Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. STAT3
3. Physiopathological roles of...
4. Physiopathological roles of...
5. Inhibitors
6. Implication of STAT3 in...
7. Conclusions and perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(22):6424-6442. doi:10.7150/thno.35528 This issue Cite
Review
Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis
Qi Chen1*, Jianjun Lv2*, Wenwen Yang1, Baoping Xu1, Zheng Wang3, Zihao Yu1, Jiawei Wu1, Yang Yang1 ![]() , Yuehu Han4
, Yuehu Han4 ![]()
1. Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education. Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China
2. School of Basic Medicine, The Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China
3. Department of Cadio-Thoracic Surgery, Wuhan General Hospital of The People's Liberation Army, 627 Wuluo Road, Wuhan 430070, China
4. Department of Cardiovascular Surgery, Xijing Hospital, The Fourth Military Medical University, 127 Changle West Road, Xi'an 710032, China
*These authors contributed equally to this work.
Received 2019-4-7; Accepted 2019-7-10; Published 2019-8-14
Abstract

Atherosclerosis is the main pathological basis of ischemic cardiovascular and cerebrovascular diseases and has attracted more attention in recent years. Multiple studies have demonstrated that the signal transducer and activator of transcription 3 (STAT3) plays essential roles in the process of atherosclerosis. Moreover, aberrant STAT3 activation has been shown to contribute to the occurrence and development of atherosclerosis. Therefore, the study of STAT3 inhibitors has gradually become a focal research topic. In this review, we describe the crucial roles of STAT3 in endothelial cell dysfunction, macrophage polarization, inflammation, and immunity during atherosclerosis. STAT3 in mitochondria is mentioned as well. Then, we present a summary and classification of STAT3 inhibitors, which could offer potential treatment strategies for atherosclerosis. Furthermore, we enumerate some of the problems that have interfered with the development of mature therapies utilizing STAT3 inhibitors to treat atherosclerosis. Finally, we propose ideas that may help to solve these problems to some extent. Collectively, this review may be useful for developing future STAT3 inhibitor therapies for atherosclerosis.
Keywords: atherosclerosis, STAT3, endothelial cell dysfunction, macrophage polarization, inflammation, immunity, inhibitors
1. Introduction
Cardiovascular and cerebrovascular diseases, especially heart attack and stroke, significantly contribute to worldwide mortality [1], causing approximately 17 million deaths per year [2]. Moreover, atherosclerosis is recognized as the original cause of most cardiovascular and cerebrovascular diseases [3-5]. As a chronic progressive inflammatory arterial wall disease, atherosclerosis is characterized by the accumulation of lipids in the intima, thickening of the arterial wall, and narrowing of the vascular cavity. The major drivers leading to atherosclerosis include hyperlipidemia, hyperglycemia, insulin resistance, hypertension, and other factors such as genetics, age, cigarette smoking, and mental status [6]. Thus, various atherosclerosis treatments have emerged that directly target the above risk factors, such as lipid-lowering medications and antiplatelet aggregation therapies. However, these treatments are not entirely effective due to incomplete knowledge of the mechanisms of and effective target sites for atherosclerosis. Therefore, the mechanism of atherosclerosis has been a popular focus of research, from which scholars hope to find novel breakthroughs, develop feasible intervention measures, and improve the overall prevention and treatment strategies for atherosclerosis. In addition, with the progress and development of basic and clinical research, study of the pathogenesis of atherosclerosis has made great strides. The pathological mechanisms of atherosclerosis are complex and can be mainly summarized as endothelial cell dysfunction, macrophage polarization, inflammation, and immune responses. Emerging studies reveal that the signal transducer and activator of transcription 3 (STAT3) may play a critical role in all these factors, which indicates that STAT3 might become a new target of atherosclerosis therapies.
STATs, consisting of seven members (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) [7-11], have dual functions in signal transduction and transcriptional regulation. STAT3, one of the seven STAT members, was initially identified by two individual groups in 1994 [12, 13] and has increasingly gained focused attention due to its significant roles in diverse biological processes, including cell proliferation, cell differentiation, cell survival, inflammation, immunity, and angiogenesis [14]. Since the gene Stat3 was first described as an oncogene in 1999 [15], STAT3 has become the research focus for several disease areas as a potential anticancer target. Previous studies have verified that STAT3 plays an essential role in various diseases, including cancers [16], myocardial ischemic injury [17], stroke [8], and obesity [18]. Recently, with the study of cardiovascular and cerebrovascular diseases becoming increasingly popular, STAT3 has been demonstrated to play roles in several cardiovascular diseases, including arteriosclerosis, cardiac hypertrophy, and heart failure [19-22]. Although atherosclerosis is considered the critical pathological basis of most cardiovascular and cerebrovascular diseases, no specific reviews are aimed at the emerging roles of STAT3 in atherosclerosis. Thus, the present review aims to fill this gap.
In this review, by summarizing the current literature, we highlight the essential roles of STAT3 in atherosclerosis and present STAT3 inhibitors that may become potential treatment agents for atherosclerosis. First, we describe the general background of STAT3, including its structure, function, and regulation. Subsequently, we discuss the pathological roles of STAT3 in atherosclerosis from three independent but related biological processes, endothelial cell dysfunction, macrophage polarization, inflammation, and immunity. Moreover, we summarize the current inhibitors of STAT3 (Table 1) and explore their implications in atherosclerosis treatments. Finally, we highlight some potential issues and propose some solutions to these issues. In conclusion, this review may contribute to the application of STAT3 as a novel target of atherosclerosis therapies.
Inhibitors of STAT3
| Indirect inhibitors of STAT3 | |||||
| Classification | Inhibitors | Year | Target site | Mode of targeting STAT3 | Reference |
| Small-molecule inhibitors | Ruxolitinib | 2012 | JAK1/2 | Phosphorylation | [182, 186] |
| Tofacitinib | 2013 | JAK3 | Phosphorylation | [185] | |
| AZD1480 | 2011 | JAK1/2 | Phosphorylation | [184] | |
| SB1578 | 2010 | JAK2 | Unknown | [182] | |
| WP1066 | 2010 | JAK2 | Phosphorylation | [183] | |
| AG490 | 1996 | JAK2 | Phosphorylation | [187] | |
| Naringenin | 2013 | SOCS3 | Unknown | [61] | |
| Flavone | 2013 | SOCS3 | Unknown | [61] | |
| Natural inhibitors | Tricin | 2014 | JAK1/2 | Phosphorylation | [189] |
| Direct inhibitors targeting the SH2 domain of STAT3 | |||||
| Classification | Inhibitors | Year | Target site | Mode of targeting STAT3 | Reference |
| Peptides and peptide-like inhibitors | PpYLKTK | 2001 | SH2 | Dimerization | [192] |
| pYLPQTV | 2003 | SH2 | Dimerization | [193] | |
| Acetyl-pYLKTKF | 2007 | SH2 | Dimerization | [194] | |
| Small-molecule inhibitors | STA-21 | 2009 | SH2 | Dimerization | [195] |
| LLL12 | 2012 | SH2 | Phosphorylation | [160] | |
| OPB-31121 | 2013 | SH2 | Phosphorylation | [196] | |
| OPB-51602 | 2015 | SH2 | Phosphorylation | [197] | |
| Stattic | 2006 | SH2 | Phosphorylation | [196] | |
| S31-201 & analogs | 2007 | SH2 | Dimerization | [196] | |
| Natural inhibitors | Curcumin | 2014 | SH2 | Phosphorylation | [198-200] |
| Cucurbitacin E | 2010 | SH2 | Phosphorylation | [221] | |
| Alantolactone | 2015 | SH2 | Unknown | [222] | |
| Cryptotanshinone | 2009 | SH2 | Unknown | [227] | |
| Piperlongumine | 2015 | SH2 | Unknown | [223] | |
| Silibinin | 2015 | SH2 | Unknown | [224] | |
| Direct inhibitors targeting DBD and other domains of STAT3 | |||||
| Classification | Inhibitors | Year | Target site | Mode of targeting STAT3 | Reference |
| Small-molecule inhibitors | C48 | 2011 | DBD | DNA binding | [229] |
| InS3-54 & analogs | 2014 | DBD | DNA binding | [222, 230] | |
| MMPP | 2017 | DBD | DNA binding | [222] | |
| Platinum compounds | IS3-295 | 2005 | DBD | DNA binding | [231, 232] |
| CPA-1 | 2004 | DBD | DNA binding | [231, 232] | |
| CPA-7 | 2004 | DBD | DNA binding | [231, 232] | |
| Platinum (IV) tetrachloride | 2004 | DBD | DNA binding | [231, 232] | |
| Natural inhibitors | Galiellalactone | 2014 | DBD | DNA binding | [233, 234] |
| Peptides | 2007 | ND | Transcriptional activity | [192] | |
| ST3-Hel2A-2 | 2013 | ND | Transcriptional activity | [235] | |
| K116 | 2018 | TAD | Dimerization | [236] | |
2. STAT3
2.1 The STAT and JAK families
Members of the STAT protein family are localized in the cytoplasm, can translocate into the nucleus to bind DNA, and dually function in signal transduction and transcriptional regulation. These proteins have been shown to participate in diverse cellular processes, including stem cell maintenance, lipid metabolism, neuron function, carcinogenesis, inflammation, and immunity [16, 23-26].
STATs range in size from 750 to 850 amino acid residues and contain the following 6 conserved domains: 1) a helical N-terminus domain (ND) that can promote binding with DNA and regulate translocation into the nucleus, 2) a coiled-coil domain (CCD) that provides the action site for transcription factors and regulatory proteins, 3) a central DNA-binding domain (DBD) that can determine the specific DNA sequence binding with STATs, 4) a linker domain (LD) that affects DNA binding stability, 5) an Src homology 2 (SH2) domain that recognizes phosphotyrosine residues and is closely related to STAT activation, and 6) a C-terminal transactivation domain (TAD) with a conserved tyrosine residue at position 705 (Tyr-705) and a serine phosphorylation site at 727 (Ser-727) (Figure 1) [22].
Structural characteristics of STATs and JAKs. STATs cover 6 domains: a helical N-terminus domain (ND); a coiled-coil domain (CCD); a central DNA-binding domain (DBD); a linker domain (LD); an Src homology 2 (SH2) domain; and a C-terminal transactivation domain (TAD) with a conserved tyrosine residue at 705 (Y705) and a serine phosphorylation site at 727 (S727). JAKs cover 4 domains: a C-terminal tyrosine kinase domain (JH1); a pseudokinase domain (JH2), which is necessary for maintaining the JAK inactive state and critical for regulating JH1 activity; a FERM (4.1 protein, ezrin, radixin, and moesin) domain; and an SH2 domain.
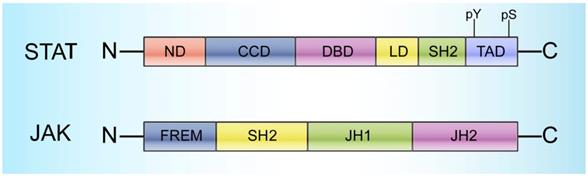
Generally, STATs are localized to the cytoplasm and are inactive; stimulation by diverse cytokines as well as growth factors can trigger their subsequent activation [27]. Janus activated kinases (JAKs) and other tyrosine kinases can activate STATs by phosphorylating the tyrosine residues on the cytoplasmic domain [28-33]. JAKs, a group of receptor-associated cytoplasmic tyrosine kinases, were initially discovered more than 20 years ago and have gained increasing attention due to their critical roles in STAT activation [34-39]. Four JAK family members have been identified to date, namely, JAK1, JAK2, JAK3, and TYK2. All four members range in size from 120-140 kDa, and each member has 7 conserved domains consisting of Janus homologies 1-7 (JH1-7) that can be mainly divided into three parts: 1) a C-terminal tyrosine kinase domain (JH1), which can activate JAKs; 2) a pseudokinase domain (JH2), which is necessary to maintain the JAK inactive state and critical for regulating JH1 activity [17, 32, 40]; and 3) a FERM (4.1 protein, ezrin, radixin, and moesin) domain and an SH2 domain in the N-terminal region (JH3-7), which are responsible for the interactions between JAKs and various cytokine receptors (Figure 1) [14].
Notably, despite considerable homology, STAT proteins share functional differences. Except for STAT5b, other STAT family members have been demonstrated to participate in atherogenesis in different ways. STAT1, STAT4, and STAT5a have been shown to play an essential role in inflammation during atherosclerosis [8, 41, 42]. STAT2 can mediate interferon (IFN) signaling exclusively and then affect atherogenesis [21]. STAT6 was found to participate in immune activity and lipid accumulation and thus contributes to atherosclerosis [20]. In addition, STAT3 is the most-studied STAT protein in atherosclerosis, not only for its effects on all the above activities but also for its roles in endothelial cell dysfunction.
2.2 The structural and functional characteristics of STAT3
As the most conserved protein in the STAT family, STAT3 is composed of the 6 conserved structural domains (ND, CCD, DBD, LD, SH2, and TAD), like other STAT family proteins (Figure 1). Among these domains, both TAD, which has conserved phosphorylation sites at Tyr705 and Ser727, and SH2 can recognize phosphotyrosine residues and are thus closely related to STAT3 activation.
