Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Methods
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Results
Discussion
Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(7):1864-1877. doi:10.7150/thno.29579 This issue Cite
Research Paper
miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease
Bin Wang1,3*, Aiqing Zhang2,3*, Haidong Wang3,4, Janet D. Klein3, Lin Tan5, Ze-Mu Wang6, Jie Du7, Nawazish Naqvi5, Bi-Cheng Liu1, Xiaonan H. Wang3 ![]()
1. Institute of Nephrology, Zhong Da Hospital, Southeast University, Nanjing, Jiangsu, 210009, China
2. Department of Pediatric Nephrology, the Second Affiliated Hospital of Nanjing Medical University, Nanjing, 210003, China
3. Renal Division, Department of Medicine, Emory University, Atlanta, GA 30322, USA
4. College of Animal Science and Veterinary Medicine, Shanxi Agricultural University, Taigu, Shanxi, 030801, China
5. Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA 30322, USA
6. Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, China
7. The Key Laboratory of Remodeling-related Cardiovascular Diseases, Beijing Anzhen Hospital Affiliated to Capital Medical University, Beijing Institute of Heart Lung and Blood Vessel Diseases, Beijing 100029, China
*Both authors contributed equally to this work.
Received 2018-8-29; Accepted 2019-1-19; Published 2019-3-7
Citation:
Wang B, Zhang A, Wang H, Klein JD, Tan L, Wang ZM, Du J, Naqvi N, Liu BC, Wang XH. miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics 2019; 9(7):1864-1877. doi:10.7150/thno.29579. https://www.thno.org/v09p1864.htm
Other stylesAbstract

Uremic cardiomyopathy and muscle atrophy are associated with insulin resistance and contribute to chronic kidney disease (CKD)-induced morbidity and mortality. We hypothesized that restoration of miR-26a levels would enhance exosome-mediated microRNA transfer to improve muscle wasting and cardiomyopathy that occur in CKD.
Methods: Using next generation sequencing and qPCR, we found that CKD mice had a decreased level of miR-26a in heart and skeletal muscle. We engineered an exosome vector that contained Lamp2b, an exosomal membrane protein gene fused with a muscle-specific surface peptide that targets muscle delivery. We transfected this vector into muscle satellite cells and then transduced these cells with adenovirus that expresses miR-26a to produce exosomes encapsulated miR-26a (Exo/miR-26a). Exo/miR-26a was injected once per week for 8 weeks into the tibialis anterior (TA) muscle of 5/6 nephrectomized CKD mice.
Results: Treatment with Exo/miR-26a resulted in increased expression of miR-26a in skeletal muscle and heart. Overexpression of miR-26a increased the skeletal muscle cross-sectional area, decreased the upregulation of FBXO32/atrogin-1 and TRIM63/MuRF1 and depressed cardiac fibrosis lesions. In the hearts of CKD mice, FoxO1 was activated, and connective tissue growth factor, fibronectin and collagen type I alpha 1 were increased. These responses were blunted by injection of Exo/miR-26a. Echocardiograms showed that cardiac function was improved in CKD mice treated with Exo/miR-26a.
Conclusion: Overexpression of miR-26a in muscle prevented CKD-induced muscle wasting and attenuated cardiomyopathy via exosome-mediated miR-26a transfer. These results suggest possible therapeutic strategies for using exosome delivery of miR-26a to treat complications of CKD.
Keywords: uremic cardiomyopathy, muscle wasting, insulin resistance, surface peptide, Lamp2b
Introduction
Chronic kidney disease (CKD) develops over time and ultimately leads to end-stage renal disease. Uremic cardiomyopathy and muscle atrophy are critical complications for CKD-related disastrous outcomes [1, 2]. Cardiomyopathy accounts for approximately 50% of CKD deaths [3]. Muscle wasting occurs in almost every type of CKD and is an independent risk factor for CKD-induced morbidity and mortality [4, 5]. Current treatment strategies offer improved quality and length of patient life but are not effective in curing or reversing either cardiomyopathy or muscle wasting associated with CKD.
Uremic cardiomyopathy is independent of primary/intrinsic cardiomyopathies and other atherosclerotic coronary-related heart disease. In fact, atherosclerotic coronary disorders are decreased in CKD patients compared with the normal population [6]. CKD-induced uremic cardiomyopathy is characterized by cardiac fibrosis and heart failure, which are closely related to insulin resistance [1]. Uremic cardiomyopathy has a higher rate of sudden cardiac death than does atherosclerotic coronary disease [7].
Insulin resistance is an acknowledged risk factor for cardiac disease related to CKD [8, 9]. During a study of 1187 first hospitalization heart failure patients, the investigator found that insulin resistance predicts and precedes the development of chronic heart failure [9]. In addition, insulin resistance is positively correlated with heart function decline [10]. A widely used antagonist of insulin resistance, metformin, can improve left ventricular function and reduce myocardial lipid accumulation and cardiac fibrosis [11]. Blocking insulin resistance with metformin results in 13% lower mortality compared with therapies to increase insulin sensitivity [12]. Insulin resistance causes heart problems through multiple mechanisms. It can downregulate sodium-calcium exchange, decrease myosin ATPase activity and upregulate angiotensin-II in the heart, resulting in reduced cardiac efficiency [13]. Insulin resistance is also a critical contributor to skeletal muscle atrophy in CKD and other metabolic diseases by decreasing protein synthesis, increasing protein degradation and limiting skeletal muscle regeneration [2, 14].
In humans, insulin regulates many metabolism processes by influencing glucose metabolism, fat synthesis and fatty acid esterification, increasing protein synthesis through control of amino acid uptake, altering DNA replication, and modifying numerous enzyme activity states [15]. Our group found that insulin resistance induces muscle atrophy by increasing protein degradation in skeletal muscle [16]. Insulin regulates protein metabolism through the insulin/IGF-1 signaling pathway, which includes the insulin-like growth factor-1 (IGF-1), insulin receptor substrate, phosphoinositide 3-kinase (PI3K), GSK/Akt and FoxO cascade [15]. Akt plays a central role in this insulin-signaling pathway. Phosphorylation of Akt at serine 473 indicates activation and leads to insulin sensitivity. Decreased phosphorylation at this site leads to insulin resistance. Akt and GSK-3β activity are reciprocal regulators of the insulin signaling pathway. Activation of GSK-3β by tyrosine-216 phosphorylation induces insulin resistance, whereas serine (9/21) phosphorylation inhibits GSK-3β, leading to increased insulin sensitivity [17]. Normally, Akt inhibits GSK-3β activity, resulting in increased insulin sensitivity [18]. Under CKD conditions, Akt activation is inhibited by uremic toxins, and the resulting increased GSK-3β activity leads to insulin resistance, impaired glucose and lipid metabolism and the development of uremic cardiomyopathy [1]. FoxO1 is another important factor that contributes to uremic muscle wasting. Activation of FoxO by de-phosphorylation induces muscle wasting along with transcriptional upregulation of E3 ubiquitin ligases, cardiac hypertrophy and fibrosis [19, 20].
Recent studies have shown that microRNAs (miRs) are important regulators of metabolism during development and disease [21]. Many miRs have been linked to regulation of the insulin signaling pathway and have led to some new treatment strategies using these miRs. miR-26a has been suggested as a therapeutic target for traditional atherosclerotic cardiac disease [22], but it is not clear whether it can limit CKD-induced muscle atrophy and uremic cardiomyopathy. A major challenge in using synthetic miRs for treatment purposes is that exogenously added miRs are quickly degraded by high levels of ribonuclease activity in plasma or are rapidly cleared by phagocytosis, renal filtration or bile excretion resulting in minimal tissue accumulation and minimal therapeutic effectiveness [23]. These drawbacks could be overcome by using exosomes as microRNA carriers. Because exosomes stabilize miRs and are non-cytotoxic and non-mutagenic to the recipient, they are longer lived compared to viral or liposome-based gene delivery vehicles [24].
Here, we show the impact of intramuscular injection of exosomes carrying miR-26a-5p on muscle atrophy and cardiac fibrosis in CKD mice. We investigated the outcome of exogenous miR-26a on CKD-induced muscle wasting, insulin signaling and heart function in uremic mice. We also used fluorescently labeled exosomes/miR-26a to track the exosomes in vivo. Our findings provide new insights into how miR-26a participates in the control of skeletal muscle mass and cardiac fibrosis.
