Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(9):2460-2474. doi:10.7150/thno.31097 This issue Cite
Research Paper
HCP5 is a SMAD3-responsive long non-coding RNA that promotes lung adenocarcinoma metastasis via miR-203/SNAI axis
Lin Jiang1,2, Ranran Wang3, Li Fang4, Xiaolu Ge1,2, Lingna Chen1,2, Ming Zhou1,2, Yanhong Zhou1,2, Wei Xiong1,2, Yerong Hu3, Xianming Tang3, Guiyuan Li1,2, Zheng Li1,2,5 ![]()
1. The Key Laboratory of Carcinogenesis of the Chinese Ministry of Health, Xiangya Hospital, Central South University, Changsha, Hunan, China
2. The Key Laboratory of Carcinogenesis and Cancer Invasion of the Chinese Ministry of Education, Cancer Research Institute and School of Basic Medical Sciences, Central South University, Changsha, Hunan, China
3. Department of Thoracic and Cardiovascular Surgery, The Second Xiangya Hospital, Central South University, Changsha, Hunan, China
4. Hunan Cancer Hospital, The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan, China
5. High Resolution Mass Spectrometry Laboratory of Advanced Research Center, Central South University, Changsha, Hunan, China
Received 2018-10-30; Accepted 2019-3-23; Published 2019-4-13
Abstract

Introduction: Transforming growth factor-beta (TGFβ) signaling plays a vital role in lung adenocarcinoma (LUAD) progression. However, the involvement of TGFβ-regulated long non-coding RNAs (lncRNAs) in metastasis of LUAD remains poorly understood.
Methods: We performed bioinformatic analyses to identify putative lncRNAs regulated by TGF-β/SMAD3 and validated the results by quantitative PCR in LUAD cells. We performed luciferase reporter and chromatin immunoprecipitation assays to demonstrate the transcriptional regulation of the lncRNA histocompatibility leukocyte antigen complex P5 (HCP5) we decided to focus on. Stable HCP5 knockdown and HCP5-overexpressing A549 cell variants were generated respectively, to study HCP5 function and understand its mechanism of action. We also confirmed our findings in mouse xenografts and metastasis models. We analyzed the correlation between the level of lncRNA expression with EGFR, KRAS mutations, smoke state and prognostic of LUAD patients.
Results: We found that the lncRNA HCP5 is induced by TGFβ and transcriptionally regulated by SMAD3, which promotes LUAD tumor growth and metastasis. Moreover, HCP5 is overexpressed in tumor tissues of patients with LUAD, specifically in patients with EGFR and KRAS mutations and current smoker. HCP5 high expression level is positively correlated with poor prognosis of patients with LUAD. Finally, we demonstrated that upregulation of HCP5 increases the expression of Snail and Slug by sponging the microRNA-203 (miR-203) and promoting epithelial-mesenchymal transition (EMT) in LUAD cells.
Conclusions: Our work demonstrates that the lncRNA HCP5 is transcriptionally regulated by SMAD3 and acts as a new regulator in the TGFβ/SMAD signaling pathway. Therefore, HCP5 can serve as a potential therapeutic target in LUAD.
Keywords: SMAD3, Long non-coding RNA, HCP5, lung adenocarcinoma, metastasis.
Introduction
The transforming growth factor-β (TGF-β) signaling regulates physiological processes including embryonic development and immune tolerance, and is dysregulated in some pathologies such as malignant tumors and pulmonary fibrosis [1-3]. TGFβ receptor (TGFβRI and TGFβRII) are activated by TGFβ isoforms and phosphorylate downstream SMAD proteins therefore called receptor-regulated SMADs (R-SMADs), including SMAD2 and SMAD3 [4]. R-SMADs interact with SMAD4 and translocate to the nucleus to activate transcriptional networks that drive phenotypic transformations of cells [5]. Activation of the TGFβ signaling causes diverse results in cancer development. In early carcinogenesis, TGFβ induces G1 cell cycle arrest and apoptosis to inhibit tumor growth. However, in advanced stages, TGFβ promotes epithelial-mesenchymal transition (EMT) and stemness, which lead to tumor metastasis, drug resistance and malignant progression [6, 7].
Lung cancer is the first cause of cancer-related deaths around the world: it is classified into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [8]. NSCLC constitutes 85% of all lung cancer cases, and includes lung adenocarcinoma (LUAD), squamous cell carcinoma and large cell lung cancer [3]. Despite the advances in molecular targeted therapy and immunotherapy, the prognosis of patients with LUAD with distant metastases at diagnosis remains unfavorable, with a 5-year survival rate of 1% [9, 10]. The TGFβ signaling is often activated in advanced LUAD and correlates with malignant progression and poor prognosis [11, 12]. Some studies have investigated the combined effect of anti-TGFβ monoclonal antibodies and small molecule kinase inhibitors or radiotherapy, while the effect of anti-TGFβ antibodies and programmed death-ligand 1 (PD-1) inhibitors is under clinical investigation (NCT02423343) [3, 12, 13].
TGFβ induces the activity of the SMAD complex, which induces the transcription of hundreds of protein-coding genes and leads to various cancer-associated pathological processes [14]. In addition, recent studies have suggested that some non-coding RNAs, especially long non-coding RNAs (lncRNAs) are involved in TGFβ signaling pathway and cancer development [15, 16]. The lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), for example, is overexpressed in tumor tissues and peripheral blood of patients with metastatic LUAD [17, 18]. The lncRNA activated by TGF-β (ATB) is induced by TGFβ1 and promotes EMT through the microRNA-200 (miR200)/Zinc finger E-box-binding homeobox (ZEB) axis in hepatocellular carcinoma [19]. However, whether lncRNAs are responsible for SMADs transcriptional regulation and are involved in the TGFβ signaling during LUAD development is still poorly understood.
Here, we describe the TGFβ-responsive lncRNA histocompatibility leukocyte antigen complex P5 (HCP5), which is transcriptional regulated by SMAD3 in LUAD cells. HCP5 promotes tumor proliferation, migration and invasion in LUAD. Additionally, HCP5 promotes EMT by enhancing the expression of Snail and Slug expression through the sponging of miR-203. Moreover, HCP5 is overexpressed in tumor tissues of patients with LUAD, specifically in patients with EGFR and KRAS mutations and current smoker. Finally, elevated HCP5 levels are correlated with poor prognosis in patients with LUAD.
Materials and Methods
Cell culture
Human lung adenocarcinoma cell lines A549, PC9, H1975, Calu3 and the normal human bronchial epithelial cell line (HBE) were cultured in RPMI-1640 (Gibco, Life Technology, Carlsbad, CA, USA). Human embryonic kidney cell line HEK293 and HEK293T cells were grown in DMEM medium (Gibco, Life Technology, Carlsbad, CA, USA). Both RPMI-1640 and DMEM medium were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and all of cell lines were cultured in a humidified chamber with 5% CO2 at 37 °C. The cells were obtained from the cell bank of Cancer Research Institute of Central South University (Hunan, China) and were tested regularly for mycoplasma using quantitative Real-time PCR (qRT-PCR).
Patients and specimens
30 pairs of LUAD samples were collected to analyze the expression of CAR10 in tumor tissue samples compared to matched para-carcinoma tissues. These 30 pairs samples were collected from diagnosed LUAD patients including 12 patients with lymph node metastasis and 18 patients without lymph node metastasis at the Second Xiangya Hospital (Changsha, China). Collection and use of tissue samples were approved by the Ethical Review Committee of Hunan Second Xiangya Hospital, and written informed consent was obtained from each patient. Clinic information were collected from patient medical records and are reported in Table S1.
Bioinformatic analysis
The goal of this study was to identify lncRNAs increased in LUAD and was affected by the TGFβ/SMAD3 signal transduction, which contributed to the metastasis of LUAD and positively related to poor prognosis. We downloaded Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) dataset GSE26858 [20], which includes the transcriptome changes in human LUAD cells A549 treated with TGFβ and a small-molecule inhibitor of SMAD3 (SIS3) at different times. Using via criteria log2FC (fold change) ≥ 1.5 or ≤ -1.5 and p value <0.05, we screened lncRNAs induced by TGF-β and downregulated upon SIS3 treatment. Using the further bioinformatics analysis combining data from the Gene Transcription Regulation Database (GTRD, http://gtrd.biouml.org/) [21] and the Gene Expression Profiling Interactive Analysis website (GEPIA, http://gepia.cancer-pku.cn/) [22], we found lncRNAs responsible to TGF-β/SMAD3 and overexpression in tissue samples of patients with LUAD. We analyzed the expression of lncRNAs between LUAD tissue samples and normal lung tissue samples using GEO dataset GSE31210. The Kaplan-Meier survival analysis of lncRNA in LUAD tissue was executed in GEO dataset GSE19188 [23]. In addition, the relationship between lncRNA expression level and EML4-ALK rearrangement, EGFR mutation, KRAS mutation and smoking state of LUAD patients was analyzed using GEO dataset GSE31210 [24, 25] and GSE10072 [26].