Moreover, STAT3 has been identified as having distinct isoforms, STAT3α, STAT3β, STAT3γ, and STAT3δ, which are considered determinants of its functional heterogeneity [43]. Recently, two of the isoforms, STAT3α and STAT3β, showed contrasting effects during the process of atherosclerosis. STAT3α, participating in the mediation of cellular responses to interleukin (IL)-6, is assumed to exert most pro-oncogenic functions. STAT3β, a spliced transcript of full-length STAT3α, replaces the 55 amino acids in TAD with seven different amino acids. In contrast to the effect of STAT3α, STAT3β can not only inhibit inflammatory cytokine synthesis but also promote the expression of certain anti-inflammatory genes [44-46]. Mice deficient in both STAT3β and apolipoprotein E (apoE) showed enhanced atherosclerotic plaque formation, most likely due to the unopposed action of STAT3α [47].
STAT3 functions as an essential signal transduction effector protein for cytokine- and hormone-induced pathways that control the development, proliferation or differentiation, and homeostasis of numerous cell types. Like other STATs, STAT3 is mostly activated by phosphorylation of its tyrosine and serine residues via signaling from upstream regulators [48, 49]. This phosphorylation event induces dimerization between two STAT3 molecules via reciprocal phosphotyrosine-SH2 interactions. Activated STAT3 dimers then translocate to the nucleus and bind to the consensus promoter sequences of their target genes to initiate transcription. Studies that modulate constitutive STAT3 activation by genetic and pharmacological approaches have verified the critical roles of STAT3 in cell proliferation, apoptosis, angiogenesis, metastasis, and immune responses [50].
However, like phosphorylated STATs, unphosphorylated STAT proteins can also translocate to and prominently exist in the nucleus in various types of mammalian cells in quiescence [51-55]. Notably, studies have shown that unphosphorylated STAT3 sustains cytokine-dependent signaling for long periods through a mechanism completely distinct from that used by phosphorylated STAT3 (p-STAT3) [55]. Yang and colleagues have shown that the formation of tyrosine-phosphorylated STAT3 (p-STAT3) is stimulated by some gp130-linked cytokines, such as IL-6, accompanied by the activation of many genes, including Stat3 itself [55]. The increase in the concentration of unphosphorylated STAT3 drives the expression of a second wave of genes, including RANTES, IL-6, IL-8, MET, and MRAS, which do not respond directly to p-STAT3 [55].
Thus, STAT3 functions in two distinct ways in cytokine-dependent transcription: by playing a role in the primary response through formation of p-STAT3 dimers and by playing a secondary role in the complete response through the action of increased amounts of unphosphorylated STAT3.
2.3 STAT3 signaling pathway is involved in atherosclerosis
2.3.1 IL-6 cytokine family/JAK2/STAT3 signaling pathway
The IL-6 family of cytokines, including IL-6, leukemia inhibitory factor (LIF), oncostatin M (OSM), neuropoietin (NP), cardiotrophin-1 (CT-1), and ciliary neurotrophic factor (CNTF), are important regulators of JAK2/STAT3 signaling pathway activation in the development of atherosclerosis [34, 56]. First, IL-6 family cytokines bind to their corresponding receptor, enabling activation of the expressed signal transductor gp130; this event is followed by the dimerization and phosphorylation of JAK2 and STAT3 [57]. Next, phosphorylated STAT3 dimers translocate into the nucleus and promote the transcription of target genes, which exert regulatory effects on various activities, such as endothelial cell injury and immune responses [58]. H2O2-induced cell apoptosis and death depend on JAK2 and STAT3 activation [59, 60]. Notably, activation of the JAK2/STAT3 pathway is closely associated with the IL-6 cytokine family, which plays an essential role in endothelial cell dysfunction during atherosclerosis. Furthermore, as an important proinflammatory cytokine, IL-6 exerts a profound influence on STAT3-mediated inflammation in atherosclerosis. In vitro studies have demonstrated that after activating the JAK2/STAT3 pathway in vascular endothelial cells, IL-6 upregulates the expression of monocyte chemotactic protein-1 (MCP-1) and exerts a series of proinflammatory effects [61]. IL-6 also participates in the differentiation of immune cells through the JAK2/STAT3 signaling pathway. In conclusion, this signaling pathway is involved in the process of atherosclerosis, and its modulation may provide an effective therapeutic option for atherosclerosis.
2.3.2 IL-10/JAK/STAT3 signaling pathway
IL-10 is an immunomodulatory cytokine that has potent anti-inflammatory activity, and its signaling pathway has been well characterized in macrophages and T lymphocytes. IL-10 signaling is mediated via a transmembrane receptor, which consists of a ligand-binding IL-10 receptor1 (IL-10R1) chain and an accessory subunit, IL-10R2 [62]. The binding of IL-10R1 to IL-10 induces conformational changes and dimerization with IL-10R2, exerting downstream effects [63]. Following activation, the IL-10 receptor regulates cellular activity via the JAK-STAT3 signaling pathway [64], resulting in recruitment of the cytoplasmic protein JAK1 followed by phosphorylation of the tyrosine at position 705 in the STAT3 molecule. p-STAT3 forms a homodimer and then translocates to the nucleus to enhance the transcriptional regulation of target genes, such as vascular endothelial growth factor A (VEGF-A), basic fibroblast growth factor-2 and placental growth factor, all major angiogenic factors [65]. Loss-of-function approaches targeting the IL-10/STAT3 pathway utilizing genetic and antibody-mediated modalities have shown decreased angiogenesis in vivo and in vitro [65]. Notably, IL-10 expression has been detected in human atherosclerotic plaques, primarily in macrophages [66]. Moreover, disruption or cell-specific overexpression of IL-10 in murine models results in modifications of the lesion size, immune cell accumulation, and cell pattern phenotype in the lesion [67-70]. Unlike the STAT3 signaling pathway induced by IL-6, the IL-10/JAK/STAT3 signaling pathway has anti-inflammatory functions in macrophages [18]. Taken together, these results suggest that this signaling pathway plays a pivotal role in the process of atherosclerosis, especially during macrophage polarization and neovascular proliferation.
2.3.3 Regulation of the STAT3 signaling pathway
STAT3 can be phosphorylated and activated by different factors, among which the activation process through JAKs is the best studied. Various types of stimuli, including cytokines and growth factors, can initiate STAT3 activation. When receptors on the membrane bind with their ligands (e.g., cytokines and growth factors) and dimerize, pairs of JAKs are recruited to the receptor domains and initiate self-activation via either auto- or transphosphorylation [18]. Then, the activated JAKs phosphorylate tyrosine residues in the intracellular receptor domains, which can be recognized by the SH2 domain of STAT3 and provide a docking site for STAT3 [21]. Consequently, the activated JAKs phosphorylate Tyr 705 of STAT3, which contributes to the binding of STAT3 molecules to the receptor domains and the homo- or heterodimerization of STAT3. Next, the homo- or heterodimers of STAT3 translocate to the nucleus, bind to specific sequences on the promoters of target genes and induce the transcription of target genes, such as SOCS3, MCP-1, and VEGF-A [33, 61, 65, 71]. The palindromic DNA core motif recognized by STAT3 dimers with 2-fold symmetry is called the GAS element (IFNγ activation site, TTCN2-4GAA) [72]. The binding affinity to GAS elements varies among STAT proteins. For example, TTC(N)3GAA is an optimal variant for STAT1, which prefers binding sites containing an intrasite spacer of three bases (CCG). On the other hand, STAT3, STAT4, and STAT5a/b prefer to bind at sites with 2- to 4-base spacers, and most high-equilibrium binding intensity interactions occur at sites with a 3-base spacer [73, 74] Interestingly, although the phosphorylation of STAT3 is important for its function, the translocation of STAT3 from the cytoplasm to the nucleus may be independent of its phosphorylation status due to the constitutive binding of STAT3 to importin α-3 [53]. Moreover, STAT3 can also be phosphorylated by some receptor tyrosine kinase (RTKs), including epidermal growth factor receptor (EGFR) and insulin-like growth factor receptor (IGFR), and other nonreceptor kinases, including Src and Abl [7].
Molecules that negatively regulate STAT3, including protein tyrosine phosphatases (PTPs), protein inhibitors of activated STAT (PIASs), and suppressor of cytokine signaling 3 (SOCS3), are critical to prevent its hyperphosphorylation. PTPs regulate the activation of STATs through direct and indirect dephosphorylation of p-STAT3. The indirect regulators, including CD45 and PTP-1B, can downregulate STAT3 activation via dephosphorylation of JAK2 [36, 37]. SH2-containing protein tyrosine phosphatase (SHP)-1, SHP-2, and PTP receptor T (PTPRT) can dephosphorylate and inactive STAT3 directly [32, 35] Notably, PTPRT specifically dephosphorylates the Tyr705 residue of STAT3 and thus regulates the cellular localization and target gene expression of STAT3 [32]. Additionally, PIAS3 inhibits the binding of dimerized p-STAT3 to DNA, eventually blocking the target gene transcription of STAT3 [17]. In particular, cytokines are important for the occurrence and development of atherosclerosis due to their participation in several pivotal signaling pathways associated with atherosclerosis. SOCS3 proteins are considered crucial in the regulation of the cytokine-JAK-STAT3 signaling pathway [75]. SOCS3 modulates JAK/STAT3 signaling in a negative feedback loop that utilizes 3 mechanisms: kinase-mediated inhibition of JAKs via the kinase inhibitory region (KIR) domain located at the C-terminus, binding site competition with STATs for initiating JAKs, and gp130 degradation via the SOCS box located at the N-terminus [13, 14]. Furthermore, prolonged phosphorylation in SOCS3 gene-deficient mouse macrophages due to IL-6 stimulation suggests that SOCS3 plays an important role in controlling the responses to IL-6 [76]. Previous studies have demonstrated that the proinflammatory cytokine IL-6 and the anti-inflammatory cytokine IL-10 share the same STAT3 signaling pathway, which induces SOCS3 expression. SOCS3 targets gp130, a receptor for IL6, but not IL-10R, which results in shortened IL-6-driven STAT3 activity, while IL-10-driven STAT3 activation is prolonged [77]. The downregulation of JAK-STAT activation secondarily induces the expression of SOCS3, which is a negative regulator of STAT3. This negative feedback through SOCS decreases cellular sensitivity to cytokines and appears to be necessary for suppressing inflammation and cellular proliferation [32, 78].
3. Physiopathological roles of STAT3 in atherosclerosis
Atherosclerosis is an intricate process involving multiple cell types, such as endothelial cells and macrophages. In Table 2, we present the essential roles of these cells in the development of atherosclerosis. Atherosclerosis can be triggered by several factors, resulting in dysfunction of the endothelium and accumulation of oxidized low-density lipoproteins (ox-LDLs) in the intima. Ox-LDLs then trigger the expression of adhesion molecules and the secretion of chemokines by endothelial cells, driving monocyte migration and adhesion to the endothelium. Afterwards, the secretion of macrophage colony-stimulating factor (M-CSF) induces the differentiation of monocytes into macrophages, where scavenger receptors recognize and take up highly ox-LDL particles, ultimately leading to foam cell formation [79]. Excessive inflammatory and immune responses communicated by these different cell types are driven by inflammatory cytokines and other inflammatory stimuli that promote associated tissue damage and contribute to local inflammation and vascular dysfunction [80-82]. Notably, STAT3 can be divided into nuclear STAT3 and mitochondrial STAT3 according to its translocation, and both are believed to play important roles in the development of atherosclerosis, including endothelial cell dysfunction, macrophage polarization, inflammation and immunity.