Results
miR-26a was decreased in the heart and skeletal muscle of CKD mice
In exploring new therapeutic targets for treating cardiomyopathy associated with CKD, we performed miRNA deep sequence analysis from heart samples of CKD mice. The data revealed 56 miRs that were altered in CKD mice compared with sham-operated pair-fed mice (Figure 1A and Table S1). These included miR-26a, which was decreased 37.2% in CKD heart. We first used qPCR to determine whether miR-26a was expressed in major organs of normal mice and found that miR-26a was mainly expressed in skeletal muscle and heart (Figure 1B). Further qPCR analysis showed that miR-26a levels were 61% lower in the cardiac muscle of CKD versus sham mice (Figure 1C). When we examined whether CKD influences miR-26a expression in skeletal muscle, we found that miR-26a was decreased by 35% in skeletal muscle of CKD vs. sham mice (Figure 1D).
Figure 1
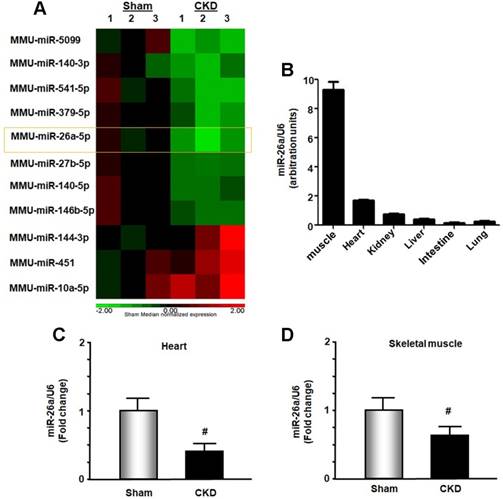
miR-26a was decreased in the heart and skeletal muscle of CKD mice. (A) Small RNA libraries were prepared using a SeqMatic tailormix miRNA sample preparation kit. The adapter-ligated libraries were then enriched using PCR amplification followed by gel enrichment for the mature miRNA library. The libraries were quantified on a Qubit® 2.0 Fluorometer using the High Sensitivity dsDNA assay. The heat map showed that miR-26a (orange square) was decreased in serum exosomes from CKD mice compared with sham mice (n = 3/group). (B) Total RNA was extracted from skeletal muscle, heart, kidney, liver, intestine and lung of normal mice. The expression of miR-26a-5p was assayed by real-time qPCR. The bar graph shows microRNA expression from each organ. The results are normalized to U6 (Bars: mean ± se; n = 6/group). (C) Total RNA was extracted from the hearts of sham and CKD mice. The expression of miR-26a-5p was assayed by real-time qPCR at 8 weeks after CKD surgery. The bar graph shows microRNA from the heart of CKD mice compared with levels in sham mice (represented by 1-fold). The results are normalized to U6 (Bars: mean ± se; n = 9/group; #p<0.05 vs. control). (D) Total RNA was extracted from skeletal muscle of sham and CKD mice. The expression of miR-26a-5p was assayed by real-time qPCR at 8 weeks after CKD surgery. The bar graph shows microRNA from the gastrocnemius muscle of CKD mice compared with levels in sham mice (represented by 1-fold). The results are normalized to U6 (Bars: mean ± se; n = 9/group; #p<0.05 vs. control).
Uremic serum increased catabolic signaling and profibrotic proteins
To further study the impact of CKD stress on miR-26a, we treated skeletal muscle satellite cells and H9C2 cardiac myoblasts with 5% mouse serum from CKD or normal control mice for 24 hours. We isolated RNA from these cells and probed for the expression of miR-26a by qPCR. Treatment with uremic serum resulted in a 31% decrease in miR-26a in skeletal muscle satellite cells (Figure 2A) and a 44% decrease in H9C2 cardiac myocytes (Figure 2B). These results suggest that a decrease in miR-26a in skeletal muscle and heart could be due to uremic toxicity. To explore the impact of uremic serum on protein changes, we measured Akt, PTEN and TGFβ1 in uremic serum-treated cells by western blot. The uremic serum decreased Akt phosphorylation and increased PTEN protein in satellite cells (Figure 2C). Similar decreases in Akt phosphorylation were also observed in H9C2 cells (Figure 2D). TGFβ1 was increased by uremic serum, suggesting increased fibrosis signaling. These results suggest that uremic serum is critical for the induction of insulin resistance and is associated with decreasing miR-26a.
Exosomes with encapsulated miR-26a and muscular surface target peptide (MSTP) surface peptide were generated
Since microRNA has a relatively short life and exosomes can stabilize microRNA [25], we used exosome-encapsulated microRNA to overexpress miR-26a in vivo (Figure S4). First, a pLamp2b/MSTP vector was constructed. This vector contained lysosomal-associated membrane protein 2b (Lamp2b), which is an exosomal membrane protein gene [26], fused with 3-muscle surface targeting peptide SKTFNTHPQSTP. This peptide has strong cardiac muscle targeting ability [27]. Second, an adenovirus containing a precursor miR-26a gene (Ad-miR-26a) was generated. Third, satellite cells were transfected with pLamp2b/MSTP and transduced with Ad-miR-26a or control virus (Ad-empty) in exosome-free medium. In this case, the exosomes secreted from satellite cells should contain miR-26a with muscle target ability. Last, the pLamp2b/MSTP-miR-26a enriched exosomes (Exo/miR-26a) and pLamp2b/MSTP-ctrl exosomes (Exo/ctrl) were isolated from the conditioned medium of cultured cells. The concentration and size of the collected exosomes were measured using a NanoSight instrument (Figure S1). The average size of the isolated exosomes was 89 ± 1.8 nm. The exosome protein marker TSG101 was absent in the transduced cells but present in the Exo/miR-ctrl and Exo/miR-26a isolated from conditioned medium (Figure S3). Real-time qPCR showed that miR-26a was increased 35-fold in Exo/miR-26a vs. Exo/ctrl (Figure S5). We labeled Exo/miR-26a with DiR (1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide), a fluorescent tag for exosome surface lipids (D12731; Invitrogen, Carlsbad, CA, USA), before intramuscular injection and imaged the legs with an In-Vivo Xtreme camera system (Bruker, Billerica, MA, USA). We observed a dose-dependent increase in fluorescence intensity in the injected TA muscles (Figure S6).
Figure 2
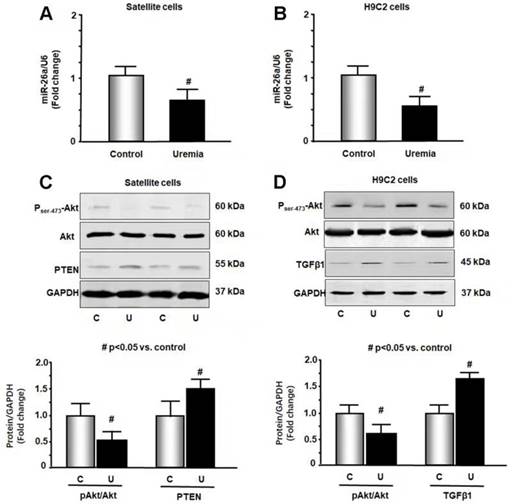
Exosome-carried mediator induces insulin resistance in skeletal and cardiac muscle cells. Cultured skeletal muscle satellite cells or cardiac H9C2 cells were treated with media containing 5% sham mouse serum (control) or 5% CKD mouse serum (uremia) for 24 hours. (A) Total RNA was extracted from satellite cells. The expression of miR-26a-5p in cells was assayed by real time qPCR. The bar graph shows miR-26a-5p from the uremic serum treatment group compared with the level in the control serum treatment group (represented by 1-fold). The results are normalized to U6 (Bars: mean ± se; n=8/group; #p<0.05 vs. control serum). (B) Total RNA was extracted from cardiac H9C2 cells. The expression of miR-26a-5p in cells was assayed by real time qPCR. The bar graph shows miR-26a-5p from the uremic treatment group compared with the levels in the control serum treatment group (represented by 1-fold). The results are normalized to U6 (Bars: mean ± se; n=8/group; #p<0.05 vs. control serum). (C) Protein was isolated from satellite cells. PTEN and Akt in cell lysates were measured by western blots from different groups of cells. The bar graph shows the fold change of each protein band compared with levels in control serum treatment group represented by 1-fold. (Bars: mean ± se; n = 8/group; #p<0.05 vs. control serum). (D) Protein was isolated from H9C2 cells. TGFβ1 and Akt in cell lysates were measured by western blots. The bar graph shows the fold change of each protein band compared with levels in control serum treatment group represented by 1-fold. (Bars: mean ± se; n = 8/group; #p<0.05 vs. control).