The online software JASPAR (http://jaspar.genereg.net/) [27] and meme-FIMO (http://meme-suite.org/) [28] were utilized to predicate SMAD3 binding sites on the HCP5 promoter. Additionally, we used miRDB database (http://www.mirdb.org/) [29] to analyze whether HCP5 is contained complementary region of the miRNA 'seed' sequence.
Antibodies and reagents
Information on antibodies used in this study is provided in Table S2. Snai1, Slug, E-cadherin and Vimentin were purchased from Cell Signaling Technology, Inc (USA). Slug was purchased from Proteintech (Chicago, USA) and Snail was purchased from OriGene (Maryland, USA). SMAD3 and p-SMAD3 were purchased from Thermo Fisher Scientific (Waltham, USA). MiR-203 mimics, inhibitor and forward primers were purchased from RiboBio Co. (Guangzhou, China). Primers of EMT-related markers for qRT-PCR analysis were synthesized by Tsingke (Beijing, China). qRT-PCR primers used are listed in Table S3.
Plasmid construction and cell transfection
For knockdown of HCP5, two specific small interference RNAs from RiboBio (Guangzhou, China) were transfected into cells with Lipofectamine 3000 (InvitrogenTM). A full-length human HCP5 sequence from Genebank (NR_040662.1) was generated by PCR and subcloned into the pCDH vector to establish the stably HCP5-overexpression cells. To obtain a stable HCP5 knockdown A549 cell line, we synthesized two small hairpin RNAs (shRNA) containing a 19-bp interfering sequence against HCP5 transcript, and cloned them into a lentiviral vector pLVTH. HCP5 siRNA, shRNA and primers used in plasmid construction are shown in Table S4.
RNA extraction and real-time PCR analysis
When cells were grown to 80% confluence on 6-well plates, total RNA was extracted using Trizol reagent (CWIO, Beijing, China) according to the manufacturer's instructions. The RNA quantity and quality were evaluated by Nanodrop ND-2000 spectrophotometer. For mRNAs and lncRNAs expression analysis, 2 μg total RNA was reverse transcribed into cDNA with RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific™, USA). For miRNAs expression analysis, 2 μg total RNA was reverse transcribed into cDNA with miDETECT A Track™ qRT-PCR Kit (RiboBio, Guangzhou, China). qRT-PCR was performed using 2x SYBR Green qPCR Master Mix (Bimake, China) according to the manufacturer's instructions on Bio-Rad CFX Connect™ Real-Time PCR Detection System (Bio-Rad, USA). GAPDH, β-actin or U6 were used as a control for mRNA qualifcation and the 2-ΔΔCt method was applied in the analysis of relative quantification of gene expression levels.
Cell invasion assay
Cell invasion assay was performed using Transwell plates (24-well plate format). Corning Costar Transwell 24-well plates (8 μm pores; Corning, USA) were coated with Matrigel (BD) on the upper surface of the filter in each of the upper chamber and placed in a cell culture hood for 3 h at 37 °C. Cells (20000 cells/well) with serum-free RPMI-1640 were seeded in the upper chambers. Cultured medium with 20% serum was placed in the lower chamber. Cells were then allowed to invade for 48 h. Then, cotton swab was used to wipe the cells on the upper surface of the filter membrane and invasive cells on the lower surface were then fixed with 1% formaldehyde solution for 15 min and stained with 0.1% of crystal violet for 15 min. The stained cells were captured using a microscope (Nikon) and counted by software Image-Pro Plus 6.0.
Wound-healing assay
Four linear scratch wounds were created by sterile 20 μL pipette tip when cells were grown to confluent monolayers. The wells were washed by D-Hanks for three times to remove detached cells followed by cultured in medium with 2% serum. After 0, 24, 36 and 48 hours we took the image on the same wound with a microscope (Nikon) and calculated the distance between the edges of the wound on both sides.
Cell Counting Kit-8
CCK8 assay was conducted in accordance with the manufacturer's instructions (Bimake, China). Cells, transfected with siHCP5 or scrambled siCtrl for 48 h, were plated at a density of 2 x 103cells/well in 96-well plates containing 200 µL of culture medium and cultivated in 37 oC incubator with 5% CO2. The stably HCP5 over-expressing cell lines of A549 and Calu3 were cultured to logarithmic growth phase, and then seeded in a 96-well plate at a density of 800 cells/well. After 0, 1, 2, 3, 4 or 5 day, 20 μL of CCK8 solution (5 mg/mL) was added to each well of the plate to measure changes in cell viability. This plate was then incubated at 37 °C for an additional 2 h. Absorbance at 450 nm was evaluated using a scanning multi-well spectrophotometer. The experiment was performed with three replicates and six parallel samples were measured each time.
Colony formation assay
First, cells transfected with HCP5-targeting siRNA for 48h or stably overexpressing HCP5 cells were trypsinized into individual cells, and about 800 cells were seeded in each well of a 6-well plate and grown for 2 weeks at 37 °C, 5% CO2, and 100% humidity without changing the medium. After the formation of the bacteria, the plate was gently washed with PBS and fixed with 1% formaldehyde solution for 10 min, and then stained with 0.1% crystal violet for 15 min. Colonies with more than 50 cells were manually counted. The number of colonies was calculated by Image-Pro Plus 6.0.
Western blot analysis
The protein of cells and tissue samples were lysed using a RIPA lysis buffer (Beyotime Biotechnology, Haimen, China) with protease inhibitor (Roche, Basel, Switzerland) and phosphatase inhibitor cocktails (Bimake, China). Followed by ultrasound for 30 s and centrifuged at 12,000 rpm for 20 min, the concentration of protein was quantified using the Pierce™ BCA Protein Assay Kit (Thermo Scientific™, USA). The total protein products (50 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a PVDF membrane (Millipore). The membrane was blocked in 5% BSA in TBS Tween 20 (TBST; 25 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature and then incubated primary antibody with 5% BSA diluted overnight at 4 °C. The membranes were washed three times in TBST buffer and incubated with horseradish peroxidase-labeled secondary antibody for 1 h at 37 °C. The signal was visualized using an ECL detection reagent (BioRad, USA) and quantified by densitometry (BioRad ChemiDoc XRS system).
In vivo assay
Female BALB/c-nude mice (4 week-old) were purchased from Hunan SJA Laboratory Animal Company (Hunan, China) and maintained under SPF conditions. Animal experiments were approved by the Institutional Animal Care and Use Committee of Central South University (Changsha, China). To confirm the role of HCP5 in the promotion of LUAD cell proliferation in vivo, we performed subcutaneous tumor mouse models. Twelve female BALB/c-nude mice were randomly divided into two groups, with six mice in each group. The shHCP5 and negative control A549 cells (1 × 106) were washed once with PBS and subcutaneously injected into nude mice (n=6). The mice were checked regularly for tumor size at three weeks after injection, and the growth curves were plotted for each group. The tumor volumes were estimated using the following formula: length × width2× 0.52. All mice were sacrificed 27 days after inoculation and tumors were isolated and photographed and tumor weights were measured. All photographed tumors were isolated from single experiments at a similar time points. Part of the subcutaneous tumors were detected the expression of HCP5 by qRT-PCR.
For in vivo metastasis assays, shHCP5 and negative control A549 cells (4 × 106) were resuspended in PBS and injected into the tail vein of the nude mice (4 mice per group). The mice were sacrificed 8 weeks after injection, and their lungs were dissected, and fixed in 10% formalin and embedded in paraffin for hematoxylin-eosin staining (HE). To further confirm the HCP5 positive impact on LUAD cells metastasis in vivo, shHCP5 and negative control A549 cells were infected with a virus containing pLenti6V5D-TOPO luciferase EGFP (CMV/luciferase 17-EGFP and BSD) and were injected into the tail veins of the nude mice (4 × 106 per mice). Optical in vivo imaging of cancer metastasis was monitored with in vivo luminescence imaging system (IVIS) at 8 weeks after injection.
