Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental and Materials
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1714-1727. doi:10.7150/thno.30716 This issue Cite
Research Paper
Exosomes from M1-Polarized Macrophages Enhance Paclitaxel Antitumor Activity by Activating Macrophages-Mediated Inflammation
Piaopiao Wang1,2, Huihui Wang1,2, Qianqian Huang1,2, Can Peng1,2,3,4,5, Liang Yao1,2, Hong Chen1,2, Zhen Qiu1,2, Yifan Wu1,2, Lei Wang1,3,4 ![]() , Weidong Chen1,2,3,4,5
, Weidong Chen1,2,3,4,5 ![]()
1. School of Pharmacy, Anhui University of Chinese Medicine, Hefei, Anhui, 230012, China
2. Institute of Drug Metabolism, Anhui University of Chinese Medicine, Hefei, Anhui, 230012, China
3. Anhui Province Key Laboratory of Chinese Medicinal Formula, Hefei, Anhui, 230012, China
4. Institute of Pharmaceutics, Anhui Academy of Chinese Medicine, Hefei, Anhui, 230012, China
5. Synergetic Innovation Center of Anhui Authentic Chinese Medicine Quality Improvement, Hefei, Anhui, 230012, China
Received 2018-10-16; Accepted 2019-1-7; Published 2019-2-28
Abstract

Objective: Exosomes (Exos) are membrane-encased vesicles derived by nearly all cell types for intercellular communication and regulation. They also received attention for their use as natural therapeutic platforms and drug delivery system. Classically activated M1 macrophages suppress tumor growth by releasing pro-inflammatory factors. This study investigated the suitability of M1-exosomes (M1-Exos) as drug carrier and their effect on the NF-κB signal pathway and further detected whether macrophages repolarization can potentiate the antitumor activities of chemotherapeutics.
Methods: M1-Exos were isolated from M1-macrophages by ultracentrifugation and characterized by transmission electron, nanoparticle tracking analysis, dynamic light scattering and western blot. Then M1-Exos were used as Paclitaxel (PTX) carriers to prepare a nano-formulation (PTX- M1-Exos). A relatively simple slight sonication method was used to prepare the drug delivery system (PTX-M1-Exos). The cytotoxicity of PTX-M1-Exos on cancer cells was detected by MTT and flow cytometry in vitro. 4T1 tumor bearing mice were used to perform the therapeutic effect of PTX-M1-Exos in vivo.
Results: The expression of caspase-3 in breast cancer cells was increased when co-incubated with macrophages in the presence of M1-Exos in vitro. The production of pro-inflammatory cytokines was increased after exposure of macrophages in M1-Exos. M1-Exos provided a pro-inflammatory environment which enhanced the anti-tumor activity via caspase-3 mediated pathway. The treatment of M1-Exos to the tumor bearing mice exhibit anti-tumor effects in vivo. Meanwhile, the treatment of PTX-M1-Exos demonstrated higher anti-tumor effects than the M1-Exos or PTX group.
Conclusion: The results in our study indicate that the M1-Exos act as the carrier to deliver PTX into the tumor tissues, and also enhance the anti-tumor effects of chemotherapeutics in tumor bearing mice.
Keywords: anti-cancer, immunological-chemo therapy, M1-macrophage, M1- exosomes, drug delivery
Introduction
Exosomes (Exos) are 50-150 nm membrane-encased vesicles derived by virtually all cell types and identified in various body fluids [1, 2]. These small vesicles contain various functional molecules including nucleic acids, proteins, and cholesterol [3, 4]. In recent years, much attention had been attracted due to their intercellular roles in communication, signal transduction, diagnosis, regulation of immune response and drug delivery [5-7]. With these features, Exos have promoted many scholars to exploit the natural vesicles as drug carries. Moreover, Exos derived from antigen-presenting cells, which can in turn activate naïve immune cells. Dendritic cells (DCs) derived Exos promote Natural killer cells (NK) activation [8-10], induce T-cells proliferation and enhance CTL cytotoxicity [11, 12], inhibit tumor cells proliferation, and promote apoptosis [13, 14].
Exos carry RNA and proteins constituents from their parental cells, and have been implicated in signal transduction, tumor immune escape, diagnoses and treatment of some diseases [15, 16]. Macrophages are heterogeneous cells which could be phenotypically polarized in the tumor microenvironment to mount specific functional processes [17] and their phenotypes represent the two extremes of the broad range of macrophage functional states [18]. Macrophages, activated by LPS, can inhibit the growth of tumor by releasing some pro-inflammatory cytokines, inducing stromal destruction, and normalizing the tumor vasculature [19-21]. We posited that Exos derived from pro-inflammatory M1-macrophage cells may be used to potentiate the anti-tumor activities of chemotherapeutics. Previous studies have proved that Exos derived from M2-macrophages induce the migration, invasion, and metastasis of cancer cells [18, 22] and confer cisplatin resistance in gastric cancer cells [23]. However, Exos secreted from activated macrophages stimulate the release of cytokines (including TNF-α, IL-6, IL-12) which cause activation of naïve recipient immune cells [24]. Additionally, classically activated M1 macrophage secreted Exos propagate pro-inflammatory signals and create a local immune stimulatory microenvironment [25, 26]. The release of pro-inflammatory cytokines, such as TNF-α, induce the apoptosis of cancer cells in tumor tissue which is enhanced by CpG DNA treatment [27]. Therefore, this study focused on whether Exos secreted from pro-inflammatory macrophages could activate the NF-κB pathway of naïve immune cells, promote the release of cytokines to establish a local inflammatory environment, and thus potentiate the ability of chemotherapy drug to kill tumor cells.
Exos are promising candidate delivery vehicles as their endogenous characteristics make them to deliver cargo in a safe and effective manner [28]. Compared to micelles, liposomes and polymeric nanoparticles, Exos as a natural delivery system which can evade phagocytosis, extend blood half-life, and exhibit optimal biocompatibility without potential long-term safety issues [29-31]. Previous studies [32, 33] have proved natural non-modified Exos also possess intrinsic tumor targeting properties. Moreover, several clinical trials have confirmed that Exos isolated from the patient's own cells are less immunogenic [4, 34, 35]. On the contrary, the potential immunogenicity and toxicity are inevitable in the artificial delivery systems [31].
In this study, Exos were isolated from M1-macrophage cells media with ultracentrifugation, then we evaluated their potential to activate the NF-κB pathway and to create a pro-inflammatory environment. In addition, we investigated whether the pro-inflammatory environment could potentiate the anti-tumor effects of chemotherapeutics such as paclitaxel. Previous study had stated that Exos secreted from classically activated macrophages enhanced the anti-cancer efficiency of the cancer vaccine through establishing a pro-inflammatory microenvironment in the lymph nodes [36]. The results of our study showed that M1-Exos potentiated the anti-tumor efficacy of chemotherapy by activating NF-κB pathway and creating a pro-inflammatory environment.
Experimental and Materials
Reagents
IFN-γ, thioglycollate Broth (70157), PKH67 (MINI67, green) and DAPI (D9542) were obtained from Sigma (Sigma-Aldrich, USA). Alix (ab186429), Anti-iNOS (ab178945) and Anti-CD63 (ab216130) were obtained from Abcam (Cambridge, MA, UK). Anti-Arg-1 (93668T) and Anti-caspase-3 (#9662) were obtained from Cell Signaling Technology (Danville, USA). PTX was got from Meilun Biotechnology Co., Ltd (Dalian, China). TRITC Phalloidin was got from Solarbio (Beijing, China).
Animals and cell culture
Balb/c mice (4-6 weeks) were purchased from the Laboratory Animal Center of Anhui Medical University (Anhui, China). All animal experiments were approved by the Institutional Animal Care and Use Committee of Anhui University of Chinese Medicine.
Murine breast cancer cells 4T1 and the macrophage cells RAW264.7 were got from Cell Bank for Type Culture Collection of Chinese Academy of Science (Shanghai, China). The 4T1 cultured with RPMI-1640 medium and RAW264.7 cultured with Dulbecco's modified eagle's media (DMEM, Hyclone, UT, USA). They all supported with 10% fetal bovine serum, 1% penicillin-streptomycin at 5% CO2 under 37°C.
Preparation of M1 and M2-Exos from cells medium
Fetal bovine serum was centrifuged at 120,000 g (4°C) for 18 h to eliminate Exos [37-39]. To prepare M1 and M2-Exos, about 2.0×106 macrophage cells RAW264.7 were seeded in 100-mm culture dish. Then cells were cultured with IFN-γ (100 ng/mL) or IL-4 (40 ng/mL) for 24 h when they reached about 60% [36]. Thereafter, M1 and M2-Exos were isolated from the conditioned media as previously described [2, 40]. First, the debris were discard by centrifugation at 300 g (10 min), 2000 g (10 min) and 10,000 g (30 min) at 4 °C and filtered through a 0.22 μm filter (Millipore, Billerica, USA). Second, media were centrifuged at 120,000 g for 2 h by an L-90K ultracentrifuge with a type Ti41 rotor (Beckman Coulter, Brea, CA, USA). Then M1 and M2-Exos pellet were re-suspended in PBS and ultra-centrifuged again at 120,000 g for 2 h to wash away the contaminating protein.
Characterization of M1 and M2-Exos
To determine the presence of M1 and M2-Exos markers, the concentrations of M1 and M2-Exos were analyzed by a Micro BCA Protein Assay kit (Thermo, USA) and western blot was performed to detect TSG101, CD9, and Alix. Transmission electron microscopy (TEM, FEI Tecnai G2 spirit, Czech) was used to visualize and examine the morphology of M1 and M2-Exos. A drop of M1 or M2-Exos (20 μL) was pipetted into 200 mesh carbon-coated copper grids. The excess fluid was dried with filter paper for 1 min, and the samples were stained with 1% uranium acetate for 1 min. Samples were fluorescent-dried for 10 min and then examined by TEM. Dynamic light scattering (DLS; Nano-zs30, Malvern, UK) was employed to measure the size, intensity-weighted z-average diameter and zeta potential of M1 and M2-Exos. Nanoparticle tracking analysis (NTA, Malvern, UK) was used to estimate the number-weight diameter and particle concentration. All samples were measured as triplicates.
Exos internalization in vitro
The membrane intercalating dye PKH67 (Sigma- Aldrich, USA) was used to label Exos [41]. As is shown in manufacturer's protocol, PKH67 (1.0 μL) was diluted into 150 μL of diluent C (PKH67 solution), 151 μL PKH67 solution was co-cultured with the Exos (150 μL), then the mixed samples were incubated for 10 min at room temperature. The reaction was terminated with 10% Exos-free FBS. Afterwards, the Exos were centrifuged at 120,000 g for 90 min to discard unincorporated dye.
To investigate the Exos internalization in cells, 2×105 4T1 cells were seeded into 35 mm glass bottom dish (Corning Costar, Sigma-Aldrich, USA) and cultured for 24 h. PKH67-labeled Exos (green fluorescence, 20 μg/mL) were added to the culture media of 4T1 cells for 2 h, 4 h, and 8 h. Buffered paraformaldehyde (4%) was used to fix the cells before mounted by 0.2% Triton X-100. After cleaning the cells with PBS. The cell nucleus were stained by DAPI (blue fluorescence) and incubated with TRITC Phalloidin (TRITC Phalloidin is a dye used to label the cytoskeleton, red fluorescence, Solarbio, China) for 10 min. Fluorescence imaging was observed by confocal laser microscope (FV 1000, Olympus, Japan) and analyzed by flow cytometry using Backman FC500 instrument for the PKH67 fluorescence transfer.
Detection of the NF-κB pathway
To detect the signal pathways associated with the naïve macrophage polarization by M1-Exos, we used the M1-Exos to stimulate the naïve macrophages cells in vitro. Naïve macrophages were seeded in a 6-well plate containing Exos-free media. M1-Exos (40 ug/mL, 80 ug/mL), M2-Exos (80 ug/mL), IFN-γ (100 ng/mL) or IL-4 (40 ng/mL) was added into the naïve macrophage cells for 12 h in vitro. Thereafter, RNA was extracted by Trizol and cDNA was obtained via RNA transcription kit. The expressions of NF-κB and i-κB were performed by RT-PCR (ABI Step-one Plus, Foster City, CA, USA) via SYBR Green (Bimake. Shanghai, China) assay according to the protocol. The primers were shown in Table S1 (Table S1).
Transwell Assays
To evaluate whether M1-Exos could enhance the activity of caspase-3 by creating an inflammatory environment, breast cancer cells and naïve macrophage cells added M1 (20 μg/mL, 40 μg/mL) or M2-Exos (40 μg/mL) co-cultured in transwell systems with 4 μm pore-size for 12 h (6-well Coring, New York, NY, USA), which allowed free diffusion of molecules between the two chambers but not cell translocation.
RT-PCR Analysis
To determine inflammatory cytokines gene expression in vitro, macrophages were harvested from the transwell assays (upper chamber), and used for RNA extraction with the Trizol assay. Then the expressions of M1 (iNOS) marker and inflammatory cytokines (IL-4, IL-6, IL-10, IL-12 and TNF-α) were performed with RT-PCR.
ELISA assay
To further quantified the release of inflammatory cytokines (IL-4, IL-12 and TNF-α). The media from transwell assays (bottom) were collected and samples were assayed using mouse IL-4, 12 and TNF-α Elisa assay (R&D System, USA) as the manufacturer's protocol.
Caspase-3 activity assay
Moreover, the breast cancer cells were also collected from the transwell assay (bottom chamber) and the activity of caspase-3 was performed with caspase-3 activity kit (Beyotime, Shanghai, China). Caspase-3 activity was detected by a SpectraMax M2 microplate reader (Molecular Devices, USA) at 405 nm.
Preparation of PTX-loaded M1-Exos
PTX and M1-Exos were mixed in the ratio of 1:6 (m/m) and sonicated using Model 505 Sonic Dismembrator with .25'' tip with the following settings: 20% amplitude, 6 cycles of 30s on/off for 3 min with a 2 min cooling period between each cycle [42, 43]. In order to make the M1-Exos membrane recovery, the mixture was co-cultured at 37°C for 1 h. Then un-encapsulated PTX was removed by centrifugation at 1000 rpm for 10 min [44, 45], and the PTX-M1-Exos pellet was centrifuged at 120,000 g. HPLC was used to measure the amount of loaded PTX in each group. PTX was quantified by a Waters 1290 HPLC systems on a C18 column.
MTT Assay
4T1 cells were seeded in 96 multi-well plates (Corning Costar, USA) at a density of 4000 cells per well in the culture medium. The different concentrations (at a PTX dose of 0, 5, 7.5, 15, 25, 30 and 40 μg/mL) of PTX, M1-Exos and PTX-M1-Exos were used to treated 4T1 cells. After 72 h, 20 μL MTT solutions (5 mg/mL) were added to each well and incubated for 4 h at 37°C, and the formazan precipitate was dissolved in DMSO. The multi-well plates were shaken and analyzed by spectrophotometry at 570 nm. The IC50 was calculated with GraphPad Prism 7.0.
Apoptosis assay
According to the manufacturer, Annexin V-FITC/PI Apoptosis Detection Kit (Mountain View, CA, USA) was used to detect the apoptosis of the tumor cells. Briefly, cells were treated and collected after being washed with PBS. Subsequently, they were re-suspended in 500 μL of binding buffer and incubated in darkness with 5 μL of Annexin V-FITC for 5 min and 5 μL of PI (propidium iodide) for another 5 min at room temperature. Finally they were evaluated by flow cytometry (Beckman Coulter FC 500, Brea, CA, USA).
Establishment of xenograft tumors
4T1 cells were harvested and suspended in medium and then 1×106 4T1 cells were inoculated into the flanks of each female BALB/c mice (about 20 g). The tumor volume was assessed by measuring the size of the xenografts every three days, and the tumor volume was calculated as V= π× width × length × height/6. To investigate the antitumor effects in vivo, once the tumor volume reached approximately 50 mm3, the mice were divided into four groups randomly (each group n=10). Thereafter, the mice were intravenously injected with PBS, PTX, M1-Exos, and PTX-M1-Exos, (at a PTX dose of 5 mg/kg) and treated every 3 days.
TUNEL staining and frozen section experiments
Apoptotic cells in tumor tissues were detected by TUNEL assay according to the standard procedure. After fixed in 4% paraformaldehyde, the tissues were stained by 50 μL TUNEL reaction mixture (Roche) for 60 min at 37°C. The cell nucleus was stained with DAPI and observed through the Olympus microscope (Olympus FV1000, Japan). For frozen sections experiments, PTX-M1-Exos (5 mg/kg of PTX concentration) were labeled with PKH67 reagent and injected to the tumor-bearing mice (i.v.). Two hours later, tissues were obtained for frozen sections experiments to measure the fluorescence intensity.
H&E staining
For H&E staining, after the mice were euthanized, tumor tissues were collected and photographed. Tissue samples were fixed in 4% formaldehyde for 24 h, and paraffin-embedded. It was sectioned (5μm sections) and stained with hematoxylin and eosin (H&E). An optical microscope was performed to analyze these slides.
Western blot
Tumor tissues were lysed with RIPA buffer which contained proteinase and phosphatase inhibitor cocktail. After centrifugation at 12,000 rpm for 30 min (4°C), the BCA assay was used to measure the protein concentration of the tumor lysates. Protein samples were separated on SDS-polyacrylamide gels and transferred onto PVDF membrane (Millipore, Bedford, MA, USA). Membranes were blocked and incubated with the anti-caspase-3 antibody overnight at 4℃. HRP-conjugated secondary antibody was used to detect the bound primary antibodies. The signals were detected by the chemiluminescence kit (Thermo, USA). The data were quantified by Image J software.
Statistical analysis
The data were expressed as mean ± SD. The significance of the difference between two groups was tested by Student's t-test. All analyses were performed with GraphPad Prism 7.0 (GraphPad Software Inc, San Diego, CA, USA). Differences were considered significant at *P<0.05, **P<0.01, *** P<0.001.
Results
Isolation, characterization and uptake of Exos derived from Macrophages
To obtain M1-Exos and M2-Exos, macrophages were treated with IFN-γ (100 ng/mL) or IL-4 (40 ng/mL) for 24h which could induce the polarization of macrophages into M1 or M2 type. The biomarkers of M1 and M2 macrophages were detected by the expression of iNOS and Arg-1(Figure 1A), after which the M1-Exos and M2-Exos were isolated from the conditioned media with ultracentrifugation. The isolation method of Exos was based on differential centrifugation as in previous studies [43, 46]. The surface markers of M1 and M2-Exos were detected as shown in Figure 1B. TEM (Figure 1C-D) was used to reveal the morphology of Exos. The TEM images showed that the Exos were well dispersed, generally spherical nanoparticles. The size was detected by DLS (Figure 1C-D).
The characterization and uptake of Exos. (A) M1-macrophage marker iNOS and M2-macrophage maker Arg-1 were analyzed by western blot. (B) M1 and M2-Exos markers Alix, TSG101, and CD9 were detected by western blot. (C-D) Morphological characterization was detected by transmission electron microscope (scale bar corresponds to 200 nm) and the size distribution was measured using dynamic light scattering. (E) Laser scanning confocal microscope recorded the uptake of PKH67-labeled Exos (green fluorescence) by 4T1 cells (blue fluorescence). (n=3, p<0.01 vs. 2 h). The data represent mean ± SD. (The Scale bar at 200 x is 50 μm, and the Scale bar at 600 x is 10 μm)
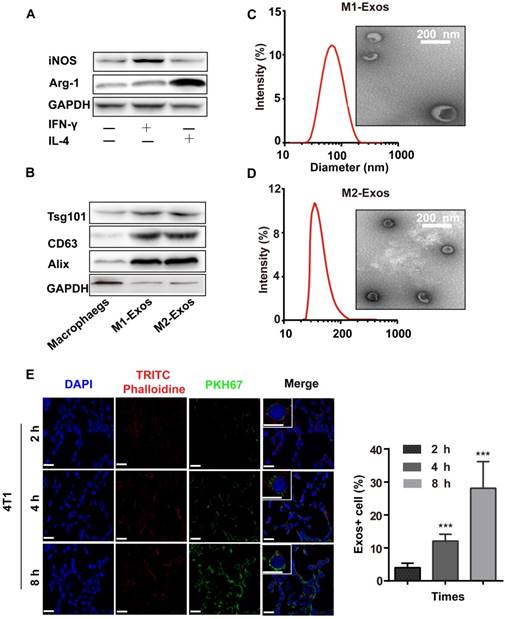
To confirm whether Exos could be taken up by cells, the purified Exos were labeled with PKH67 (green fluorescence), which was a fluorescent membrane dye. Thereafter, the labled-M1-Exos were centrifuged for 2 h to remove free dye. After incubation with PKH67-labeled Exos (2 h, 6 h and 8 h), a punctuated pattern of fluorescence was observed in 4T1 cells, with increasing signal over time. This implied that the M1-Exos internalization was time-dependent [2, 46] (Figure 1E and 1F). Moreover, the results of flow cytometry (Figure S1) further confirmed this conclusion.
M1-Exos activated the NF-κB pathway of naïve macrophages and promoted the release of cytokines to establish a local inflammatory environment
To detect the signaling pathway associated with the macrophage polarization by M1-Exos, we used the M1-Exos obtained from the IFN-γ-activated macrophages, to stimulate the macrophage cells in vitro. As shown in Figure 2A, M1-Exos can activated NF-κB pathway with dose-dependent manner. Two concentrations of M1-Exos (20 μg/mL and 40μg/mL) were determined based on exosomal protein concentration. After a 12 h incubation, RNA was extracted. Then RT-PCR was used to detect the expression of NF-κB and i-κB at mRNA level. The results showed that M1-Exos (40 ug/mL) increased the expression of NF-κB and i-κB compared with the control group. Previous study [25] had shown that Exos from LPS-stimulated macrophages induce the activation of NF-κB with dose-dependent way. Similarly, the results of our study revealed the Exos secreted from IFN-γ-stimulated macrophage induced the activation of NF-κB pathway.
M1-Exos caused activation of NF-κB signal pathway, induction of cytokines expression in naïve macrophages, and enhanced the activities of caspase-3 in cancer cells. (A) The miRNA expressions level of NF-κB and i-κB were quantified by RT-PCR. (B) M1-macrophaegs maker and pro-inflammatory cytokines were detected at mRNA level by RT-PCR. (C) The expressions of pro-inflammatory cytokines in the co-cultured system were detected by ELISA. (D) The expression of caspase-3 was determined in vitro. All data were representative of at least three (n=3) independent experiments for each experimental group and was analyzed by Student's test. (n=3; *P<0.05 **P<0.01, *** P<0.001vs. the control group).
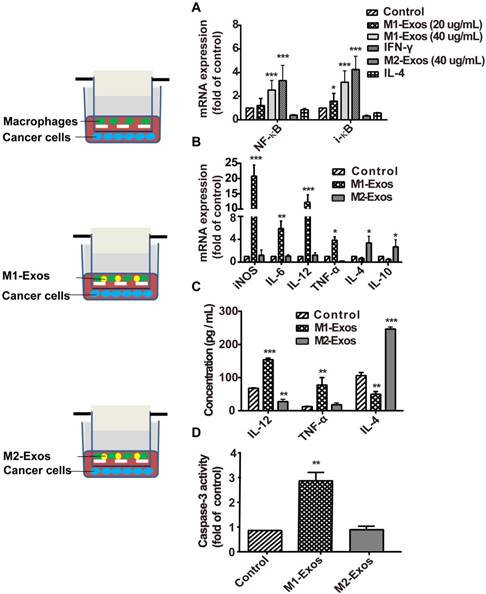
Moreover, it has been reported that Exos, isolated from IFN-γ-stimulated macrophages, which can potentiate the antitumor activity of cancer vaccine by creating a pro-inflammatory microenvironment in the lymph node [36]. To investigate whether the M1-Exos induce the release of inflammatory cytokines and enhance the therapeutic effect of chemotherapeutics, we measured the level of apoptosis of 4T1 cancer cells in co-cultured with macrophages, in presence of M1-Exos or M2-Exos. First, macrophages were isolated from the above mentioned co-cultures and RT- PCR was performed to detect the expression profiles of inflammatory cytokines. The analysis showed (Figure 2B) that M1-Exos up-regulated the expression of inflammatory cytokines iNOS, IL-6, and IL-12 and down-regulated the expression levels of IL-4 and IL-10. However, M2-Exos down-regulated the expression levels of IL6, TNF-α, IL-12 and up-regulated the expression of IL4 and IL-10 M2 phenotypes of macrophages. Similarly, the significantly increasing in the production of inflammatory cytokines (IL-4 and TNF-α) were detected by ELISA when macrophages co-cultured with M1-Exos. However, the M2-Exos treated groups showed opposite results (Figure 2C). The cytokines expression indicated that naïve macrophages can be induced into a pro-inflammatory form to produce more Th1 cytokines.
Second, cancer cells apoptosis was detected with caspase-3 activity assay kit (Figure 2D). We found that the activity of caspase-3 was significantly increased in the cancer cells incubated with macrophages together with M1-Exos compared with cancer cells co-cultured with either macrophages or M2-Exos (P<0.05; Figure 2D).
The formulation of PTX-loaded M1-Exos and that anti-tumor effects in vitro
As Exos from M1-polarized macrophages increased the activities of caspase-3 in cancer cells by creating a pro-inflammatory, we selected it as the carrier for deliver PTX to tumor tissues. Also, TEM (Figure 1C and Figure 3 A and B) was used to reveal the morphology of naïve M1-Exos, sonicated M1-Exos and PTX-M1-Exos, and sonication (Figure 3A) did not affect the morphology of M1-Exos [42, 43]. The size was detected by DLS (Figure 3A) and NTA (Figure S 2A-C). The mean size of naïve M1-Exos, sonicated M1-Exos, and PTX-M1- Exos was 75.3 nm, 142.9 nm, and 172.8 nm, respectively. Naïve M1-Exos, sonicated M1-Exos and PTX-M1-Exos were negatively charged in NaCl solution (Figure 3A).
The formulation of PTX-loaded M1-Exos and that anti-tumor effects in vitro. (A)The morphological, size, protein marker and zeta potential of sonicated-M1-Exos and PTX-M1-Exos. (B) Cell apoptosis was detected by flow cytometry at 24 h in each group, and (C) Statistical analysis of data. The data shown are mean ± SD (n=3). (D) 4T1 cells were incubated with medium (Control), M1-Exos, free PTX, and PTX-M1-Exos for 24 h at different concentrations of PTX and cell viability was assessed using the MTT assay. PTX-M1-Exos (IC50, 4.30 μg/mL) showed higher cytotoxicity compared to free PTX (IC50, 17.49 μg/mL). (n=6; *P<0.05 **P<0.01, *** P<0.001vs. the control group).
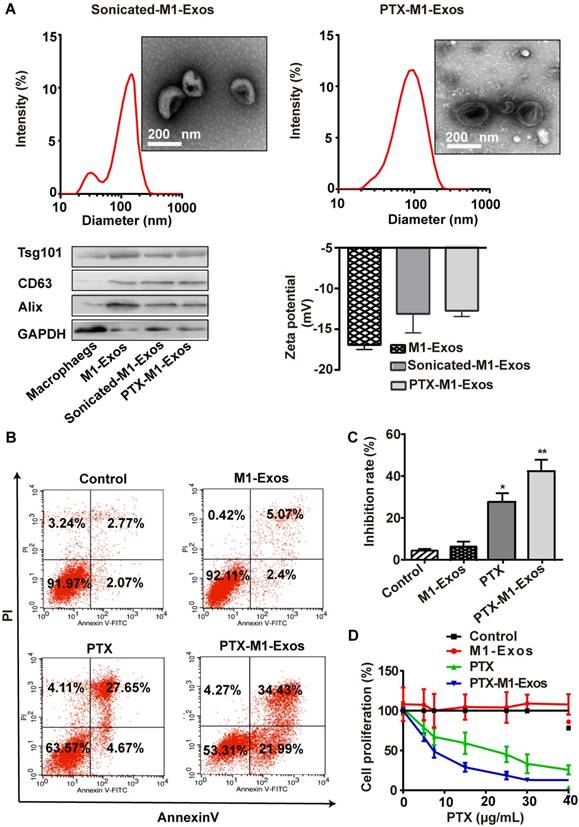
Compared with naïve M1-Exos (75.3 nm), the size of PTX-M1-Exos (172.8 nm) was slightly increased. The change of the size could be attributed partly to the incorporation of PTX into the lipid bilayer of Exos, the surface adsorption due to hydrophobic interaction. Previous studies also found that the size of Exos was increased after drug loading into Exos [47-49]. The morphology and surface markers protein of PTX-M1-Exos were no altered (Figure 3A), which further revealed the suitability of the loading conditions.
The loading capacity of PTX-M1-Exos formulation was analyzed by HPLC (Figure S3). The concentration of PTX in the M1-Exos was obtained from the standard curve generated. The average PTX loading efficiency after sonication was 19.55±2.48%. But the mean loading efficiency after simple mixing was 4.85±1.65%. During sonication, researchers found a formation of transient pores or even reformation of Exos upon sonication allowed diffusion of some substances from the surrounding media into Exos [50]. Moreover, sonication does not affect the membrane-bound proteins significantly (Figure 3A) [42, 43].
We further detected the capacity of PTX-M1-Exos to influence cancer cells survival rate. MDA-MB-231, MCF-7, 4T1, A549, Hep G2 and Hela cells were incubated with media, free-M1 or M2-Exos, free PTX, PTX-M1-Exos and PTX-M2-Exos for 24 h followed by measurement of cell viability by MTT assay. PTX-M1-Exos inhibited cell proliferation with an efficiency way compared with the free PTX group (Figure S4) (Figure S4), however, no significant inhibition effects were observed in the blank-M1-Exos treated group. M2-exosomes promote cancer cells proliferation compared with control, however, the inhibitory effects of PTX-M2-Exos were equal to those of PTX (Figure S5). To further detect the pharmacological impact of PTX encapsulated into M1-Exos, the flow cytometry was used to detect the apoptosis of tumor cells with the treatment of PTX-M1-Exos (Figure 3B, 3C and S6).
Next, the anti-proliferative effects of PTX and PTX-M1-Exos on 4T1 cells were determined at different concentrations of PTX (at a PTX dose of 0, 10, 20, 30 and 40 μg/mL) (Figure 3D). Compared with the free PTX (IC50 17.49 μg/mL), the PTX-M1-Exos (IC50 4.30 μg/mL) had better anti-proliferative effects on 4T1 cells (Figure 3D). PTX-M1-Exos also exhibited dose-dependent anti-proliferative effects compared to PTX alone (Figure 3D). Following the treatment with PTX and PTX-M1-Exos, the percentage of apoptotic cells was increased compared with control group. Indeed, previous study [51] had demonstrated that Exos consisted of phospholipid bilayers, which may directly fuse the target cell plasma membrane leading to increase the cellular internalization of the encapsulated drugs. These data revealed that M2-Exos could promote the activities of cancer cell, however, M1-Exos itself does not induce apoptosis in cancer cells, which further confirm the superiority of M1-Exos as a drug carrier.
M1-Exos enhance efficacy of PTX In vivo
Encouraged by the above results, we then determined the antitumor effects of PTX-M1-Exos treatment in vivo. 4T1 cells were injected subcutaneously in the flanks of female Balb/c mice to establish the tumor model as in a previous study [52]. The tumor-bearing mice were divided into four groups randomly when the tumor volume about 50 mm3 (each group n=10) and then intravenously injected with therapeutic agents. Then, the anti-tumor activities of PTX-M1-Exos in 4T1 tumor bearing mice were detected. Firstly, tumor volume was measured every 3 days (Figure 4A). In comparison with the PBS groups, tumor volumes were increased gradually (p<0.05) following the treatment with M1-Exos and PTX for 27 days. Interestingly, the significant anti-tumor effects (p<0.001) were observed in the PTX-M1-Exos treated group at 27 days (Figure 4B). Secondly, compared with the PBS and PTX groups, animals in M1-Exos and PTX-M1-Exos groups did not show obvious weight loss during the study period (Figure 4C). The survival rate of mice that treated with PTX-M1-Exos was increased compared with the control (Figure 4D). Taken together, these datasets indicated a strong anti-tumor effect in the PTX-M1-Exos groups.
Detection of anti-tumor effects of M1-Exos, PTX and PTX-M1-Exos in tumor-bearing mice. (A) Representative images of the tumor tissues in each group. (B) Tumor volumes in each group were detected. (n=10 in each group) (C) The survival rates in each group. (D) Body weight changes in each group. (E) Western blot analysis of apoptosis-related protein caspase-3 in tumor tissues. (F) The expression level of caspase-3/β-actin. (G) Representative TUNEL staining for apoptotic cells in the xenograft tumor tissues, the green fluorescence represents apoptotic cells. (H) The quantification of the apoptosis in the tumor tissues. (n=6 in each group). Values are expressed as mean ± SD (n=6; *P<0.05, **P<0.01, ***P<0.001vs. the control group. Scale bar=100 μm).
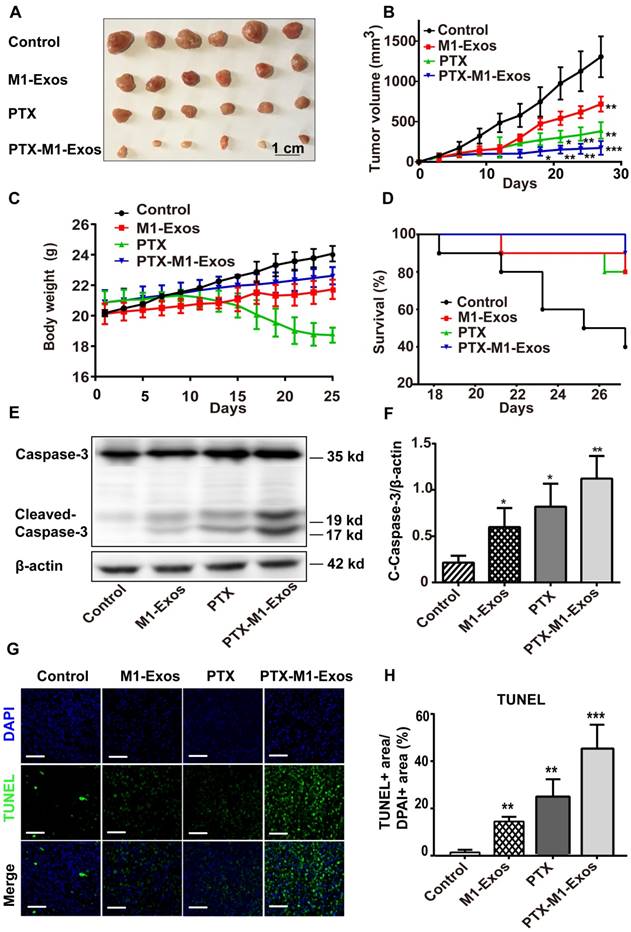
In order to confirm the effect of PTX-M1-Exos on tumor cells further, the apoptosis related proteins were detected via western blot. Thus, we evaluated the expression level of c-caspase3 in the tumor tissues. Compared with M1-Exos and free PTX treated groups, PTX-M1-Exos treated groups showed a significant inhibitory effect on apoptosis (Figure 4E). The expression of caspase-3 protein increased in M1-Exos, PTX and PTX-M1-Exos treated group (5.2 fold, 3.8 fold, and 2.8 fold, respectively), compared with the control group (Figure 4F).
Meanwhile, the apoptosis of tumor tissues were detected by the TUNEL (terminal deoxynucleotidyl transferase UTP nick end labeling) assay. The results (Figure 4G and 4H) showed that much more positive expression of TUNEL was found in the PTX-M1-Exos treated group, which further confirmed that the PTX-loaded into M1-Exos suppressed tumor growth.
The M1-Exos co-localization in vivo
Exos are small vesicles present in many tissues in vivo. To detect the distribution of Exos in vivo, frozen section experiments were carried out on the tissues. The representative fluorescence of the stained tissues was observed with a fluorescence microscope (Figure 5A and 5B). The frozen sections and tumor tissues from liver, spleen and lung showed strong green fluorescence compared with control group. This implied that the DIO-labeled Exos remained in the lung, liver and spleen. Most importantly, the DIO-labeled M1-Exos infiltrated into the tumor tissues which indicated that M1-Exos stimulated the macrophage in tumor tissues.
The distribution of Exos in vivo. (A) Representative fluorescence images showing bio-distributions of M1-Exos in tumor tissues and organs (heart, spleen, liver, lung, and kidney) 2 h after injection (i.v.) of Dio-labeled Exos (50 μg) into 4T1 tumor-bearing-mice and (B) calculation of accumulative fluorescence signals in major organs and tumor tissue. (n=3 per group; *P<0.05, **P<0.01, ***P<0.001vs. the control group, Student's-test). (Scale bar=100 μm).
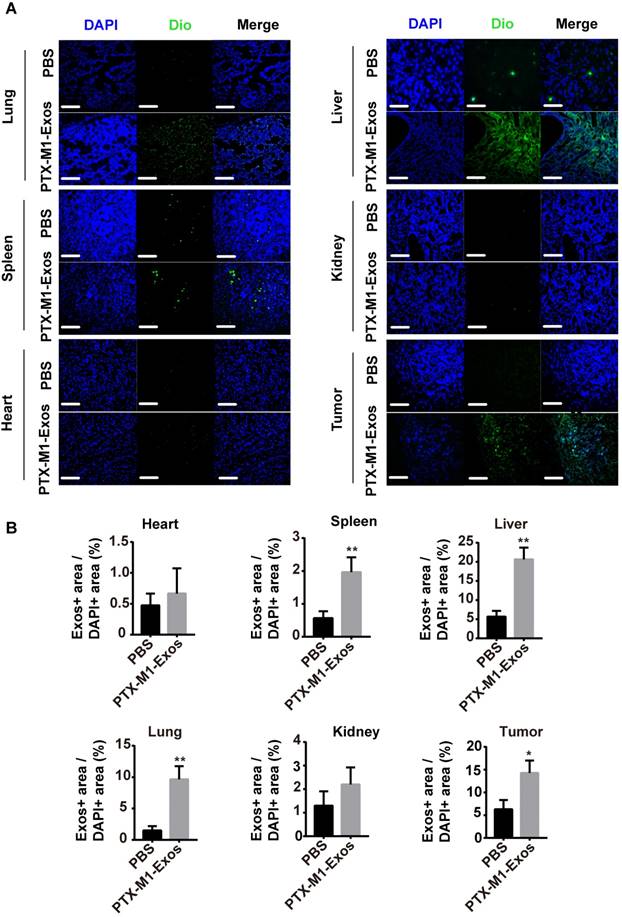
Hence, the distribution of Exos may be important in the treatment of cancer. M1-Exos stimulated the macrophage polarization in vivo. Since some macrophage are present in the spleen, M1-Exos may stimulate these macrophages in spleen in vivo. Previously study [53] had reported, tumor tissues have been shown to possess macrophages and M1-Exos can reverted the M2-type macrophages into M1-type macrophages in tumor tissues.
H&E staining
Chemotherapy is often associated with weight loss, which is a common side effect in treated mice [54]. The potential toxicity of different formulations was assessed in Balb/c mice. Compared with the PBS treated groups, mice in M1-Exos and PTX-M1-Exos groups did not show obvious weight loss (Figure 4C), indicating that there was no overt toxicity caused by the treatments. Zrrvogel found that Exos derived from DCs can eradicate tumors in a murine model [55]. Interestingly, DC-derived Exos have passed Phase I and Phase II in unresectable clinical trials for non-small-cell lung carcinoma [56, 57]. In this study, the pathological changes of various organs were evaluated by H&E staining assay. As shown in Figure 6, compared with control, M1-Exos and PTX-M1-Exos did not induce cell degeneration or necrosis in major organs, suggesting a reasonable safety margin in their application.
Histological examination by staining of major organs. H&E was used to stain the tissues obtained from major organs. The representative specimens were examined at 20x magnification. No noticeable abnormality was found in the heart, liver, spleen, lung, or kidney. (Scale bar=100 μm).
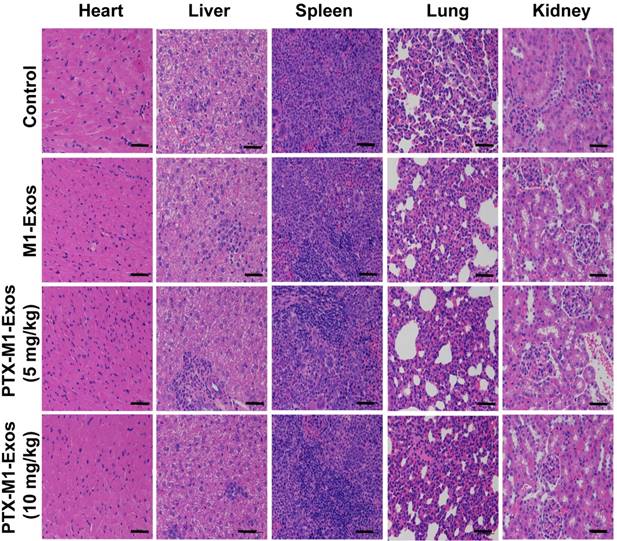
The nanoparticle PTX-M1-Exos showed high anti-tumor effect with low systemic toxicity. This is PTX chemotherapy and the biological therapy provided an excellent complementary treatment. In addition, the therapeutic effect of PTX may influence the mitosis of the nucleus. M1-Exos are endogenous substances hence are more effective delivering PTX into cells. Overall, the multifunctional nano-platform has demonstrated a high potential for application in a chemo/-biological combination therapy.
Discussion
Macrophages are the primary cellular component in the innate immune system which play key functions including regulation of inflammation, phagocytosis, tissue remodeling, metabolism and proliferation [58]. It is well known that nanoparticles are internalized by tumor-associated macrophages (TAMs) in the tumor microenvironment [59-63]. Besides, preclinical study had demonstrated the therapeutic and bioactivity of macrophages [64]. However, the in vivo effects of two forms of macrophages M1-type and M2-type are two-faced. The M1-type macrophage could secrete pro-inflammatory and exhibit anti-tumor effects, while the M2-type macrophage enhances tumor growth and metastasis. In tumor-bearing mice, the M1-type macrophage exhibited anti-tumor effects via regulating the tumor microenvironment. IFN-γ can induce naïve macrophage into the pro-inflammatory and cytotoxic M1 phenotype which activates anti-tumor immunity [65]. Activated M1-macrophages secrete various inflammatory cytokines (such as IL-1β, TNF-γ, and IL-6), thereby triggering resistance to intracellular parasites and tumor [66, 67]. Especially, previous study indicates that differential modulation of the chemokine system integrates polarized macrophages in pathways responsible for resistance to tumors, immune regulation, tissue repair and remodeling [68]. The Exos possess some characteristics of their parent cells and have attracted much attention with regard to their roles in intercellular communication and signal transduction [5, 6]. Previous studies have revealed that stem cells could be induced into specific lineage by Exos which isolated from differentiated cells [69]. In this study, the Exos were secreted from IFN-γ induced macrophages. Our results shown that the NF-κB pathway was activated by M1-Exos, which was consistent with the results of a previous study that LPS-induced macrophage derived Exos activated the NF-κB pathway [25]. Then, we co-cultured cancer cells with naïve macrophages in the presence of M1-Exos. The mRNA expression of M1-macrophages marker and the pro-inflammatory cytokines was significantly increased [36]. Furthermore, the activity of caspase-3 in cancer cells was significantly increased.
Although PTX represents an important class of antitumor agents and plays an important role in the treatment of various malignant tumors, such as human breast cancer [48], the dose-dependent toxic effect of PTX that has hampered the use of PTX in clinic. Recently, as natural nano-sized drug carriers, Exos have been used to deliver chemotherapy drugs to specific tissues or cell types in vivo [2], especially in the tumor tissues [34]. In this study, M1-Exos were employed to deliver the chemotherapeutic drug PTX to tumor tissues in mice. In this way, they not only acted as carriers but also activated the NF-κB pathway, thereby, potentiating the antitumor effects of drug. Drug loading was accomplished by mixing a PTX solution with the Exos as described previously for other various agents [46, 48]. The size of PTX-M1-Exos (172.8 nm) was modestly increased compared to M1-Exos (75.3 nm). Evidence suggests that may be PTX is partially embedded in the bilayer of phospholipids [48]. The morphology of Exos did not change after PTX loading into the naïve Exos which further confirms the suitability of the loading conditions. A modest decreasing in zeta potential was observed from -17 mV to -12 mV after the loading of PTX which is consistent with the results of a previous study [48].
In vitro, the PTX-M1-Exos co-cultured with MDA-MB-231, MCF-7, 4T1, Hela, Hep G2 and A549 cancer cells showed higher cell cytotoxicity compared with free PTX. Additionally, M1-Exos alone showed no toxicity to these cancer cells which is in line with the findings of another study [53]. In vivo, the 4T1 tumor bearing mice model were established to explore the therapeutic efficacy of PTX-M1-Exos administered through tail intravenous injection. PTX-M1-Exos formulation was tested at a dose of 5 mg/kg based on a previous study [70]. At this dose, the tumor growth was markedly inhibited (p<0.001). The inhibitory effects of M1-Exos alone were almost equal to those of PTX and they were statistically significant (p<0.01). Significant inhibition was produced by M1-Exos alone according to the results of in vivo experiment (Figure 4), and this was attributed to the classically activated M1 macrophages. These results indicate that the M1-Exos not only delivered a higher amount of drug to the tumor sites compared with the free PTX [48], but also inhibited tumor growth by activating macrophages. Our results are consistent with previous study [36], which Exos from IFN-γ-induced macrophages potentiated the effects of a cancer vaccine by creating a pro-inflammatory microenvironment.
For PTX conventional therapy, dose-dependent toxic effects are inevitable. Here, we investigated major organ toxicity induced by PTX-M1-Exos. Treatment with M1-Exos and paclitaxel (5 mg/kg) separately showed no overt toxicity in major organs. Moreover, PTX-M1-Exos, even at a two-fold higher dose (10 mg/kg) compared with the therapeutic dose, did not show any overt organic toxicity.
Systemic administration application of the PTX delivery system suppressed tumor growth significantly, and no obvious toxicity was observed via H&E staining. Our results and other studies [24, 58, 59] show that Exos are superior carriers for specific delivery of therapeutic drugs to tumors. Recently, Choo et al. [53], found that M1-macrophage secreted Exos-mimetic nano-vesicles will caused repolarize repolarization of M2 tumor-associated macrophages (TAM) to M1-macrophages. The polarized M1-macrophages could release pro-inflammatory cytokines which induce antitumor immune response. However, the potential influence of macrophages secreted endogenous nucleic acids and proteins among other components within Exos on the immune system needs to be further investigated and the possibility of clinical use of Exos requires more experimental support.
Conclusions
This work describes a standard and rapid method of isolating Exos from activated M1-type macrophages by ultracentrifugation and their application in the delivery of drugs to tumor cells. When breast cancer cells co-cultured with macrophages in the presence of M1-Exos, the expression level of caspase-3 and pro-inflammatory Th1-type cytokine were increased. The results showed that the anti-tumor activity of M1-Exos was due to the polarization of macrophages and release of pro-inflammatory cytokines. The anti-tumor effects of PTX was significantly improved when PTX was loaded into M1-Exos. Furthermore, M1-Exos-based chemotherapy enhanced the anti-tumor effects in tumor-bearing mice via regulating apoptosis in vivo. Thus, M1-nacrophages derived Exos may be an effective approach to treat cancer cells in the future.
Abbreviations
Exos: exosomes; M1-Exos: M1-exosomes; M2-Exos: M2-exosomes; PTX: paclitaxel; DCs: Dendritic cells; NK cells: Natural killer cells; CTL cytotoxicity: cytotoxicity T lymphocyte; FBS: fetal bovine serum; TEM: transmission electron microscopy; DLS: dynamic light scattering; NTA: nanoparticle tracking analysis; HPLC: high performance liquid chromatography; MTT: 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl- 2-H-tetrazolium bromide, thiazolyl blue tetrazolium bromide; TUNEL: terminal deoxynucleotidyl transferase UTP nick end labeling; H&E: hematoxylin-eosin staining; TAMs: tumor associates macrophages.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by Natural Science Foundation of Anhui University of Chinese Medicine (2018zrzd04) and National Natural Science Foundation of China (81773988). We show our appreciation to Guoqi Zhu for his help in data analysis.
Author contributions
Piaopiao Wang and Lei Wang designed the study. Piaopiao Wang performed most experimental work and data analyses. Huihui Wang and Zhen Qiu contributed animal model. Liang Yao provided assistance. Weidong Chen edited the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kourembanas S. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol. 2015;77:13-27
2. Yuan D, Zhao Y, Banks WA, Bullock KM, Haney M, Batrakova E. et al. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials. 2017;142:1-12
3. Lunavat TR, Jang SC, Nilsson L, Park HT, Repiska G, Lasser C. et al. RNAi delivery by exosome-mimetic nanovesicles - Implications for targeting c-Myc in cancer. Biomaterials. 2016;102:231-238
4. Gyorgy B, Hung ME, Breakefield XO, Leonard JN. Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu Rev Pharmacol Toxicol. 2015;55:439-464
5. Simons M, Raposo G. Exosomes-vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21:575-581
6. Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581-593
7. Zhou R, Chen KK, Zhang J, Xiao B, Huang Z, Ju C. et al. The decade of exosomal long RNA species: an emerging cancer antagonist. Mol Cancer. 2018;17:75
8. Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S. et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One. 2009;4:e4942
9. Sobo-Vujanovic A, Munich S, Vujanovic NL. Dendritic-cell exosomes cross-present Toll-like receptor-ligands and activate bystander dendritic cells. Cell Immunol. 2014;289:119-127
10. Simhadri VR, Reiners KS, Hansen HP, Topolar D, Simhadri VL, Nohroudi K. et al. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS One. 2008;3:e3377
11. Guan S, Li Q, Liu P, Xuan X, Du Y. Umbilical cord blood-derived dendritic cells loaded with BGC823 tumor antigens and DC-derived exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumor immunity in vitro and in vivo. Cent Eur J Immunol. 2014;39:142-151
12. Xie Y, Wu J, Xu A, Ahmeqd S, Sami A, Chibbar R. et al. Heterologous human/rat HER2-specific exosome-targeted T cell vaccine stimulates potent humoral and CTL responses leading to enhanced circumvention of HER2 tolerance in double transgenic HLA-A2/HER2 mice. Vaccine. 2018;36:1414-1422
13. Wang J, Wang Z, Mo Y, Zeng Z, