Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1728-1740. doi:10.7150/thno.30915 This issue Cite
Research Paper
MMP-2-Controlled Transforming Micelles for Heterogeneic Targeting and Programmable Cancer Therapy
Zihua Wang1,2,4, Yuehua Wang2, Xiangqian Jia2, Qiuju Han2, Yixia Qian2, Qian Li4, Junfeng Xiang4, Qian Wang5, Zhiyuan Hu1,2,3, Weizhi Wang2,6 ![]()
1. Center for Neuroscience Research, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian Province, 350108, China.
2. CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology of China, Beijing 100190, China;
3. Sino-Danish College, University of Chinese Academy of Sciences, Beijing 100049, P. R. China;
4. Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China;
5. Department of Diagnostic Imaging National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College
6. School of Life Science, Beijing Institute of Technology, Beijing, 100081, China
Received 2018-10-24; Accepted 2018-12-30; Published 2019-2-28
Abstract

Herein, through the active-peptide-functionalization, we developed a nanoscale micelles system (named HEKM) which consists of tumor microenvironment-regulated shape-changing with specific recognition abilities for enhanced cellular targeting, internalization and therapy of heterogeneic tumors. As a result, HEKMs could recognize and bind the tumor heterogeneity marker EGFR-HER2 complex, which led to an enhanced tumor targeting effect. In particular, HEKMs could self-assemble into nanorods under normal physiological conditions while transform into nanospheres in the tumor extracellular microenvironment by a sensitive response to matrix metalloproteinase-2 (MMP-2). The nanorods could prolong the blood circulation time while the nanospheres could accelerate tissue penetration in tumors. In vivo dual-modal targeted imaging was realized by FRET-fluorophore conjugation and gadolinium loading in HEKMs. Tumor cell apoptosis was achieved by proapoptotic element integration. The in vitro and in vivo studies both demonstrated that these rationally designed, shape-changing and targeting micelles could achieve maximized drug efficacy and minimum side effects.
Keywords: specific targeting peptide, MMP-2-controlled, shape transformation, dual-modal imaging, cancer therapy
Introduction
Nanoparticle (NP)-based tumor theranostics have ushered in new opportunities for personalized nanomedicine due to their rational design and functionalization. Despite the great improvements in drug transport, NPs still face some challenges, such as instability in blood circulation or lack of specific targeting of the tumor lesion, that limit the delivery efficiency and inevitably causes side effects during tumor treatment [1-3]. Many reported studies have shown that the shapes of NPs have great effects on their internalization behavior and endocytic pathways involving particle circulation, tissue penetration and cell uptake efficiency [4, 5]. For instance, rod-shaped NPs have a long circulation time, leading to a sustained accumulation of drugs at a lesion site, while spherical-NPs have deep cell penetration abilities [6, 7].Therefore, researchers are dedicated to developing a variety of transformable NPs, and using the advantages of different drug carrier shapes.
Most current studies have focused on the influence of the geometrical shape on tumor tissue internalization [4, 8]. Therefore, smart materials with tumor stimuli-responsive shape-changing ability are desirable for potential applications in precise detection, imaging and drug delivery [9-11]. If the geometrical shape of the nanomaterials could be controllably regulated by tumor markers and the tumor microenvironment, a promising tumor-targeted recognition and drug delivery system with enhanced cellular internalization could be developed [12, 13]. Peptide-based nanomaterials have attracted intensive research interest in recent years because of their affinity, specificity, biocompatibility, bioactivity, fast responsiveness, and ease of design and synthesis [14, 15]. Affinity peptides towards biomarkers could play important roles in NP functionalization for specific recognition and binding. Furthermore, peptides and their derivatives with programmable designed molecular structures could participate in the formation of nanostructures, such as nanofibers, nanomicelles, and nanospheres, with regular highly ordered structures [16-19]. Therefore, a tumor-regulated and affinity peptide-based nanosystem could be a promising approach to demonstrate the abovementioned ideal feature of controllable drug delivery and targeted imaging for tumor theranostics [20, 21].
In this study, we developed a nanoscale system, named HEKM, which consists of tumor microenvironment-regulated rod-to-sphere shape-changing micelles with specific recognition abilities for enhanced cellular targeting, internalization and therapy of heterogeneic tumors. HEKMs consist of a micelle backbone and three functional elements (Figure 1 A). The first element is a recognition element, which is a peptide that contains two targeting segments of the EGFR-HER2 complex and mitochondria, respectively [22, 23]. The second element is the imaging element, which contains a matrix metalloproteinase-2 (MMP-2)-regulated fluorescence resonance energy transfer (FRET) probe to turn on with near-infrared (NIR) fluorescence imaging and a gadolinium probe (Gd) to achieve magnetic resonance imaging (MRI). The third element is the therapeutic element, which includes the mitochondrial damage segment KLA and the chemotherapy drug DOX (doxorubicin), both of which evoke cancer cell apoptosis to achieve a synergetic antitumor treatment. HEKMs were initially self-assembled as nanorods. In the circulatory system, the nanorod were disinclined to enter the reticuloendothelial system (RES) so that prolonged blood circulation could be achieved (Figure 1B). When HEKMs circulated around the tumor area, the EGFR-HER2 complex (a complex of epidermal growth factor receptors 1 and 2) recognition element could bind to the tumor specifically, and the uptake of the NPs in the tumor was greatly enhanced. Simultaneously, the MMP-2 substrate element was sheared in the tumor matrix, which changed the HEKM self-assembly behavior and led to the rod-to-sphere shape transformation. The nanospherical micelles were small enough to accumulate in the tumor for deep penetration. Based on the tumor-regulated smart micelle system, multimodal imaging was achieved for precise imaging, and programable cell apoptosis was achieved for effective therapy.
Schematic representation of the HEKM smart micelle system for tumor theranostics by tumor environment-regulated geometrical shape transformation. (A) The chemical structure of HEKM building blocks that contain a peptide (including an EGFR-HER2 complex targeting segment, an MMP-2 responsive linker and a mitochondrial proapoptotic segment), the amphiphilic molecule DSPE-PEG and the FRET signal pair BHQ-3/Cy5.5. (B) The process of MMP-2-triggered the nanorod-to-nanosphere transformation of HEKMs, which enhanced the stability in blood circulation and facilitated internalization into cancer cells.