Dual luciferase reporter assay
To explore how SMAD3 regulated HCP5 transcription in lung cancer cells. Using the human genomic DNA, different SMAD3 binding sites on the HCP5 promoter, which were predicted by the website, were amplified by PCR and cloned into the PGL3-basic vector (Promega). These reporter gene plasmids included H1 (-2000 ~ +200), H2 (-2000 ~ -1370), H3 (-1369 ~ -1150), H4 (-1149 ~ -844) and H5 (-843 ~+200). HEK-293 cells were seeded in 24-well plates (1 × 105cells per well) and co-transfected with 500 ng luciferase reporter gene plasmids, 500 ng pcDNA3.1-SMAD3 or control plasmid, and 50 ng renilla luciferase reporter plasmid pRL-TK. Luciferase activity was measured 48 h after the transfection with Dual-Luciferase Reporter Assay System (Promega). Data were normalized against values of co-transfected renilla luciferase.
In addition, to evaluate the interaction between miR-203 and HCP5, A549 cells were transfected with psiCHECK2-based constructs containing HCP5 Wild-Type (HCP5 WT) and HCP5-Mutant (HCP5 Mut). After 48 h, firefly and renilla luciferase activity was examined by the Dual-Luciferase Reporter Assay System, and renilla activity was used to normalize firefly activity as well. Cells transfected with scrambled miRNA (a synthesized RNA showing no homology to any human mRNA sequence) served as negative controls.
Chromatin immunoprecipitation
The experiments were performed according to previous reports [30]. A549 cells(4 × 106) that were transfected with pcDNA3.1-SMAD3 were crosslinked with 1% formaldehyde solution, quenched with glycine (0.125 M) and washed with cold 1×PBS for three times. Cells were scraped with cold 1×PBS (containing protease inhibitors), and centrifuged at 500×g for 4 min at 4 °C. The cells were lysed with 300-500 μL SDS lysate for 20 min and mixed once every 5 min. Subsequently, Cells were sonicated for 15 min (sonication for 5s pause for 5 s) to shear DNA to lengths between 300 and 500 bp on ice. Stained chromatin was cultured overnight with anti-SMAD3 monoclonal antibody (Thermo Fisher, USA) or normal mouse IgG (Santa Cruz Blotechnology, USA). The beads were washed with washing buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.0, 150 mM NaCl, 1% NP40, 1% NaDOC). The qRT-PCR is then performed using R1 regions (including H3 fragments) specific primers: 5'-GATGACTATGGGGTGAGGGG-3' (sense), 5'-TATGGAGATGAGGTGTGCCG -3' (anti-sense).
Biotinylated RNA Pull-down assay
The entire experimental process should be performed in the RNase-free conditions. Biotin- labeled HCP5 and antisense-HCP5 control transcripts were executed respectively by in vitro transcription with T7 RNA polymerase in the presence of the Biotin RNA Labeling Mix (Roche, Mannheim, Germany), which was treated with Ribonuclease Inhibitor and RNase-free DNase I (Takara, Japan). 3 μg of purified biotin-labeled transcript were incubated with cell lysates from 2 × 107 A549 cells for one hour at room temperature. Dynabeads™ M-270 Streptavidin (InvitrogenTM) was then added to isolate the biotin-labeled RNA. The enrichment of HCP5 and miR-203 were detected by qRT-PCR analysis.
RNA fluorescent in situ hybridization
Cy3-labeled HCP5 probe and FAM-labeled miR-203 were purchased from GenePharma (Shanghai, China). A549 cells (1 × 103) were seeded on glass coverslips in the 6-well plate and grown overnight at 37 °C. Then, we washed cells twice with 1 × PBS and fixed them in 4% paraformaldehyde for 15 min. Permeabilization and hybridization followed by washing with sodium citrate buffer (SSC) according to the manufacturer's instructions of the FISH Kit (GenePharma, Shanghai, China). The nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI, GenePharma, China) and finally images were obtained by confocal microscope (Leica TCS SP8 × & MP). The sequences of RNA-FISH probes are listed in Table S5.
Statistical analysis
GraphPad Prism (version 5.01, La Jolla, CA, USA) software was used for statistical analyses. Significant differences between any two groups of data were made using Student's t-test. One-way analysis of variance was used when assessing significant differences between multiple sets of data. The cumulative overall survival (OS) was calculated using the Kaplan-Meier method, and the log-rank test was used to analyze differences in the survival times. All data were determined in triplicate and are representative of at least two separate experiments. All data are shown as mean ± S.E.M. Differences were considered significant if P < 0.05. *P<0.05, **P<0.01, ***P<0.001.
Results
Identification of HCP5 as a TGFβ/SMAD3-induced lncRNA
To identify lncRNAs responsive to TGFβ/SMAD3, we used the GSE26858 profiling data analysis, which includes the transcriptome changes in human LUAD cells A549 treated with TGFβ and a small-molecule inhibitor of SMAD3 (SIS3) at different times (Figure S1A). According to the threshold of log2 fold change (FC) ≥ 1.5 or ≤ -1.5 and p-value < 0.05, we identified 48 lncRNAs induced by TGF-β and downregulated upon SIS3 treatment (Figure S1B). Using the further bioinformatics analysis combining data from the GTRD and GEPIA, we selected ten lncRNAs, which were potentially simultaneously regulated by TGF-β/SMAD3 and overexpressed in LUAD. The hierarchical clustering analysis of these lncRNAs in the negative control (NC), TGF-β- and TGF-β/SIS3-treated groups is shown in Figure S1C.
To validate these data, we conducted qRT-PCR assays in A549 cells treated with SIS3 and TGF-β. TGF-β induced the upregulation of phosphorylated (p)-SMAD3 and N-cadherin and the downregulation of E-cadherin, while SIS3 had the opposite effect on their expression, indicating that our model was reliable (Figure 1A). We validated three lncRNAs (HCP5, Antisense Growth-Associated Long Non-Coding RNA [HAGLR] and MIR181A2HG) whose expression increased after TGF-β treatment and decreased upon SIS3 treatment, indicating that they were regulated by the TGFβ/SMAD3 signaling (Figure 1B).
HCP5 is a TGFβ/SMAD3 induced lncRNA. (A) A549 cells were treated with or without 10 μM SIS3 for 30 min and subsequently treated with or without 5 ng/mL TGF-β for 1 h. The levels of E-cadherin, N-cadherin, SMAD3 and p-SMAD3 were determined by western blotting. (B) The levels of ten lncRNA were detected by qRT-PCR in A549 cells treated as indicated above. The three lncRNAs with the most obvious changes are in a red frame. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (C-E) HCP5 (C), HAGLR (D) and MIR181A2HG (E) expression in human LUAD tissues and normal lung tissues analyzed in the GSE31210 dataset. (F-H) The expression level of HCP5, HAGLR and MIR181A2HG was verified by qRT-PCR in four LUAD lines, human bronchial epithelial (HBE) and human renal epithelial cell line 293 (HEK293). (I) The relative expression of HCP5 in 30 paired LUAD tissues and adjacent lung tissues. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (J) Kaplan-Meier analysis of the overall survival in patients with LUAD based on the levels of HCP5 expression using the GSE19188 dataset (p < 0.05). (K) HCP5 expression in EGFR and KRAS mutations and EML4-ALK rearrangement LUAD patients was analyzed in GSE31210 dataset. *P < 0.05, **P < 0.01, ***P< 0.001, NS: no statistical significance.