miR-26a-5p attenuated CKD-induced muscle atrophy
The CKD experimental procedures are shown in Figure 3A. To examine whether enhancing miR-26a in the muscles might prevent muscle wasting, Exo/miR-26a and Exo/ctrl were injected into the TA muscle of CKD mice once per week beginning immediately following the second CKD surgery. Skeletal muscle and heart were harvested 8 weeks after initiation. The exogenously added Exo/miR-26a replenished miR-26a in skeletal muscle to control levels (Figure 3B) as well as limited CKD-induced mouse body weight and muscle weight loss (Table 1). This result was supported by the improved muscle cross section area. Consistent with our previous studies [2], the muscle cross section area was decreased by CKD (Figure 3C). Conversely, overexpressing miR-26a caused a shift to a large cross sectional area when compared with results in CKD muscles injected with the Exo/ctrl (Figure 3D). To examine the possible mechanisms by which overexpression of Exo/miR-26a attenuates muscle mass loss in CKD mice, we examined the proteins related to the insulin-IGF-1/Akt/FoxO signaling pathway. The active form of Akt, phosphorylated at serine-473, was decreased in CKD, indicating decreased insulin sensitivity. Similarly, CKD decreased the amounts of serine-21/ 9-phosphorylated GSK-3β and tyrosine-24-phosphorylated FoxO1, resulting in their activation and increasing insulin resistance. Consequent to FoxO1 activation, FBXO32/atrogin-1 and TRIM63/MuRF1, two FoxO-targeted E3 ubiquitin ligases, were increased (Figure 3E). Provision of miR-26a in TA muscle reversed all these changes (Figure 3) and should lessen insulin resistance, which suggests a mechanism for limiting skeletal muscle loss.
Figure 3
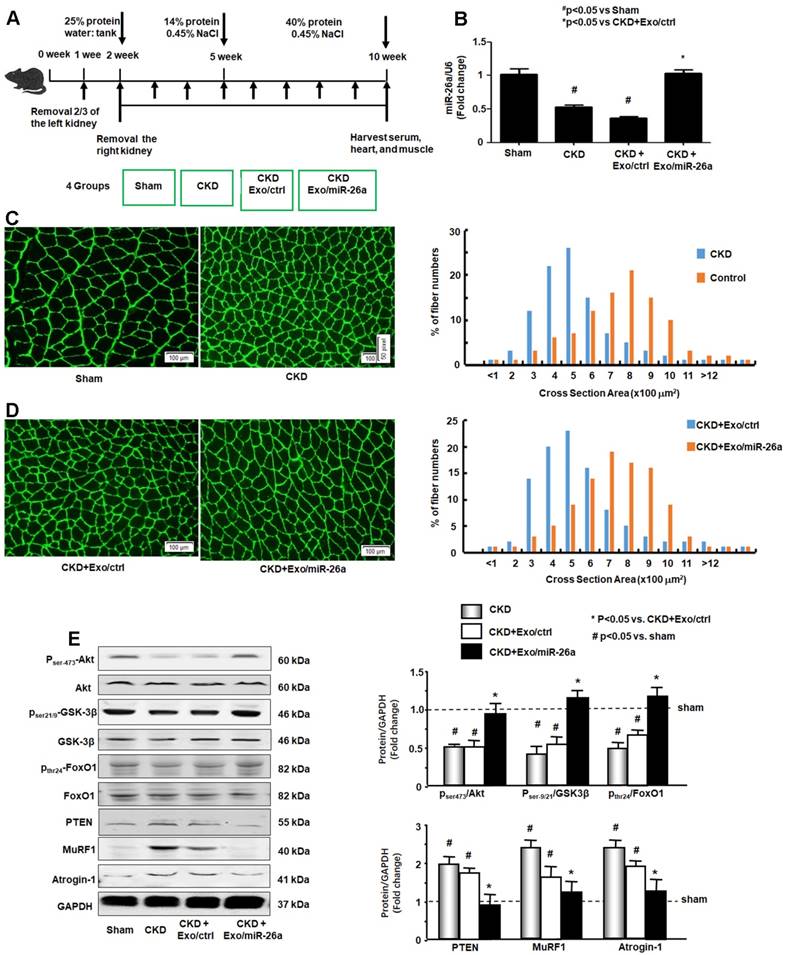
Provision of miR-26a-5p in skeletal muscle attenuated CKD-induced muscle atrophy. (A) Experiment procedure: Mice were divided into 4 groups: sham, CKD without exosome treatment, CKD with intramuscular injection of exosome-encapsulated microRNA control (Exo/ctrl) and CKD with intramuscular injection of exosome-encapsulated miR-26a (Exo/miR-26a) mice, once per week. (B) Total RNA was isolated from TA muscle of sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice. The expression of miR-26a-5p in muscle was assayed by real time qPCR. The bar graph shows the expression levels of the three miR-26a in each group compared with levels in the sham mice (represented by 1-fold). The results are normalized to U6. (Bars: mean ± se; n=9/group; #p<0.05 vs. sham and *p<0.05 vs. CKD+Exo/ctrl). (C) The representative cross-sectional area of TA muscle of sham and CKD mice. Cryosections of TA muscles were immunostained with anti-laminin antibody. The bar graph shows the frequency distribution of fiber cross-sectional areas (µm2) in sham (orange) and CKD (blue) mice (n=6/group). (D) The representative cross-sectional area of TA muscle of CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice. The bar graph shows the frequency distribution of fiber cross-sectional areas in CKD plus Exo/Ctrl (blue) and CKD with Exo/miR-26a (orange) mice (n=6/group). (E) Akt, p-Akt, GSK-3β, pGSK-3β, FoxO1, pFoxO1, PTEN, TRIM63/MuRF1 and FBXO32/atrogin-1 were measured by western blot in CKD mice with or without miR-26a. The bar graph shows the fold change of each protein band (bottom panel) or ratio of phospho-protein to total protein (top panel) compared with levels in sham mice (represented by a line at 1-fold). GAPDH was used as a loading control (Bars: mean ± se; n=9/group; *p<0.05 vs. CKD and #p<0.05 vs. sham).
Intramuscular injection of Exo/miR-26a attenuated uremic cardiomyopathy in CKD mice
Since Exo/miR-26a contains a muscle-specific surface peptide with the potential to target cardiac muscle [27], we hypothesized that these exosomes would carry miR-26a to the heart. To verify whether skeletal muscle injection of Exo/miR-26a could transfer miR-26a to the heart or other distant organs, we stained Exo/miR-26a with 1 μmol/l fluorescent tracer DiR before intramuscular injection, and one week later, we imaged dissected organs with the In-Vivo Xtreme camera system. Dissected organs from uninjected normal mice (right group), sham mice injected with Exo/miR-26a (center group) and CKD mice injected with Exo/miR-26a (left group) are shown in Figure S7. In the injected CKD mice, fluorescence was observed not only in injected TA muscle but also in contralateral uninjected TA muscle and heart (Figure 4A). In the injected sham mice, fluorescence was observed in the same organs; however, the labeled exosome fluorescence intensity was higher in CKD hearts than in sham hearts (Figure 4B). The expression of miR-26a was higher in CKD hearts than in hearts from sham-operated mice (Figure S8), which suggests that the uremic heart has a higher capacity to recruit Exo/miR-26a. We measured the expression of miR-26a in the hearts of CKD mice with Exo/miR-26a or Exo/MiR-ctrl intramuscular injections. The expression of miR-26a was increased 1.8-fold in CKD mouse heart from the Exo/miR-26a-injected animals versus the mice receiving Exo/ctrl injection (Figure 4C). We next determined if Exo/miR-26a was increased in the circulation. We found a 2.1-fold increase in miR-26a in serum exosomes from CKD mice injected with Exo/miR-26a versus Exo/ctrl administration (Figure 4D). To discover whether circulating exosomes that are enriched in miR-26a have the potential to attenuate insulin resistance and inhibit fibrosis, we measured FoxO1 phosphorylation and TGFβ1 in serum exosomes. FoxO1 phosphorylation was decreased, and TGFβ1 and PTEN were increased in the exosomes from CKD mouse serum. These changes were blunted in serum exosomes from CKD mice that were treated with Exo/miR-26a (Figure 4E). Treatment of CKD mice with Exo/ctrl did not show the beneficial blunting of these protein responses. These data suggest that 1) damaged heart has a tendency to recruit more exosomes/miR than a healthy heart; 2) intramuscular injection of Exo/miR-26a can increase miR-26a expression in the heart; and 3) in serum, miR-26a-enriched exosomes carry signaling proteins that can limit insulin resistance.