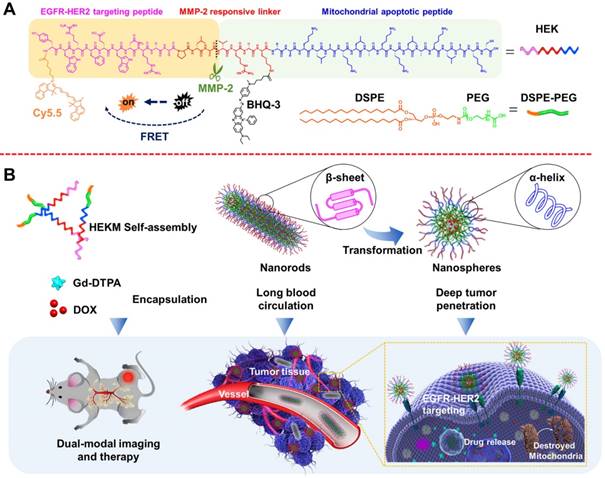
Materials and Methods
Materials
Wang resin (loading 0.34 mmol/g) was obtained from Rapp Polymere (Germany), 9-fluorenylmethoxy carbonyl (Fmoc)-conjugated amino acids and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from GL Biochem (China). BHQ-3 was obtained from LGC Biosearch Technologies (USA). Piperidine is from Sigma. 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)2000]-maleimide (DSPE -PEG2000-MAL) was purchased from Nanosoft Biotechnology LLC (USA). JC-1(5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolycarbocyanine iodide) fluorescent dye, Cy5.5 succinimide ester, MitoTracker Orange and LysoTracker Green DND-26 were both obtained from Invitrogen (USA). Gadolinium-triamine penta-acetic acid (Gd-DTPA) was purchased from Magnevist™ (Germany). The human breast cancer cell line MDA-MB-468 and SKBR-3 as well as the human embryonic kidney cell line 293T were purchased from the Infrastructure of Cell Line Resources, Chinese Academy of Medical Sciences. Dulbecco's-modified Eagle's medium (DMEM, high glucose), RPMI-1640 medium, trypsin-EDTA (0.25%), fetal bovine serum (FBS) were purchased from GE Healthcare Life Sciences (USA). All cell lines were cultured with 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 U/mL, Gibco) in a humidified atmosphere containing 5% CO2 at 37 ºC.
Synthesis of the HEK peptide
The peptide YWRFEWNRGGPLGVRGKGGD(KLAKLAK)2C was synthesized using the standard solid-phase Fmoc peptide synthesis method. Wang resin was used as the solid phase support. The Fmoc group on the N-terminus was deprotection by piperidine (20% v/v). A Kaiser test (ninhydrin, phenol, VC 1:1:1 v/v) was confirmed Fmoc deprotection. Amino acid Coupling was achieved by NMM (0.4 M) and HBTU (the same number of moles as the amino acid) in anhydrous DMF. A Black Hole Quencher-3 (BHQ-3) quenchable fluorophore was attached to the primary amine of the lysine adjacent to the cleavable sequence. The 1-(4,4-dimethyl-2,6-dioxocyclohexylidene) (Dde)-protecting group was selectively removed from the Fmoc-Tyr(tBu)-Trp(Boc)-Arg(Pbf)-Phe-Glu(OtBu)-Trp(Boc)-Asn-Arg(Pbf)-Gly-Gly-Pro-Leu-Gly-Val-Arg(Pbf)-Gly-Lys(Dde)-Gly-Gly-Lys(Boc)-Leu-Ala-Lys(Boc)-Leu-Ala-Lys(Boc) -Lys(Boc)-Leu-Ala-Lys(Boc)-Leu-Ala-Lys(Boc)-Cys(Trt)-Wang resin by adding 2% hydrazine in DMF. After selective lysine deprotection, BHQ-3 succinimide ester in anhydrous DMF was added, the sample was covered with tin foil and reacted overnight, followed by washing of the peptide resin. Subsequently, the last Fmoc group was removed from the peptide resin by piperidine, and the NIR dye Cy5.5 succinimide ester (2 mg) in DMF (0.1 mL) and DIEA (8 μL) were coupled to the N-terminus of the HEK peptide resin. The Cy5.5-HEK was then removed from the resin using TFA (92.5%, v/v), EDT (2.5%, v/v) TIPS (2.5%, v/v) and H2O (2.5%, v/v) for 30 min in an ice bath and then at room temperature for 4 h. The crude peptides were then precipitated in cold anhydrous diethyl ether, washed with ethyl ether, collected by centrifugation and dried under vacuum. Finally, the peptides were purified using a Waters high liquid chromatography (HPLC) system (Waters e2695) on a C18 column. The molecular weight was evaluated by MALDI-TOF-MS. The control peptide NF (Sequence: YCy5.5EDQTPNHGGPLGVRG KBHQ-3GGD(KLAKLAK)2C) was synthesized in a similar method.
Preparation and characterization of DSPE-PEG2000-HEK and micelles
Briefly, DSPE-PEG2000-MAL was used to conjugate HEK through the coupling of the thiol group (-SH) with the maleimide group. HEK and DSPE-PEG2000-MAL (1:2, w/w) were dissolved in deionized water and stirred continuously at room temperature for 48 h. Then, the reaction mixture was purified by dialysis (cut-off 3500 Da) and lyophilized. Doxorubicin-loaded micelles (HEKMsDOX) were prepared from the HEK-DSPE-PEG2000, DSPE-PEG2000, Gd-DTPA and doxorubicin mixture (1:8:1, w/w/w) and dissolved in a chloroform/methanol mixture (4:1) in a round-bottom flask by a film dispersion method. Gd-DTPA was incorporated as 10% (w/w) micelles. The HEKMs and NFMs micelles were prestained with 1% uranyl acetate and analyzed by transmission electron microscopy (TEM, Tecnai G220 S-TWIN). Hydrodynamic size was determined by dynamic light scattering using a Zetasizer 5000 (Malvern Instruments, U.K.). The loading content (LC) and the encapsulation efficiency (EE) of the micelles were analyzed by UV-Vis spectrometer (Shimadzu SPD-10AVP, Japan).
Cellular uptake and subcellular localization
Cellular uptake was investigated using CLSM and flow cytometry. SKBR-3, MDA-MB-468 and 293T cells at a concentration of approximately 1×105 cells mL-1 were separately seeded into culture dishes and cultured at 37 °C and 5% CO2. The three cell lines were treated with HEKMs and NFMs (20 μg/mL) for different periods of time at 37 °C. To observe the subcellular localization, the cells were rinsed with PBS. Subsequently, 100 nM Hoechst 33342, LysoTracker Green DND-26 and MitoTracker Orange dissolved in DMEM were added to stain the cell nuclei, lysosomes and mitochondria, respectively, for 30 min at 37 °C. The treated cells were analyzed using a ZEISS LSM710 confocal laser scanning microscope. For flow cytometry analysis, the cells were treated in a similar way.
Mitochondrial targeting and cytotoxicity
SKBR-3 and MDA-MB-468 cells were incubated with 50 μg/mL HEKMDOX in cell culture dishes. After incubation 4, 8 and 12 h of at 37 °C, the JC-1(10 μg/mL) were incubated with mitochondria for 30 minutes.After washing the cells with PBS for 3 times, the cells were observed with confocal microscope. The cells in each well were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) at 4 °C overnight. After washing the cells with PBS 3 times, the cells were further stained with 4% osmium tetroxide for 30 min, then rinsing the cells with ddH2O. The cells were dehydrated in different gradients of ethanol (30, 50, 70 and 90%) at room temperature, followed by rinsing 3 times with 100% ethanol. After dehydration, the cells were embedded in epoxy resin and stored at 37 °C for 12 h and 60°C for 48 h, respectively. Finally, the cells were stained with uranyl acetate (4%) and lead citrate (2%) for 15 min respectively, and then observed by transmission electron microscopy.