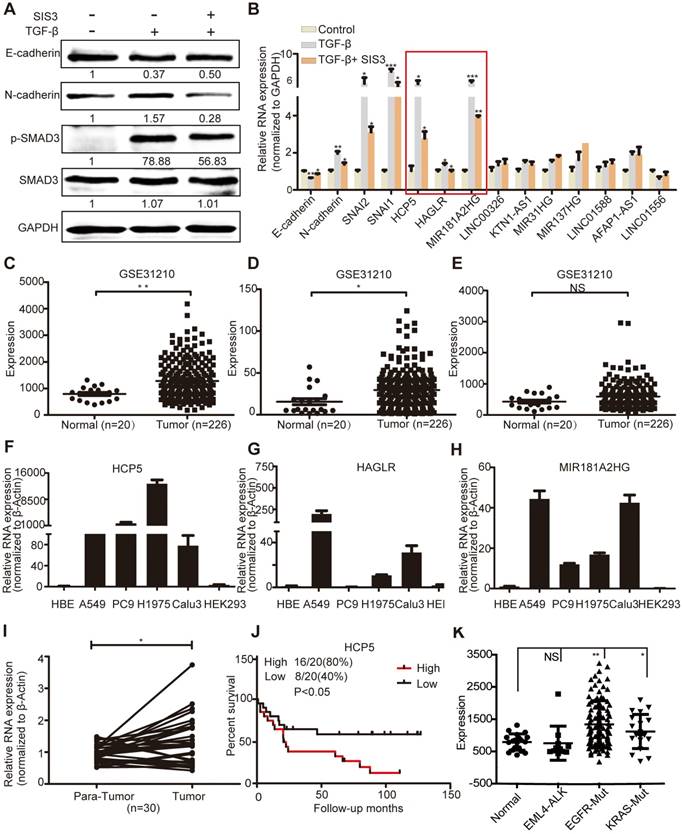
GSE31210 analysis showed that the levels of HCP5 and HAGLR were significantly higher in LUAD tissues, while MIR181A2HG was not significantly different between tumoral and non-tumoral tissues (Figure 1C-E). Compared with immortalized lung bronchial epithelial cells HBE and the human renal epithelial cell line 293, HCP5 shown obviously more abundant in all lung adenocarcinoma cell lines than HAGLR and MIR181A2HG (Figure 1F-H). Furthermore, using 30 pairs of LUAD tissue samples and paired healthy tissue, we confirmed that HCP5 was more expressed in LUAD compared to the adjacent lung tissue (Figure 1I). GSE19188 analysis showed that high expression of HCP5 positively correlated with poor prognosis of patients with LUAD (Figure 1J). Further analysis of GSE31210 showed that high expression of HCP5 was in tumor tissue of EGFR and KRAS mutations LUAD patients, but not EML4-ALK rearrangement patients (Figure 1K). Interestingly, we found that HCP5 is highly expression in patients with current smoking and not former smokers (Figure S1D, E). Thus, we focused on HCP5 transcriptional regulation and its functional relationship with EMT and metastasis in LUAD.
HCP5 is a direct transcriptional target of SMAD3
To explore the mechanism of HCP5 upregulation in LUAD, we analyzed LUAD expression profiles and found that SMAD3 is highly expressed in LUAD and positively correlates with HCP5 (Figure S2A, B). Overexpression of SMAD3 in A549 cells increased HCP5 levels about 2.5 folds, whereas knockdown of SMAD3 decreased HCP5 expression by half (Figure 2A, B). Because SMAD3 is a transcription factor, we hypothesized that SMAD3 binds the promoter of HCP5 and activates its transcription. Using the online software JASPAR (http://jaspar.genereg.net/) and meme-FIMO (http://meme-suite.org/), we predicted three major SMAD3 binding sites on the HCP5 promoter, located at -1,278/-1,219 (site 1), -1,200/-1,191 (site 2), and -883/-874 (site 3) nucleotides from the transcription starting site (Figure S2C).
HCP5 is a direct transcriptional target of SMAD3. (A, B) The expression of HCP5 was detected by qRT-PCR in SMAD3-silenced (A) or -overexpressing (B) A549 cells. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (C) Schematic representation of five HCP5 promoter reporters. The red dots represent the three binding sites of SMAD3 to the HCP5 promoter as predicted by the JASPAR and meme-FIMO databases. (D) Dual-luciferase reporter assays detected the activity of the five HCP5 promoter fragments described in (C) in SMAD3-overexpressing A549 cells; luciferase activity was normalized to Renilla. The H3 promoter fragment exhibits the highest activity. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (E) Schematic illustrations showing the predicted locations of two SMAD3-binding sites (SMAD3-BR) in HCP5 promoter and the amplified regions (R1, including H3) of PCR for ChIP assays. (F) ChIP-PCR assays using antibodies specific for SMAD3 to prove that SMAD3 binds to HCP5 promoter. *P < 0.05, **P < 0.01, ***P < 0.001.
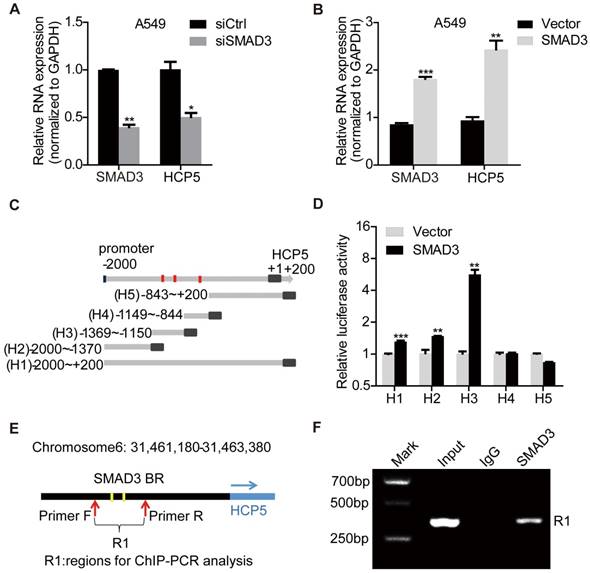
To confirm HCP5 was a transcriptional target of SMAD3, the promoter region of HCP5 (-2,000 to +200) was cloned into the pGL3-Basic luciferase reporter plasmid to generate the so-called H1 plasmid. Four different reporter from the H1 reporter were also generated, each containing a different portion of HCP5 (Figure 2C): H2 (-2,000/-1,370), H3 (-1,369/-1,150), H4 (-1,149/-844), H5 (-843/+200). These reporters were co-transfected with pcDNA3.1 (empty vector used as the control) or pcDNA3.1-SMAD3 plasmid into HEK-293 cells and luciferase activities were determined. We found that SMAD3 strongly increased the luciferase activity of H3 (Figure 2D). These data suggested that SMAD3 binds the promoter region between -1369/-1150 bp including two putative HCP5 binding sites and in such way mediates the activation of HCP5. To further prove the interaction between the H3 fragment and SMAD3, we performed chromatin immunoprecipitation (ChIP) assays in A549 cells, using primer sets that amplified the H3 fragment (Figure 2E). The chromatin pulled down by an anti-SMAD3 antibody indicated the occupancy of SMAD3 on HCP5 promoter sequence (Figure 2F). These findings showed that SMAD3 directly regulates HCP5 transcription in LUAD.
HCP5 promotes LUAD cells proliferation and invasion
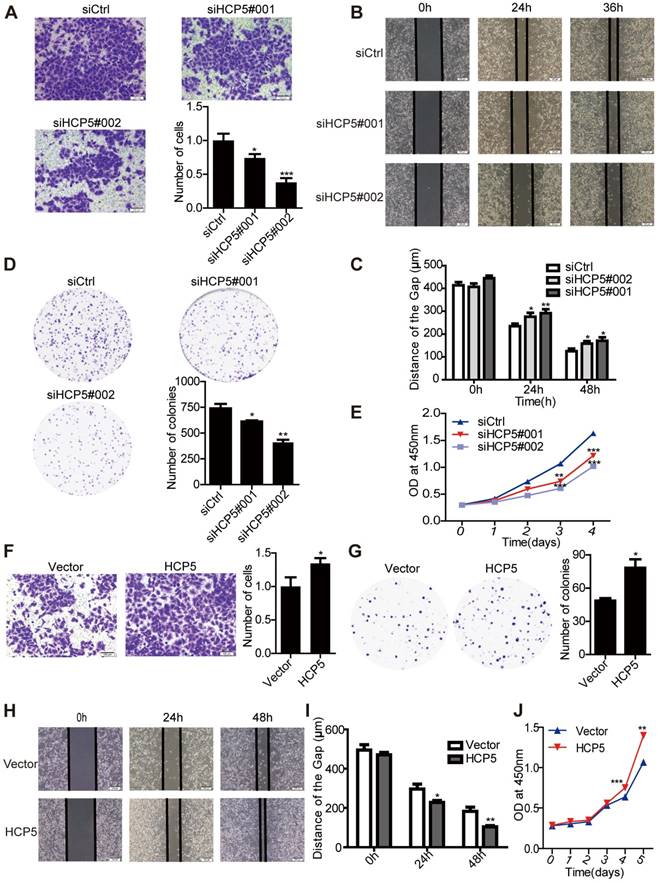
To investigate the function of HCP5 in LUAD tumorigenesis, we knocked down the expression of HCP5 by transfecting two small interfering RNAs against HCP5 (siHCP5#001 and siHCP5#002) in A549 and Calu3 cells. As expected, endogenous HCP5 was obviously suppressed (Figure S3A, C). Transwell and wound-healing assays demonstrated that HCP5 silencing reduced the invasion and migratory capability of A549 cells (Figure 3A-C). Additionally, HCP5 knockdown inhibited cell growth and colony formation of A549 cells (Figure 3D, E). Conversely, increasing the expression of HCP5 has opposite effect on the invasion and proliferation capabilities of A549 cells (Figure 3F-J). Similar results were obtained in Calu3 cells (Figure S3E-J). Together, these data indicated that HCP5 plays an important role in LUAD tumorigenesis and metastasis.