Roles of different cell types in the development of atherosclerosis
| Cell type | Stage of atherosclerosis in which they participate | Trigger factor | Effects of STAT3 | Effect on the development of atherosclerosis | Reference |
|---|---|---|---|---|---|
| Endothelial cells | Mainly in the initial stage of atherosclerosis | ROS or sustained STAT3 phosphorylation | Causing endothelial cell dysfunction, including activation of RhoA, rearrangement of microfilaments and microtubules, upregulation and overexpression of adhesion molecules | Vascular endothelial cell dysfunction is the initiator of and key link to ensuing atherosclerosis. Promoting accumulation of lipids, adhesion of inflammatory cells, and proliferation of VSMCs. | [84, 88, 90, 91, 93-95] |
| Macrophages | Almost in all stages of atherosclerosis | TNF-α; IL-1β | Affecting the number of macrophages in the microenvironment by regulating monocyte-to-macrophage differentiation, participating in M1 phenotype polarization | Expressing scavenger receptors, taking up high ox-LDL particles, leading to foam cell formation. Macrophages can adopt different functional phenotypes according to the atherosclerotic environment. M1 is a proinflammatory phenotype that leads to atherosclerosis, while M2 is an anti-inflammatory phenotype that maintains the stability of atherosclerotic lesions | [65, 79, 101, 103, 108-113] |
| VSMCs | In the progressive stage of atherosclerosis | Ang-II; atherogenic factors including PDGF-BB, endothelin-1, thrombin, and IL-1 | Promoting Ang-II- and PDGF-induced VSMC proliferation and migration; regulating the phenotypic switch of VSMCs | Excessive proliferation and migration of abnormal VSMCs are major causes of the development of cardiovascular diseases, including atherosclerosis; excessive synthesis of extracellular matrix, thickening of arterial walls, and development of atherosclerotic plaques | [126-128, 131-136, 140] |
| CD4+ T cells | In all stages of atherosclerosis | IL-6 | Sustaining prolonged cytokine production; contributing to the differentiation of CD4+ T cells; regulating the Th17/Treg ratio | CD4+ T cells, especially Th1, Th17, and Treg cells, participate throughout the entire atherosclerosis process, playing important roles in the rupture of atherosclerotic plaques and Th17/Treg cell imbalance, leading to atherosclerosis | [121, 142-144, 154, 157-160] |
| DCs | In the late stage of atherosclerosis | IL-6; IL-12 | Inhibiting IL-12 cytokine production | Promoting antigen presentation and T cell activation in the lesion; Th cell polarization by IL-12 secretion | [161-165] |
3.1 Endothelial cell dysfunction
Endothelial cells play central roles in the functions of the cardiovascular system [83]. Endothelial cell dysfunction causes the accumulation of lipids, inflammatory cells, and coagulation materials as well as vascular smooth muscle cell (VSMC) proliferation, thus promoting atherosclerotic plaque formation [84]. Numerous clinical studies have shown that vascular endothelial cell dysfunction is the initiator of and key link to ensuing atherosclerosis [84]. Furthermore, endothelial cell dysfunction is closely related to injuries induced by various types of hazards (smoking, hyperlipidemia, oxygen free radicals, etc.) [85]. Oxygen free radicals, collectively known as reactive oxygen species (ROS), are the main cause of endothelial cell injury [85]. Increased ROS levels are due to an imbalance between their production and elimination and characteristic of oxidative stress. Thus, the injury caused by ROS is classified as oxidative stress injury (OSI) and can increase endothelial permeability, promote leukocyte adhesion, and change endothelial gene expression.
Recently, an increasing number of studies have demonstrated the high activation of the JAK2/STAT3 signaling pathway in the OSI of various cell types (glioma cells, VSMCs, endothelial cells, etc.), which suggests the essential role of this pathway in the modulation of oxidative stress responses [86, 87]. Ivan H.W. Ng and colleagues have shown that in a murine embryonic fibroblast (MEF) model system, H2O2 may cause OSI in endothelial cells through the IL-6/JAK2/STAT3 signaling pathway, the high activation of which allows STAT3 to maintain a phosphorylated state [88]. Under normal conditions, cytokine-induced STAT3 phosphorylation is rapid and transient. T cell protein tyrosine phosphatase (TC-PTP) [89] and SOCS3 play important roles in the negative regulation of STAT3 activation, which limits sustained STAT3 phosphorylation. In H2O2-simulated oxidative stimulation, the activation rate of SOCS3 slows down and the STAT3 phosphatase TC-PTP (TC45) mislocalizes to the cytoplasm, both of which weaken their inhibition of STAT3 phosphorylation [88]. These results show that STAT3 does not dephosphorylate in time in instances of endothelial cell injury.
Sustained STAT3 phosphorylation also leads to the abnormal expression of adhesion molecules [90]. Adhesion molecules are currently believed to induce inflammation and thus play an important role in the development of atherosclerosis [91]. In the early stage of atherosclerosis, adhesion molecules mainly promote monocyte migration, and these cells further adhere to the endothelium [91, 92]. As atherosclerosis progresses, adhesion molecules can promote a cascade of mononuclear cells migrating to the lesions, T lymphocyte activation, and interactions between different cells [91]. Afterwards, adhesion molecules mediate more cells into plaques, further prompting plaque development and affecting plaque stability [91].
There are 3 main adhesion molecules related to atherosclerosis: intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and platelet endothelial cell adhesion molecule-1 (PECAM-1) [91]. Activation of the IL-6/JAK2/STAT3 signaling pathway can lead to the upregulation and overproduction of ICAM-1 and VCAM-1 in endothelial cells [90]. The phosphorylation of STAT3 can activate Ras homolog gene family member A (RhoA), which is an essential regulator, to rearrange the microfilaments and microtubules as well as inhibit the phosphorylation of endothelial nitric oxide synthase (eNOS) [93-95]. eNOS phosphorylation can suppress the effect of matrix metallopeptidase 9 (MMP-9), thus downregulating the expression of ICAM-1 and VCAM-1 [90]. Thus, the suppression of eNOS promotes the expression of ICAM-1 and VCAM-1, which can cause more cells to adhere to and affect the stability of plaques (Figure 2).
Schematic diagram of the impact of STAT3 on endothelial cell dysfunction and some STAT3 inhibitors during this process. In the stimulation of ROS, the transcription rate of SOCS3 slows down, and STAT3 phosphatase TC-PTP (TC45) mislocalises to the cytoplasm, both of which weaken their inhibition of STAT3 phosphorylation and promote the hyperphosphorylation of STAT3. The hyperphosphorylation of STAT3 can activate RhoA which can rearrange the microfilaments and microtubules through its direct effect or through phosphorylating FAK indirectly. RhoA could inhibit the phosphorylation of eNOS thus promoting the effect of MMP-9 to improve the expression of ICAM-1 and VCAM-1. The combination of these mechanisms leads to dysfunction of endothelial cells. The whole process involves five classes of STAT3 inhibitors including indirect inhibitors and direct inhibitors that play an essential role in the phosphorylation, dimerization, DNA-binding, and transcriptional activity of STAT3. ROS, reactive oxygen species; SOCS3, suppressor of cytokine signaling 3; TC-PTP, T-cell protein tyrosine phosphatase; RhoA, Ras homolog gene family member A; eNOS, endothelial nitric oxide synthase; MMP-9, matrix metallopeptidase 9; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1.
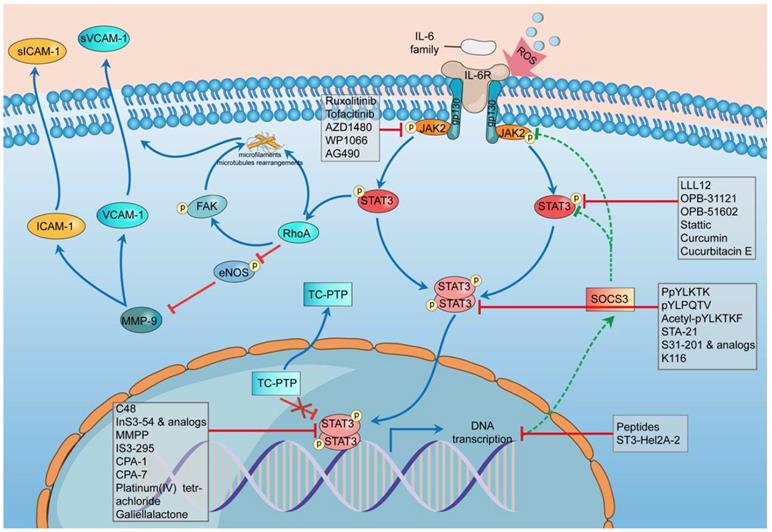
In the stimulation of ROS, the transcription rate of SOCS3 slows down, and STAT3 phosphatase TC-PTP (TC45) mislocalizes to the cytoplasm, both of which weaken their inhibition of STAT3 phosphorylation and promote the hyperphosphorylation of STAT3 [88]. The hyperphosphorylation of STAT3 can activate RhoA, which can rearrange the microfilaments and microtubules through its direct effect or indirectly phosphorylate FAK [96-98]. RhoA could inhibit the phosphorylation of eNOS, thus promoting the effect of MMP-9 on improving the expression of ICAM-1 and VCAM-1 [96, 99, 100]. The combination of these mechanisms leads to endothelial cell dysfunction.
One important manifestation of endothelial cell dysfunction is the abnormal expression of adhesion molecules, which can be caused by ROS through the IL-6/JAK2/STAT3 signaling pathway. This pathway can be highly activated by ROS, leading to the sustained phosphorylation of STAT3 and the further overrelease of ICAM-1 and VCAM-1. Excess adhesion molecules induce inflammatory responses and aggravate endothelial cell dysfunction, thus prompting the development of atherosclerosis [91].
3.2 Macrophage polarization
Macrophages are now thought to be present in all stages of atherosclerosis, from the initiation and expansion of lesions to necrosis or rupture, and have become a clinical manifestation of atherosclerosis [101]. Macrophages can release a diverse repertoire of inflammatory mediators and construct an inflammatory environment in the atherosclerotic neointima [102], leading to the proximal exacerbation of arterial damage. Notably, the number of macrophages mainly depends on the infiltration and differentiation of monocytes. Thus, monocyte-to-macrophage differentiation is a critical event that accentuates atherosclerosis by promoting an inflammatory environment within the vessel wall [103]. STAT3 participates in regulating monocyte-to-macrophage differentiation, and inhibition of STAT3 activity suppresses both inflammation and monocyte-to-macrophage differentiation [103].
Macrophages can respond to various environmental stimuli by adopting one of several functional phenotypes, such as the classically activated macrophage phenotype (M1), the alternatively activated macrophage phenotype (M2), and the Mox macrophage phenotype [104-106]. Although they are all secrete inflammatory molecules and factors that further regulate lipoprotein retention, functional differences exist in these different macrophage phenotypes [107]. M1 macrophages can produce proinflammatory cytokines, including IL-6, IL-12, and tumor necrosis factors (TNFs), as well as toxic agents, such as nitric oxide (synthesized by inducible NO synthase, iNOS) and free oxidative radicals, thus promoting increased and sustained inflammation and leading to an acute atherothrombotic vascular event [108-113]. M2 macrophages are anti-inflammatory macrophages identified by the expression of molecules such as IL-10, arginase 1 (Arg1), and Mrc1 (also known as CD206) and are involved in tissue repair, enhancing plaque stability during atherosclerosis [114-117]. Jinjin Cui and colleagues found that the JAK2/STAT3 signaling pathway is involved in M1 polarization [118]. Moreover, STAT3 signaling is an essential determinant of the alternative M2 phenotype [65]. Targeted inhibition of both IL-10 receptor-mediated signaling and STAT3 activation in macrophages reverses the aging phenotype [65]. Given the crucial roles of various macrophage phenotypes in the pathogenesis and progression of atherosclerosis, intervening with macrophage polarization represents a potentially effective therapeutic strategy for atherosclerosis. Therefore, STAT3 may become a potent target to treat atherosclerosis via regulating the polarization of macrophages.
3.3 Inflammation
Basic and clinical studies have shown that unchecked chronic inflammation is responsible for many deadly cardiovascular diseases, including atherosclerosis [61]. Atherosclerosis is closely correlated with inflammation and exhibits diverse inflammatory behaviors at different stages. In the early stage of atherosclerosis, inflammation is mainly associated with mononuclear macrophage infiltration and increased secretion of proinflammatory cytokines, including IL-6, TNF-α and IL-1β, while in the progressive stage of atherosclerosis, it mainly manifests as massive VSMC proliferation [61, 119, 120]. Additionally, studies have found that p-STAT3 mainly localizes in the endothelial nucleus of the inflammatory response region of atherosclerotic plaques but not in the noninflammatory response region, which strongly indicates that STAT3 activation is involved in the atherosclerotic inflammation.
3.3.1 Early stage of atherosclerosis: cytokine secretion
Human atherosclerotic plaques contain various inflammatory cell types, including monocytes, macrophages, and mast cells. Recruitment of these inflammatory cells by adhesion molecules (ICAM-1 and VCAM-1) and E-selectin in endothelial cells is the critical step for inducing the formation of atherosclerotic plaques [121, 122]. This stage is characterized by the secretion of some cytokines, mainly IL-6, TNF-α and IL-1β.
TNF-α and IL-1β have been demonstrated to induce the massive IL-6 expression and aggravate vascular inflammation, thus promoting the formation of atherosclerotic plaques [123]. Together with TNF-α and IL-1β, IL-6 is thought to participate in B cell maturation and T cell differentiation and drive acute inflammatory response [61]. However, sustained IL-6 production plays a role in the chronic low-level inflammation associated with atherosclerosis [124, 125]. Moreover, IL-6 has been detected in atherosclerotic plaques, and it mainly functions by the JAK/STAT3 signaling pathway in atherosclerosis [61, 119]. Binding of the IL-6/IL-6R complex to gp130 results in activation of the JAK/STAT3 signaling pathway and induction of proinflammatory IL-6-responsive genes, including MCP-1 and ICAM-1, inducing a series of proinflammatory effects [61, 71].