CKD mouse heart showed significant uremic cardiomyopathy, as evidenced by the increased ratio of heart weight to body weight (Table 2). Exogenous miR-26a reduced CKD-induced increases in the heart/body weight ratio. CKD mice also had significantly increased cardiac fibrosis vs. sham mice, as demonstrated by Masson's trichrome staining. Collagen deposits (indicated by blue staining in heart sections) were increased 43-fold in the hearts of CKD mice, and intervention with miR-26a in CKD mice reduced the elevated collagen accumulation by 50% compared to CKD with Exo/miR-ctrl (Figure 5A). The interstitial matrix protein marker fibronectin is a signature for myofibroblast activation and was significantly increased in the heart of CKD mice. The provision of miR-26a to CKD mice decreased the amount of fibronectin (Figure 5B) and also partially limited the CKD-induced increase in two fibrotic protein markers, collagen 1A1 and connective tissue growth factor (CTGF), in the heart (Figure 5C). These results suggest that intramuscular injection of Exo/miR-26a can decrease cardiac fibrosis in CKD mice.
Table 1
Physiological parameters - body and muscle weights and BUN in various cohorts
| Sham | CKD | CKD+Exo/ ctrl | CKD+Exo/ miR-26a | |
|---|---|---|---|---|
| Baseline B.W. (g) | 22.6 ± 1.9 | 22.7 ± 1.8 | 22.5 ± 2.3 | 22.9 ± 1.6 |
| Terminal B.W. (g) | 24.2 ± 2.5 | 19.5 ± 3.8* | 20.5 ± 2.3* | 22.9 ± 2.6 |
| TA weight (mg) | 35.1 ± 1.1 | 28.7 ± 0.4* | 28.8 ± 0.5* | 32.1 ± 0.6# |
| Soleus weight (mg) | 10.5 ± 0.1 | 8.7 ± 0.2 | 8.1 ± 0.5* | 9.8 ± 0.8*# |
| EDL weight (mg) | 10.3 ± 0.2 | 8.9 ± 0.6 | 8.2 ± 0.7* | 9.7 ± 0.3*# |
| BUN (mg/dL) | 21.3 ± 4.3 | 88.9 ± 11.6* | 86.2 ± 8.7* | 72.3 ± 6.5* |
Data are presented as the mean ± se; P < 0.05 is significant (*vs. sham, #vs. CKD), n=12/group; B.W.: body weight; TA: tibialis anterior; EDL: extensor digitorum longus; BUN: blood urea nitrogen
Table 2
Cardiac function evaluated by echocardiography
| Sham (n=5) | CKD (n=5) | CKD+Exo/ ctrl (n=5) | CKD+Exo/ miR-26a (n=5) | |
|---|---|---|---|---|
| Heart weight (g) | 0.14±0.01 | 0.17±0.03 | 0.18±0.04 | 0.15±0.01 |
| Heart/Body weight | 5.78±0.51 | 8.72±0.86# | 8.78±0.71# | 6.55±0.49* |
| IVS-d (mm) | 0.51±0.0 | 0.48±0.0 | 0.49±0.0 | 0.48±0.0 |
| LVEDD (mm) | 3.71±0.1 | 4.01±0.0# | 4.11±0.0# | 3.83±0.0* |
| FW-D (mm) | 0.50±0.0 | 0.47±0.0 | 0.46±0.0 | 0.48±0.0 |
| IVS-s (mm) | 1.05±0.0 | 1.10±0.0 | 1.09±0.0 | 1.07±0.0 |
| LVESD (mm) | 2.68±0.1 | 3.19±0.1# | 3.23±0.2# | 2.86±0.1* |
| FW-S (mm) | 0.74±0.0 | 0.73±0.0 | 0.74±0.0 | 0.74±0.0 |
| LV Vol-d | 58.94±3.1 | 70.66±1.6# | 71.33±1.8# | 62.33±2.5* |
| LV Vol-s | 26.98±2.3 | 40.97±2.3# | 41.58±2.7# | 32.69±2.1* |
| %EF | 54.59±2.2 | 42.20±2.2# | 41.78±2.1# | 49.87±2.4* |
| FS | 27.85±1.5 | 20.49±1.2# | 19.55±1.1# | 25.61±1.4* |
| LV Mass | 56.79±2.7 | 60.84±1.7 | 61.0±1.8 | 57.89±2.6 |
#P < 0.05 vs. Sham; *P < 0.05 vs. CKD+Exo-ctrl; IVS-d: interventricular septum diameter; LVEDD: left ventricular end-diastolic diameter; FW-D: free wall in diastole; IVS-s: interventricular septum in systole; LVESD: left ventricular end-systolic diameter; FW-S: free wall in systole; LV Vol-d: left ventricular volume in diastole; LV Vol-s: left ventricular volume in systole; %EF: % ejection fraction; FS: fractional shortening; LV Mass: left ventricular mass
Figure 4
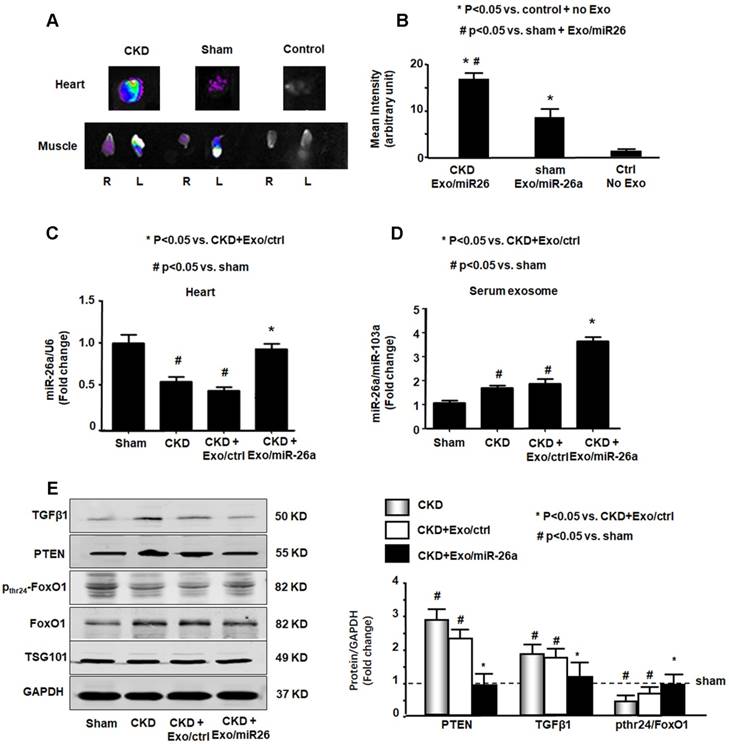
miR-26a was increased in the heart of mice with intramuscular injection of Exo/miR-26a. (A) Exosomes were labeled with 1 μmol/l fluorescent lipophilic tracer DiR. Mice were injected in skeletal muscle with Exo/miR-26a. Fluorescent organ images were acquired using a Bruker Small Animal Optical Imaging System. Normal mice (right) were not injected; sham mice (middle) and CKD mice (left) were injected with Exo/miR-26a in the left TA muscle. The fluorescence in the heart and TA muscle was assessed at 7 days after injection. (B) The fluorescence intensity of the heart was acquired by a Bruker Small Animal Optical Imaging System at 7 days after injection. The control was no exosome injection. The bar graph shows fluorescence intensity in the heart. (Bars: mean ± se; n=6/group; *p<0.05 vs. control, #p<0.05 vs. sham). (C) Total RNA was isolated from the heart of sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice. The expression of miR-26a-5p was assayed by real time qPCR. The bar graph shows miR-26a expression in hearts from each group compared with levels in sham hearts (represented at 1-fold). The results are normalized to U6. (Bars: mean ± se; n = 6/group; #p<0.05 vs. sham, #p<0.05 vs. CKD + Exo/ctrl). (D) Total RNA was isolated from serum exosomes of sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice. The expression of miR-26a-5p was assayed by real time qPCR. The bar graph shows miR-26a expression in exosomes from each group compared with levels in sham exosomes (represented at 1-fold). The results are normalized to miR-103. (Bars: mean ± se; n = 6/group; #p<0.05 vs. sham, #p<0.05 vs. CKD + Exo/ctrl). (E) Exosomes were isolated from the serum of sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice and lysed in RIPA buffer. The insulin signaling- and fibrosis-related proteins phosphorylated FoxO1, PTEN and TGF-β1 were measured by western blot. TSG101 is an exosome protein marker. All protein band densities are normalized to the appropriate GAPDH control. The bar graph shows total PTEN, total TGF-β1 and the ratio of phospho-FoxO1 to total FoxO protein. The fold change is compared to levels in the control plus Exo/miR-ctrl (represented by a line at 1-fold). (Bars: mean ± se; n=6/group; *p<0.05 vs. CKD+Exo//ctrl and #p<0.05 vs. sham).