HCP5 regulates LUAD cell proliferation and metastasis. (A) Transwell assays to investigate the invasion ability of HCP5-silenced and control A549 cells. Scale bar: 100 μm. (B, C) Wound-healing assay investigate the migratory ability of HCP5 knockdown and control A549 cells. Scale bar: 200 μm. (D) Silencing of HCP5 inhibits the colony formation of A549 cells. (E) Cell Counting Kit-8 (CCK-8) assays were performed in A549 cells silenced for HCP5. (F) Transwell assay to investigate the invasion ability of HCP5-overexpressing and control A549 cells. Scale bar: 100 μm. (G) Overexpression of HCP5 promotes the colony formation of cells. (H, I) Wound-healing assay investigate the migratory ability of HCP5 overexpression and control A549 cells. Scale bar: 200 μm. (J) Cell Counting Kit-8 (CCK-8) assays were performed in A549 cells overexpressing HCP5. All data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). *P< 0.05, **P < 0.01, ***P< 0.001.
To further estimate the oncogenic role of HCP5 in LUAD, we constructed a lentiviral vector harboring a short hairpin RNA targeting HCP5, established stable knockdown A549 cells. We chose shHCP5-2 in which expression of HCP5 was down-regulated almost 70% (Figure S4A). Stably HCP5-silenced A549 cells (shHCP5) or control cells (shCtrl) were subcutaneously injected into nude mice. We found that HCP5 knockdown obviously inhibited the tumor growth and produced significantly smaller tumors compared with control mice (Figure 4A-D).
HCP5 knockdown inhibits cell proliferation and metastasis in nude mice. (A) Mice were subcutaneously injected with cells stably silenced for HCP5 (shHCP5) or control cells (shCtrl). Images of the tumor lumps from the indicated groups at the endpoint of the experiment. n = 6 mice per group. (B) Tumor formation in mice treated as in (A) was monitored at the indicated time points. Data are shown as the mean ± SEM (two-tailed Student's t-test); n = 6 mice per group. (C) Tumor weights in mice treated as in (A) were recorded. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (D) QRT-PCR for HCP5 expression in xenograft tumor tissues of shHCP5 or shCtrl group. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (E) shHCP5 or shCtrl A549 cells were intravenously injected into nude mice and lung tissues were isolated after eight weeks. Representative lung images showing metastases in the lung. Arrows indicate the metastatic nodules. (F) Microscopic images of lung tissue sections stained by hematoxylin and eosin. Left: Scale bar = 200 μm. Right: Scale bar = 50 μm. (G) The number of metastatic nodules in the lungs is shown as the mean ± S.E.M.; n=4 mice per group (two-tailed Student's t-test). *P< 0.05, **P < 0.01, ***P < 0.001.
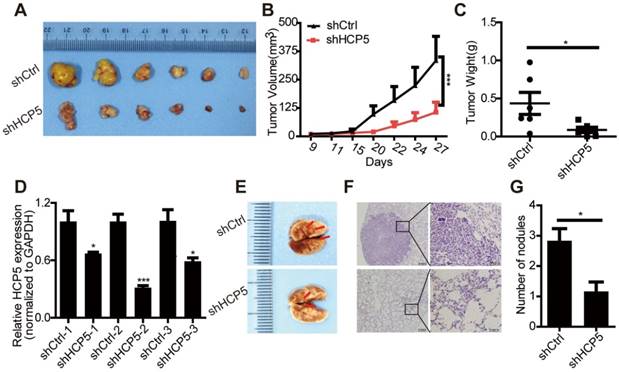
In addition, we used a tail vein injection model to explore the effect of HCP5 on tumor metastasis in vivo. We injected either shCtrl or shHCP5 A549 cells (4 × 106) into nude mice via the tail vein. Eight weeks after injection, we found that A549 shHCP5 cells produced significantly less lung metastases compared with A549 shCtrl cells (Figure 4E). Further histological examination of the lungs showed less lung nodules in mice injected with A549 shHCP5 cells than in those injected with A549 shCtrl cells (Figure 4F, G). To further examine the influence of HCP5 had on metastasis in vivo, shHCP5 or shCtrl A549 cells that were labeled with EGFP-luciferase were intravenously injected into nude mice. Bioluminescence was examined via an in vivo luminescence imaging system after 8 weeks. The results revealed that A549 cells with low expression of HCP5 had fewer metastatic sites compared with the control group (Figure S4C, D). These data showed that HCP5 knockdown significantly inhibits proliferation and metastasis of LUAD in vivo.
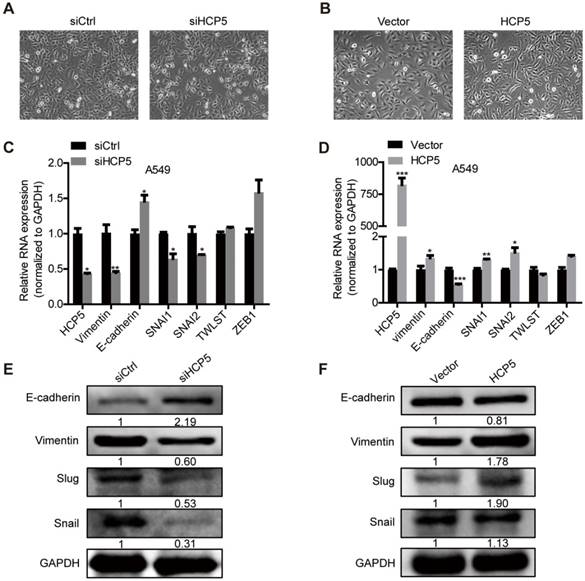
HCP5 positively regulates EMT via the miR-203/SNAI axis
EMT plays an important role in tumors metastasis. We observed that silencing of HCP5 induced epithelial-like morphological features and overexpression of HCP5 induced a mesenchymal-like phenotype in lung cancer cells (Figure 5A, B).To explore how HCP5 regulates lung cancer cell invasion, we examined the expression of EMT-related genes in A549 cells. QRT-PCR assays revealed that knockdown of HCP5 increased the expression of the epithelial marker E-cadherin (CDH1) and downregulated the mesenchymal marker vimentin (VIM), while HCP5 overexpression induced opposite effects. We also examined the effect of HCP5 on the expression of EMT associated transcription factors and found HCP5 silencing reduced the expression of Snail (SNAI1) and Slug (SNAI2) but not that of TWIST and ZEB-1. Western blot analysis also confirmed that HCP5 silencing or overexpression induced the expression changes of E-cadherin, vimentin, Snail and Slug (Figure 5C-F). GSE19188 analysis showed that high expression of SNAI2 rather than SNAI1 obviously negatively correlated with poor prognosis of patients with LUAD(Figure S5A and B). We also evaluated overall survival of LUAD patients via the combined index of HCP5 and SNAI1/2 expression. We found that overexpression of both HCP5 and SNAI2 suggest the worst prognosis for LUAD patients (Figure S5C, D).
HCP5 promotes EMT of A549 cells. (A, B) Phase-contrast microscopy images of A549 silenced for or overexpressing HCP5. (C, D) The mRNA levels of EMT markers (CDH1, VIM) and transcription factors (SNAI1, SNAI2, TWIST and ZEB1) in indicated A549 silenced for or overexpressing HCP5. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (E, F) Western-blot analysis of the EMT markers (E-cadherin, Vimentin), Snail and Slug expression in A549 cells silenced for or overexpressing HCP5. *P < 0.05, **P < 0.01, ***P< 0.001.
Analysis of the miRDB database (http://www.mirdb.org/) showed that HCP5 contained one region complementary to the 'seed' region of miR-203 (Figure S6A). Nuclear/cytoplasmic separation experiments in A549 cells showed that HCP5 is distributed in the cytoplasm and nuclear compartments, which suggests that HCP5 may interact with miR-203 (Figure S6B). Furthermore, we transfected miR-203 mimics into A549 cells and western blot assays revealed that the expression of Snail, Slug and vimentin were increased and E-cadherin was reduced. In contrast, miR-203 inhibitors induced opposite effects (Figure S6C-F). We hypothesized that HCP5 acts as a competitive endogenous RNA for miR-203 and further stabilizes the expression of SNAI1 and SNAI2 to promote EMT of lung cancer cells.