3.3.2 Progressive stage of atherosclerosis: VSMC proliferation and migration
Excessive proliferation and migration of abnormal VSMCs are major causes of atherosclerosis development [126]. In response to atherogenic factors secreted by endothelial cells and leukocytes, including platelet-derived growth factor (PDGF)-BB, endothelin-1, thrombin, and IL-1, VSMCs proliferate massively, migrate into the intima, and modify the inflammatory phenotype in atherosclerosis and thus are the main cell type associated with arterial intimal thickening [127]. VSMCs can be classified into two major phenotypes: one includes fully differentiated, contractile cells responsible for vasodilation and vasoconstriction, and the other consists of migratory, proliferative cells activated during growth or injury [128]. Notably, switching of the two phenotypes from the former to the latter is known to be vital for stable plaque formation [128]. However, VSMC apoptosis leads to drastic vessel remodeling, with increased inflammation and coagulation and thinning of the fibrous cap, making the plaque more prone to rupture [129, 130]. Thus, reducing the excessive proliferation of VSMCs, improving their functions, and preventing their apoptosis could substantially slow the development of atherosclerosis.
Accumulating studies have demonstrated that activated STAT3 participates in the proliferation of VSMCs. After inhibiting STAT3 in human aortic VSMCs cells, cell proliferation is obviously reduced, while activating STAT3 via LIF enhances cell proliferation [131]. Angiotensin (Ang)-II promotes oxidative stress and mediates VSMC proliferation, thus playing an essential role during this period. The JAK2/STAT3 signaling pathway, a well-known contributor of oxidative stress, is closely associated with the Ang-II type 1 receptor and nuclear transcriptional changes, such as the activated transcription of early growth response genes, eventually resulting in VSMC proliferation [132-135]. Moreover, a study has verified that Ang-converting enzyme 2 (ACE2), a specific Ang-II-degrading enzyme, can attenuate VSMC proliferation via suppressing the activation of the JAK2/STAT3/SOCS3 signaling pathway [136].
The transcript levels of PDGF-BB in human atherosclerotic plaques are increased compared to those in normal arteries [137]. Moreover, as the most potent mitogen, PDGF-BB has been shown to induce VSMC migration and intimal hyperplasia in the arteries of carotid injury model rats, mice, and porcine in vivo [138]. Importantly, accumulating evidence supports an important role for the activation of STAT3 in PDGF-BB-induced VSMC proliferation and migration [139]. A previous study demonstrated that PDGF-BB-induced VSMC motility requires activation of the JAK2/STAT3 signaling pathway [140]. PDGF-BB can promote the tyrosine phosphorylation of JAK2 and STAT3 in a time-dependent manner [140]. However, the dominant negative mutant-dependent suppression of JAK2 and STAT3 can block PDGF-BB-induced VSMC migration [140]. These results indicate that the JAK2/STAT3 pathway plays an important role in PDGF-BB-induced VSMC migration.
Phenotypic switching is also a pivotal step underlying many VSMC-related pathological conditions, especially atherosclerosis. Liao and colleagues have demonstrated that the JAK/STAT3 signaling pathway is a central regulator of the VSMC phenotypic switch [131]. These researchers found that knockdown of endogenous STAT3 enhances the VSMC contractile phenotype by promoting the association of the myocardin/serum response factor-CArG complex. In contrast, the activated STAT3 signaling pathway suppresses the expression of VSMC-specific contractile protein genes and is thus positively correlated with the synthetic VSMC phenotype [131]. Therefore, the phenotypic switch of VSMCs can be controlled by modulation of the JAK/STAT3 signaling pathway. Inhibition of STAT3 activation can prevent the VSMC contractile phenotype from switching to the inflammatory phenotype, eventually slowing the progression of atherosclerosis.
3.4 Immunity
Immune cells, especially CD4+ T cells, play a central role in the physiological process of atherosclerosis based on their vital roles in the cellular immune network. CD4+ T cells are divided into various cell types according to different effectors, including Th1, Th2, Th9, Th17, Th22, T follicular helper (Tfh), and regulatory T (Treg) cells [141].
After the discovery of Th17 cells, several groups addressed their potential contribution to atherogenesis [142]. Moreover, elevated numbers of Th17 cells, which produce the proinflammatory molecule IL-17A, are associated with autoimmune diseases and have been observed in atherosclerotic lesions [143]. Recently, Th17 cell processes were verified to be closely related to the occurrence and development of atherosclerosis, and STAT3 is the key regulator of Th17 cell differentiation through IL-6 induction [121, 144]. The combined stimulation of transforming growth factor (TGF)-β and IL-6 can initiate the differentiation of CD4+ T cells into Th17 cells in mice [145]. IL-6 upregulates the expression of IL-21 through the STAT3 pathway, which then increases the expression of the IL-23 receptor and the retinoic acid-related orphan receptor (ROR)γt [146, 147]. In cooperation with STAT3, RORγt promotes the expression of IL-17 and inhibits the expression of forkhead transcription factor p3 (Foxp3) [148, 149]. In the early stage of atherosclerosis, IL-6 inhibits Foxp3 and promotes the expression of RORγt by activating STAT3 [150]. In the intermediate stages, IL-21 secreted by the cell itself promotes the expression of the RORγt and IL-23 receptors through activation of STAT3, resulting in a positive feedback effect [151-153]. During the later stages, IL-23 also promotes the expression of IL-22 and inhibits the effects of IL-10 through STAT3, enabling the complete differentiation of Th17 cells [150]. Furthermore, IL-6-mediated mitochondrial Ca2+ sustains the production of two cytokines (IL-21 and IL-4) known to be regulated by IL-6 in CD4+ cells [154]. Thus, mitochondrial STAT3 can sustain prolonged cytokine production and contribute to the differentiation of CD4+ T cells in atherosclerosis.
Treg cells provide protection against autoimmunity and are regarded as promising targets of clinical therapies to treat various diseases caused by autoimmunity, including atherosclerosis [155, 156]. Treg cells can modulate several processes involved in the development of atherosclerosis. For example, Tregs can inhibit proatherogenic T cells, dendritic cell (DC) activation and migration, macrophage inflammation, foam cell formation, EC activation and affect cholesterol metabolism [157]. STAT3 mutations disenable Treg cells to produce IL-17, indicating that IL-17 secretion and Treg cell functions depend on STAT3 [158]. Additionally, the Th17/Treg cell imbalance plays key roles in atherosclerosis in apoE(-/-) mice [159]. STAT3 can regulate the Th17/Treg ratio, which is closely related to the amount of IL-6 and the number of Treg cells [160].
A comparison of DCs in different intima showed that the atherosclerotic intima contains significantly more DCs than the normal intima, suggesting that DCs play an active role in the initial stage of atherosclerosis [161]. Studies have demonstrated that DCs distributed in atherosclerotic lesions are mainly responsible for antigen presentation and T cell activation in the lesion [102]. IL-6 secreted by mature DCs plays an important role in the differentiation of CD4+ T cells into Th17 cells by the IL-6/JAK2/STAT3 pathway [162]. Additionally, T cell activation and Th cell polarization are shaped by costimulatory molecule engagement and exposure to a specific cytokine milieu, with Th1 cells critically depending on IL-12 secretion from DCs. Studies have shown that STAT3 inhibits IL-12 cytokine production in DCs and increases STAT3 mRNA expression but decreases IL-12 transcript expression in advance compared with that in early lesions [163-165]. These data indicate that STAT3 in DCs plays an essential role in the development of atherosclerotic lesions.
3.5 STAT3 in mitochondria
In addition to its well-established roles in the nucleus during the progression of atherosclerosis, STAT3 is also present in mitochondria and contributes to the regulation of the electron transport chain (ETC) activity. As a major source of cellular ROS, mitochondrial-derived reactive oxygen species (mtROS) are natural byproducts of the ETC [166]. Importantly, several lines of evidence indicate that excessive mtROS-induced oxidative damage occurs in atherosclerotic lesions in both animal models and humans, indicating that excessive mtROS is associated with atherosclerosis progression [166]. Although not required for mitochondrial function, mitochondrial STAT3 can directly interact with ETC complexes, enhancing ETC activities independent of nuclear activity and eventually contributing to enhanced oxidative phosphorylation (OXPHOS) and ATP production [18, 167]. Additionally, mitochondrial STAT3 is a regulator of ETC and Ca2+ homeostasis and affects the mitochondrial production of ATP and ROS, thus playing a crucial role in atherosclerosis.
4. Physiopathological roles of other STATs in atherosclerosis
In addition to STAT3, other STATs, especially STAT1 and STAT2, also play essential roles in atherosclerosis. STAT1 has been identified as a regulator of foam cell formation and atherosclerotic lesion development in an intraperitoneal inflamemation model and an atherosclerosis-susceptible bone marrow transplantation mouse model [168]. Moreover, increased STAT1 activity results in VSMC proliferation and neointimal hyperplasia, while deficient STAT1 in the bone transplantation mouse model reduces macrophage apoptosis and plaque necrosis [169, 170]. STAT1 also elevates the expression of chemokines and promotes oxidative stress and tissue injury by stimulating the NADPH oxidase gene and protein expression [171, 172]. Moreover, apoF is a sialoglycoprotein component of high-density lipoproteins (HDL) and LDL fractions in human serum. Although the exact role of STAT2 in atherosclerosis has not been reported, genetic manipulation of the apoF/Stat2 locus supports an important role for STAT2-dependent type I IFN signaling and gene expression in atherosclerosis [173].
5. Inhibitors
The above findings provide convincing evidence that STAT3 can be a novel therapeutic target for atherosclerosis. Therefore, intensive efforts have been devoted to developing STAT3 inhibitors. Currently, there are two strategies for inhibiting the STAT3 signaling pathway: indirect and direct STAT3 inhibitors. The former strategy can block molecules upstream of the STAT3 signaling pathway and indirectly inhibit the signal transduction functions of STAT3, for example, by inhibiting the function of JAKs, Src, Abl, and Lyn [174-181]. The latter strategy can be divided into several types according to different target domains, including the SH2 domain, DBD, ND, and TAD. Here, we summarize important STAT3 inhibitors targeting the JAK2/STAT3 signaling pathway and STAT3 structural domains.
5.1 Indirect inhibitors
Since the substitution of valine with phenylalanine at amino acid 617 (V617F) within the JH2 'kinase-like' domain of JAK2 was demonstrated to result in an overactivation of JAK2, inhibitors targeting JAK2 specifically have become the focus of studies. Over the years, numerous JAK2 inhibitors have been designed, including ruxolitinib, tofacitinib, AG490, AZD1480, SB1578, and WP1066 [182-185]. These inhibitors inhibit the JAK2/STAT3 signaling pathway in a similar way, which indicates that they may function by suppressing immune and inflammatory responses during the development of atherosclerosis. Notably, ruxolitinib and tofacitinib have been approved by the FDA for the treatment of myelofibrosis and rheumatoid arthritis [186]. However, their potential application in the treatment of atherosclerosis has not been investigated. AG490 has been widely used as a JAK2 inhibitor in cardiovascular research but has no clinical application [187]. The most promising novel substance is WP1066, which achieved outstanding results in the treatment of malignant and vascular diseases. In preclinical studies, WP1066 was not only shown to significantly impact the prevention of tumor angiogenesis but also to successfully prevent neointima formation and to contribute to plaque stability in atherosclerosis [182, 183, 188]. However, these kinase inhibitors, which are similar in structure to those of other kinases, play a major role in the enzymatic catalytic center and thus often exert off-target effects. Recently, flavonoids have become a research hotspot due to their potential to inhibit STAT3 activity. Surprisingly, researchers have demonstrated that tricin, a bioflavonoid expressed at significantly high levels in rice bran of Njavara, can significantly inhibit the activation of both STAT1 and STAT3 via the downregulation of upstream phosphorylation enzymes such as JAK1 and JAK2, exerting a potent anti-inflammatory effect [189]. Additionally, the heterocyclic small molecules naringenin and flavone exhibit the novel combined functions of inhibiting IL-6-promoted STAT3 and inducing SOCS3 in vascular endothelial cells. Nevertheless, their mechanism of inhibition contrasts with that of structurally related traditional JAK inhibitors, which inhibit both IL-6-promoted STAT3 activation and SOCS3 induction independently [61]. Taken together, these results suggest that indirect inhibitory strategies through traditional JAK inhibitors have relatively large adverse reactions and need further improvement. However, emerging natural inhibitors, such as tricin, are indirect STAT3 inhibitors that could be used to treat atherosclerosis and have fewer side effects than previously used inhibitors. Moreover, the newly identified mechanism by which flavonoids (naringenin and flavone) inhibit STAT3 may provide an avenue for developing novel therapies for atherosclerosis based on the induction of negative STAT3 regulators.
5.2 Direct inhibitors
5.2.1 Inhibitors targeting the SH2 domain
There has been recent interest in the direct inhibition of STAT3, and this approach has mainly focused on targeting the SH2 domain [190, 191]. The SH2 domain, which contains a pocket that binds to another STAT3 protein, is an important domain by which STAT3 maintains its biological functions. After phosphorylation at the Tyr705 site of the STAT3 protein, the SH2 domain binds two activated STAT3 monomers to form a dimer, translocates into the nucleus, and binds to DNA to initiate the transcription of specific target genes. Inhibition of the SH2 domain results in impairment of not only STAT3 activation and dimerization but also the subsequent nuclear translocation and expression of STAT3-regulated genes.