miR-26a limited insulin resistance resulting in improved cardiac function
Insulin/IGF-1 signaling pathway-related proteins were evaluated by western blot. The phosphorylation levels of ser-21/9 in GSK-3β and thr-24 in FoxO1 were decreased by CKD, resulting in increased insulin resistance. The increase of pser-473-Akt by Exo/miR-26a suggested increasing insulin sensitivity (Figure 6A). Exogenous miR-26a increased both pser-21/9-GSK-3β and pthr-24-FoxO1, limiting insulin resistance. In addition, we determined whether overexpression of miR-26a might downregulate FoxO1 expression since FoxO1 was predicted as a target of miR-26a-5p by in silico analysis (TargetScan, PITA, miRanda). We performed a luciferase reporter analysis in cultured skeletal muscle satellite cells using a reporter construct in which the luciferase coding sequence was fused to the 3'-UTRs (position 3358 - 3364 nt) of human FoxO1 (pLuc.miR-26a/FoxO1-3'UTR). miR-26a markedly repressed luciferase activity in cells transfected with pLuc.miR-26a/FoxO1-3'UTR (Fox/3'UTR). Mutation of the miR-26a target sites abrogated miR-26a-induced repression of luciferase activity (Figure 6B). These data suggest that FoxO1 is a direct target of miR-26a.
Figure 5
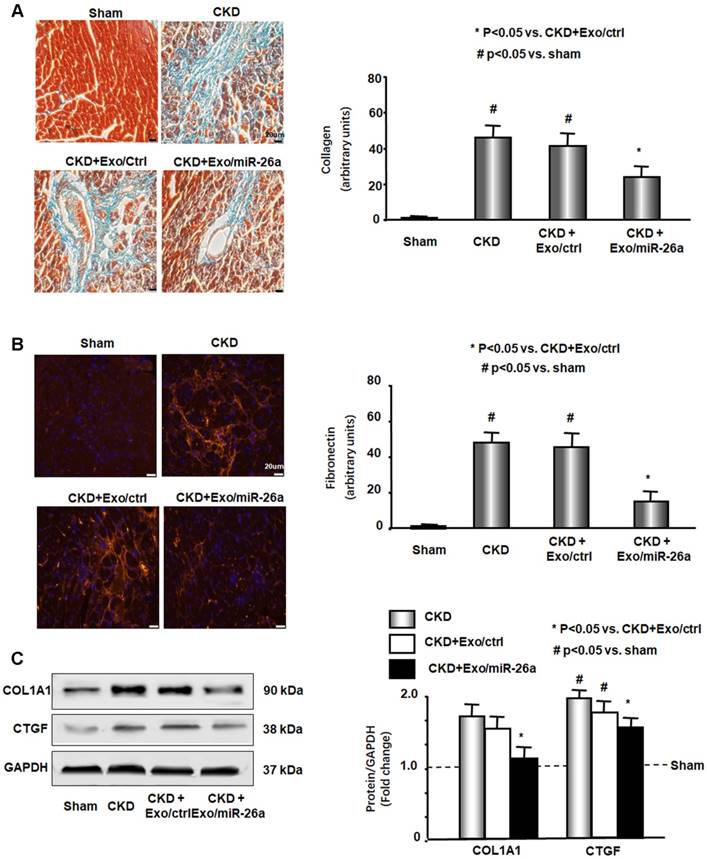
Intramuscular injection of Exo/miR-26a attenuated uremic cardiomyopathy in CKD mice. (A) Representative Masson's Trichrome staining of hearts from sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice. The bar graph shows collagen (blue in staining) measured at 8-weeks using the Micro-suite Five Biological Software (Olympus, Melville, NY, USA). The results show the fold change compared to sham levels, represented as a dotted line at 1-fold (Bars: mean ± se; n = 6/group; #p<0.05 vs. sham, *p<0.05 vs. CKD with Exo/ctrl). (B) Representative cryosections of heart from the four different treatment groups were immunostained with a fibronectin antibody. The bar graph shows fibronectin measured at 8-weeks. Fold change reflects a comparison with sham levels, represented as a dotted line at 1-fold (Bars: mean ± se; n = 6/group; #p<0.05 vs. sham, *p<0.05 vs. CKD with Exo/ctrl). (C) Protein was isolated from the heart of sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice. The fibrosis-related protein CTGF and collagen1A1 in heart lysates were measured by western blots from different groups of mice. The bar graph shows the fold change from sham mice, represented by a line at 1-fold. (Bars: mean ± se; n = 9/group; #p<0.05 vs. sham, *p<0.05 vs. CKD plus Exo/ctrl).
Figure 6
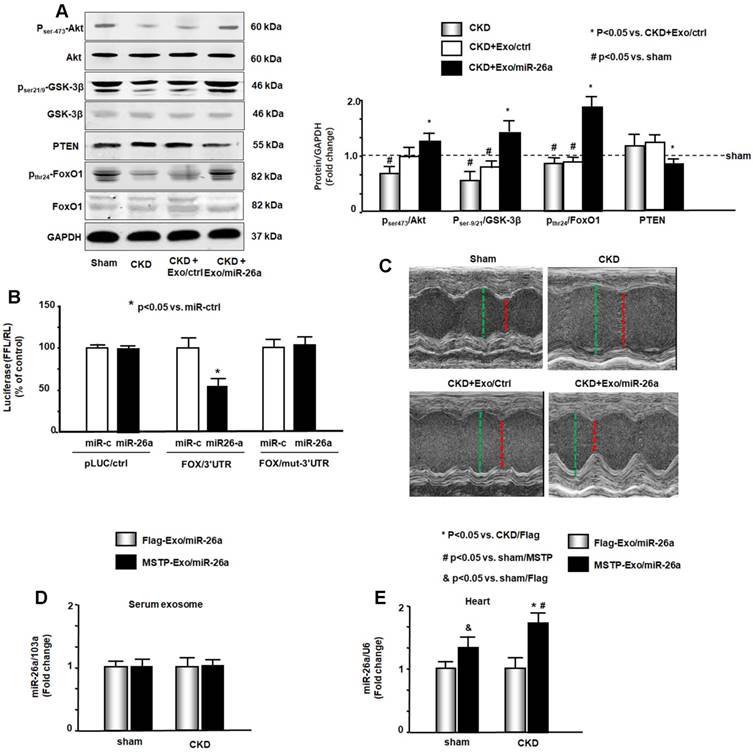
miR-26a limits insulin resistance by targeting FoxO1, leading to improved cardiac function. (A) Insulin signaling protein markers Akt, p-Akt, GSK-3β, pGSK-3β, PTEN, FoxO1, and pFoxO1 were measured by western blot in the heart of sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice. The bar graph shows the fold change of each individual protein band or ratio of phospho-protein to total protein compared with levels in sham mice (represented by a line at 1-fold). GAPDH was used as a loading control (Bars: mean ± se; n=9/group; #p<0.05 vs. sham, *p<0.05 vs. CKD plus Exo/ctrl). (B) Binding of miR-26a to the 3'-UTR of FoxO1 inhibits FoxO1 translation. H9C2 cells were transfected with luciferase pLuc-ctrl vector or the vector containing the 3'-UTR of FoxO1 (pMIR-FoxO1/3358 - 3364) or a vector containing a mutated 3'-UTR of FoxO1 (pMIR-mut-FoxO1). Cells were cotransfected with renilla luciferase as a transfection control. Cells were then transduced by adenovirus containing miR-26a precursor RNA (miR-26a) and control virus (miR-ctrl). Luciferase activity in cells that received the pLuc-ctrl (no target 3'UTR) and miR-ctrl (sequence unrelated to miR-26a) was designated as 100%. The response to miR-26a is expressed as a percent relative to the control. Bars present the results from triplicate determinations. Data: mean ± se; n=9; #p<0.05 vs. FoxO/3UTR + miR-ctrl. (C) Representative echocardiographic evaluations of cardiac function in sham, CKD, CKD plus Exo/ctrl and CKD plus Exo/miR-26a (Exo/miR-26a) mice are shown. Echocardiography was performed on lightly anesthetized mice (under 1-2% isoflurane, in oxygen) using a Vevo 3,100 ultrasound system (VisualSonics). The green line represents left ventricular end-diastolic dimensions, and the red line represents left ventricular end-systolic dimensions. Detailed information is provided in table 2. (D) Comparison of miR-26a expression in Flag-Exo/miR-26a- and MSTP-Exo/miR-26a-injected mice. RNA was isolated from serum exosomes from sham and CKD mice treated with Flag-Exo/miR-26a and MSTP-Exo/miR-26a. The expression of miR-26a-5p was assayed by real-time qPCR. The bar graph shows the expression of miR-26a from each cohort compared with levels in sham plus Flag-Exo/miR-26a injection mice (represented at 1-fold). The results are normalized to miR-103a. (Bars: mean ± se; n=6/group). (E) RNA was isolated from the hearts of sham and CKD mice. The expression of miR-26a-5p was assayed by real-time qPCR. The expression of miR-26a from each cohort compared with levels in sham plus Flag-Exo/miR-26a-injected mice (represented at 1-fold). The results are normalized to U6. (Bars: mean ± se; n=6/group; *p<0.05 vs. CKD + Flag-Exo/miR-26a, #p<0.05 vs. sham + MSTP-Exo/miR-26a, &p<0.05 vs. Flag-Exo/miR-26a).