Transfection of miR-203 mimics or inhibitors attenuated or increased HCP5 expression compared with controls respectively (Figure 6A, B). To identify whether HCP5 is a target of miR-203, we constructed luciferase reporters with the wild-type (HCP5 WT) or mutated (HCP5 Mut) miR-203-binding region of HCP5 upstream the luciferase coding region. Notably, miR-203 mimics attenuated the activity of the HCP5 Wild-Type reporter, but not that of the HCP5 Mutant reporter (Figure 6C); miR-203 inhibitors has opposite effect (Figure 6D). Conversely, downregulation of HCP5 increased the expression of miR-203, while the expression of miR-203 decreased upon HCP5 overexpression (Figure S6G, H). RNA Pull Down experiment showed that biotin-labeled HCP5 can enrich more miR-203 compare with antisense-HCP5 control (Figure 6E). We also confirmed that HCP5 and miR-203 were co-localized in cytoplasm by FISH assay (Figure 6F).
HCP5 positively regulates EMT via the miR-203/SNAI axis. (A, B) The expression of HCP5 in A549 cells transfected with miR-203 mimics or inhibitors or negative control oligonucleotides. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (C, D) miR-203 mimics inhibit (C), whereas miR-203 inhibitors promote (D) Luciferase activity in A549 cells cotransfected with miR-203 mimics or inhibitors and luciferase reporters containing HCP5 or mutant transcript. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (E) MiR-203 was specifically pulled down by biotin-labelled HCP5 compared with HCP5-antisense in A549 cells. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (F) FISH assay HCP5 (red) is co-localized with miR-203 (green). The arrow indicates the approximate location. (G-I) The mRNA (G, H) or protein levels(I) of Slug, Snail, vimentin and E-cadherin when HCP5 knockdown A549 cells transfected with miR-203 inhibitors or HCP5-overexpressing A549 cells transfected with miR-203 mimics. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (J) Transwell assays to investigate the invasion ability of HCP5 knockdown A549 cells transfected with miR-203 inhibitors or HCP5-overexpressing A549 cells transfected with miR-203 mimics. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). *P< 0.05, **P< 0.01, ***P < 0.001.
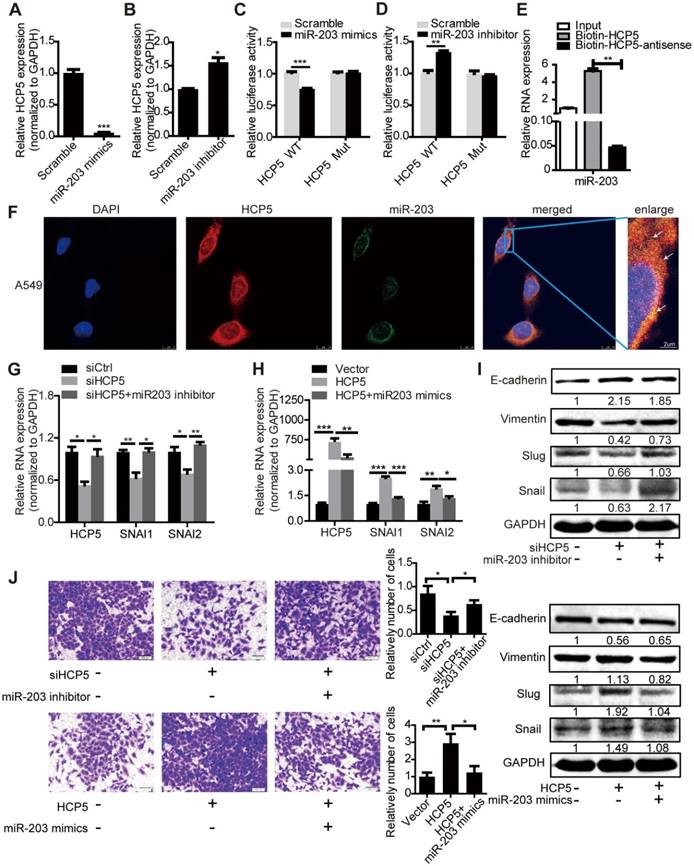
To explore whether HCP5 stabilized SNAI1 and SNAI2 expression through miR-203, we transfected miR-203 inhibitors into shHCP5 A549 cells or co-transfected miR-203 mimics and HCP5 into A549 cells, and detected the expression of SNAI1 and SNAI2. QRT-PCR and western blot assays showed that the knockdown of miR-203 abolished the inhibition of Slug and Snail expression induced by HCP5 knockdown. Consistently, miR-203 mimics abolished the up-regulatory effect of HCP5 on these factors. Notably, E-cadherin and vimentin expression showed opposite and similar changes, respectively, in these groups (Figure 6G-I). Furthermore, we investigated changes in the invasion ability of A549 cells induced by changes in the expression of HCP5 and miR-203 in Transwell assays. MiR-203 inhibitors abolished the inhibitory effect of HCP5 knockdown on cell migration. On the other hand, HCP5 overexpression induced cell migration, and, miR-203 mimics partly reversed this effect (Figure 6J). These results indicated that HCP5 positively regulates EMT via the miR-203/SNAI axis to promote metastasis of lung cancer cells.
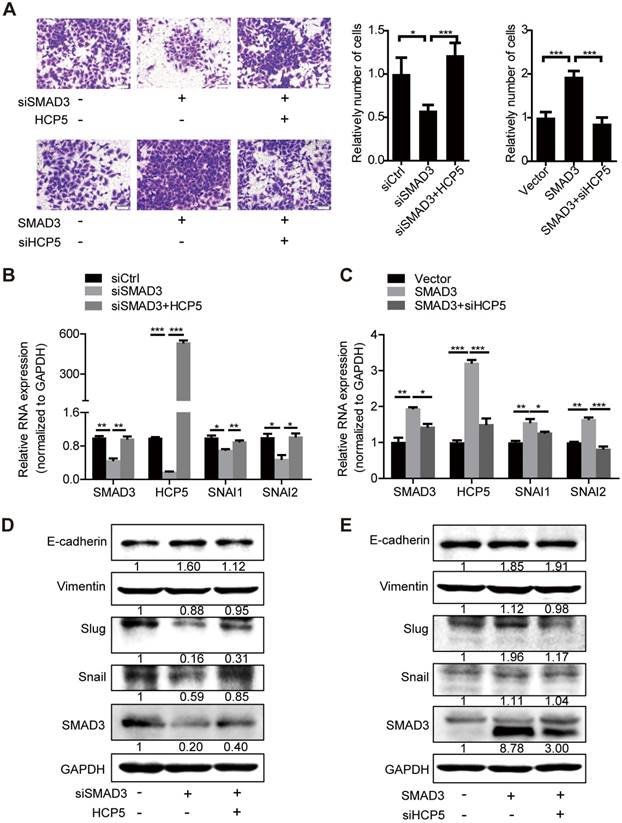
HCP5 is required for the TGF-β/SMAD3 induced invasive phenotype in A549 cells
To further investigate whether HCP5 is necessary for SMAD3-regulated EMT processes, we transfected HCP5 into SMAD3-silenced A549 cells. Transwell assays showed that the invasive ability of the cells was weakened upon SMAD3 silencing into A549 cells. However, this phenomenon was abolished by overexpression of HCP5. In addition, knockdown of HCP5 inhibited the invasive abilities of cells overexpressing SMAD3 (Figure 7A). Consistently, qRT-PCR and western blot assays showed that overexpression of HCP5 abolished the inhibition of the expression of vimentin, Slug and Snail and the upregulation of E-cadherin induced by SMAD3 knockdown. HCP5 silencing abolished the effect of SMAD3 on the expression of these markers (Figure 7B-E). Considering the biphasic functions of the TGF-β/SMAD signaling, we evaluated whether HCP5 affects the expression of the TGF-β/SMAD cytostatic targets p15, p21 and p57. Interestingly, overexpression HCP5 decreased the expression of the tumor suppressors p15 and p57 expression and had no affection on p21 expression. In contrast, HCP5 silencing led to higher p15 and p57 expression (Figure S6I). These results demonstrate that HCP5 plays a vital role in the EMT induced by the TGF-β/SMAD3 signaling in LUAD cells.