Peptide and peptide-like STAT3 inhibitors, including PpYLKTK (P: proline, L: leucine, K: lysine, T: threonine) [192], pYLPQTV (V: valine) [193], and acetyl-pYLKTKF (F: phenylalanine) [194], mainly target the SH2 domain. These inhibitors can impede the binding of two STAT3 monomers, thus preventing the dimerization and nuclear translocation of STAT3 proteins and inhibiting the binding of STAT3 to DNA.
STA-21 (deoxytetrangomycin) and its structurally optimized analog LLL12 can bind to the SH2 domain, blocking STAT3 dimerization and DNA binding [160, 195]. Furthermore, OPB-31121 and OPB-51602 have been demonstrated to interact with the SH2 domain of STAT3 with high affinity [196]. Cell culture assays showed that OPB-31121 and OPB-51602 inhibited the phosphorylation of both Tyr705 and Ser727. Moreover, both of these proteins were able to uniquely bind to the STAT3 SH2 domain, which does not overlap with the binding site for other STAT3 inhibitors. Their distant binding pocket and potent affinity for STAT3 SH2 make OPB-31121 and OPB-51602 the most promising candidates for further development [197]. Phase I/II studies on the use of STA-21 for the treatment of psoriatic lesions and on OPB-31121 and OPB-51602 for the treatment of advanced cancers have been completed, revealing that all of these compounds are effective inhibitors of STAT3 phosphorylation and have acceptable safety profiles. Although they have not been clinically used for atherosclerosis treatment, further clinical development of these compounds is expected in the future. Other notable small-molecule STAT3 inhibitors, including Stattic, S3I-201 and S3I-201 analogs, bind to the STAT3 SH2 domain and inhibit STAT3 activity. Notably, molecular docking studies show that S3I-201, LLL12, and Stattic have highly conserved pTyr-SH2 binding pockets, allowing the primary targeting of STATs but not STAT3 specifically [72].
Another group of inhibitors targeting the SH2 domain of STAT3 are derivatives from natural compounds. Curcumin, a naturally derived phytochemical from plants such as turmeric (Curcuma longa), has many pharmacological activities, including antioxidant, anticarcinogenic, antiobesity, antiarthritic, analgesic, hepatoprotective, antiangiogenesis, and anti-inflammatory properties [198-213]. Previous studies have shown that curcumin may exert antiatherosclerotic effects by regulating a broad spectrum of factors linked to inflammation and oxidative stress [214-216]. Additionally, curcumin affects lipid metabolism, subsequently alleviating hyperlipidemia and atherosclerosis [201]. Notably, curcumin has been shown to inhibit STAT3 phosphorylation and block the STAT3 signaling pathway in various types of cell types [217, 218]. Based on this new insight, curcumin also represents a potential therapeutic option for atherosclerosis as an effective STAT3 inhibitor. Resveratrol is another essential natural inhibitor of STAT3 that has the ability to attenuate some detrimental atherosclerotic processes, such as transportation of LDL, oxidation of lipids, and invasion of macrophages [219, 220]. Furthermore, resveratrol has been tested in preclinical studies and clinical trials for the treatment of atherosclerosis [219]. Preclinical studies showed promising results for the use of resveratrol to treat atherosclerosis, revealing that its antioxidant and anti-inflammatory properties may mediate its antiatherogenic effect [219]. Nevertheless, clinical trials investigating the effect of resveratrol on the plasma lipid profiles in human subjects have not been as clear [219]. Notably, in some experimental conditions, curcumin and resveratrol may activate rather than inhibit STAT3 functions, and the acute activation of STAT3 by these drugs during stroke and myocardial infarction promotes cell survival. Additionally, cucurbitacin E [221], alantolactone [222], piperlongumine [223], and silibinin [224] all bind the STAT3 SH2 domain and thus play essential roles in STAT3 inhibition. However, most of these agents are not STAT3-specific, as they are capable of binding the SH2 domains of all STAT proteins. Smaller compounds, such as resveratrol, were shown to predominantly target only the pTyr-binding cavity, while higher molecular weight compounds, including curcumin and cucurbitacin E, bound additional SH2 cavities [72].
These inhibitors play important roles in endothelial cell dysfunction, anti-inflammation and immunity, which means that they may be promising treatment agents for atherosclerosis. Unfortunately, these inhibitors still have some negative qualities. Although peptides and peptide-like compounds have high biological activities, they are easily metabolically inactivated and have low bioavailability, which limits their further development in medical arenas. Additionally, most inhibitors targeting the SH2 domain are not STAT3-specific, which makes it difficult to rule out the roles of other STATs in atherosclerosis [72]. Furthermore, due to the difficulty of developing small molecules capable of disrupting protein-protein interactions over a large surface area that still retain drug-like properties, only a limited number of SH2 domain inhibitors have reached preclinical and clinical trials [225-228].
5.2.2 Inhibitors targeting the DBD
The DBD is an important binding region between the STAT3 protein and DNA and has a relatively high specificity. This domain can induce the expression of target genes via the binding of STAT3 to target gene promoters.
To date, few small molecules have been reported as STAT3 DBD inhibitors due to the lack of an efficient in vitro screening assay, hindering the drug discovery process. C48 was first found to alkylate glutathione sulfhydryl on a STAT3 cysteine (C468), a unique ammonia acid residue in the STAT3 DBD region [229]. Afterwards, two small-molecule inhibitors targeting the DBD, inS3-54 and inS3-54a18, were shown to inhibit the STAT3 dimer from binding DNA [230]. Of the analogs tested, inS3-54a18, A26, and A69 were identified to be more active than the parent inS3-54. Moreover, several platinum compounds, including IS3-295, CPA-1, CPA-7, and platinum (IV) tetrachloride, have the same effect [231, 232]. Galiellalactone, a natural product, was identified as an inhibitor of the DNA-binding activity of STAT3, thus impeding STAT3 downstream gene expression through the IL-6/STAT3 signaling pathway [233]. A study found that galiellalactone likely binds the DBD of STAT3 to block the activity of STAT3 both in vitro and in vivo [234]. Collectively, inhibitors targeting the DBD mainly affect the endothelial cell dysfunction in atherosclerosis. By suppressing the expression of target genes, these inhibitors can help return SOCS3 expression to normal levels. Thus, the sustained phosphorylation of STAT3 can be controlled by SOCS3, and endothelial cell dysfunction can be reduced.
Because DBDs are flat with similarities among different isoforms of the same transcription factor family, they are generally considered “undruggable”. Although substantial progress has been made regarding inhibitors targeting the DBD of STAT3, these inhibitors still face challenges similar to those related to the therapeutic utilization of the SH2 domain described above.
5.2.3 Inhibitors targeting other domains
Inhibitors targeting ND and TAD can mediate the binding of STAT3 dimers and regulate DNA transcription, which may be useful for treating atherosclerosis by suppressing endothelial cell dysfunction, inflammation, and abnormal immune cell differentiation. Researchers have synthesized a highly selective STAT3 ND inhibitor, ST3-Hel2A-2, that effectively activates the expression of the proapoptotic gene CHOP and thus induces apoptosis in tumor cells [235]. Moreover, researchers found that in addition to the SH2 domain of the STAT3 protein, some sites (D171, N175, Q202 and M213) in the TAD domain can inhibit STAT3 dimerization and regulate DNA transcription. From the TAD, researchers were able to precisely screen for the allosteric active small molecule K116 that can bind to TAD and inhibit the activity of STAT3 [236].
6. Implication of STAT3 in atherosclerosis
As we have illustrated above, individuals with abnormal STAT3 activity are prone to atherosclerosis. Thus, targeting the STAT3 pathway has become a popular therapeutic approach in the treatment of atherosclerosis. Currently, an increasing number of STAT3 inhibitors have been identified and shown effects on not only inflammatory or proliferative diseases but also on diseases caused by vascular cell function [237-241].
Inhibitors approved by the FDA, including ruxolitinib and tofacitinib, suggest that STAT3-inhibiting strategies may offer promising developments in clinical fields [237-241]. Recently, Johnson et al. provided the first evidence that inhibitors of STAT3 activation protect against Ang-II-induced oxidative stress, endothelial dysfunction, and hypertension in mice [242]. Oxidative stress and endothelial dysfunction are key steps in the process of atherosclerosis, as we have presented above, which means that STAT3 inhibitors may block the development of atherosclerosis and thus be novel therapeutic options. WP1066, which showed outstanding results for treating vascular diseases, is expected to be the most promising novel therapeutic agent in the near future. In preclinical studies, WP1066 successfully prevented neointima formation and contributed to plaque stability in atherosclerosis [182, 183, 188]. The same results have been found for a number of STAT3-inhibiting natural compounds, including capsaicin, curcumin, cryptotanshinone and resveratrol, which have numerous clinical implications [227]. Collectively, STAT3 inhibitors, which block oxidative stress and vascular dysfunction, could serve as therapeutic agents for atherosclerosis.
7. Conclusions and perspectives
Previously, substantial evidence has provided support for the hypothesis that STAT3 is a prominent regulator of various cancers, ischemic injury, and obesity. However, the role of STAT3 in the regulation of atherosclerosis has not been clearly illustrated, and atherosclerosis is still a threat to humans. In this review, we provided a basic overview of STAT3 and showed its pathological roles in atherosclerosis. After summarizing previous studies, we illustrated how aberrant STAT3 activation contributes to endothelial cell dysfunction, macrophage polarization, inflammation, and immunity and may thus become an essential modulator during atherosclerosis. Therefore, inhibitors targeting the STAT3 signaling pathway are expected to be promising therapeutic methods for the treatment of atherosclerosis. We then summarized the current STAT3 inhibitors, which are indirect and direct inhibitors, among which we may find novel therapies for atherosclerosis.
However, the road leading from STAT3 inhibitors to mature therapies for atherosclerosis remains long, and the therapeutic application of STAT3 inhibitors has some limitations. First, STAT3 inhibitors are toxic and potent. From a security standpoint, natural products, including curcumin and resveratrol, are good candidates for STAT3 inhibition because of their noticeable dose-limiting toxicity [243]. However, their relatively poor bioavailability and potency limit their application in the treatment of various diseases, including cancers and atherosclerosis [243]. As described above, several STAT3 inhibitors, especially those targeting the SH2 domains, are not STAT3-specific, which weakens their effectiveness to some extent. Moreover, somatic mutations in STAT3, which may affect its binding site, render the inhibitors described above ineffective and affect the outcomes of clinical studies. Unfortunately, studies considering somatic mutations are complex, and no study has yet addressed this problem. In the future, understanding the relationship between somatic mutations and STAT3 inhibitors and identifying more effective inhibitors presents a difficult challenge. Although numerous direct STAT3 inhibitors have been developed and several STAT3 inhibitors have completed Phase I/II clinical trials, targeting STAT3 as an atherosclerosis therapy remains frustratingly elusive. Therefore, there is an urgent need to reassess ongoing strategies and to develop clinically useful drugs. Additionally, the occurrence and development of atherosclerosis is a complicated process in which STAT3 plays a critical but not solo role, and the inhibitors that simply target the STAT3 pathway may thus not be sufficiently potent. Therefore, combinations of STAT3 inhibitors with other targeted therapeutics may be promising. To determine the most promising combinations, we must have a thorough understanding of the pathways participating in atherosclerosis and the interactions among them.
Moreover, current STAT3 inhibitors are not sensitive to the different isoforms of STAT3. Four isoforms of STAT3 have been identified, STAT3α, STAT3β, STAT3γ, and STAT3δ, which all have different functions. STAT3α and STAT3β have been verified to have contrasting effects during the process of atherosclerosis [237], which suggests that STAT3 inhibitors must discriminate among distinct isoforms to preserve their functions.
Finally, it is important to consider any negative bodily effects when holistically inhibiting STAT3 activation, as STAT3 is found to have positive effects on some processes, including cardiac protection, wound healing, and angiogenesis [244-247]. Early studies have shown that activation of STAT3 plays important roles in myocardial recovery from myocarditis-induced damage in adult mammalian hearts [244]. Furthermore, activated STAT proteins, including STAT3, upregulate cardioprotective genes, such as bcl-xL and VEGF, playing an important role in the maintenance of cardiac function [245, 247-249]. STAT family proteins also reportedly function as regulators of angiogenic growth factors, and their signaling in cardiac myocytes can control vessel growth during cardiac remodeling [247]. Moreover, increased expression of STAT3 in combination with other factors, including decreased miR-17-5p expression, can lead to wound healing [246]. However, when inhibiting cardiac STAT3 by its dominant negative form in zebrafish, cardiomyocyte proliferation after ventricular amputation is decreased by ~80%, resulting in insufficient heart regeneration [244, 250]. Importantly, cardiomyocyte-specific ablation of the STAT3 gene was shown to suppress the frequency of cycling cardiomyocytes in the recovery period without influencing the inflammatory status, resulting in impaired tissue repair and cardiac dysfunction [244]. Therapies aimed at systemic STAT3 inhibition under conditions that are already associated with reduced STAT3 activity in organs may further promote heart failure [38]. Thus, for the treatment of single atherosclerotic lesions, a local application might be a favorable option given that local application using drug-eluting stents or balloons may exert potent effects with negligible systemic side effects.