Cardiac function was evaluated by echocardiography (Table 2 & Figure 6C). In CKD mice, left ventricle (LV) end-diastolic diameter (LVEDD), LV end-systolic chamber dimensions (LVESD), and left ventricular volume in diastole and systole (LV vol-d and LV vol-s) were all significantly increased in the hearts of CKD mice. These changes were accompanied by a decrease in the percentage of ejection fraction (%EF) and fractional shortening (FS) in CKD mice. Provision of miR-26a improved all of these CKD-induced changes in cardiac function parameters. The echocardiographic results suggest that miR-26a intervention improves cardiac function.
To demonstrate that MSTP is important in Exo/miR-26a accumulation in the heart, we generated additional exosomes encapsulated miR-26a without MSTP (Flag-Exo/miR-26a). We injected both Flag-Exo/miR-26a and MSTP-Exo/miR-26a into the TA muscles of CKD mice and measured miR-26a expression in circulation exosomes and in the heart. The expression of miR-26a in serum exosomes from the two groups of mice was not significantly different (Figure 6D). However, the expression of miR-26a in the cardiac muscle of MSTP-Exo/miR-26a-injected mice was significantly higher than in hearts from Flag-Exo/miR-26a-injected mice (Figure 6E). Interestingly, the miR-26a that accumulated in the CKD heart was significantly higher than that in the sham-operated heart. These data indicate that MSTP plays an important role in targeting microRNA to the injured heart. To show that the fluorescence accumulated in the various tissues was an indication of Exo/miR-26a or Exo/miR-ctrl, not just nonspecific spreading of unattached dye, we injected fluorescent lipophilic DiR without exosomes into TA muscle and looked for fluorescence throughout the body. A signal was detected at 1, 2 and 4 weeks after DiR injection in vivo. Analysis of organs ex vivo showed that fluorescence was confined to muscle and not detected in other organs, including the kidney at all times (Figure S9).
Discussion
In this study, we showed that under CKD stress conditions, skeletal muscle atrophy and uremic cardiomyopathy are related to a decrease in miR-26a. When we injected exosomes encapsulating miR-26a with the muscle-targeting peptide MSTP into skeletal muscle of CKD mice, we observed attenuated skeletal muscle wasting but also decreased evidence of cardiomyopathy. Our data indicate that overexpression of miR-26a achieves these benefits by limiting insulin resistance. In addition, we determined that FoxO1 is a direct target of miR-26a.
Our findings that exogenous application of miR-26a increases muscle cross section area, diminishes CKD-induced cardiac fibrosis and improves heart function strongly suggest that these miR-related changes are involved in the limitation of insulin resistance. Many systems and renal-specific factors contribute to muscle atrophy and uremic cardiomyopathy in CKD, including metabolic acidosis, inflammation, and increased oxidant stress [1, 28]. However, insulin resistance is recognized as a major and common mechanism causing both muscle wasting and cardiomyopathy in CKD [1]. Our previous studies proved that the downregulation of insulin/IGF-1 signaling is the major cause of muscle wasting in CKD because it increases accelerated protein degradation by activating the ubiquitin proteasome pathway [16, 29]. In the current study, we found that several proteins closely related to the insulin/IGF-1 cascade were altered by miR-26a-5p. Activation of Akt by miR-26a upregulates the insulin/IGF-1 signaling pathway. Inactivation of FoxO1 and GSK-3β by miR-26a lessens insulin resistance. Other investigators have also observed that obesity-induced decreases in miR-26a attenuate insulin sensitivity [30], overexpression of miR-26a inhibits GSK-3β activation [31], and miR-26a has important implications for cardiovascular repair [32].
In non-CKD pathologies, the cardiac response to increased phospho-Akt is to undergo postnatal coronary angiogenesis and cardiac hypertrophy [33]. However, in the heart of CKD animals, we and others have found that phospho-Akt protein is decreased and FoxO is activated [34]. Importantly, CKD promotes the development of specific forms of cardiomyopathy that are independent of increased coronary artery disease or hypertension. In CKD pathology, decreased phospho-Akt and increased FoxO1 can cause increased insulin resistance with a disadvantageous outcome. In an ischemia reperfusion injury model, Rota et al. showed that nuclear-targeted overexpression of Akt1 did not induce cardiac hypertrophy but instead increased the number of cardiomyocytes, increased contractility and protected against ischemia-reperfusion injury [35].
Insulin resistance can induce cardiac fibrosis, which is another consequence of CKD. In the uremic heart, increased expression of profibrosis mediators such as TGFβ and CTGF causes increased collagen levels and results in interstitial fibrosis, which contributes to diastolic dysfunction, ventricular stiffness, and cardiac dysrhythmias [36]. Our study demonstrates that miR-26a intervention reduces CTGF abundance and restricts collagen deposition in the heart. Koga et al. showed that miR-26a inhibits TGFβ by directly targeting CTGF [37]. This is consistent with our findings that show that cardiac fibrosis is inhibited by miR-26a.
Studies have found that miR-26a has multiple targets that are involved in fibrosis and insulin resistance, such as GSK-3β, PTEN, CTGF and collagen I [31, 38, 39]. In this study, we identified a novel target of miR-26a, FoxO1. The following is evidence to prove this outcome: provision of miR-26a in skeletal muscle decreased FoxO1 protein in vivo (Figure 3E and Figure 6A). Using a luciferase reporter assay, we found direct binding of miR-26a to the 3'-UTR of FoxO1 (Figure 6B). Both our FoxO1 target results and the target studies from us and others provide reasonable evidence that intervention with miR-26a can limit insulin resistance and fibrosis. All microRNAs have the potential for off target effects due to the multiplicity of their targets. According to the literature, miR-26a suppresses cell proliferation in esophageal cancer cells and tenon capsule fibroblasts [40]. In addition, one study found that miR-26a decreases inflammation-mediated tumorigenesis and metastasis of cancer cells [41]. Our results indicating that muscle size and mass increased in response to miR-26a suggest that this miR is not inhibiting or suppressing cell proliferation, i.e., it does not show the same trend as the previously observed off-target effects. However, we cannot rule out the possibility that miR-26a could affect other tissues that are not the subject of our investigations.
Another important outcome of these studies is the determination that the use of a muscle-specific targeting peptide is a good approach to direct microRNA to skeletal muscle and heart. Exosomes are carrier vehicles. Both pre-miRNAs and mature miRNAs are located in exosomes and have been determined to be quite stable [42]. In a clinical trial, patients tolerated exosome injection for up to 21 months [43]. In this study, we constructed a new exosome targeting vector to promote the accumulation of exosome-packaged miRs to specific destinations. Using a muscle-specific target peptide, SKTFNTHPQSTP, in the structure of the exosomes/microRNA caused them to preferentially move to skeletal muscle and heart. This vector contains Lamp2b and MSTP. Since Lamp2b is ubiquitously expressed on the surface of exosomes, it facilitates the fusion of target peptides to the surface of exosomes and endows exosomes with targeting ability [27]. Interestingly, we also found that an injured organ has a higher capacity to recruit exosomes/miR than do normal organs because miR-26a expression is much higher in CKD hearts than in normal sham control hearts after matched intramuscular injections of Exo/miR-26a (Figure 4A and Figure 4B). We believe that the damaged tissue creates a hierarchy for the recruitment of exosomes. The CKD heart has damage, giving it higher priority to collect therapeutic exosomes than healthy heart in sham mice. This could involve the response of injured organs to induce secretion of inflammatory cytokines, leading to increased capillary permeability. Any increased vascular permeability could result in increased exosome uptake. Another potential mechanism for increased exosome uptake is fusion. Fusion efficiency is enhanced in an acidic environment. A hallmark of CKD is tissue acidosis, which could increase exosome uptake [44]. The mechanisms of these phenomena require future studies.