In vitro drug release and cytotoxicity assay
One milligram of DOX-loaded micelles was dispersed uniformly in 10 ml of medium in a dialysis bag (cut-off 3500 Da). The dialysis bag was immersed in 50 mL of the release medium at 37 °C and placed a shaker with 1000 rpm. The released DOX was analyzed by a UV spectrophotometer (ex: 485 nm; em: 590 nm). The cytotoxicity of HEKMDOX, NFMDOX, HEKM, and free DOX was evaluated by an MTT assay. SKBR-3, MDA-MB-468 and 293T cells were seeded in 96-well plates at a density of 5000 cells per well and cultured for 24 h at 37 °C. Then, the cells were treated with 100 µL different concentrations free DOX, NF-DOX and HEKM-DOX. The materials and cells were incubated for 8 h and then the medium was replaced with fresh medium culture for 24h. MTT solution (0.5 mg/mL) was added to each well for another 4 h at 37 °C. Then, the solution was removed, and 150 μL dimethyl sulfoxide (DMSO) was added to each well. After 10 min of vibrational mixing, the optical density (OD) at a wavelength of 570 nm was measured using an ELISA reader. The cytotoxicity is expressed by the following formula: cell viability=OD sample /OD control × 100%, OD is the optical density,n=4.
Western blot assay
SKBR-3 cells and tumor tissues were treated with cell lysis buffer. Cell lysates were collected at 12,000 rpm at 4°C in a microcentrifuge and determine the protein concentration for each sample lysate (BCA assay). According to the standard western blot procedures, proteins were separated by SDS-PAGE and transferred onto PVDF (polyvinylidene fluoride) membranes (Merck Millipore, DE). Block the membrane for 1 h at room temperature using blocking buffer andincubate the membrane with MMP-2 and β-actin primary antibody in blocking buffer. Afte wash the membrane in three washes of PBST, 5 min each. The membranes incubate with the HRP conjugated secondary antibody in blocking buffer at room temperature for 1 h. Wash the membrane and acquire image using darkroom for chemiluminescence.
In vivo imaging
Female BALB/c nude mice (6 weeks of age) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). All animal experimental procedures were approved by the Institutional Animal Care and Use Committee. Xenograft tumors were established by the subcutaneous (S.C.) injection of 5×106 cells/mL SKBR-3 cells to the left hind leg of the mice. When the tumor size reached approximately 90 mm3, HEKMs, NFMs and free DOX were individually injected intravenously into the tail vein of the xenograft mice at a dose of 5.0 mg/kg using a Maestro in vivo spectrum imaging system for detection. After in vivo detection, the mice were sacrificed. The tumor, heart, liver, spleen, lung and kidney were excised for in vitro imaging. For MRI imaging, mice were treated in a similar manner. The MRI study was performed using a Bruker Biospec 7 T MRI scanner (Bruker Corp., Billerica, MA, USA). Images were acquired using a FLASH sequence with respiratory gating before injection. After preinjection baseline MR imaging, HEKMsGd or NFMsGd were injected at a dose of 5.0 mg/kg (Gd3+ dose: 0.02 mM). The T1-weighted FLASH sequence was acquired at different timepoints.
In vivo therapeutic study
When the tumor size reached approximately 90 mm3, the tumor-bearing mice were divided into five groups, and each group was injected in the tail vein with PBS, free DOX, HEKMDOX, NFMDOX and KLA (n=5 per group), respectively. The dose of DOX that was intravenously administered to the mice was 5 mg/kg body weight every four days. During the treatment, the tumor size and body weight of each mouse were measured every other day. After the in vivo antitumor treatment, the mice were sacrificed, and the major organs (tumor, heart, liver, spleen, lung, and kidney) were collected for histological analysis. To evaluate the levels of apoptosis in cells and tissues, the tumors and major organs were embedded in paraffin, sectioned and detected by a TUNEL (TdT-mediated dUTP nick end labeling) assay. Furthermore, these paraffin sections were stained with hematoxylin and eosin (H&E) for histopathology analysis.
Results and Discussion
Preparation and characterization of HEKMs
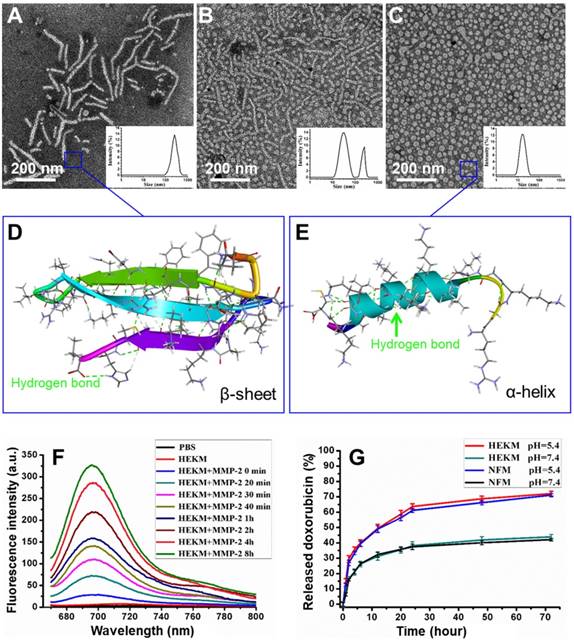
To construct a three-dimensional nanostructure, the amphipathic molecule 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) was used to build the framework of the micelles. Then, a series of functionalizations was performed. First, HMP, a specific targeting peptide of the EGFR-HER2 complex, which is an important biomarker for tumor heterogeneity, was used as one recognition element. Since HMP and one of the therapeutic elements (KLA) are both amino acid sequences, we constructed the segments as one peptide with an enzyme-triggered linker (PLGVRGK). The amino acids of the KLA mitochondrial damage segment were replaced by a D-enantiomer to avoid degradation by proteases and to improve the in vivo stability [24]. The linker was the specific substrate of MMP-2, which is highly expressed at the tumor site. The whole peptide was named HEK (Figure 1A). HEK was synthesized using the Fmoc solid-phase peptide synthesis (SPPS) strategy (Figure S1) [25]. To construct a nanoscale system, the DSPE-PEG2000 hydrophilic tail was conjugated with the HEK peptide to produce the amphiphilic self-assembled micelles, named HEKMs. HEK was efficiently cleaved into two fragments, mediated by MMP-2, which could be indicated by monitoring the process using HPLC and mass spectrometry (Figure S2 and 3). Then, the morphologies of HEKMs during the MMP-2 response were observed by transmission electron microscopy (TEM). As shown in Figure 2A, HEKMs were first self-assembled into nanorod with a transverse diameter of 20 nm and lengths in the range of 50-300 nm. When HEKMs were incubated with MMP-2 for 2 h, a large number of spherical nanoparticles were observed (Figure 2B), which suggested the shape transformation of HEKMs in the presence of the enzyme. When HEKMs were incubated with MMP-2 for 4 h, the transformation from nanorods to nanospheres was nearly complete (Figure 2C). Dynamic light scattering (DLS) analysis was also conducted. Only one peak at approximately 253.5 nm was observed at the primary stage, suggesting that a uniform assembly was formed. However, two peaks (approximately 226.7 and 33.7 nm) were observed in the presence of MMP-2. As time progressed, all of the particles changed into smaller ones, and their diameter was ~35 nm which was the suitable size for deep tumor tissue penetration [26]. The molecular structure of the HEK peptide before and after enzyme cleavage was modeled by molecular simulation using Discovery Studio. The Generalized Born implicit solvent model was used in the simulation. The simulation systems were energy minimized using 5000 steps of the steepest descent and followed by 5000 steps of the conjugate gradient method. The peptides are shown as stick modes, and the secondary structures are shown as ribbons. It was shown that there were secondary structure transformations of HEK from a β-sheet to an α-helix during the MMP-2-responsive process. First, the β-sheet formation of the peptide remained in a stable state (potential energy -1830.64 kcal/mol) by the hydrogen bond (green dot line in Figure 2D). It has been reported that the β-sheet region and the stearic acid tail are likely to promote hydrophobic collapse and lead to cylindrical nanorod formation[27]. HMP plays an important role because of changes in the secondary structure after cleavage. The shape transformation may result from the hydrophobicity of HEKMs decreasing after MMP-2 cleavage since the HMP segment was removed. The arrangement of the building blocks in HEKMs changed from a hydrophobic-hydrophilic-hydrophobic formation to a hydrophilic-hydrophobic formation [27]. Additionally, the vicinity of charged groups (lysines) likely generated a strong electrostatic repulsion rather than cohesive interactions, leading to the formation of spherical micelles rather than rods [7, 28, 29]. Due to the disturbance in the balance of electrostatic repulsions and hydrogen-bonding interactions, the HEKM self-assembly behavior could change [30, 31]. A new α-helix (potential energy -1378.15 kcal/mol) formed that could facilitate cell penetration (Figure 2E). To investigate the secondary structure conformational change of HEK after cleaved, the circular dichroism (CD) spectra of the HEK was analyzed using standard CONTINLL algorithms. The result was shown in Figure S4. In the initial stage, HEK formed β-sheet secondary structures in PBS (negative bands at 216 nm and positive bands at 195 nm), which suggested that the formation of HEKM nanofiber was driven by the strong hydrophobic chain and intermolecular H-bonds interactions. But in the presence of MMP-2, along with the time the HEK conformational gradually transformed from β-sheet to α-helix, an obvious H-bonded α-helix structure (negative bands at 208 nm and at 222 nm) was confirmed. In addition, the reduction of the β-sheet structure produces a synergistic effect to promote the morphology transformation of HEKM. As a control, a scrambled peptide (NF)-functionalized micelle (NFM) was prepared for comparison with HEKMs. However, the morphology of NFM did not change significantly in the presence of MMP-2 (Figure S5). This finding indicates that the specific shape transformation of HEKMs might be regulated by the actual tumor microenvironment and could benefit both the long circulation in blood and deep tissue penetration in tumors, which could facilitate drug delivery. To construct an integrated delivery system for diagnosis and therapy, drugs and signal molecules were encapsulated or conjugated with HEKMs. In our design, Gd-diethylenetriamine penta-acetic acid (Gd-DTPA), for T1 detection of MRI, was first loaded in the hydrophobic cavity of the micelles at a ratio of 10% (w/w). The chemotherapy drug DOX was also encapsulated into the micelles through hydrophobic interactions with a mass ratio of 1:10. The NIR dye Cy5.5 was conjugated on the N-terminus of HMP, and the fluorescence-quenching molecule BHQ-3 was conjugated on the MMP-2 linker segment of the lysine side chain to alter the fluorescence of Cy5.5 through FRET. When the peptide was exposed to MMP-2, the PLGVRGK segment was cleaved between V and R, causing the fluorescence of Cy5.5 (Figure 1). Based on this design, we successfully prepared an integrated micelle system. The FRET quenching efficiency of the BHQ-3/Cy5.5 pair was first evaluated at the molecular level. As shown in Figure 2F, the red fluorescence of Cy5.5 in the probe was originally quenched, resulting in a low intrinsic fluorescence intensity. Compared with the negligible fluorescence recovery without enzymes, an approximate 13-fold fluorescence enhancement was observed at 8 h after MMP-2 treatment. None of the controls demonstrated any significant changes in the fluorescence intensity. Then, we evaluated the properties of HEKMs affected by the acidity. We first drew the drug release profile by a dialysis method at both pH 5.4 and 7.4. As shown in Figure 2G, the DOX release rate at pH 5.4 was much greater (74%) than that at pH 7.4 (37%). We then monitored the HEKM morphology in different conditions. Therefore, HEKMs were active and disassemble in acidic environments, and drug release might have been triggered and accelerated not only by the tumor microenvironment but also by endogenous lysosomal acidic conditions after HEKM internalization (Figure S6). The micelle delivery system showed a control and release ability in a pH-dependent manner due to the isoelectric points (PIs) and solubility of DOX at mild acidic environment [32]. Therefore, HEKMs were active and disassemble in acidic environments, and drug release might have been triggered and accelerated not only by the tumor microenvironment but also by endogenous lysosomal acidic conditions after HEKM internalization.
Characterizations and drug release behaviors of HEKMs. (A) DLS and TEM images of HEKMs without MMP-2. (B) DLS and TEM images of HEKMs incubated with MMP-2 for 2 h. (C) DLS and TEM images of HEKMs incubated with MMP-2 for 4 h. The scale bar indicates 200 nm. (D) Molecular simulation of the secondary structures of HEKMs before MMP-2 cleavage. (E) Molecular simulation of the secondary structures of HEKMs after cleavage. (F) Fluorescence emission spectra of HEKMs incubated with MMP-2 (10 nM) in PBS (pH 7.4) from 0 h to 8 h. (G) In vitro drug release profiles of doxorubicin from HEKMs at pH 5.4 and pH 7.4; NFMs served as the control.