The time to adopt treatment is also essential, as earlier treatment times are correlated with increased effectiveness. However, due to low penetrance in diseased regions, identifying the stage of atherosclerosis is difficult. Thus, the effectiveness of such therapies also depends on advances in diagnostic methods. After overcoming these challenges, the inhibition of STAT3 has the potential to be a valuable therapeutic strategy for atherosclerosis.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81871607, 81700236, 81600306, and 81500263) and Natural Science Foundation of Shaanxi Province (2018JM3042).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jiang S, Li T, Ji T, Yi W, Yang Z, Wang S. et al. AMPK: Potential Therapeutic Target for Ischemic Stroke. Theranostics. 2018;8:4535-51
2. Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38-360
3. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801-9
4. Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S-60S
5. Nigro J, Osman N, Dart AM, Little PJ. Insulin resistance and atherosclerosis. Endocr Rev. 2006;27:242-59
6. Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N. et al. Heart disease and stroke statistics-2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25-146
7. Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res Cardiol. 2007;102:279-97
8. Liang Z, Wu G, Fan C, Xu J, Jiang S, Yan X. et al. The emerging role of signal transducer and activator of transcription 3 in cerebral ischemic and hemorrhagic stroke. Prog Neurobiol. 2016;137:1-16
9. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311-28
10. Pan J, Fukuda K, Saito M, Matsuzaki J, Kodama H, Sano M. et al. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res. 1999;84:1127-36
11. Qi QR, Yang ZM. Regulation and function of signal transducer and activator of transcription 3. World J Biol Chem. 2014;5:231-9
12. Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T. et al. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63-71
13. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95-8
14. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798-809
15. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C. et al. Stat3 as an oncogene. Cell. 1999;98:295-303
16. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736-46
17. Yang Y, Hu W, Di S, Ma Z, Fan C, Wang D. et al. Tackling myocardial ischemic injury: the signal transducer and activator of transcription 3 (STAT3) at a good site. Expert Opin Ther Targets. 2017;21:215-28
18. Hu W, Lv J, Han M, Yang Z, Li T, Jiang S. et al. STAT3: The art of multi-tasking of metabolic and immune functions in obesity. Prog Lipid Res. 2018;70:17-28
19. Knight RA, Scarabelli TM, Stephanou A. STAT transcription in the ischemic heart. JAKSTAT. 2012;1:111-7
20. Mohan CD, Bharathkumar H, Bulusu KC, Pandey V, Rangappa S, Fuchs JE. et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J Biol Chem. 2014;289:34296-307
21. Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E. et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation. 2012;126:418-29
22. Santoni M, Massari F, Del Re M, Ciccarese C, Piva F, Principato G. et al. Investigational therapies targeting signal transducer and activator of transcription 3 for the treatment of cancer. Expert Opin Investig Drugs. 2015;24:809-24
23. Dinasarapu AR, Gupta S, Ram Maurya M, Fahy E, Min J, Sud M. et al. A combined omics study on activated macrophages-enhanced role of STATs in apoptosis, immunity and lipid metabolism. Bioinformatics. 2013;29:2735-43
24. Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C. et al. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374-90
25. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017;18:374-84
26. Zarkower D. chinmo Mutant fly testis stem cells switching sex farewell to maleness. Dev Cell. 2014;31:385-7
27. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP. et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochimica et biophysica acta. 2014;1845:136-54
28. Dai X, Ahn KS, Kim C, Siveen KS, Ong TH, Shanmugam MK. et al. Ascochlorin, an isoprenoid antibiotic inhibits growth and invasion of hepatocellular carcinoma by targeting STAT3 signaling cascade through the induction of PIAS3. Mol Oncol. 2015;9:818-33
29. Firmbach-Kraft I, Byers M, Shows T, Dalla-Favera R, Krolewski JJ. tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene. 1990;5:1329-36
30. Kawamura M, McVicar DW, Johnston JA, Blake TB, Chen YQ, Lal BK. et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci U S A. 1994;91:6374-8
31. Lee JH, Kim C, Sethi G, Ahn KS. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget. 2015;6:6386-405
32. Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene. 2000;19:5662-79
33. Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell. 1992;70:313-22
34. Brosius FC, Tuttle KR, Kretzler M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia. 2016;59:1624-7
35. Klemm JD, Schreiber SL, Crabtree GR. Dimerization as a regulatory mechanism in signal transduction. Annu Rev Immunol. 1998;16:569-92
36. Min X, Ungureanu D, Maxwell S, Hammaren H, Thibault S, Hillert EK. et al. Structural and Functional Characterization of the JH2 Pseudokinase Domain of JAK Family Tyrosine Kinase 2 (TYK2). J Biol Chem. 2015;290:27261-70
37. Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON. et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18:971-6
38. Wilks AF. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc Natl Acad Sci U S A. 1989;86:1603-7
39. Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zurcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057-65
40. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415-21
41. Ai D, Jiang H, Westerterp M, Murphy AJ, Wang M, Ganda A. et al. Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circ Res. 2014;114:1576-84
42. Qiu H, Lizano P, Laure L, Sui X, Rashed E, Park JY. et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation. 2011;124:406-15
43. Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G. et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 2001;409:349-54
44. Frisdal E, Lesnik P, Olivier M, Robillard P, Chapman MJ, Huby T. et al. Interleukin-6 protects human macrophages from cellular cholesterol accumulation and attenuates the proinflammatory response. J Biol Chem. 2011;286:30926-36
45. Khan JA, Cao M, Kang BY, Liu Y, Mehta JL, Hermonat PL. AAV/hSTAT3-gene delivery lowers aortic inflammatory cell infiltration in LDLR KO mice on high cholesterol. Atherosclerosis. 2010;213:59-66
46. Marino F, Orecchia V, Regis G, Musteanu M, Tassone B, Jon C. et al. STAT3beta controls inflammatory responses and early tumor onset in skin and colon experimental cancer models. Am J Cancer Res. 2014;4:484-94
47. Haghikia A, Hoch M, Stapel B, Hilfiker-Kleiner D. STAT3 regulation of and by microRNAs in development and disease. JAKSTAT. 2012;1:143-50
48. Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X. et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5:401-9
49. Yoo JY, Huso DL, Nathans D, Desiderio S. Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell. 2002;108:331-44
50. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651-62
51. Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19:4111-22
52. Iyer J, Reich NC. Constitutive nuclear import of latent and activated STAT5a by its coiled coil domain. FASEB J. 2008;22:391-400
53. Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci U S A. 2005;102:8150-5
54. Vinkemeier U. Getting the message across, STAT! Design principles of a molecular signaling circuit. J Cell Biol. 2004;167:197-201
55. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396-408
56. Richard AJ, Stephens JM. Emerging roles of JAK-STAT signaling pathways in adipocytes. Trends Endocrinol Metab. 2011;22:325-32
57. Gjurich BN, Taghavie-Moghadam PL, Ley K, Galkina EV. L-selectin deficiency decreases aortic B1a and Breg subsets and promotes atherosclerosis. Thromb Haemost. 2014;112:803-11
58. Kang JW, Lee SM. Melatonin inhibits type 1 interferon signaling of toll-like receptor 4 via heme oxygenase-1 induction in hepatic ischemia/reperfusion. J Pineal Res. 2012;53:67-76
59. Ponnusamy M, Pang M, Annamaraju PK, Zhang Z, Gong R, Chin YE. et al. Transglutaminase-1 protects renal epithelial cells from hydrogen peroxide-induced apoptosis through activation of STAT3 and AKT signaling pathways. Am J Physiol Renal Physiol. 2009;297:F1361-70
60. Yu HM, Zhi JL, Cui Y, Tang EH, Sun SN, Feng JQ. et al. Role of the JAK-STAT pathway in protection of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress in PC12 cells. Apoptosis. 2006;11:931-41
61. Wiejak J, Dunlop J, Mackay SP, Yarwood SJ. Flavanoids induce expression of the suppressor of cytokine signalling 3 (SOCS3) gene and suppress IL-6-activated signal transducer and activator of transcription 3 (STAT3) activation in vascular endothelial cells. Biochem J. 2013;454:283-93
62. Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J Interferon Cytokine Res. 1999;19:563-73
63. Reineke U, Schneider-Mergener J, Glaser RW, Stigler RD, Seifert M, Volk HD. et al. Evidence for conformationally different states of interleukin-10: binding of a neutralizing antibody enhances accessibility of a hidden epitope. J Mol Recognit. 1999;12:242-8
64. Finbloom DS, Winestock KD. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J Immunol. 1995;155:1079-90
65. Nakamura R, Sene A, Santeford A, Gdoura A, Kubota S, Zapata N. et al. IL10-driven STAT3 signalling in senescent macrophages promotes pathological eye angiogenesis. Nat Commun. 2015;6:7847
66. Mallat Z, Heymes C, Ohan J, Faggin E, Leseche G, Tedgui A. Expression of interleukin-10 in advanced human atherosclerotic plaques: relation to inducible nitric oxide synthase expression and cell death. Arterioscler Thromb Vasc Biol. 1999;19:611-6
67. Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H. et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ Res. 2002;90:1064-71
68. Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK. et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10-7
69. Potteaux S, Esposito B, van Oostrom O, Brun V, Ardouin P, Groux H. et al. Leukocyte-derived interleukin 10 is required for protection against atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:1474-8
70. Han X, Kitamoto S, Wang H, Boisvert WA. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. 2010;24:2869-80
71. Sands WA, Woolson HD, Milne GR, Rutherford C, Palmer TM. Exchange protein activated by cyclic AMP (Epac)-mediated induction of suppressor of cytokine signaling 3 (SOCS-3) in vascular endothelial cells. Mol Cell Biol. 2006;26:6333-46
72. Szelag M, Piaszyk-Borychowska A, Plens-Galaska M, Wesoly J, Bluyssen HA. Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease. Oncotarget. 2016;7:48788-812
73. Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M. et al. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem. 2001;276:6675-88
74. Bonham AJ, Wenta N, Osslund LM, Prussin AJ 2nd, Vinkemeier U, Reich NO. STAT1:DNA sequence-dependent binding modulation by phosphorylation, protein:protein interactions and small-molecule inhibition. Nucleic Acids Res. 2013;41:754-63
75. Choe JY, Park KY, Park SH, Lee SI, Kim SK. Regulatory effect of calcineurin inhibitor, tacrolimus, on IL-6/sIL-6R-mediated RANKL expression through JAK2-STAT3-SOCS3 signaling pathway in fibroblast-like synoviocytes. Arthritis Res Ther. 2013;15:R26
76. Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN. et al. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546-50
77. Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN. et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012;36:239-50
78. Nakagawa T, Iida S, Osanai T, Uetake H, Aruga T, Toriya Y. et al. Decreased expression of SOCS-3 mRNA in breast cancer with lymph node metastasis. Oncol Rep. 2008;19:33-9
79. Lusis AJ. Atherosclerosis. Nature. 2000;407:233-41
80. Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297-329
81. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868-74
82. Wang Z, Hu W, Lu C, Ma Z, Jiang S, Gu C. et al. Targeting NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome in Cardiovascular Disorders. Arterioscler Thromb Vasc Biol. 2018;38:2765-79
83. Tao RR, Huang JY, Shao XJ, Ye WF, Tian Y, Liao MH. et al. Ischemic injury promotes Keap1 nitration and disturbance of antioxidative responses in endothelial cells: a potential vasoprotective effect of melatonin. J Pineal Res. 2013;54:271-81
84. Menghini R, Casagrande V, Cardellini M, Ballanti M, Davato F, Cardolini I. et al. FoxO1 regulates asymmetric dimethylarginine via downregulation of dimethylaminohydrolase 1 in human endothelial cells and subjects with atherosclerosis. Atherosclerosis. 2015;242:230-5
85. Forstermann U, Xia N, Li H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ Res. 2017;120:713-35
86. Carballo M, Conde M, El Bekay R, Martin-Nieto J, Camacho MJ, Monteseirin J. et al. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem. 1999;274:17580-6
87. Tawfik A, Jin L, Banes-Berceli AK, Caldwell RB, Ogbi S, Shirley A. et al. Hyperglycemia and reactive oxygen species mediate apoptosis in aortic endothelial cells through Janus kinase 2. Vascul Pharmacol. 2005;43:320-6