HCP5 is required for the TGF-β/SMAD3-induced invasive phenotype in A549 cells. (A) Transwell assays to investigate the invasion ability of SMAD3 knockdown A549 cells transfected with HCP5 or SMAD3-overexpressing A549 cells transfected with siHCP5. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). (B-E) The mRNA (B, C) or protein levels (D, E) of Slug, Snail, vimentin and E-cadherin when SMAD3 knockdown A549 cells transfected with HCP5 or SMAD3-overexpressing A549 cells transfected with siHCP5. Data are shown as the mean ± S.E.M. of three independent experiments (two-tailed Student's t-test). *P< 0.05, **P < 0.01, ***P< 0.001.
Discussion
The TGF-β/Smad canonical signaling facilitates tumor progression by inducing EMT and metastasis [31, 32]. In this study, we demonstrated that the lncRNA HCP5 is induced by TGF-β/SMAD3 in LUAD. HCP5 is located at 6P21.33 and has sequence homologies to retroviral genes [33]. Our results suggested that HCP5 is upregulated in tumor tissues and positively correlates with poor prognosis in patients with LUAD. Although several lncRNAs that regulate lung cancer progression through the TGF-β/Smad pathway have been identified [34-36], HCP5 is distinguished by its direct transcriptional regulation by SMAD3. Several findings supported this conclusion: The expression of SMAD3 and HCP5 positively correlated in LUAD tissues; the overexpression or knockdown of SMAD3 changed HCP5 levels; finally, ChIP and luciferase assays, confirmed the direct binding of SMAD3 to HCP5 promoter.
Studies exploring the role of HCP5 in cancer are limited. A recent study has reported that a single-nucleotide polymorphism (SNP; rs2244546) in HCP5 is considered a susceptibility locus for hepatitis C virus (HCV)-associated hepatocellular carcinoma [33]. HCP5 is upregulated in glioma and follicular thyroid carcinoma tissues to promote tumor progression [37, 38]. In this study, we demonstrated the oncogenic role of HCP5 in LUAD: HCP5 knockdown inhibited cell proliferation, migration and invasion in A549 and Calu3 cells and HCP5 had opposite effects. Furthermore, silencing of HCP5 also inhibited tumor growth in a xenograft murine model and metastasis formation in a lung metastasis model of A549 cells in nude mice. Silencing of HCP5 induced epithelial-like morphological features in A549 cells, accompanied by downregulation of the mesenchymal marker vimentin and upregulation of the epithelial marker E-cadherin, and HCP5 was associated with opposite results.
TGF-β induces the nuclear translocation of the SMAD complex, resulting in the expression of EMT-associated transcription factors including SNAI, ZEB and TWIST [39-41]. Interestingly, we observed that the ectopic expression of HCP5 was sufficient to increase the expression of SNAI1 and SNAI2 but not that of ZEB1 and TWIST. Consistently, HCP5 silencing attenuated SNAI1 and SNAI2 expression and upregulated CDH1. Notably, we found that HCP5, like SNAI1/2, contains miR-203 response elements. MiR-203 expression was downregulated in NSCLS and negatively correlated to lymphatic metastasis and clinical TNM stages [42]. Moreover, some studies have reported that both of SNAI1 and SNAI2 are targets of miR-203 in hepatocellular carcinoma and malignant glioma [43, 44]. Indeed, HCP5 sequesters miR-203 acting as a molecular sponge, and therefore stabilizes Snail and Slug, in turn, promote invasion of lung cancer cells. Thus, HCP5 augments the expression of Snail and Slug by sponging miR-203 and amplifies the TGF-β/SMAD signaling that promotes metastasis of LUAD.
The TGF-β/SMAD signaling has biphasic functions in cancer progression depending on the cellular context [40]. We show here that overexpression of HCP5 counteracts the inhibitory effect on cell invasion caused by SMAD3 downregulation and silencing of HCP5 inhibits cell invasion even upon overexpression of SMAD3. Notably, HCP5 decreased the expression of the tumor suppressors p15 and p57 and increased the expression of Slug and Snail in A549 cells. Previous studies have shown that 14-3-3ζ [45] interacts with the SMAD partners from p53 to GLI family zinc finger 2 (Gli2) to promote breast cancer bone metastasis and paraspeckle component 1 (PSPC1) [40] favors the binding of SMADs to the promoters of pro-metastatic transcription factors to promote tumor expansion. Whether HCP5 acts as a master regulator of TGF-β/SMAD dichotomous functions needs to be further investigated.
Our results revealed that HCP5 is overexpressed in tumor tissues of LUAD patients with EGFR and KRAS mutations and current smoker. EGFR mutation and KRAS mutations are very common in LUAD, present in about 20% and 25% respectively [46]. Recent study reported that a synonymous mutation in EGFR (c.2361G>A) induced decreasing of lncRNA EGFR-AS1 expression to enhance sensitivity to EGFR TKI in squamous-cell carcinoma [47]. The smoking prevalence in women younger than 40 years increased obviously and cigarette smoke induced deregulation of lncRNAs in HBE cells and lung cancer cells [48, 49]. Further studies are needed to investigate the relationship of HCP5 with oncogenic mutations of LUAD and cigarette smoke extract induced malignant transformation.
In conclusion, HCP5 is induced by TGF-β and is directly regulated by SMAD3, which, in turn, is a positive regulator of the TGF-β/SMAD signaling via the miR-203/SNAI axis. Our studies demonstrate that overexpression of HCP5 in LUAD is important for promoting EMT and metastasis and may be a potential therapeutic target for LUAD treatment.
Abbreviations
AUC: area under the curve; CCK-8: Cell Counting Kit-8; CDH1: E-cadherin gene; ChIP: chromatin immunoprecipitation; EMT: epithelial-mesenchymal transition; FC: fold change; GAPDH: glyceraldeyde 3-phosphate dehydrogenase; GEPIA: Gene Expression Profiling Interactive Analysis website; Gli2: GLI family zinc finger 2; GTRD: Gene Transcription Regulation Database; HAGLR: Antisense Growth-Associated Long Non-Coding RNA; HBE: human bronchial epithelial; HCP5: histocompatibility leukocyte antigen complex P5; HCV: hepatitis C virus; lncRNA: long non-coding RNA; LUAD: lung adenocarcinoma; MALAT: metastasis-associated lung adenocarcinoma transcript 1; miR: microRNA; NC: negative control; NSCLC: non-small cell lung cancer; P: phosphorylated; PD-1: programmed death-ligand 1; PSPC1: paraspeckle component 1; qRT-PCR: quantitative Real-time PCR; R-Smads: receptor-regulated Smads; ROC: receiver operating characteristics; SCLC: small cell lung cancer; shCtrl: shRNA control; shHCP5: short hairpin RNA targeting HCP5; siHCP5: small interference RNA targeting HCP5; siCtrl: siRNA control; SIS3: small-molecular inhibitor of SMAD3; SMAD3-BR: SMAD3-binding sites; SNAI1: Snail gene; SNAI2: Slug gene; SNP: single-nucleotide polymorphism; TGFβ: transforming growth factor-beta; TGFβR: TGFβ receptor; VIM: vimentin gene; ZEB: Zinc finger E-box-binding homeobox; ZO-1: Tight Junction Protein 1; T: human LUAD tissues; N: matched para-carcinoma tissues; GEO: Gene Expression Omnibus; DMSO: dimethyl sulfoxide; OS: overall survival.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China(81602030 to R.R.W); the Overseas Expertise Introduction Project for Discipline Innovation (111-2-12 to G.Y. Li); the Open-End Fund for the Valuable and Precision Instruments of Central South University (CSUZC2014044 to Z.L); and the Undergraduate Training Program for Innovation and Entrepreneurship (CSUZY20181285 to Z.L).