Internalization and Subcellular Distribution of HEKMs in the Cytoplasm
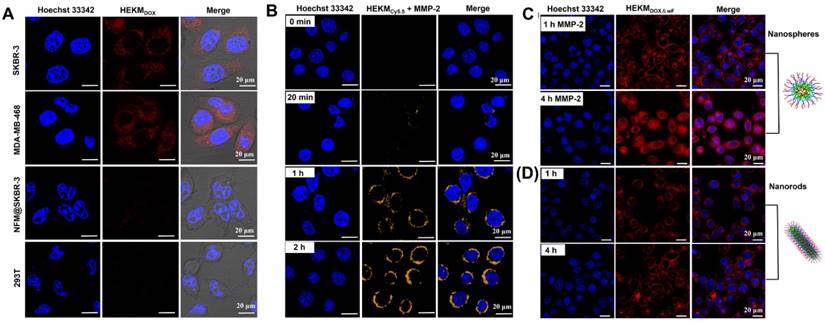
We then investigated the internalization of HEKMs at the cellular level. First, the specific cell recognition and binding ability was monitored by confocal laser scanning microscopy (CLSM). The human breast cancer cell lines SKBR-3 and MDA-MB-468 cells (both cell types have high expression of EGFR-HER2) were used as positive cells, and the human embryonic kidney 293T cell line was used as a control. DOX-encapsulated HEKMs (HEKMsDOX) were incubated with the abovementioned cells. Figure 3A shows the endocytosis of HEKMsDOX (red channel). With the assistance of the HMP targeting peptide, HEKMsDOX could effectively bind to both SKBR-3 and MDA-MB-468 cell membranes. As a control, both HEKMDOX-treated 293T and NFMDOX-treated SKBR-3 cells showed inconspicuous fluorescence, indicating low uptake without cell-targeting ligand-receptor recognition. Next, the MMP-2-responsive behavior of HEKMs in cells was assessed. As shown in Figure 3B, without MMP-2, the FRET element of HEKMs was quenched by BHQ-3, and there was no fluorescence observed (0 min). When the cells were incubated with MMP-2, the peptide was cleaved, and the FRET process was activated, which caused Cy5.5 to fluoresce. As the incubation time with MMP-2 increased, the fluorescence intensity of the Cy5.5 signal increased. It was shown that HEKMs had a sensitive response to MMP-2. Finally, we evaluated the shape transformation effect on the efficiency of cellular uptake in the absence of MMP-2. We constructed HEKMsDOX without FRET (wF) signaling molecules HEKMsDOX&wF to avoid fluorescence channel overlap. With the aid of MMP-2, the morphological transformation occurred, and small spherical particles with a greater penetration efficiency were formed. Therefore, a large number of HEKMsDOX&wF entered the cytoplasm and then gradually diffused into the nucleus (Figure 3C). With increasing time, HEKMsDOX&wF gradually diffused from the cytoplasm and penetrated the mitochondria. In contrast, most of the fluorescence signal was observed on the membrane without MMP-2 treatment (Figure 3D). According to these results, we concluded that the rod-to-sphere shape change accelerated intracellular uptake and that micelles could enter the cytoplasm and corresponding organelles in a specific and efficient manner under enzymatic regulation. Furthermore, we used flow cytometry to evaluate the cellular uptake of HEKMs in SKBR-3 and MDA-MB-468 cells (Figure S7). The results were consistent with the CLSM images, indicating that the rod-to-sphere shape change promoted the cellular uptake of HEKMs to EGFR-HER2-positive cells. Next, we evaluated the subcellular distribution of HEKMs. Figure S8 showed the colocalization of HEKMsDOX in mitochondria and lysosomes (indicated by the mitochondrial tracker MitoTracker Orange in the yellow channel and the lysosomal tracker Lysotracker Green in the green channel). It was confirmed that HEKMs entered lysosomes within 2 h of incubation. After 4 h of incubation, HEKMsDOX escaped from the lysosomes, and DOX was released because a fairly intense red emission was observed in the whole nucleus and cytoplasm region. Finally, due to the presence of the KLA mitochondrial recognition and damage element, which is favorable for efficient endosomal escape, HEKM exhibited endosomal escape and was localized in the mitochondria. In contrast, NFMs were only distributed in lysosomes and the cytoplasm HEKMs were (Figure S9). HEKM confirmed to have a programmable track in subcellular tissue, which started from the cell membrane to the lysosome and then from the lysosome to mitochondria and nuclei.
Confocal images of cellular internalization and subcellular distribution of HEKMs. (A) CLSM imaging of EGFR-HER2-positive and -negative cellular internalization of targeting HEKMsDOX incubate for 4h. The red fluorescence from the emission of DOX channel. (B) In the presence of MMP-2, the FRET probe was activated, and the fluorescence signal of Cy5.5 gradually increased in SKBR-3 cells at different timepoints. (C) In the presence of MMP-2, SKBR-3 cells were treated with HEKMs for 1 and 4 h. The red fluorescence from the emission of DOX channel. (D) SKBR-3 cells were treated with HEKMsDOX&wF without MMP-2 for 1 and 4 h. The red fluorescence from the emission of DOX channel. The scale bar indicates 20 μm.
Mitochondrial Targeting and Cytotoxicity
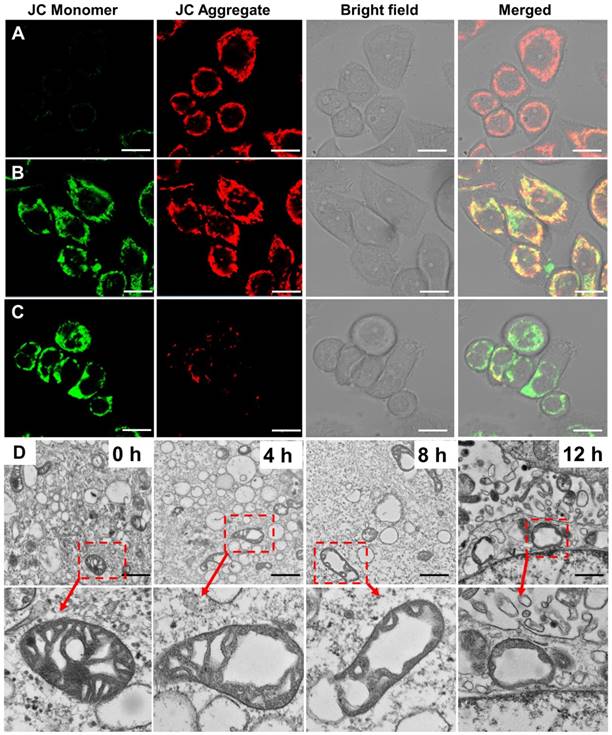
In our design, one of the functions of HEKMs is mitochondrial apoptosis-based therapy. Mitochondrial damage is caused by the mitochondrial membrane-targeting element KLA. We evaluated mitochondrial activity during HEKM treatment in SKBR-3 cells. Loss of mitochondrial membrane potential can lead to mitochondrial damage or dysfunction [33, 34]. We used a commercial dye JC-1 to monitor mitochondrial morphological changes during HEKM treatment. The JC-1 dye will undergo reversible fluorescence changes with the state of membrane potential. JC-1 aggregates and emits red fluorescence at high mitochondrial membrane potential levels, indicating normal mitochondria. However, JC-1 aggregates dissociate into monomers at low mitochondrial membrane potential levels and emit green fluorescence, indicating depolarization and damage to mitochondria [35]. Figure 4A-C shows representative confocal images of the JC-1 assays. At the first 4 h of incubation of HEKMs and SKBR-3 cells, most of the JC-1 dye was in an aggregated state. With the increase in the incubation time (8 h), the JC-1 signal changed, and green fluorescence increased, indicating the loss of mitochondrial membrane potential and damage to mitochondria. After 12 h of HEKM incubation, the red fluorescence nearly disappeared, while the green fluorescence greatly increased, indicating that the mitochondria were substantially destroyed. In contrast, HEKM-treated 293T cells did not cause obvious mitochondrial damage, further indicating that HEKM internalization is mediated by the targeting markers on the cell membrane (Figure S10). In contrast, for NFM-treated SKBR-3 cells, the JC-1 dye always showed red fluorescence, which was similar to the control cells without any treatment, indicating that NFMs could not induce cell apoptosis (Figure S11). Furthermore, TEM imaging was performed to directly demonstrate the morphology of the mitochondria after micelle treatment. After 8 h of incubation with HEKMs, the mitochondria in the SKBR-3 cells showed abnormal swelling, severe cristae disruptions and a vacuolization shape, indicating substantial mitochondrial damage (Figure 4D) [36]. However, normal cristae structures and healthy mitochondrial morphology were observed in the NFM group (Figure S12). These results indicated that HEKMs could enter cells through a receptor-mediated endocytosis pathway and effectively damage mitochondria, which causes high cytotoxicity specifically towards EGFR-HER2-positive tumor cells [37]. To further assess the cytotoxicity efficiency, a 3-(4,5-dimeth-ylthiazol-2-yl)-2,5-diphenylte-trazolium bromide (MTT) assay was used to evaluate cell viability. HEKMS showed much greater cytotoxicity to EGFR-HER2-positive cancer cells than to normal cells, while NFMS exhibited almost no differences among the cells. When the rod-to-sphere transformation took place, HEKMDOX showed higher pharmaceutical effect (Figure S13). This was ascribed to the multifunctionalities of HEKMs, which can first recognize EGFR-HER2 on the membrane, then the lysosome, followed by the mitochondria, finally destroying the mitochondria as well as whole tumor cells.