88. Ng IH, Yeap YY, Ong LS, Jans DA, Bogoyevitch MA. Oxidative stress impairs multiple regulatory events to drive persistent cytokine-stimulated STAT3 phosphorylation. Biochimica et biophysica acta. 2014;1843:483-94
89. Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. 2008;13:4925-32
90. Wang Z, Ni L, Wang J, Lu C, Ren M, Han W. et al. The protective effect of melatonin on smoke-induced vascular injury in rats and humans: a randomized controlled trial. J Pineal Res. 2016;60:217-27
91. Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170:191-203
92. Price DT, Loscalzo J. Cellular adhesion molecules and atherogenesis. Am J Med. 1999;107:85-97
93. Kirsch T, Beese M, Wyss K, Klinge U, Haller H, Haubitz M. et al. Aldosterone modulates endothelial permeability and endothelial nitric oxide synthase activity by rearrangement of the actin cytoskeleton. Hypertension. 2013;61:501-8
94. Rao J, Ye Z, Tang H, Wang C, Peng H, Lai W. et al. The RhoA/ROCK Pathway Ameliorates Adhesion and Inflammatory Infiltration Induced by AGEs in Glomerular Endothelial Cells. Sci Rep. 2017;7:39727
95. Renault-Mihara F, Okano H. STAT3-regulated RhoA drives reactive astrocyte dynamics. Cell Cycle. 2017;16:1995-6
96. Wei Z, Jiang W, Wang H, Li H, Tang B, Liu B. et al. The IL-6/STAT3 pathway regulates adhesion molecules and cytoskeleton of endothelial cells in thromboangiitis obliterans. Cell Signal. 2018;44:118-26
97. Lu WJ, Tseng SC, Chen S, Tighe S, Zhang Y, Liu X. et al. Senescence Mediated by p16(INK4a) Impedes Reprogramming of Human Corneal Endothelial Cells into Neural Crest Progenitors. Sci Rep. 2016;6:35166
98. Zheng S, Huang J, Zhou K, Xiang Q, Zhang Y, Tan Z. et al. Progesterone enhances vascular endothelial cell migration via activation of focal adhesion kinase. J Cell Mol Med. 2012;16:296-305
99. Seo J, Cho DH, Lee HJ, Sung MS, Lee JY, Won KJ. et al. Citron Rho-interacting kinase mediates arsenite-induced decrease in endothelial nitric oxide synthase activity by increasing phosphorylation at threonine 497: Mechanism underlying arsenite-induced vascular dysfunction. Free Radic Biol Med. 2016;90:133-44
100. Deem TL, Cook-Mills JM. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood. 2004;104:2385-93
101. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508-19
102. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341-55
103. Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64:2028-41
104. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71-82
105. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953-64
106. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR. et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737-46
107. Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res. 2016;118:653-67
108. Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25:1101-4
109. Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209-12
110. Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E. et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A. 2004;101:4560-5
111. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723-37
112. Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H. et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121:985-97
113. Fang S, Xu Y, Zhang Y, Tian J, Li J, Li Z. et al. Irgm1 promotes M1 but not M2 macrophage polarization in atherosclerosis pathogenesis and development. Atherosclerosis. 2016;251:282-90
114. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593-604
115. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V. et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001;194:809-21
116. Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology. 2010;215:788-95
117. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17:109-18
118. Cui J, Zhang F, Cao W, Wang Y, Liu J, Liu X. et al. Erythropoietin alleviates hyperglycaemia-associated inflammation by regulating macrophage polarization via the JAK2/STAT3 signalling pathway. Mol Immunol. 2018;101:221-8
119. Bruunsgaard H, Pedersen M, Pedersen BK. Aging and proinflammatory cytokines. Curr Opin Hematol. 2001;8:131-6
120. Calabro P, Limongelli G, Pacileo G, Di Salvo G, Golino P, Calabro R. The role of adiposity as a determinant of an inflammatory milieu. J Cardiovasc Med (Hagerstown). 2008;9:450-60
121. Kleemann R, Verschuren L, Morrison M, Zadelaar S, van Erk MJ, Wielinga PY. et al. Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis. 2011;218:44-52
122. Yang XP, Irani K, Mattagajasingh S, Dipaula A, Khanday F, Ozaki M. et al. Signal transducer and activator of transcription 3alpha and specificity protein 1 interact to upregulate intercellular adhesion molecule-1 in ischemic-reperfused myocardium and vascular endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1395-400
123. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-25
124. Lee JM, Kim SR, Yoo SJ, Hong OK, Son HS, Chang SA. The relationship between adipokines, metabolic parameters and insulin resistance in patients with metabolic syndrome and type 2 diabetes. J Int Med Res. 2009;37:1803-12
125. Schuett H, Oestreich R, Waetzig GH, Annema W, Luchtefeld M, Hillmer A. et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:281-90
126. Chistiakov DA, Orekhov AN, Bobryshev YV. Vascular smooth muscle cell in atherosclerosis. Acta Physiol (Oxf). 2015;214:33-50
127. Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007:45 Suppl A:A25-32
128. Kolodgie FD, Burke AP, Farb A, Gold HK, Yuan J, Narula J. et al. The thin-cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol. 2001;16:285-92
129. Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD. et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075-80
130. Badimon L, Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med. 2014;276:618-32
131. Liao XH, Wang N, Zhao DW, Zheng DL, Zheng L, Xing WJ. et al. STAT3 Protein Regulates Vascular Smooth Muscle Cell Phenotypic Switch by Interaction with Myocardin. J Biol Chem. 2015;290:19641-52
132. Shibata R, Kai H, Seki Y, Kato S, Wada Y, Hanakawa Y. et al. Inhibition of STAT3 prevents neointima formation by inhibiting proliferation and promoting apoptosis of neointimal smooth muscle cells. Hum Gene Ther. 2003;14:601-10
133. Heeneman S, Sluimer JC, Daemen MJ. Angiotensin-converting enzyme and vascular remodeling. Circ Res. 2007;101:441-54
134. Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P. et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature. 1995;375:247-50
135. Seki Y, Kai H, Shibata R, Nagata T, Yasukawa H, Yoshimura A. et al. Role of the JAK/STAT pathway in rat carotid artery remodeling after vascular injury. Circ Res. 2000;87:12-8
136. Song B, Jin H, Yu X, Zhang Z, Yu H, Ye J. et al. Angiotensin-converting enzyme 2 attenuates oxidative stress and VSMC proliferation via the JAK2/STAT3/SOCS3 and profilin-1/MAPK signaling pathways. Regul Pept. 2013;185:44-51
137. Cagnin S, Biscuola M, Patuzzo C, Trabetti E, Pasquali A, Laveder P. et al. Reconstruction and functional analysis of altered molecular pathways in human atherosclerotic arteries. BMC Genomics. 2009;10:13
138. Jun MY, Karki R, Paudel KR, Sharma BR, Adhikari D, Kim DW. Alkaloid rich fraction from Nelumbo nucifera targets VSMC proliferation and migration to suppress restenosis in balloon-injured rat carotid artery. Atherosclerosis. 2016;248:179-89
139. Park HS, Quan KT, Han JH, Jung SH, Lee DH, Jo E. et al. Rubiarbonone C inhibits platelet-derived growth factor-induced proliferation and migration of vascular smooth muscle cells through the focal adhesion kinase, MAPK and STAT3 Tyr(705) signalling pathways. Br J Pharmacol. 2017;174:4140-54
140. Neeli I, Liu Z, Dronadula N, Ma ZA, Rao GN. An essential role of the Jak-2/STAT-3/cytosolic phospholipase A(2) axis in platelet-derived growth factor BB-induced vascular smooth muscle cell motility. J Biol Chem. 2004;279:46122-8
141. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445-89
142. Taleb S, Tedgui A, Mallat Z. IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb Vasc Biol. 2015;35:258-64
143. van Bruggen N, Ouyang W. Th17 cells at the crossroads of autoimmunity, inflammation, and atherosclerosis. Immunity. 2014;40:10-2
144. Wessely R, Hengst L, Jaschke B, Wegener F, Richter T, Lupetti R. et al. A central role of interferon regulatory factor-1 for the limitation of neointimal hyperplasia. Hum Mol Genet. 2003;12:177-87
145. Chang H, Zhao F, Xie X, Liao Y, Song Y, Liu C. et al. PPARalpha suppresses Th17 cell differentiation through IL-6/STAT3/RORgammat pathway in experimental autoimmune myocarditis. Exp Cell Res. 2019;375:22-30
146. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS. et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358-63
147. Wei L, Laurence A, Elias KM, O'Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605-10
148. Wang C, Liu X, Peng Q, Fang L, Wang C, Li Z. [Role of Foxp3/Treg and RORgammat/Th17 cell imbalance in rat model of chronic obstructive pulmonary disease]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2014;26:860-4
149. Ivanov II, McKenzie BS Zhou L, Tadokoro CE Lepelley A, Lafaille JJ et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121-33
150. Chen Z, Laurence A, O'Shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin Immunol. 2007;19:400-8
151. Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936-46
152. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T. et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967-74
153. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L. et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480-3
154. Yang R, Lirussi D, Thornton TM, Jelley-Gibbs DM, Diehl SA, Case LK. et al. Mitochondrial Ca(2)(+) and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife. 2015:4
155. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849-59
156. Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE. et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39:329-37
157. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. 2015;35:280-7
158. Afzali B, Mitchell PJ, Edozie FC, Povoleri GA, Dowson SE, Demandt L. et al. CD161 expression characterizes a subpopulation of human regulatory T cells that produces IL-17 in a STAT3-dependent manner. Eur J Immunol. 2013;43:2043-54
159. Chen YL, Jian Y, Liu MJ, Zhong L, Zhang F, Yang WF. et al. [Role of the Th17/Treg functional imbalance on the development of atherosclerosis in apo E knockout mice]. Zhonghua Xin Xue Guan Bing Za Zhi. 2013;41:416-21
160. Park JS, Kwok SK, Lim MA, Kim EK, Ryu JG, Kim SM. et al. STA-21, a promising STAT-3 inhibitor that reciprocally regulates Th17 and Treg cells, inhibits osteoclastogenesis in mice and humans and alleviates autoimmune inflammation in an experimental model of rheumatoid arthritis. Arthritis Rheumatol. 2014;66:918-29
161. Butcher MJ, Galkina EV. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front Physiol. 2012;3:44
162. Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F. et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol. 2007;19:695-702
163. Chaudhari SM, Sluimer JC, Koch M, Theelen TL, Manthey HD, Busch M. et al. Deficiency of HIF1alpha in Antigen-Presenting Cells Aggravates Atherosclerosis and Type 1 T-Helper Cell Responses in Mice. Arterioscler Thromb Vasc Biol. 2015;35:2316-25
164. Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C. et al. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol. 2010;184:2638-45
165. Nefedova Y, Cheng P, Gilkes D, Blaskovich M, Beg AA, Sebti SM. et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol. 2005;175:4338-46
166. Wang Y, Tabas I. Emerging roles of mitochondria ROS in atherosclerotic lesions: causation or association? J Atheroscler Thromb. 2014;21:381-90
167. Yang R, Rincon M. Mitochondrial Stat3, the Need for Design Thinking. Int J Biol Sci. 2016;12:532-44
168. Agrawal S, Febbraio M, Podrez E, Cathcart MK, Stark GR, Chisolm GM. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation. 2007;115:2939-47
169. Torella D, Curcio A, Gasparri C, Galuppo V, De Serio D, Surace FC. et al. Fludarabine prevents smooth muscle proliferation in vitro and neointimal hyperplasia in vivo through specific inhibition of STAT-1 activation. Am J Physiol Heart Circ Physiol. 2007;292:H2935-43
170. Arzt L, Halbwedl I, Gogg-Kamerer M, Popper HH. Signal Transducer and Activator of Transcription 1 (STAT1) Knock-down Induces Apoptosis in Malignant Pleural Mesothelioma. Pathol Oncol Res. 2017;23:595-605
171. Chmielewski S, Olejnik A, Sikorski K, Pelisek J, Blaszczyk K, Aoqui C. et al. STAT1-dependent signal integration between IFNgamma and TLR4 in vascular cells reflect pro-atherogenic responses in human atherosclerosis. PLoS One. 2014;9:e113318
172. Manea A, Tanase LI, Raicu M, Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30:105-12
173. Lagor WR, Fields DW, Bauer RC, Crawford A, Abt MC, Artis D. et al. Genetic manipulation of the ApoF/Stat2 locus supports an important role for type I interferon signaling in atherosclerosis. Atherosclerosis. 2014;233:234-41
174. Antonarakis ES, Heath EI, Posadas EM, Yu EY, Harrison MR, Bruce JY. et al. A phase 2 study of KX2-391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013;71:883-92
175. Ferrajoli A, Faderl S, Van Q, Koch P, Harris D, Liu Z. et al. WP1066 disrupts Janus kinase-2 and induces caspase-dependent apoptosis in acute myelogenous leukemia cells. Cancer Res. 2007;67:11291-9
176. Gangadhar TC, Clark JI, Karrison T, Gajewski TF. Phase II study of the Src kinase inhibitor saracatinib (AZD0530) in metastatic melanoma. Invest New Drugs. 2013;31:769-73
177. Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I. et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435-44
178. Li SQ, Cheuk AT, Shern JF, Song YK, Hurd L, Liao H. et al. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS One. 2013;8:e76551
179. Oyaizu T, Fung SY, Shiozaki A, Guan Z, Zhang Q, dos Santos CC. et al. Src tyrosine kinase inhibition prevents pulmonary ischemia-reperfusion-induced acute lung injury. Intensive Care Med. 2012;38:894-905
180. Sun X, Li B, Xie B, Xu Z, Chang G, Tao Y. et al. DCZ3301, a novel cytotoxic agent, inhibits proliferation in diffuse large B-cell lymphoma via the STAT3 pathway. Cell Death Dis. 2017;8:e3111
181. Yu LM, Di WC, Dong X, Li Z, Zhang Y, Xue XD. et al. Melatonin protects diabetic heart against ischemia-reperfusion injury, role of membrane receptor-dependent cGMP-PKG activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864:563-78
182. Daniel JM, Dutzmann J, Bielenberg W, Widmer-Teske R, Gunduz D, Hamm CW. et al. Inhibition of STAT3 signaling prevents vascular smooth muscle cell proliferation and neointima formation. Basic Res Cardiol. 2012;107:261
183. Horiguchi A, Asano T, Kuroda K, Sato A, Asakuma J, Ito K. et al. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br J Cancer. 2010;102:1592-9
184. Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M. et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71:6601-10
185. Zgheib A, Pelletier-Bonnier E, Levros LC Jr, Annabi B. Selective JAK/STAT3 signalling regulates transcription of colony stimulating factor-2 and -3 in Concanavalin-A-activated mesenchymal stromal cells. Cytokine. 2013;63:187-93
186. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF. et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799-807
187. Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z. et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645-8
188. Demyanets S, Kaun C, Rychli K, Pfaffenberger S, Kastl SP, Hohensinner PJ. et al. Oncostatin M-enhanced vascular endothelial growth factor expression in human vascular smooth muscle cells involves PI3K-, p38 MAPK-, Erk1/2- and STAT1/STAT3-dependent pathways and is attenuated by interferon-gamma. Basic Res Cardiol. 2011;106:217-31
189. Shalini V, Jayalekshmi A, Helen A. Mechanism of anti-inflammatory effect of tricin, a flavonoid isolated from Njavara rice bran in LPS induced hPBMCs and carrageenan induced rats. Mol Immunol. 2015;66:229-39
190. Fletcher S, Drewry JA, Shahani VM, Page BD, Gunning PT. Molecular disruption of oncogenic signal transducer and activator of transcription 3 (STAT3) protein. Biochem Cell Biol. 2009;87:825-33
191. Morlacchi P, Robertson FM, Klostergaard J, McMurray JS. Targeting SH2 domains in breast cancer. Future Med Chem. 2014;6:1909-26
192. Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E. et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276:45443-55
193. Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg Med Chem Lett. 2003;13:633-6
194. Chen J, Nikolovska-Coleska Z, Yang CY, Gomez C, Gao W, Krajewski K. et al. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via 'click chemistry'. Bioorg Med Chem Lett. 2007;17:3939-42
195. Miyoshi K, Takaishi M, Nakajima K, Ikeda M, Kanda T, Tarutani M. et al. Stat3 as a therapeutic target for the treatment of psoriasis: a clinical feasibility study with STA-21, a Stat3 inhibitor. J Invest Dermatol. 2011;131:108-17
196. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235-42
197. Brambilla L, Genini D, Laurini E, Merulla J, Perez L, Fermeglia M. et al. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Mol Oncol. 2015;9:1194-206
198. Bendell JC, Hong DS, Burris HA 3rd, Naing A, Jones SF, Falchook G. et al. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74:125-30
199. Oh DY, Lee SH, Han SW, Kim MJ, Kim TM, Kim TY. et al. Phase I Study of OPB-31121, an Oral STAT3 Inhibitor, in Patients with Advanced Solid Tumors. Cancer Res Treat. 2015;47:607-15
200. Okusaka T, Ueno H, Ikeda M, Mitsunaga S, Ozaka M, Ishii H. et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol Res. 2015;45:1283-91
201. Ganjali S, Blesso CN, Banach M, Pirro M, Majeed M, Sahebkar A. Effects of curcumin on HDL functionality. Pharmacol Res. 2017;119:208-18
202. Shin SK, Ha TY, McGregor RA, Choi MS. Long-term curcumin administration protects against atherosclerosis via hepatic regulation of lipoprotein cholesterol metabolism. Mol Nutr Food Res. 2011;55:1829-40
203. Jang EM, Choi MS, Jung UJ, Kim MJ, Kim HJ, Jeon SM. et al. Beneficial effects of curcumin on hyperlipidemia and insulin resistance in high-fat-fed hamsters. Metabolism. 2008;57:1576-83
204. Wang S, Moustaid-Moussa N, Chen L, Mo H, Shastri A, Su R. et al. Novel insights of dietary polyphenols and obesity. J Nutr Biochem. 2014;25:1-18
205. Panahi Y, Kianpour P, Mohtashami R, Jafari R, Simental-Mendia LE, Sahebkar A. Efficacy and Safety of Phytosomal Curcumin in Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Drug Res (Stuttg). 2017;67:244-51
206. Mirzaei H, Shakeri A, Rashidi B, Jalili A, Banikazemi Z, Sahebkar A. Phytosomal curcumin: A review of pharmacokinetic, experimental and clinical studies. Biomed Pharmacother. 2017;85:102-12
207. Lelli D, Sahebkar A, Johnston TP, Pedone C. Curcumin use in pulmonary diseases: State of the art and future perspectives. Pharmacol Res. 2017;115:133-48
208. Esmaily H, Sahebkar A, Iranshahi M, Ganjali S, Mohammadi A, Ferns G. et al. An investigation of the effects of curcumin on anxiety and depression in obese individuals: A randomized controlled trial. Chin J Integr Med. 2015;21:332-8
209. Panahi Y, Ghanei M, Hajhashemi A, Sahebkar A. Effects of Curcuminoids-Piperine Combination on Systemic Oxidative Stress, Clinical Symptoms and Quality of Life in Subjects with Chronic Pulmonary Complications Due to Sulfur Mustard: A Randomized Controlled Trial. J Diet Suppl. 2016;13:93-105
210. Panahi Y, Badeli R, Karami GR, Sahebkar A. Investigation of the efficacy of adjunctive therapy with bioavailability-boosted curcuminoids in major depressive disorder. Phytother Res. 2015;29:17-21
211. Sahebkar A. Molecular mechanisms for curcumin benefits against ischemic injury. Fertil Steril. 2010;94:e75-6 author reply e7
212. Iranshahi M, Sahebkar A, Hosseini ST, Takasaki M, Konoshima T, Tokuda H. Cancer chemopreventive activity of diversin from Ferula diversivittata in vitro and in vivo. Phytomedicine. 2010;17:269-73
213. Teymouri M, Pirro M, Johnston TP, Sahebkar A. Curcumin as a multifaceted compound against human papilloma virus infection and cervical cancers: A review of chemistry, cellular, molecular, and preclinical features. Biofactors. 2017;43:331-46
214. Wongcharoen W, Phrommintikul A. The protective role of curcumin in cardiovascular diseases. Int J Cardiol. 2009;133:145-51
215. Momtazi AA, Derosa G, Maffioli P, Banach M, Sahebkar A. Role of microRNAs in the Therapeutic Effects of Curcumin in Non-Cancer Diseases. Mol Diagn Ther. 2016;20:335-45
216. Momtazi AA, Shahabipour F, Khatibi S, Johnston TP, Pirro M, Sahebkar A. Curcumin as a MicroRNA Regulator in Cancer: A Review. Rev Physiol Biochem Pharmacol. 2016;171:1-38
217. Alexandrow MG, Song LJ, Altiok S, Gray J, Haura EB, Kumar NB. Curcumin: a novel Stat3 pathway inhibitor for chemoprevention of lung cancer. Eur J Cancer Prev. 2012;21:407-12
218. Liu Y, Wang X, Zeng S, Zhang X, Zhao J, Zhang X. et al. The natural polyphenol curcumin induces apoptosis by suppressing STAT3 signaling in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2018;37:303
219. Zordoky BN, Robertson IM, Dyck JR. Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Biochimica et biophysica acta. 2015;1852:1155-77
220. Chu LM, Lassaletta AD, Robich MP, Sellke FW. Resveratrol in the prevention and treatment of coronary artery disease. Curr Atheroscler Rep. 2011;13:439-46
221. Dong Y, Lu B, Zhang X, Zhang J, Lai L, Li D. et al. Cucurbitacin E, a tetracyclic triterpenes compound from Chinese medicine, inhibits tumor angiogenesis through VEGFR2-mediated Jak2-STAT3 signaling pathway. Carcinogenesis. 2010;31:2097-104
222. Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H. et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene. 2016;35:783-92
223. Bharadwaj U, Eckols TK, Kolosov M, Kasembeli MM, Adam A, Torres D. et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene. 2015;34:1341-53
224. Bosch-Barrera J, Menendez JA. Silibinin and STAT3: A natural way of targeting transcription factors for cancer therapy. Cancer Treat Rev. 2015;41:540-6
225. Botta A, Sirignano E, Popolo A, Saturnino C, Terracciano S, Foglia A. et al. Identification of Lead Compounds as Inhibitors of STAT3: Design, Synthesis and Bioactivity. Mol Inform. 2015;34:689-97
226. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem Biol. 2016;11:308-18
227. Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12:611-29
228. Zhang X, Sun Y, Pireddu R, Yang H, Urlam MK, Lawrence HR. et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73:1922-33
229. Buettner R, Corzano R, Rashid R, Lin J, Senthil M, Hedvat M. et al. Alkylation of cysteine 468 in Stat3 defines a novel site for therapeutic development. ACS Chem Biol. 2011;6:432-43
230. Huang W, Dong Z, Wang F, Peng H, Liu JY, Zhang JT. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem Biol. 2014;9:1188-96
231. Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J Biol Chem. 2005;280:32979-88
232. Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB. et al. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol Cancer Ther. 2004;3:1533-42
233. Weidler M, Rether J, Anke T, Erkel G. Inhibition of interleukin-6 signaling by galiellalactone. FEBS Lett. 2000;484:1-6
234. Don-Doncow N, Escobar Z, Johansson M, Kjellstrom S, Garcia V, Munoz E. et al. Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J Biol Chem. 2014;289:15969-78
235. Timofeeva OA, Tarasova NI, Zhang X, Chasovskikh S, Cheema AK, Wang H. et al. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc Natl Acad Sci U S A. 2013;110:1267-72
236. Huang M, Song K, Liu X, Lu S, Shen Q, Wang R. et al. AlloFinder: a strategy for allosteric modulator discovery and allosterome analyses. Nucleic Acids Res. 2018;46:W451-W8
237. Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res. 2015;106:365-74
238. Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D. et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370:2377-86
239. Papp KA, Menter A, Strober B, Langley RG, Buonanno M, Wolk R. et al. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a Phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol. 2012;167:668-77
240. Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S. et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367:616-24
241. Wollenhaupt J, Silverfield J, Lee EB, Curtis JR, Wood SP, Soma K. et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J Rheumatol. 2014;41:837-52
242. Johnson AW, Kinzenbaw DA, Modrick ML, Faraci FM. Small-molecule inhibitors of signal transducer and activator of transcription 3 protect against angiotensin II-induced vascular dysfunction and hypertension. Hypertension. 2013;61:437-42
243. Zhao C, Li H, Lin HJ, Yang S, Lin J, Liang G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol Sci. 2016;37:47-61
244. Miyawaki A, Obana M, Mitsuhara Y, Orimoto A, Nakayasu Y, Yamashita T. et al. Adult murine cardiomyocytes exhibit regenerative activity with cell cycle reentry through STAT3 in the healing process of myocarditis. Sci Rep. 2017;7:1407
245. Oshima Y, Fujio Y, Nakanishi T, Itoh N, Yamamoto Y, Negoro S. et al. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc Res. 2005;65:428-35
246. Yang ZG, Awan FM, Du WW, Zeng Y, Lyu J, Wu D. et al. The Circular RNA Interacts with STAT3, Increasing Its Nuclear Translocation and Wound Repair by Modulating Dnmt3a and miR-17 Function. Mol Ther. 2017;25:2062-74
247. Funamoto M, Fujio Y, Kunisada K, Negoro S, Tone E, Osugi T. et al. Signal transducer and activator of transcription 3 is required for glycoprotein 130-mediated induction of vascular endothelial growth factor in cardiac myocytes. J Biol Chem. 2000;275:10561-6
248. Fujio Y, Kunisada K, Hirota H, Yamauchi-Takihara K, Kishimoto T. Signals through gp130 upregulate bcl-x gene expression via STAT1-binding cis-element in cardiac myocytes. J Clin Invest. 1997;99:2898-905
249. Osugi T, Oshima Y, Fujio Y, Funamoto M, Yamashita A, Negoro S. et al. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J Biol Chem. 2002;277:6676-81
250. Fang Y, Gupta V, Karra R, Holdway JE, Kikuchi K, Poss KD. Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc Natl Acad Sci U S A. 2013;110:13416-21
Author contact
![]() Corresponding authors: Yang Yang MD., PhD., and Yuehu Han., PhD. Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Telephone: +86 13379217366; Email address: yang200214yycom (Yang Yang) and yuehuhanxijingcom (Yuehu Han)
Corresponding authors: Yang Yang MD., PhD., and Yuehu Han., PhD. Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, Faculty of Life Sciences, Northwest University, 229 Taibai North Road, Xi'an 710069, China. Telephone: +86 13379217366; Email address: yang200214yycom (Yang Yang) and yuehuhanxijingcom (Yuehu Han)