Contributions
Zheng Li and Ranran Wang conceived and designed the study. Lin Jiang, Xiaolu Ge, Lingna Chen, Yerong Hu, Xianming Tang performed experiments, with additional technical guidance and expertise from Ming Zhou, Yanhong Zhou, Wei Xiong and Guiyuan Li. Lin Jiang and Fang li performed computational analysis and statistical analyses. Zheng Li and Lin Jiang wrote the manuscript, with extensive input from all authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Eser PO, Janne PA. TGFbeta pathway inhibition in the treatment of non-small cell lung cancer. Pharmacol Ther. 2018;184:112-30
2. Yu JR, Tai Y, Jin Y, Hammell MC, Wilkinson JE, Roe JS. et al. TGF-beta/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015;29:250-61
3. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST. et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479-99
4. Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659-93
5. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-84
6. Massague J. TGFbeta in Cancer. Cell. 2008;134:215-30
7. Miyazono K, Katsuno Y, Koinuma D, Ehata S, Morikawa M. Intracellular and extracellular TGF-beta signaling in cancer: some recent topics. Front Med. 2018;12:387-411
8. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
9. Novello S, Barlesi F, Califano R, Cufer T, Ekman S, Levra MG. et al. Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27:v1-v27
10. Gibson AJW, Li H, D'Silva A, Tudor RA, Elegbede AA, Otsuka S. et al. Comparison of Clinical Characteristics and Outcomes in Relapsed Versus De Novo Metastatic Non-Small Cell Lung Cancer. Am J Clin Oncol. 2019;42:75-81
11. Huber-Ruano I, Raventos C, Cuartas I, Sanchez-Jaro C, Arias A, Parra JL. et al. An antisense oligonucleotide targeting TGF-beta2 inhibits lung metastasis and induces CD86 expression in tumor-associated macrophages. Ann Oncol. 2017;28:2278-85
12. Marini KD, Croucher DR, McCloy RA, Vaghjiani V, Gonzalez-Rajal A, Hastings JF. et al. Inhibition of activin signaling in lung adenocarcinoma increases the therapeutic index of platinum chemotherapy. Sci Transl Med. 2018;10:eaat3504
13. Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M. et al. Fibroblast-specific inhibition of TGF-beta1 signaling attenuates lung and tumor fibrosis. J Clin Invest. 2017;127:3675-88
14. Qiu P, Feng XH, Li L. Interaction of Smad3 and SRF-associated complex mediates TGF-beta1 signals to regulate SM22 transcription during myofibroblast differentiation. J Mol Cell Cardiol. 2003;35:1407-20
15. Ge X, Li GY, Jiang L, Jia L, Zhang Z, Li X. et al. Long noncoding RNA CAR10 promotes lung adenocarcinoma metastasis via miR-203/30/SNAI axis. Oncogene. 2019 [Epub ahead of print]
16. Wang J, Shao N, Ding X, Tan B, Song Q, Wang N. et al. Crosstalk between transforming growth factor-beta signaling pathway and long non-coding RNAs in cancer. Cancer Lett. 2016;370:296-301
17. Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM. et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031-41
18. Schmidt LH, Spieker T, Koschmieder S, Schaffers S, Humberg J, Jungen D. et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol. 2011;6:1984-92
19. Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF. et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer cell. 2014;25:666-81
20. Zhang Y, Handley D, Kaplan T, Yu H, Bais AS, Richards T. et al. High throughput determination of TGFbeta1/SMAD3 targets in A549 lung epithelial cells. PLoS One. 2011;6:e20319
21. Yevshin I, Sharipov R. GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res. 2017;45:D61-d7
22. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98-w102
23. Shi YX, Yin JY, Shen Y, Zhang W, Zhou HH, Liu ZQ. Genome-scale analysis identifies NEK2, DLGAP5 and ECT2 as promising diagnostic and prognostic biomarkers in human lung cancer. Sci Rep. 2017;7:8072
24. Tu CY, Cheng FJ, Chen CM, Wang SL, Hsiao YC, Chen CH. et al. Cigarette smoke enhances oncogene addiction to c-MET and desensitizes EGFR-expressing non-small cell lung cancer to EGFR TKIs. Mol Oncol. 2018;12:705-23
25. Peng F, Wang R, Zhang Y, Zhao Z, Zhou W, Chang Z. et al. Differential expression analysis at the individual level reveals a lncRNA prognostic signature for lung adenocarcinoma. Mol Cancer. 2017;16:98
26. Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A. et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3:e1651
27. Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I. et al. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102-6
28. Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017-8
29. Lv J, Liu J, Guo L. Bioinformatic analyses of microRNA-targeted genes and microarray-identified genes correlated with Barrett's esophagus. Cell Cycle. 2018;17:792-800
30. Long MD, Singh PK, Russell JR, Llimos G, Rosario S, Rizvi A. et al. The miR-96 and RARgamma signaling axis governs androgen signaling and prostate cancer progression. Oncogene. 2019;38:421-444
31. Ali A, Zhang P, Liangfang Y, Wenshe S, Wang H, Lin X. et al. KLF17 empowers TGF-beta/Smad signaling by targeting Smad3-dependent pathway to suppress tumor growth and metastasis during cancer progression. Cell Death Dis. 2015;6:e1681
32. Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D. et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov. 2012;2:248-59
33. Lange CM, Bibert S, Dufour JF, Cellerai C, Cerny A, Heim MH. et al. Comparative genetic analyses point to HCP5 as susceptibility locus for HCV-associated hepatocellular carcinoma. J Hepatol. 2013;59:504-9
34. Hao Y, Yang X, Zhang D, Luo J, Chen R. Long noncoding RNA LINC01186, regulated by TGF-beta/SMAD3, inhibits migration and invasion through Epithelial-Mesenchymal-Transition in lung cancer. Gene. 2017;608:1-12
35. Zhao JJ, Hao S, Wang LL, Hu CY, Zhang S, Guo LJ. et al. Long non-coding RNA ANRIL promotes the invasion and metastasis of thyroid cancer cells through TGF-beta/Smad signaling pathway. Oncotarget. 2016;7:57903-18
36. Wang P, Luo ML. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-beta/Smad3 pathway. Sci Transl Med. 2018;10:eaat2039
37. Liang L, Xu J, Wang M, Xu G, Zhang N, Wang G. et al. LncRNA HCP5 promotes follicular thyroid carcinoma progression via miRNAs sponge. Cell Death Dis. 2018;9:372
38. Teng H, Wang P, Xue Y, Liu X, Ma J, Cai H. et al. Role of HCP5-miR-139-RUNX1 Feedback Loop in Regulating Malignant Behavior of Glioma Cells. Mol Ther. 2016;24:1806-22
39. Budi EH, Duan D, Derynck R. Transforming Growth Factor-beta Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017;27:658-72
40. Yeh HW, Hsu EC. PSPC1 mediates TGF-beta1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol. 2018;20:479-91
41. Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem. 2008;283:33437-46
42. Tang R, Zhong T, Dang Y, Zhang X, Li P, Chen G. Association between downexpression of MiR-203 and poor prognosis in non-small cell lung cancer patients. Clin Transl Oncol. 2016;18:360-8
43. Zheng XB, Chen XB, Xu LL, Zhang M, Feng L, Yi PS. et al. miR-203 inhibits augmented proliferation and metastasis of hepatocellular carcinoma residual in the promoted regenerating liver. Cancer Sci. 2017;108:338-46
44. Chang JH, Hwang YH, Lee DJ, Kim DH, Park JM, Wu HG. et al. MicroRNA-203 Modulates the Radiation Sensitivity of Human Malignant Glioma Cells. Int J Radiat Oncol Biol Phys. 2016;94:412-20
45. Xu J, Acharya S, Sahin O, Zhang Q, Saito Y, Yao J. et al. 14-3-3zeta turns TGF-beta's function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of Smad partners from p53 to Gli2. Cancer cell. 2015;27:177-92
46. Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyo D. et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24:1559-67
47. Tan DSW, Chong FT, Leong HS, Toh SY, Lau DP, Kwang XL. et al. Long noncoding RNA EGFR-AS1 mediates epidermal growth factor receptor addiction and modulates treatment response in squamous cell carcinoma. Nat Med. 2017;23:1167-75
48. Li X, Zheng M, Pu J, Zhou Y, Hong W, Fu X. et al. Identification of abnormally expressed lncRNAs induced by PM2.5 in human bronchial epithelial cells. Biosci Rep. 2018;38:BSR20171577
49. Lin H, Zhang X, Feng N, Wang R, Zhang W, Deng X. et al. LncRNA LCPAT1 Mediates Smoking/ Particulate Matter 2.5-Induced Cell Autophagy and Epithelial-Mesenchymal Transition in Lung Cancer Cells via RCC2. Cell Physiol Biochem. 2018;47:1244-58
Author contact
![]() Corresponding author: Zheng Li, Cancer Research Institute, Central South University, 110 Xiangya Road, 410078, Changsha, China. E-mail: lizhengedu.cn.
Corresponding author: Zheng Li, Cancer Research Institute, Central South University, 110 Xiangya Road, 410078, Changsha, China. E-mail: lizhengedu.cn.