The detection of mitochondrial membrane potential and morphological changes in HEKM-treated cultured cells. (A-C) The detection of the mitochondrial membrane potential using a JC-1 assay on SKBR-3 cells treated with HEKMs for 4, 8 and 12 h, respectively. The scale bar indicates 20 μm. (D) TEM images showing the representative mitochondrial morphologies of SKBR-3 cells treated with HEKMs for 0, 4, 8 and 12 h. The scale bar indicates 500 nm. The magnified images of the areas are shown as arrowheads below each image. Scale bar is 100 nm.
In vivo Dual-modal Tumor Imaging using MRI/NIRF
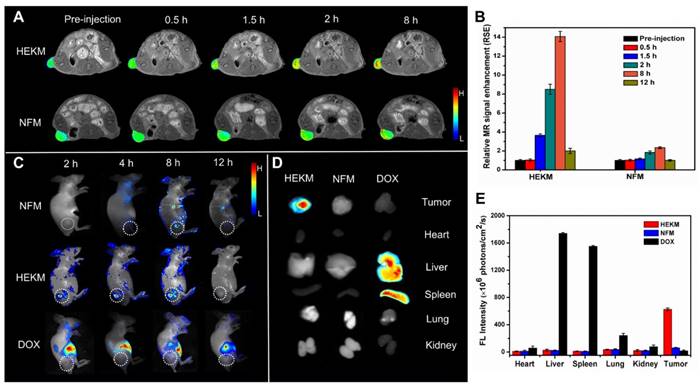
Next, we evaluated the performance of HEKMs by in vivo tumor imaging in EGFR-HER2-positive xenograft tumors. To achieve precise imaging, we used MRI to detect HEKM distribution in vivo. The contrast agent was prepared by the encapsulation of Gd-DTPA in HEKMs (HEKMsGd). MR imaging before and after the intravenous micelle injection was monitored using the T1 mapping quantification. As shown in Figure 5A, before intravenous injection, the tumors were barely detectable, whereas approximately 0.5 h after HEKM injection, the T1-weighted MRI exhibited remarkable tumor local signal enhancement. In particular, up to 8 h after injection, there was a clear signal at the tumor site. The relative MR signal enhancement of the HEKM group was determined to be 14 times greater than that of the NFM group (Figure 5B). Furthermore, Gd3+ in tumor tissues was detected by ICP-MS (Inductively coupled plasma mass spectrometry). The results showed that the concentration of Gd delivered by HEKM was twice as high as that of NFM (Figure S14), indicating that the shape transformation could facilitate the deep tumor penetration [38]. To achieve dual-modal imaging, in vivo whole-body near-infrared fluorescence (NIRF) imaging was performed. The BHQ-3/Cy5.5 FRET pair acted as a switchable NIRF signal element. Cy5.5 at the HEKM terminal was initially in the fluorescence “off” state due to BHQ-3 quenching. Western blot assay was carried out to confirm that the MMP-2 was expressed in rather SKBR3-cell inoculated tumor tissues than the SKBR3-cells (Figure S15). Subsequently, when the PLGVRGK segment was triggered by MMP-2 (50 nmol/L) in the tumor microenvironment, a FRET-based switch turned “on”. As shown in Figure 5C, 2 h after the injection, tumors treated with HEKMs began to fluoresce. At approximately 8 h, the flrescence intensity reached the highest level. Then, the fluorescence intensity gradually decreased following metabolism (12 h). In contrast, fluorescence was undetectable from the beginning to the end of the observation period in NFM-treated mice or the free-DOX-treated mice. These results indicate that the enzyme-triggered shape transformation and fluorescence switching play an important role in the recognition and detection process. Since MMP-2 was expressed at low levels in normal tissues, fluorescence was hard to activate in healthy organs [39]. Only the smart HEKMs could remain stable in blood circulation while causing a series of responsive processes, specifically in heterogeneous tumor sites. Furthermore, to examine the relative biodistribution of these nanomicelles in organs, the mice were sacrificed, and the major organs were collected for ex vivo NIRF imaging at 24 h post-injection of micelles. In accordance with the in vivo observations, strong fluorescence was detected only in the tumors. The fluorescence signal was nearly undetectable in the liver, lung, spleen, kidney or heart (Figure 5D and E) [40]. The dramatic difference between the HEKM- and NFM-treated tumor groups was confirmed, which indicated high HEKM uptake by EGFR-HER2-positive tumors. The enzyme-triggered property of HEKMs was able to achieve dual-model tumor imaging with high sensitivity and specificity.
In vivo dual-modal imaging of signal molecule-loaded micelles. (A) Representative T1-weighted MR imaging of Gd contrast agent-loaded micelles in nude mice bearing SKBR-3 xenograft tumors. The images were acquired at different timepoints post-injection of HEMKsGd and NFMsGd using a FLASH sequence. (B) The MR signal intensity at different timepoints after injection of HEMKsGd and NFMsGd (n=5). (C) Representative in vivo NIRF imaging of nude mice bearing SKBR-3 tumors after the intravenous injection of HEKMs/NFMs and free DOX (ex: 678 nm; em: 692 nm). (D) Representative ex vivo NIRF imaging of SKBR-3 tumors, which indicated the biodistribution of HEKMs/NFMs at 24 h after injection (ex: 678 nm; em: 692 nm). (E) Average fluorescence signals of tumors and major organs at 24 h after injection (n=5).
