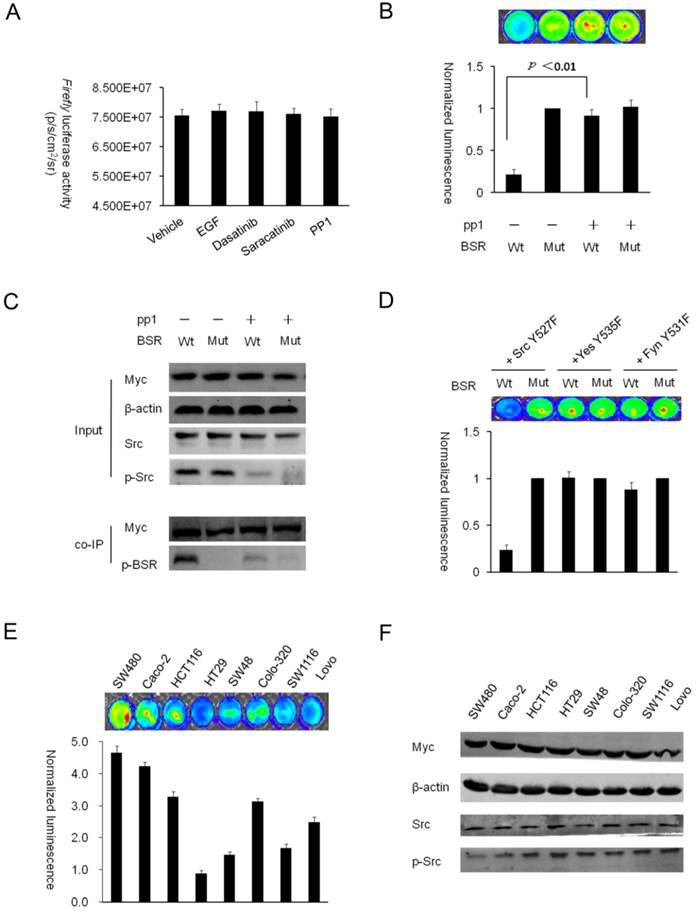
The Firefly luciferase requires ATP to catalyze the oxidation of luciferin, and the kinase phosphorylation also consumes ATP. These data demonstrated that the change of ATP levels following the kinase activity did not induce increase or decrease of the firefly luciferase activity. For the BSR reporter, the bioluminescence intensity showed a nearly 5-fold difference (p < 0.01) between the wild-type and mutant reporters; treatment with the Src selective inhibitor PP1 could reverse the Src-induced bioluminescence decrease. In contrast, HT29-BSRmut cells showed no significant change in bioluminescence activity in response to the PP1 treatment (Fig. 2B). A corresponding difference in phosphorylation of BSR between the wild-type and the mutant was also observed when the immunoprecipitated BSR was probed with phosphotyrosine antibody. Immunoprecipitation also revealed a decrease in phosphorylation of BSRwt in response to the PP1 treatment (Fig. 2C). These results further confirmed that Src-directed tyrosine phosphorylation occurred at the putative Src phosphorylation sites (Tyr 662 and 664) of consensus substrate peptide in the reporter. However, because the steady-state Src activity is low in HEK293T cells, the Src reporter showed little or no change in the complementation activity of the luciferase (data not shown).
To further validate the specificity of the reporter for Src kinase, we generated stable HEK293T cell lines expressing the BSRwt or BSRmut. The activated mutant of Src family kinase (Src Y527F, Yes Y535F or Fyn Y531F) was transfected in HEK293T-BSR cells. The results demonstrated that only HEK293T-BSRwt cells transfected with Src Y527F showed a robust nearly 5-fold Src-mediated decrease of bioluminescence compared with HEK293T-BSRmut cells, indicating BSR has the specificity for the Src reporter in mammalian cells (Fig. 2D). Next, to image the steady-state level of Src phosphorylation in cultured cells, we transiently transfected panels of colorectal cancer cells with BSRwt. The results revealed low Src activity in SW480 and Caco-2 cell lines and high Src activity in HT29, SW48, and SW1116 cell lines. The normalized complemented luciferase activity (Fluc/Gluc) in SW480-BSRwt was the highest -- about 5.3-fold higher than that in HT29-BSRwt (Fig. 2E). The bioluminescence was also correlated with the differential steady-state levels of Src phosphorylation in all cell lines, as revealed by Western blot (Fig. 2F).
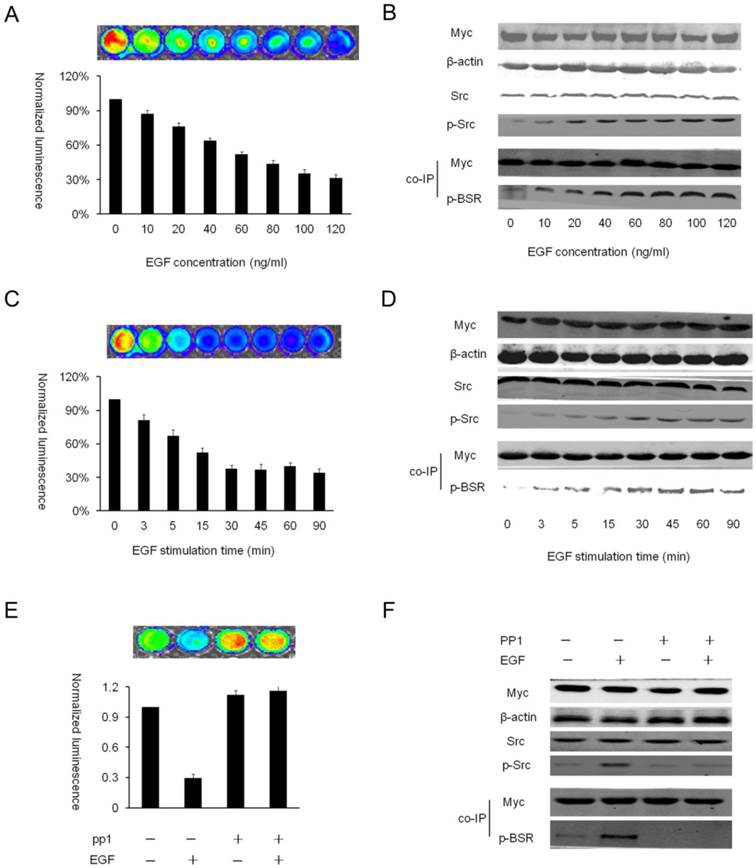
Dynamic imaging of the kinetics of EGF-induced Src activation in living cells
Treatment of cells with EGF can trigger Src activity through the corresponding receptor EGFR [14]. To validate the activation of Src kinase activity by the BSR (rather than inhibition), we examined the effects of EGF stimulation on BSR bioluminescence activity. For the dose-dependent assay, HEK293T-BSRwt cells were serum-starved overnight and stimulated with increasing concentrations of EGF for 1 hour. The results indicated a dose-dependent decrease of bioluminescence, which correlated with the increase of Src phosphorylation (Fig. 3A-B). Then, for the time-dependent assay, HEK293T-BSRwt cells were serum-starved overnight and stimulated with 100ng/ml of EGF for the indicated times. The bioluminescence activities were detected at different times. The results demonstrated that HEK293T-BSRwt cells underwent a significant EGF-mediated reduction of luciferase activity (about 62.1%) after 30 min of treatment. The reduction in luciferase activity correlated with the observed increase in Src phosphorylation over this time period, but not for total Src, as confirmed by Western blotting (Fig. 3C-D). In contrast, HEK293T-BSRmut cells showed little change in the bioluminescence activity (data not shown). After confirming the decrease of bioluminescence activity induced by Src activation, we further explored whether this reduction could be blocked by PP1, a selective inhibitor of Src kinase. As expected, the results demonstrated that the decrease in luciferase activity induced by EGF stimulation was largely eliminated in cells pretreated with PP1. Western blot analysis of these samples indicated that the increase in bioluminescence activity correlated with a decrease in Src activity (Fig. 3E-F). These results further confirmed that the reporter was specific for Src kinase and that the observed decrease of BSR luciferase activity was caused by the Src-dependent conformation change resulting in the separation of the luciferase fragments.
Dose- and time-dependent imaging of Src activity in response to specific inhibitors in cultured cells
The utility of the reporter system was further evaluated in testing the efficacy of Src inhibitors in cultured cells. We used HT29-BSR cells for more detailed analyses of the Src reporter in cell-based assays under various experimental conditions. We also generated stable HT29 cell line expressing the full-length Firefly luciferase for the control of direct drug effect on the enzyme activity. The results showed no significant changes (data were shown in Fig. 2A). In dose response studies, HT29-BSRwt cells were treated with increasing concentrations of two inhibitors under clinical investigation (dasatinib or saracatinib) for 2h following which bioluminescence activity in cells was monitored with IVIS spectrum system. Luciferase activity increased in a dose-dependent manner for both inhibitors, but saracatinib induced a greater increase in complemented Firefly luciferase activity than dasatinib. Bioluminescence was maximally induced 3.3 ± 0.18 fold over untreated controls by 150nM dasatinib and 4.5 ± 0.24 fold by 2uM saracatinib. These luciferase signal increases were correlated with the decrease of endogenous phospho-Src, but not with total Src, as detected by Western blotting (Fig. 4A-B).
In vitro characterization of EGF-induced activation of BSR (A) HEK293T-BSRwt cells were serum-starved overnight, stimulated with increased concentration of EGF for 1h, and then subjected to bioluminescence imaging. A pseudocolor data of Firefly luciferase imaging represents of four separate experiments performed with quadruple culture wells. The changes in normalized bioluminescence activity (Fluc/Gluc) compared with vehicle-treated value were expressed as the percentage change. (B) Cells from (A) were analyzed by Western blotting using the antibody specific for phospho-tyrosine, phospho-Src and total Src, β-actin and myc as controls. (C) HEK293T-BSRwt cells were serum-starved overnight, stimulated with 100ng/ml of EGF for the indicated times, and then subjected to bioluminescence imaging. (D) Cells from (C) were analyzed by Western blotting using the antibody specific for phospho-tyrosine, phospho-Src and total Src, β-actin and myc as controls. (E) Bioluminescence imaging of the effect of PP1 on EGF-induced activation of Src phosphorylation. HEK293T-BSRwt cultured on 48-well plates were serum-starved overnight, pretreated with vehicle or PP1 (10uM) for 2 hours, and then stimulated with vehicle or EGF (100ng/ml) for 30min. The changes in normalized bioluminescence activity (Fluc/Gluc) over vehicle treatment levels (line 1) were determined and reported as fold induction. (F) Cells from (E) were analyzed by Western blotting using antibodies specific for phospho-tyrosine, phospho-Src and total Src, β-actin and myc as controls.
BSR reporter in response to Src inhibitors in vitro (A and B) Dose response studies. HT29-BSRwt cells cultured on 48-well plates were treated with increasing concentrations of dasatinib or saracatinib for 2h. The changes in normalized bioluminescence activity (Fluc/Gluc) were plotted as fold induction over vehicle-treated values. Western blotting analysis with phospho-tyrosine, phospho-Src, and total Src-specific antibodies was performed to confirm the inhibition of Src activity. (C and D) Time course studies, HT29-BSRwt cells cultured on 48-well plates were treated with dasatinib (150nM) or saracatinib (2uM) for the indicated times. The changes in normalized activity (Fluc/Gluc) were plotted as fold induction over vehicle-treated values. Cells were analyzed by Western blotting using antibodies specific for phospho-tyrosine, phospho-Src, and total Src, β-actin and myc were used as controls.
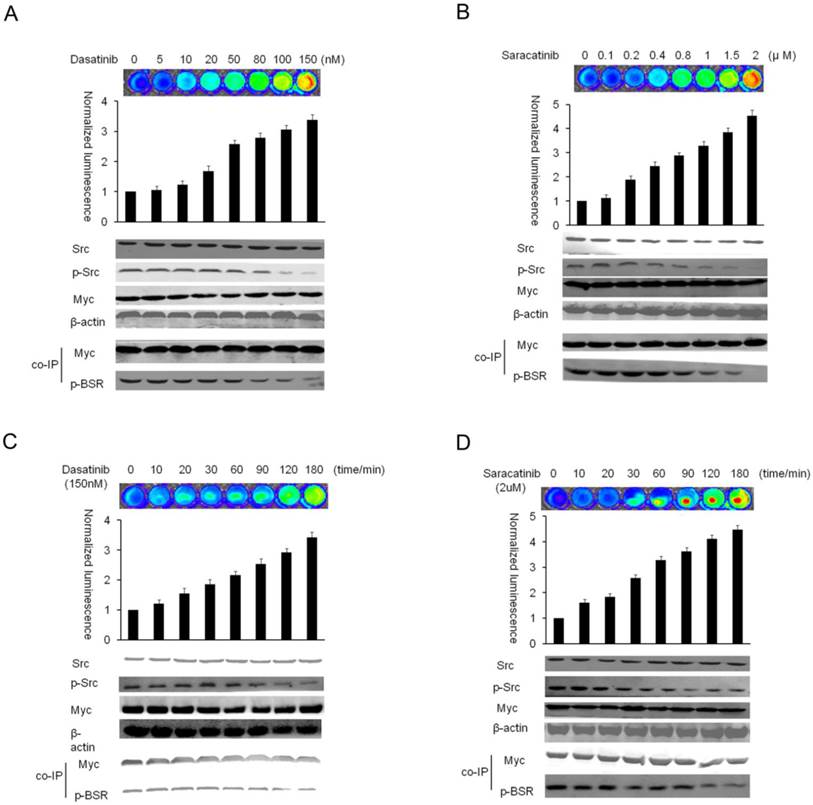
In time course studies, Src reporter cells were pretreated with 150nM dasatinib and 2uM saracatinib at various times. Luciferase activity increased in a time-dependent manner for both inhibitors; however, an early increase was observed in bioluminescent activity following the inhibition with saracatinib. Western blot analysis indicated that the activation of reporter correlated with phosphorylation of endogenous Src (Fig. 4C-D). Thus, the activatable design renders the reporter as a novel molecular tool for the measurement of Src inhibition. Our results demonstrated that saracatinib showed earlier induction and reached a higher plateau. This may be caused by the higher affinity of saracatinib for Src kinase [37]. When the normalized luciferase activity of the vehicle control was set as 0% and the maximum increased activity of drug treatments as 100%, the EC50 of dasatinib for Src activity inhibition in our intracellular context was about 42.36nM. The inhibitory EC50 of phospho-Src, calculated by Western blotting using phospho-specific antibodies, was reported to be about 20nM [38]. This difference may be due to different cell lines and experimental conditions, because the EC50 value estimated by the relative band intensities (% of vehicle control) in our Western blotting analysis was about 40.45nM. For saracatinib, the EC50 values were 0.89uM as measured by the luciferase activity and 1.04uM by Western blotting (Fig. S1). These results demonstrated that the BSR reporter exhibits similar or even higher sensitivity compared with Western blot.
Noninvasive imaging of Src activity in mice
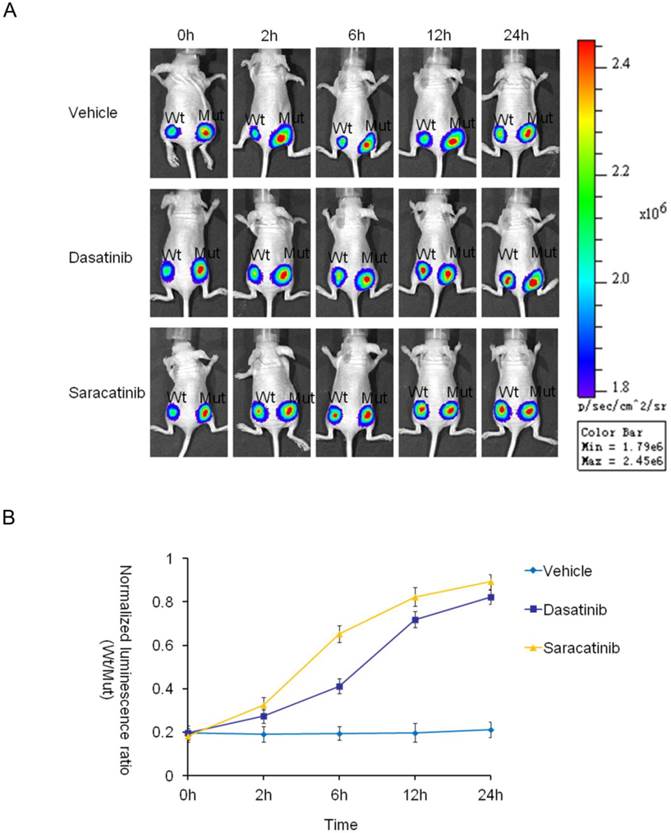
The ability of an agent to modulate its target's activity in vivo depends on multiple factors. The pharmacokinetics and bioavailability of the drug in the whole body and in the tumor itself have a substantial impact on the optimal dosing and administration schedule required for maximal target modulation [28]. So far, it has not been possible to measure the Src activity in vivo due to the lack of appropriate technologies. Bioluminescence imaging is one of the most widely used imaging technologies for interrogating cellular and molecular events in rodent models of human biology [39,40]. We have demonstrated that the BSR reporter efficiently detects Src tyrosine kinase inhibition in vitro. We therefore hypothesized that this reporter could be further developed into a model system to measure the inhibition of Src activity in vivo. To test this concept, we implanted HT29-BSRwt or HT29-BSRmut cells into the bilateral flanks of nude mice to establish human tumor xenograft models. The HT29-BSRmut on the right flank was used as a control. When the tumors reached a volume of 40 mm3, the mice were intraperitoneally treated with vehicle alone, dasatinib (20mg/kg) or saracatinib (25mg/kg). Serial imaging was performed at 0, 2, 6, 12, and 24 hours following drug administration and compared with the corresponding mutant control bioluminescence. These results demonstrated that the bioluminescence activity of BSRwt remained essentially flat over a 24-h period in vehicle-treated mice. On the other hand, in mice treated with saracatinib, the bioluminescence activity of BSRwt increased within the first 2-h and reached a peak at approximately 12 hours after treatment with about 4.5-fold increase compared to pretreatment. As for dasatinib, the bioluminescence slowly increased and reached about 4.2-fold at approximately 24 hours after treatment (Fig. 5). These results demonstrate that the BSR reporter is a novel molecular tool to measure Src inhibition in vivo. This technology also facilitated the assessment of pharmacokinetics and pharmacodynamics of drugs in mice that may be of relevance in humans. This function provides an immediate and sequential opportunity to optimize therapeutic routes of drug administration for maximal tumor control and minimal normal tissue toxicity.
High-throughput screening against a kinase inhibitor library using BSR
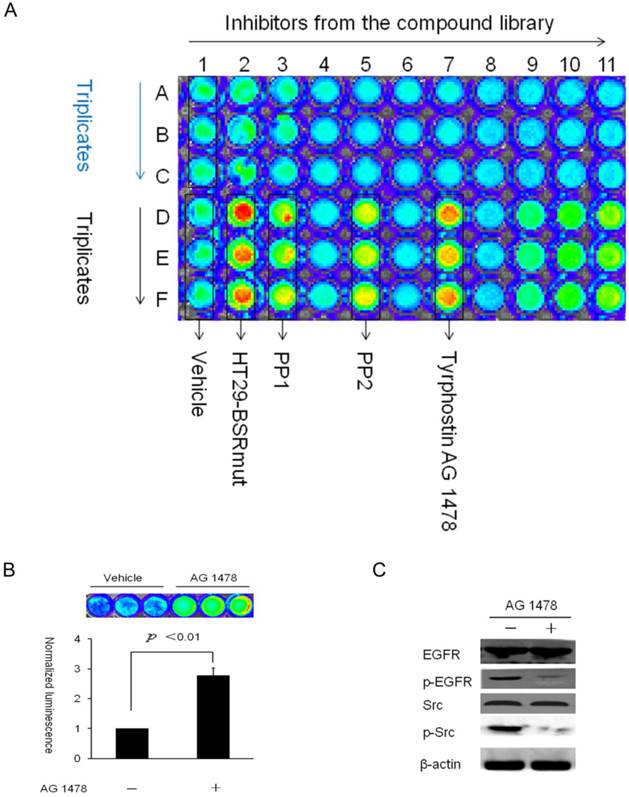
To examine the performance of the reporter for high-throughput drug screening, bioluminescence activities of HT29-BSRwt cells were screened against a kinase inhibitor library. This compound library was obtained from National Compound Resource Center in China, containing 84 small molecular weight kinase inhibitors. Src kinase inhibitors as well as upstream modulators of Src kinase activity would lead to increase in complemented Firefly luciferase activities in HT29-BSRwt cells. HT29-BSRmut cells were used as a negative control for compounds that led to changes in bioluminescence signals independent of inhibition of Src kinase activity (data not shown). The application of the bioluminescent activatable reporter for HTS has demonstrated that variability in seeding densities minimally affects the signal-to-noise ratio, thereby enabling flexibility in cell plating [30]. In our experiment, each screening plate also included intra-plate controls (vehicle treatment and HT29-BSRmut) to assess the quality of the assay protocol in a 96-well plate format. Among all the 84 kinase inhibitors screened, a compound (Tyrphostin AG1478) led to significant bioluminescence activity increase compared with the vehicle control treatment (about 3.4-fold) comparable with the known Src-specific Kinase inhibitors (PP1 and PP2, about 4.5-fold and 3.7-fold, respectively) (Fig. 6A). To further validate the effect of Tyrphostin AG1478 on Src kinase, HT29-BSRwt and HT29-BSRmut cells cultured in 24-well plates were treated with Tyrphostin AG 1478 (0.1mM) for 60min prior to the BLI analysis. The results showed that Tyrphostin AG 1478 led to significant increase (about 2.78 fold) in complemented Firefly luciferase activities in HT29-BSRwt cells (Fig. 6B). Since Tyrphostin AG1478 is a highly specific inhibitor of the EGFR tyrosine kinase, it is possible that the effect of AG1478 on Src activity is caused by the inhibition of EGFR (Fig. 6C).
Monitor two kinase activities in one subject by monitoring of Firefly and Renilla luciferase complementation assays
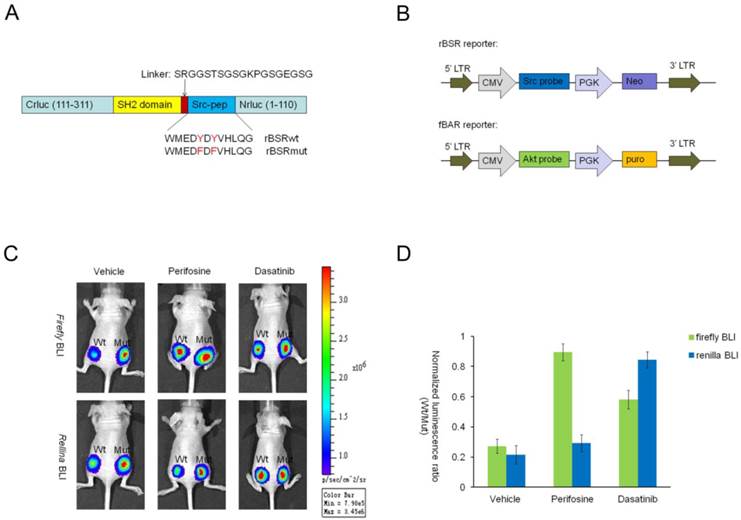
Complex signaling networks, characterized by cross-talk between various components, underlie the malignant phenotype of cancer. Therefore, it is important to develop a multi-reporting system to simultaneously monitor the activities of various kinases both in vitro and in vivo. Since Firefly and Renilla luciferases have different substrate specificities and emission spectra [41], we hypothesized that it is possible to apply imaging of both enzymes in the same model based on split luciferase complementation strategy. To test this concept, a new Src reporter based on split Renilla luciferase complementation assay (indicated as rBSR) was constructed by replacing the Firefly luciferase fragments with Renilla luciferase fragments. In our preliminary experiment, we noticed that higher luciferase activity was reconstructed when C-terminal fragment of Renilla luciferase was place on the N-terminal of the reporter. Therefore, the structure of the Renilla Src reporter was Crluc-Sh2-peptide-Nrluc (Fig. 7A).
Noninvasive imaging of inhibition of Src kinase activity in mice (A) Xenograft models were established by subcutaneous injections in the bilateral flanks of nude mice with HT29-BSRwt (left) and HT29-BSRmut (right) cells. The mutant was used as a control for each of the mice. BLI activity of pretreated and intraperitoneally treated mice with vehicle control (20% DMSO in PBS), dasatinib, (20mg/kg) or saracatinib (25mg/kg) was monitored at various times. Representative images of serial Firefly luciferase bioluminescence imaging at indicated times of treatment are shown. (B) Graph represents induction of bioluminescence by vehicle, dasatinib, or saracatinib at each time point. The value was calculated using the mutant reporter as a control at each time point for each of the mice. For each xenograft, we first calculated the normalized bioluminescence activity by the ratio of Firefly luciferase activity and Gaussia luciferase activity. We then calculated the normalized bioluminescence activity ratio of the wild-type and mutant values at the same time point in the same mouse.
High-throughput screening against a kinase inhibitor library using BSR (A) A representative pseudocolor image in the HTS assays. HT29-BSRwt cells cultured in 96-well plates were treated with 30uM of each compound for 60min in triplicates (for example: 1A~1C, 1D~1F, 2A~2C…). In this assay, PP1, PP2, and Tyrphostin AG 1478 induced significant increases in the bioluminescence activity compared to the vehicle control. (B) Further validation of the effect of Tyrphostin AG 1478 on Src activity with BSR in 24-well plates. (C) Western blotting analysis of the phosphorylation of EGFR and Src kinases after Tyrphostin AG 1478 treatment in HT29 cells.
The bioluminescent AKT reporter (indicated as fBAR), which was developed as reported by Zhang et al. [28], was chosen as the other kinase reporter based on split Firefly luciferase complementation assay (Fig. 7B). Since MDA-MB-435S cell line has high levels of Src kinase and Akt kinase [42,43], we generated stable MDA-MB-435S cell lines expressing both rBSR and fBAR after G418 and puromycin selections. Cells expressing the wild-type or the mutant reporters were implanted into the bilateral flanks of nude mice to establish xenograft models. The mutant on the right flank was used as a control. Firefly and Renilla luciferase bioluminescence imagings were concurrently performed after treatment with vehicle control, perifosine (30mg/kg), or dasatinib (20mg/kg) for 6 hours. The results demonstrated that the bioluminescence activities for both reporters increased when administrated with the corresponding inhibitors. Interestingly, treatment of Src inhibitor dasatinib induced significant increases of both Renilla luciferase and Firefly luciferase activities by about 2.14-fold and 3.92-fold, respectively. In contrast, the Akt inhibitor only significantly activated the fBAR reporter about 3.29-fold compared to pretreatment (Fig. 7C-D). This effect may be caused by the cross talks between the signaling pathways and also the fact that Akt operates downstream of the Src signaling pathway.
Discussion
Extensive preclinical evidence warrants targeting Src as a therapeutic approach for cancer. The common methods for the elucidation of Src kinase activity, such as radioimmunoassay (RIA) and Western blot, require destroying large amounts of cells or tissue, and are not accessible within the relevant cellular microenvironment. The advent of optical-based molecular imaging paradigm makes it possible to visualize, characterize, and quantitatively measure biological processes at the cellular and molecular levels in living subjects. Previous studies have utilized FRET-based reporter molecules for non-invasive imaging of Src kinase [14,15,44]. However, it is difficult to employ these reporters in living animals due to their high degree of autofluorescence and poor signal penetration depth through biologically heterogeneous tissues [45]. Therefore, in this manuscript, we described a novel activatable reporter based on split luciferase complementation.
Luciferase based complementation assay has been widely applied to visualize the phosphorylation-dependent interactions. This has been achieved through either an inter-molecular or an intra-molecular complementation strategy. For inter-molecular reporters, also called bimolecular reporters, the luciferase is split into two non-functional fragments (Nluc and Cluc), each of which is fused to one of the two binding partners. When the binding partners interact in a phosphorylation-dependent manner, the two luciferase fragments are brought into proximity, leading to luciferase reconstitution [46]. For the intra-molecular complementation strategy, the single-chain reporter is constructed by sandwiching a conformationally responsive unit between the split luciferase fragments. This conformationally responsive unit usually consists of a kinase-specific substrate and a phosphoaminoacid-binding domain. The reporter design predicts that in the absence of kinase phosphorylation the N- and C- terminal luciferase domains interact and reconstitute enzyme activity. In contrast, the conformational changes induced by the phosphorylation event can sterically prevent the complementation [28].
Monitoring two kinase activities in a single mouse using both Firefly and Renilla luciferase complementation assays. (A) Schematic representation of the bioluminescent Src reporter based on Renilla luciferase complementation strategy (rBSR). N- and C-terminal Firefly luciferase domains in the BSR reporter were replaced by C- and N-terminal Renilla luciferase domains, respectively. (B) The lentiviral vector structures of Src reporter based on split Renilla luciferase complementation assay (rBSR) and Akt reporter based on split Firefly luciferase complementation assay (fBAR). (C) MDA-MB-435S cells expressing both rBSR and fBAR were used to establish xenograft tumors by subcutaneous injections in the bilateral flanks of nude mice with 435S-rBSRwt/fBARwt cells on the left and 435S-rBSRmut/fBARmut1cells on the right cells (1x106 cells/sample). Firefly and Renilla luciferase bioluminescence imagings were performed after treatment with vehicle control, perifosine (30mg/kg) or dasatinib (20mg/kg) for 6 hours. (D) The histogram of Firefly and Renilla luciferase bioluminescence imaging described in C. The value was calculated using the mutant as control at each time point for each of the mice.
We first attempted the inter-molecular complementation strategy by fusing the Firefly luciferase fragments to the Src consensus substrate peptide and the SH2 phosphopeptide binding domain, respectively, expecting to yield a large dynamic range from low to high bioluminescence. By using a mechanistically similar strategy, other investigators have successfully monitored the phosphorylation events of IRS-1 [47], Cdc25C [46], c-Myc [48] and PKA [49]. However, in our experience, the inter-molecular Src reporters showed marginal bioluminescence increase (data not shown). We also attempted targeting the reporter to the cytoplasm with a nuclear export signal (NES) [49] or using different configuration of the fused protein at different dissection sites [50]; none of these strategies resulted in a satisfactory bioluminescence signal. This can be explained by the too short length of the Src substrate peptide to interact with the SH2 domain. Alternatively, upon interaction of the phosphorylated Src substrate peptide and SH2 domain, the configuration may not be suitable for the complementation and reconstitution of luciferase. It is also known that the geometry of Nluc and Cluc in the interacting protein complex can largely influence how the active site is reconstituted [50].
For the kinase-activatable reporter, another important aspect is to ensure the specificity. In our study, we chose the peptide derived from p130cas as the Src consensus substrate. This peptide has previously been demonstrated to be specifically phosphorylated by activated Src [14]. It is of note that SH2 domain also plays an important role in the design of the intra-molecular activatable reporter. We have shown that Shc-SH2 domain (aa 374-465) has a better dynamic range than Src-SH2 domain (aa 167-267), which was used for FRET-Src reporter [14]. This phenomenon may be due to the different binding affinity for p-Tyr-containing peptides. A plausible explanation is that the SH2 phosphotyrosine binding domain competes with phosphatase and prevents tyrosine dephosphorylation of the substrate peptide [51]. The SH2 domain of Shc can also interact with the YDYV motif [36] but displays a relatively low binding affinity for p-Tyr-containing peptides compared with the SH2 domain of c-Src [52]. A similar phenomenon was reported in the process of developing FRET RhoA sensor [53].
The Src reporter we describe here represents a novel tool for the dynamic measurement of Src activity without labor-intensive and time-consuming tasks for tissue analysis and longitudinal studies of biological processes. Our study has illustrated the general utility of the bioluminescent Src reporter for the analysis of kinase inhibitors undergoing clinical studies in cancer cells cultures and xenografts of mice. We demonstrated that inhibition of Src kinase activity resulted in activation of reporter's bioluminescence activity in a dose- and time-dependent manner. Also, the simplicity, cost-effectiveness, high sensitivity, and wide dynamic range of bioluminescent cell-based assays make them highly attractive for the development of high throughput screening to accelerate drug discovery and development of Src inhibitors. Contrary to current early stage drug discovery research depending on various in vitro biochemical and mass spectrometry protocols, mammalian cell-based functional screening assays have shown promise in predicting the in vivo dynamics. Furthermore, compared to other molecular imaging modalities, such as FRET, bioluminescent imaging is more suitable for simultaneously monitoring two molecules in one subject [19]. In our study, we employed two molecular bioluminescent reporters based on Firefly and Renilla luciferase complementation with different substrate specificities and emission spectra [41] to investigate two signaling pathways (Src kinase and Akt kinase) in one subject. This work opens up new opportunities to evaluate diverse aspects of complicated biological processes with molecular imaging in living subjects.
We have furnished an important example of the power of the luciferase-based optical imaging reporter in studying sophisticated signal transduction pathways and drug efficacy. Unlike the fluorescence complementation, luciferase complementation does not assemble irreversibly. Thus, the BSR reporter can detect not only the phosphorylation but also the subsequent dephosphorylation. Phosphorylation induces the decrease of bioluminescence, whereas, dephosphorylation increases the bioluminescence. In the continuous biological processes of living cells, the phosphorylation and dephosphorylation are in dynamic balance. And the activity change of BSR was due to a shift in the phosphorylation/dephosphorylation balance. However, it is noteworthy that monitoring molecular events in a single cell by bioluminescent reporters, requires a complicated and expensive bioluminescence microscope [54,55].
Conclusion
We have developed a novel, inter-molecular, Src-activatable reporter based on split-luciferase complementation. The reporter allows non-invasive, real time, and dynamic imaging as well as quantification of Src kinase activity in living cells and in vivo. It will greatly facilitate the investigation of Src phosphorylation events and the evaluation of anti-Src drug efficacies in preclinical models. It will also provide an approach to identify lead compounds targeted to Src kinase from libraries using cell-based, high-throughput screening.
Supplementary Material
Supplementary Figure S1.
Abbreviations
BLI: bioluminescence imaging; FRET: fluorescence resonance energy transfer; FLIM: fluorescence lifetime imaging microscopy; HTS: high-throughput screening; NES: nuclear export signal; Fluc: Firefly luciferase; Gluc: Gaussia luciferase; Rluc: Renilla luciferase; BSR: bioluminescent Src reporter; Src-pep: Src consensus substrate peptide; EGF: epidermal growth factor; rBSR: bioluminescent Src reporter based on Renilla luciferase; fBAR: bioluminescent Akt reporter based on Firefly luciferase.
Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program, 2011CB935800), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT, Grant No. IRT1272) of China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636-42
2. Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metast Rev. 2003;22:337-58
3. Roskoski R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res. 2015;94:9-25
4. Allgayer H, Boyd DD, Heiss MM. et al. Activation of Src kinase in primary colorectal carcinoma. Cancer. 2002;94:344-51
5. Fizazi K. The role of Src in prostate cancer. Ann Oncol. 2007;18:1765-73
6. Morgan L, Gee J, Pumford S. et al. Elevated Src kinase activity attenuates Tamoxifen response in vitro and is associated with poor prognosis clinically. Cancer Biol Ther. 2009;8:1550-58
7. Morton JP, Karim SA, Graham K. et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139:292-303
8. Nam A-R, Nam H-J, Kang KH. et al. Evaluation of Src as a therapeutic target and development of biomarkers of Src inhibitor in cancer. Cancer Res. 2014;74:747-747
9. Creedon H, Brunton VG. Src kinase inhibitors: promising cancer therapeutics? Crit Rev Oncog. 2012;17:145-59
10. Puls LN, Eadens M, Messersmith W. Current status of Src inhibitors in solid tumor malignancies. Oncologist. 2011;16:566-78
11. Gangadhar TC, Clark JI, Karrison T. et al. Phase II study of the Src kinase inhibitor saracatinib (AZD0530) in metastatic melanoma. Invest New Drugs. 2013;31:769-73
12. Conway JRW, Carragher NO, Timpson P. Developments in preclinical cancer imaging: innovating the discovery of therapeutics. Nat Rev Cancer. 2014;14:314-28
13. Niu G, Chen X. Molecular imaging with activatable reporter systems. Theranostics. 2012;2:413-23
14. Wang Y, Botvinick EL, Zhao Y. et al. Visualizing the mechanical activation of Src. Nature. 2005;434:1040-45
15. Seong J, Lu S, Ouyang M. et al. Visualization of Src activity at different compartments of the plasma membrane by FRET imaging. Chem Biol. 2009;16:48-57
16. Ouyang M, Huang H, Shaner NC. et al. Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer Res. 2010;70:2204-12
17. Lu S, Ouyang M, Seong J. et al. The spatiotemporal pattern of Src activation at lipid rafts revealed by diffusion-corrected FRET imaging. PLoS Comput Biol. 2008;4:e1000127
18. Nobis M, McGhee EJ, Morton JP. et al. Intravital FLIM-FRET imaging reveals dasatinib-induced spatial control of Src in pancreatic cancer. Cancer Res. 2013;73:4674-86
19. Aoki K, Kiyokawa E, Nakamura T. et al. Visualization of growth signal transduction cascades in living cells with genetically encoded probes based on Förster resonance energy transfer. Philos Trans R Soc Lond B Biol Sci. 2008;363:2143-51
20. Sung M-K, Huh WK. Bimolecular fluorescence complementation analysis system for in vivo detection of protein-protein interaction in Saccharomyces cerevisiae. Yeast. 2007;24:767-75
21. Padilla-Parra S, Tramier M. FRET microscopy in the living cell: different approaches, strengths and weaknesses. Bioessays. 2012;34:369-76
22. Willmann JK, van Bruggen N, Dinkelborg LM. et al. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7:591-607
23. Badr CE, Tannous BA. Bioluminescence imaging: progress and applications. Trends Biotechnol. 2011;29:624-33
24. Troy T, Jekic-McMullen D, Sambucetti L. et al. Quantitative comparison of the sensitivity of detection of fluorescent and bioluminescent reporters in animal models. Mol Imaging. 2004;3:9-23
25. Zhang Y, Xia H, Ge X. et al. CD44 acts through RhoA to regulate YAP signaling. Cell Signal. 2014;26:2504-13
26. Conti E, Franks NP, Brick P. Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure. 1996;4:287-98
27. Zhou S, Shoelson SE, Chaudhuri M. et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767-78
28. Zhang L, Lee KC, Bhojani MS. et al. Molecular imaging of Akt kinase activity. Nat Med. 2007;13:1114-19
29. Chan CT, Paulmurugan R, Reeves RE. et al. Molecular imaging of phosphorylation events for drug development. Mol Imaging Biol. 2009;11:144-58
30. Khan AP, Schinske KA, Nyati S. et al. High-throughput molecular imaging for the identification of FADD kinase inhibitors. J Biomol Screen. 2010;15:1063-70
31. Khan AP, Contessa JN, Nyati MK. et al. Molecular imaging of epidermal growth factor receptor kinase activity. Anal Biochem. 2011;417:57-64
32. Zhang L, Virani S, Zhang Y. et al. Molecular imaging of c-Met tyrosine kinase activity. Anal Biochem. 2011;412:1-8
33. Nyati S, Schinske K, Ray D. et al. Molecular imaging of TGFβ-induced Smad2/3 phosphorylation reveals a role for receptor tyrosine kinases in modulating TGFβ signaling. Clin Cancer Res. 2011;17:7424-39
34. Zhou M, Ravichandran KS, Olejniczak ET. et al. Structure and ligand recognition of the phosphotyrosine binding domain of Shc. Nature. 1995;378:584-92
35. Windham TC, Parikh NU, Siwak DR. et al. Src activation regulates anoikis in human colon tumor cell lines. Oncogene. 2002;21:7797-807
36. Miyake I, Hakomori Y, Misu Y. et al. Domain-specific function of ShcC docking protein in neuroblastoma cells. Oncogene. 2005;24:3206-15
37. Green TP, Fennell M, Whittaker R. et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009;3:248-61
38. Obr A, Röselová P, Grebeňová D. et al. Real-time analysis of imatinib- and dasatinib-induced effects on chronic myelogenous leukemia cell interaction with fibronectin. PLoS One. 2014;9:e107367
39. Contag CH. In vivo pathology: seeing with molecular specificity and cellular resolution in the living body. Annu Rev Pathol Mech Dis. 2007;2:277-305
40. Prescher JA, Contag CH. Guided by the light: visualizing biomolecular processes in living animals with bioluminescence. Curr Opin Chem Biol. 2010;14:80-89
41. Paulmurugan R, Gambhir SS. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal Chem. 2003;75:1584-89
42. Chen Y, Rodrik V, Foster DA. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene. 2004;24:672-79
43. Niu G, Carter WB. Human epidermal growth factor receptor 2 regulates angiopoietin-2 expression in breast cancer via AKT and mitogen-activated protein kinase pathways. Cancer Res. 2007;67:1487-93
44. Nobis M, McGhee EJ, Herrmann D. et al. Monitoring the dynamics of Src activity in response to anti-invasive dasatinib treatment at a subcellular level using dual intravital imaging. Cell Adh Migr. 2014;8:478-86
45. Amitai G, Callahan BP, Stanger MJ. et al. Modulation of intein activity by its neighboring extein substrates. Proc Natl Acad Sci U S A. 2009;106:11005-10
46. Luker KE, Smith MCP, Luker GD. et al. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci U S A. 2004;101:12288-93
47. Kaihara A, Kawai Y, Sato M. et al. Locating a protein-protein interaction in living cells via split Renilla luciferase complementation. Anal Chem. 2003;75:4176-81
48. Fan-Minogue H, Cao Z, Paulmurugan R. et al. Noninvasive molecular imaging of c-Myc activation in living mice. Proc Natl Acad Sci U S A. 2010;107:15892-97
49. Herbst KJ, Allen MD, Zhang J. Luminescent kinase activity biosensors based on a versatile bimolecular switch. J Am Chem Soc. 2011;133:5676-79
50. Leng W, Pang X, Xia H. et al. Novel split-luciferase-based genetically encoded biosensors for noninvasive visualization of Rho GTPases. PLoS One. 2013;8:e62230
51. Rotin D, Margolis B, Mohammadi M. et al. SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase C gamma. EMBO J. 1992;11:559-67
52. Zhou M, Meadows RP, Logan TM. et al. Solution structure of the Shc SH2 domain complexed with a tyrosine-phosphorylated peptide from the T-cell receptor. Proc Natl Acad Sci U S A. 1995;92:7784-88
53. Yoshizaki H, Ohba Y, Kurokawa K. et al. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J Cell Biol. 2003;162:223-32
54. Misawa N, Kafi AK, Hattori M. et al. Rapid and high-sensitivity cell-based assays of protein-protein interactions using split click beetle luciferase complementation: an approach to the study of G-protein-coupled receptors. Anal Chem. 2010;82:2552-60
55. Takakura H, Hattori M, Takeuchi M. et al. Visualization and quantitative analysis of G protein-coupled receptor-beta-arrestin interaction in single cells and specific organs of living mice using split luciferase complementation. ACS Chem Biol. 2012;7:901-10
Author contact
![]() Corresponding author: Feng Bi, E-mail: bifengcom.cn.
Corresponding author: Feng Bi, E-mail: bifengcom.cn.