In conclusion, exogenous miR-26a not only attenuated skeletal muscle atrophy but also ameliorated uremic cardiomyopathy by targeting multiple mRNAs. These findings suggest that miR-26a could be useful as a therapeutic agent for increasing insulin sensitivity and inhibiting muscle wasting and uremic cardiomyopathy.
Methods
CKD mouse model and treatment: The experiments were approved by the Emory University IACUC (protocol 4000152). Mice (C57BL/6J) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). The CKD model was obtained through a two-step 5/6 nephrectomy [14]. We started Exo/miR-26a or Exo/control tibialis anterior (TA) injection after the second CKD surgery (40 µg of exosomes each time, once per week for a total of 8 weeks).
Generation of exosome-encapsulated miR-26a and exosome purification: A pLamp2b/MSTP vector was constructed by the Genomics core of Emory University. Satellite cells were grown to 60% confluence in DMEM/F12 culture medium containing 20% fetal bovine serum [14]. The Lamp2b/MSTP vector was transfected into satellite cells using the Effectene transfection reagent (Qiagen, Valencia, CA, USA). Six hours after transfection, the cells were transduced with Ad-miR-26a (adenovirus containing miR-26a processor sequences) to produce exosome-encapsulated miR-26a (Exo/miR-26a). Control cells were transduced with Ad-empty for production of MSTP-exosome-control (Exo/ctrl). Exosome-free medium was used to replace the growth medium to allow exosome secretion for 48 hours. Last, the pLamp2b/MSTP-miR-26a enriched exosomes (Exo/miR-26a) and pLamp2b/MSTP-ctrl exosomes (Exo/ctrl) were isolated from the conditioned medium of cultured cells and resuspended in PBS. Exosomes were purified by several centrifugation and filtration steps as described previously [45]. Exosome size and concentration were analyzed using a NanoSight instrument and an electro-microscope (Figure S1 and Figure S2). The presence of the exosomal marker protein TSG101 was determined by western blot (Figure S3). Exosome distribution images were taken with the Bruker Small Animal Optical Imaging System (In-Vivo Xtreme II; Billerica, MA, USA).
Real-time quantitative PCR (qPCR): To measure microRNA, total RNA was extracted using Tri-Reagent (Molecular Research Inc., Cincinnati, OH, USA). For synthesis of cDNA, 10 ng of total RNA that was enriched in small RNAs was reverse transcribed using an NCode miRNA cDNA synthesis kit (Exiqon, Vedbaek, Denmark). The expression of microRNA was measured as described [46]. Primers were purchased from Exiqon. The mouse U6 gene was used as the standard for evaluating the tissue expression of individual miRNAs. miR-103 was used to evaluate serum microRNA levels. The response or change in expression levels of individual miRs was calculated as the difference between the threshold values of the sham and CKD genes (ΔΔcq) [47, 48].
miRNA-Seq library preparation and sequencing was performed by the Genomics core of Yerkes National Primate Research Center of Emory University. The method is provided in “A detailed, expanded Methods” (supplement).
Luciferase Reporter Assay and Transfection: The luciferase reporter constructs containing the luciferase coding sequence fused to the 3'-UTRs of FoxO1 (pLuc.miR-26a/FoxO1-3'UTR) were generated by the Emory Integrated Genomics Core. The transfection and luciferase assay was described before and is provided in “A detailed, expanded Methods” (supplement) [49].
Western blot analysis and histology staining: Proteins were lysed in RIPA buffer. Detection of protein expression by western blot was performed according to established protocols [50]. The western blot, immunofluorescence staining procedure and antibodies are listed in “A detailed, expanded Methods” (supplement). The histological analysis was performed in a blinded manner to avoid bias. All immunohistochemical analyses were repeated at least three times, and representative images are shown.
Echocardiographic Evaluations of Cardiac Function: Echocardiography was performed on lightly anesthetized mice (under 1-2% isoflurane, in oxygen) using a Vevo 3,100 ultrasound system (VisualSonics, Toronto, CA) as described previously [51]. LV dimensions were obtained using parasternal long-axis views by two-dimensional-sided M-mode imaging. The cursor was positioned perpendicular to the interventricular septum and at the level of the papillary muscles and posterior wall of the LV. An M-mode image obtained at a sweep speed of 100 mm/s was used to determine diastolic and systolic LV wall thicknesses, LV end-diastolic dimensions (LVDD) and LV end-systolic chamber dimensions (LVSD). Systolic function was calculated from LV dimensions as fractional shortening (FS), as follows: FS = (LVDD LVSD)/LVDD. Recording of echocardiographic images was performed in random order with respect to the treatment or control animals. The acquisition of images and evaluation of data were performed by independent operators who were blinded to the treatment.
Statistical analysis: Data are presented as the mean ± se. To identify significant differences between two groups, comparisons were made using the t-test. Differences with P values < 0.05 were considered significant. For a comparison of more than two groups, one-way ANOVA was performed with a post hoc analysis using the Student-Newman-Keuls test. Differences with P values < 0.05 were considered significant.
Abbreviations
BUN: blood urea nitrogen; CKD: chronic kidney disease; CTGF: connective tissue growth factor; DiR: 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide; EDL: extensor digitorum longus; EF: ejection fraction; FS: fractional shortening; IGF-1: insulin-like growth factor-1; Lamp2b: lysosomal-associated membrane protein 2b; LV: left ventricle; LVEDD: LV end-diastolic diameter; LVESD: LV end-systolic chamber dimensions; LV vol-d and LV vol-s: left ventricular volume in diastole and systole; MSTP: muscular surface target peptide; PI3K: phosphoinositide 3-kinase; TA: Tibialis anterior.
Acknowledgements
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health under Award Number R01 AR060268 and American Heart Association Discover and Innovation Grants supported by Bayer Group (17IBDG33780000) to X.H.W; The National Natural Science Foundation of China (81700618), the Natural Science Foundation of Jiangsu Province (BK20181487), China Young Nephrologist Research Funding and the Fundamental Research Funds for the Central Universities (2242018K41089) to B.W; The Natural Science Foundation of Jiangsu Province (BK20161071) to A.Q.Z; and The National Natural Science Foundation of China (31772690) to H.D.W; National Key Research Program (2018YFC1314000) to B.C.L. This research project was also supported in part (microRNA deep sequencing) by the Genomics core of Yerkes National Primate Research Center under Award number NIH ORIP/OD P51OD011132 and in part (luciferase constructs) by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities under NIH Award Number UL1TR000454. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH or the US Government.
We thank Drs. Matthew Wood and Yiqi Seow from the University of Oxford, UK, for providing the pLamp2b-Flag vectors and control vectors.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Semple D, Smith K, Bhandari S, Seymour AM. Uremic cardiomyopathy and insulin resistance: a critical role for akt? J Am Soc Nephrol. 2011;22:207-15
2. Wang XH, Mitch WE. Mechanisms of muscle wasting in chronic kidney disease. Nature reviews Nephrology. 2014;10:504-16
3. Patient mortality and survival. United States Renal Data System. Am J Kidney Dis. 1998;32:S69-80
4. Avram MM, Mittman N. Malnutrition in uremia. Seminars in nephrology. 1994;14:238-44
5. Wang XH, Mitch WE. Muscle wasting from kidney failure-a model for catabolic conditions. Int J Biochem Cell Biol. 2013;45:2230-8
6. Rostand SG, Kirk KA, Rutsky EA. Dialysis-associated ischemic heart disease: insights from coronary angiography. Kidney Int. 1984;25:653-9
7. Kottgen A, Russell SD, Loehr LR, Crainiceanu CM, Rosamond WD, Chang PP. et al. Reduced kidney function as a risk factor for incident heart failure: the atherosclerosis risk in communities (ARIC) study. J Am Soc Nephrol. 2007;18:1307-15
8. Shinohara K, Shoji T, Emoto M, Tahara H, Koyama H, Ishimura E. et al. Insulin resistance as an independent predictor of cardiovascular mortality in patients with end-stage renal disease. J Am Soc Nephrol. 2002;13:1894-900
9. Ingelsson E, Sundström J, Arnlöv J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294:334-41
10. Suskin N, McKelvie RS, Burns RJ, Latini R, Pericak D, Probstfield J. et al. Glucose and insulin abnormalities relate to functional capacity in patients with congestive heart failure. Eur Heart J. 2000;21:1368-75
11. Cittadini A, Napoli R, Monti MG, Rea D, Longobardi S, Netti PA. et al. Metformin prevents the development of chronic heart failure in the SHHF rat model. Diabetes. 2012;61:944-53
12. Masoudi FA, Inzucchi SE, Wang Y, Havranek EP, Foody JM, Krumholz HM. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation. 2005;111:583-90
13. Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57:3297-306
14. Wang XH, Hu Z, Klein JD, Zhang L, Fang F, Mitch WE. Decreased miR-29 suppresses myogenesis in CKD. J Am Soc Nephrol. 2011;22:2068-76
15. Dimitriadis G, Mitrou P, Lambadiari V, Maratou E, Raptis SA. Insulin effects in muscle and adipose tissue. Diabetes Res Clin Pract. 2011;93(Suppl 1):S52-9
16. Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology. 2006;147:4160-8
17. Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269:14566-74
18. Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR. et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:897-910
19. Xu J, Li R, Workeneh B, Dong Y, Wang X, Hu Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012;82:401-11
20. Wang XH, Du J, Klein JD, Bailey JL, Mitch WE. Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int. 2009;76:751-9
21. Wang XH. MicroRNA in myogenesis and muscle atrophy. Curr Opin Clin Nutr Metab Care. 2013;16:258-66
22. Icli B, Dorbala P, Feinberg MW. An emerging role for the miR-26 family in cardiovascular disease. Trends Cardiovasc Med. 2014;24:241-8
23. Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145-55
24. O'Loughlin AJ, Woffindale CA, Wood MJ. Exosomes and the emerging field of exosome-based gene therapy. Curr Gene Ther. 2012;12:262-74
25. Chevillet JR, Kang Q, Ruf IK, Briggs HA, Vojtech LN, Hughes SM. et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci U S A. 2014;111:14888-93
26. Hung ME, Leonard JN. Stabilization of exosome-targeting peptides via engineered glycosylation. J Biol Chem. 2015;290:8166-72
27. Seow Y, Yin H, Wood MJ. Identification of a novel muscle targeting peptide in mdx mice. Peptides. 2010;31:1873-7
28. Parfrey PS, Foley RN. The clinical epidemiology of cardiac disease in chronic renal failure. J Am Soc Nephrol. 1999;10:1606-15
29. Zhou Q, Du J, Hu Z, Walsh K, Wang XH. Evidence for adipose-muscle cross talk: opposing regulation of muscle proteolysis by adiponectin and Fatty acids. Endocrinology. 2007;148:5696-705
30. Fu X, Dong B, Tian Y, Lefebvre P, Meng Z, Wang X. et al. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest. 2015;125:2497-509
31. Mohamed JS, Lopez MA, Boriek AM. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3β. J Biol Chem. 2010;285:29336-47
32. Icli B, Wara AK, Moslehi J, Sun X, Plovie E, Cahill M. et al. MicroRNA-26a regulates pathological and physiological angiogenesis by targeting BMP/SMAD1 signaling. Circ Res. 2013;113:1231-41
33. Dharaneeswaran H, Abid MR, Yuan L, Dupuis D, Beeler D, Spokes KC. et al. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ Res. 2014;115:238-51
34. Sindhu RK, Ehdaie A, Vaziri ND, Roberts CK. Effects of chronic renal failure on caveolin-1, guanylate cyclase and AKT protein expression. Biochim Biophys Acta. 2004;1690:231-7
35. Rota M, Boni A, Urbanek K, Padin-Iruegas ME, Kajstura TJ, Fiore G. et al. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res. 2005;97:1332-41
36. Mall G, Rambausek M, Neumeister A, Kollmar S, Vetterlein F, Ritz E. Myocardial interstitial fibrosis in experimental uremia-implications for cardiac compliance. Kidney Int. 1988;33:804-11
37. Koga K, Yokoi H, Mori K, Kasahara M, Kuwabara T, Imamaki H. et al. MicroRNA-26a inhibits TGF-beta-induced extracellular matrix protein expression in podocytes by targeting CTGF and is downregulated in diabetic nephropathy. Diabetologia. 2015;58:2169-80
38. Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH. et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327-37
39. Wei C, Kim IK, Kumar S, Jayasinghe S, Hong N, Castoldi G. et al. NF-κB mediated miR-26a regulation in cardiac fibrosis. J Cell Physiol. 2013;228:1433-42
40. Bao H, Jiang K, Meng K, Liu W, Liu P, Du Y. et al. TGF-β2 induces proliferation and inhibits apoptosis of human tenon capsule fibroblast by miR-26 and its targeting of CTGF. Biomed Pharmacother. 2018;104:558-65
41. Kwon Y, Kim Y, Eom S, Kim M, Park D, Kim H. et al. MicroRNA-26a/-26b-COX-2-MIP-2 loop regulates allergic inflammation and allergic inflammation-promoted enhanced tumorigenic and metastatic potential of cancer cells. J Biol Chem. 2015;290:14245-66
42. Li Y, Shen Z, Yu XY. Transport of microRNAs via exosomes. Nat Rev Cardiol. 2015;12:198
43. Escudier B, Dorval T, Chaput N, André F, Caby MP, Novault S. et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005;3:10
44. Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A. et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem. 2009;284:34211-22
45. Zhang A, Li M, Wang B, Klein JD, Price SR, Wang XH. miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J Cachexia Sarcopenia Muscle. 2018;9:755-70
46. Su Z, Hu L, Cheng J, Klein JD, Hassounah F, Cai H. et al. Acupuncture plus low-frequency electrical stimulation (Acu-LFES) attenuates denervation-induced muscle atrophy. J Appl Physiol (1985). 2016;120:426-36
47. Hu L, Klein JD, Hassounah F, Cai H, Zhang C, Xu P. et al. Low-frequency electrical stimulation attenuates muscle atrophy in CKD-A potential treatment strategy. J Am Soc Nephrol. 2015;26:626-35
48. Su Z, Klein JD, Du J, Franch HA, Zhang L, Hassounah F. et al. Chronic kidney disease induces autophagy leading to dysfunction of mitochondria in skeletal muscle. Am J Physiol Renal Physiol. 2017;312:F1128-40
49. Wang X, Chinsky JM, Costeas PA, Price SR. Acidification and glucocorticoids independently regulate branched-chain alpha-ketoacid dehydrogenase subunit genes. Am J Physiol Cell Physiol. 2001;280:C1176-83
50. Wang B, Zhang C, Zhang A, Cai H, Price SR, Wang XH. MicroRNA-23a and microRNA-27a mimic exercise by ameliorating CKD-induced muscle atrophy. J Am Soc Nephrol. 2017;28:2631-40
51. Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J. et al. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell. 2014;157:795-807
Author contact
![]() Corresponding author: Xiaonan H. Wang, MD, Renal / Medicine, WMB Room 338C, M/S 1930/001/1AG, 1639 Pierce Dr., Emory University, School of Medicine, Atlanta, GA, 30322-0001. E-mail: xwang03edu; Tel. (404) 727-8654 (L) (404) 727-1798 (O); Fax. (404) 727-3425
Corresponding author: Xiaonan H. Wang, MD, Renal / Medicine, WMB Room 338C, M/S 1930/001/1AG, 1639 Pierce Dr., Emory University, School of Medicine, Atlanta, GA, 30322-0001. E-mail: xwang03edu; Tel. (404) 727-8654 (L) (404) 727-1798 (O); Fax. (404) 727-3425
Citation styles
APA
Wang, B., Zhang, A., Wang, H., Klein, J.D., Tan, L., Wang, Z.M., Du, J., Naqvi, N., Liu, B.C., Wang, X.H. (2019). miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics, 9(7), 1864-1877. https://doi.org/10.7150/thno.29579.
ACS
Wang, B.; Zhang, A.; Wang, H.; Klein, J.D.; Tan, L.; Wang, Z.M.; Du, J.; Naqvi, N.; Liu, B.C.; Wang, X.H. miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics 2019, 9 (7), 1864-1877. DOI: 10.7150/thno.29579.
NLM
Wang B, Zhang A, Wang H, Klein JD, Tan L, Wang ZM, Du J, Naqvi N, Liu BC, Wang XH. miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics 2019; 9(7):1864-1877. doi:10.7150/thno.29579. https://www.thno.org/v09p1864.htm
CSE
Wang B, Zhang A, Wang H, Klein JD, Tan L, Wang ZM, Du J, Naqvi N, Liu BC, Wang XH. 2019. miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics. 9(7):1864-1877.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.