In vivo therapy by the integrated smart micelles
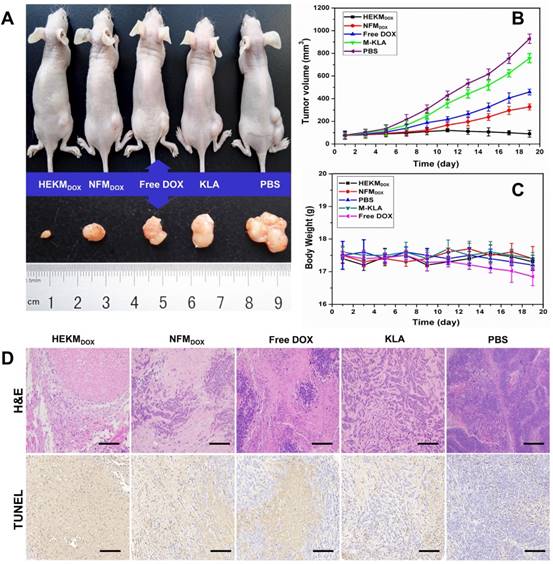
Next, we evaluated the in vivo antitumor efficacy of the DOX-encapsulated HEKMs in a mouse model. When the tumor size reached approximately 90-100 mm3, the mice were divided into five groups, and each mouse was injected intravenously through the tail vein under one of the following experimental conditions: HEKMDOX, NFMDOX, free DOX, KLA peptide and PBS (n=5 per group). The dose of DOX was 5 mg/kg body weight every four days. The progress of tumor growth was monitored for 19 days, tumor size and body weight of each mouse were recorded daily and normalized during the treatment. The tumor inhibition efficiency was summarized as plots of tumor volume for the different groups. The satisfactory therapeutic effect of HEKMsDOX was also confirmed directly by the representative tumor photograph and tumor volume. The tumors in the HEKMDOX-treated group were all effectively suppressed (Figure 6A and B). The HEKM-mediated combination therapy inhibited tumor growth more efficiently than NFMs or free DOX, which could be attributed to increased tumor distribution of HEKMs by specific targeting and deep tumor penetration by the rod-to-sphere shape transformation due to MMP-2 regulation. Moreover, KLA-caused mitochondria-dependent apoptosis and DOX-induced DNA damage facilitated the elimination in a synergistic manner. Additionally, the nanorod configuration of the micelles prevented recognition by phagocytes, which resulted in reduced RES phagocytosis and prolonged blood circulation [41]. As shown in Figure S16, similar pharmacokinetic behaviors were observed in NFMDOX and HEKMDOX, which exhibited a relative long circulation time than free DOX. The nanorods-like NPs increased the t1/2 of HEKMDOX to 11.5 h relative to free DOX (2.6 hours). Furthermore, HEKMDOX kept a high concentration in the bloodstream even up to 12 h. The reason is that the flexibility and length of rod-shaped NPs enable it to be consistent with the blood flow in the blood, reducing the metabolic clearance and vascular collisions. Long circulation and shape transformation would enhance internalization of HEKM in tumor cells, which improve therapeutic effect. These results clearly suggested a cumulative and synergetic antitumor effect of the HEKMDOX-based integrated therapy. Furthermore, almost no change in mouse body weight was observed when the mice were treated with HEKMsDOX, indicating negligible systemic cytotoxicity. Notably, the mice treated with free DOX showed a decreasing trend in weight, which indicated drug toxicity (Figure 6C). Furthermore, hematoxylin-eosin (H&E) staining of the tumor revealed membrane lysis in the HEKMDOX-treated group, which implied that HEKMsDOX caused notable apoptosis of the tumor tissues. Apoptosis was also confirmed by Tdt-mediated dUTP nick end labeling (TUNEL). Noticeable DNA damage was observed because the cell shrinkage happened in the HEKMDOX treated group (Figure 6D). The toxicity and side effects of heart, liver, spleen, lung and kidney were evaluated by histological sections. The biosafety of HEKMs was confirmed since it caused negligible organ damage (Figure S17). From the in vivo and in vitro results, we concluded that HEKMs with a tumor microenvironment-regulated geometrical shape transformation could perform well in tumor therapy and could achieve maximized drug efficacy and minimum side effects.
Evaluating the therapeutic efficacy of HEKMsDOX in SKBR-3 tumor-bearing nude mice. (A) Representative photograph of the mice as well as the corresponding tumor treatment in each group (i.e., HEKMDOX, NFMDOX, free DOX, KLA and PBS). (B) Tumor volume curves of each mouse treated with HEKMsDOX, NFMsDOX, free DOX, KLA and PBS monitored for 19 days (n=5). (C) Body weight of each mouse treated with HEKMsDOX, NFMsDOX, free DOX, KLA and PBS monitored for 19 days (n=5). (D) H&E and TUNNEL assays of tumor organs collected after sacrificing the mice. The scale bar indicates 100 µm.
Conclusion
We designed and constructed a tumor microenvironment-regulated shape-changing micelles, HEKMs that could be used as smart drug delivery materials. HEKMs could self-assemble into cylindrical nanorods with prolonged retention time in blood circulation and transformed into nanospheres with penetration ability in tumor sites regulated by MMP-2, which is highly expressed in tumor sites. HEKM-based signaling elements and therapeutic drugs were added to the system to achieve MRI/NIRF dual-modal imaging and synergistic therapy. This smart material could not only recognize the EGFR-HER2 tumor specifically but also initiate the programmable therapy process among the organelles. However, there are certainly challenge of this approach in real clinical studies. For example, the self-assembled nano material would suffer from a series of physiological processes in the complex organisms. How to executive functions ideally in vivo was far from been verified for all the well-designed smart nano materials. The smart system may be potentially applied as a new strategy for combating tumor heterogeneity.
Abbreviations
HER2: human epidermal growth factor receptor 2; EGFR: epidermal growth factor receptor; FRET: fluorescence resonance energy transfer; MMP-2: matrix metalloproteinase-2; MRI: magnetic resonance imaging; RES: reticuloendothelial system; TEM: transmission electron microscopy; DLS: dynamic light scattering; Gd-DTPA: gadolinium-triamine penta-acetic acid; CLSM: confocal laser scanning microscopy; JC-1: 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolycarbocyanine iodide; FBS: fetal bovine serum; BHQ-3: black hole quencher-3; TUNEL: TdT-mediated dUTP nick end labeling; PBS: phosphate buffer saline; DOX: doxorubicin; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (21775031, 81801766, 31870992), Beijing Natural Science Foundation (2172056 and L172035), Youth Innovation Promotion CAS, Key Research Program of the Chinese Academy of Sciences (KFZD-SW-210).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lim E-K, Kim T, Paik S, Haam S, Huh Y-M, Lee K. Nanomaterials for Theranostics: Recent Advances and Future Challenges. Chem Rev. 2015;115:327-94
2. Ling D, Park W, Park S-j, Lu Y, Kim KS, Hackett MJ. et al. Multifunctional Tumor pH-Sensitive Self-Assembled Nanoparticles for Bimodal Imaging and Treatment of Resistant Heterogeneous Tumors. J Am Chem Soc. 2014;136:5647-55
3. Chen H, Zhang W, Zhu G, Xie J, Chen X. Rethinking cancer nanotheranostics. Nat Rev Mater. 2017;2:17024
4. Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T. et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249-55
5. Venkataraman S, Hedrick JL, Ong ZY, Yang C, Ee PLR, Hammond PT. et al. The effects of polymeric nanostructure shape on drug delivery. Adv Drug Deliver Rev. 2011;63:1228-46
6. Wang Y, Wang D, Fu Q, Liu D, Ma Y, Racette K. et al. Shape-Controlled Paclitaxel Nanoparticles with Multiple Morphologies: Rod-Shaped, Worm-Like, Spherical, and Fingerprint-Like. Mol Pharm. 2014;11:3766-71
7. Moyer TJ, Kassam HA, Bahnson ESM, Morgan CE, Tantakitti F, Chew TL. et al. Shape-Dependent Targeting of Injured Blood Vessels by Peptide Amphiphile Supramolecular Nanostructures. Small. 2015;11:2750-5
8. Li X, Jin B, Gao Y, Hayward DW, Winnik MA, Luo Y. et al. Monodisperse Cylindrical Micelles of Controlled Length with a Liquid-Crystalline Perfluorinated Core by 1D "Self-Seeding". Angew Chem Int Ed. 2016;55:11392-6
9. Huang P, Gao Y, Lin J, Hu H, Liao H-S, Yan X. et al. Tumor-Specific Formation of Enzyme-Instructed Supramolecular Self-Assemblies as Cancer Theranostics. ACS Nano. 2015;9:9517-27
10. Zhang K, Yang P-P, Zhang J-P, Wang L, Wang H. Recent advances of transformable nanoparticles for theranostics. Chinese Chem Lett. 2017;28:1808-16
11. Hu Q, Chen Q, Gu Z. Advances in transformable drug delivery systems. Biomaterials. 2018;178:546-58
12. Anchordoquy TJ, Barenholz Y, Boraschi D, Chorny M, Decuzzi P, Dobrovolskaia MA. et al. Mechanisms and Barriers in Cancer Nanomedicine: Addressing Challenges, Looking for Solutions. ACS Nano. 2017;11:12-8
13. Qian Y, Wang W, Wang Z, Jia X, Han Q, Rostami I. et al. pH-Triggered Peptide Self-Assembly for Targeting Imaging and Therapy toward Angiogenesis with Enhanced Signals. ACS Appl Mater & Interfaces. 2018;10:7871-81
14. David A. Peptide ligand-modified nanomedicines for targeting cells at the tumor microenvironment. Adv Drug Deliver Rev. 2017;119:120-42
15. Walsh TR, Knecht MR. Biointerface Structural Effects on the Properties and Applications of Bioinspired Peptide-Based Nanomaterials. Chem Rev. 2017;117:12641-704
16. Eskandari S, Guerin T, Toth I, Stephenson RJ. Recent advances in self-assembled peptides: Implications for targeted drug delivery and vaccine engineering. Adv Drug Deliver Rev. 2017;110-111:169-87
17. Han K, Zhang J, Zhang W, Wang S, Xu L, Zhang C. et al. Tumor-Triggered Geometrical Shape Switch of Chimeric Peptide for Enhanced in Vivo Tumor Internalization and Photodynamic Therapy. ACS Nano. 2017;11:3178-88
18. Qian Y, Wang Y, Jia F, Wang Z, Yue C, Zhang W. et al. Tumor-microenvironment controlled nanomicelles with AIE property for boosting cancer therapy and apoptosis monitoring. Biomaterials. 2019;188:96-106
19. Yang Z, Song J, Tang W, Fan W, Dai Y, Shen Z. et al. Stimuli-Responsive Nanotheranostics for Real-Time Monitoring Drug Release by Photoacoustic Imaging. Theranostics. 2019;9:526-36
20. Hu X-X, He P-P, Qi G-B, Gao Y-J, Lin Y-X, Yang C. et al. Transformable Nanomaterials as an Artificial Extracellular Matrix for Inhibiting Tumor Invasion and Metastasis. ACS Nano. 2017;11:4086-96
21. Zhang C, Liu LH, Qiu WX, Zhang YH, Song W, Zhang L. et al. A Transformable Chimeric Peptide for Cell Encapsulation to Overcome Multidrug Resistance. Small. 2018;14:1703321
22. Wang Z, Wang W, Geng L, Hu Z. Distinguishing of tumor cell-targeting peptide ligands through a color-encoding microarray. Lab Chip. 2015;15:4512-6
23. Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD. et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032-8
24. Toft DJ, Moyer TJ, Standley SM, Ruff Y, Ugolkov A, Stupp SI. et al. Coassembled Cytotoxic and Pegylated Peptide Amphiphiles Form Filamentous Nanostructures with Potent Antitumor Activity in Models of Breast Cancer. ACS Nano. 2012;6:7956-65
25. Coin I, Beyermann M, Bienert M. Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nat Protocols. 2007;2:3247-56
26. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
27. Standley SM, Toft DJ, Cheng H, Soukasene S, Chen J, Raja SM. et al. Induction of Cancer Cell Death by Self-assembling Nanostructures Incorporating a Cytotoxic Peptide. Cancer Res. 2010;70:3020-6
28. Sato K, Ji W, Palmer LC, Weber B, Barz M, Stupp SI. Programmable Assembly of Peptide Amphiphile via Noncovalent-to-Covalent Bond Conversion. J Am Chem Soc. 2017;139:8995-9000
29. Moyer TJ, Finbloom JA, Chen F, Toft DJ, Cryns VL, Stupp SI. pH and Amphiphilic Structure Direct Supramolecular Behavior in Biofunctional Assemblies. J Am Chem Soc. 2014;136:14746-52
30. Wang J, Liu K, Xing R, Yan X. Peptide self-assembly: thermodynamics and kinetics. Chem Soc Rev. 2016;45:5589-604
31. Gilroy JB, Gädt T, Whittell GR, Chabanne L, Mitchels JM, Richardson RM. et al. Monodisperse cylindrical micelles by crystallization-driven living self-assembly. Nat Chem. 2010;2:566
32. Wei T, Liu J, Ma H, Cheng Q, Huang Y, Zhao J. et al. Functionalized nanoscale micelles improve drug delivery for cancer therapy in vitro and in vivo. Nano Lett. 2013;13:2528-34
33. Kwon HJ, Cha MY, Kim D, Kim DK, Soh M, Shin K. et al. Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimer's Disease. ACS Nano. 2016;10:2860-70
34. Mo R, Sun Q, Xue J, Li N, Li W, Zhang C. et al. Multistage pH-Responsive Liposomes for Mitochondrial-Targeted Anticancer Drug Delivery. Adv Mater. 2012;24:3659-65
35. Rao L, Bu L-L, Xu J-H, Cai B, Yu G-T, Yu X. et al. Red Blood Cell Membrane as a Biomimetic Nanocoating for Prolonged Circulation Time and Reduced Accelerated Blood Clearance. Small. 2015;11:6225-36
36. Chen W-H, Xu X-D, Luo G-F, Jia H-Z, Lei Q, Cheng S-X. et al. Dual-Targeting Pro-apoptotic Peptide for Programmed Cancer Cell Death via Specific Mitochondria Damage. Sci Rep. 2013;3:3468
37. Agemy L, Friedmann-Morvinski D, Kotamraju VR, Roth L, Sugahara KN, Girard OM. et al. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Natl Acad Sci U S A. 2011;108:17450-5
38. Hu X, Liu G, Li Y, Wang X, Liu S. Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals. J Am Chem Soc. 2015;137:362-8
39. Li H, Yu SS, Miteva M, Nelson CE, Werfel T, Giorgio TD. et al. Matrix Metalloproteinase Responsive, Proximity-activated Polymeric Nanoparticles for siRNA Delivery. Adv Funct Mater. 2013;23:3040-52
40. Yhee JY, Kim SA, Koo H, Son S, Ryu JH, Youn I-C. et al. Optical Imaging of Cancer-Related Proteases Using Near-Infrared Fluorescence Matrix Metalloproteinase-Sensitive and Cathepsin B-Sensitive Probes. Theranostics. 2012;2:179-89
41. Yu M, Zheng J. Clearance Pathways and Tumor Targeting of Imaging Nanoparticles. ACS Nano. 2015;9:6655-74
Author contact
![]() Corresponding author: Weizhi Wang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545752; Fax: +86-10-82545752; E-mail: wangwzcn
Corresponding author: Weizhi Wang, National Center for Nanoscience and Technology, Beijing 100190, China. Phone: +86-10-82545752; Fax: +86-10-82545752; E-mail: wangwzcn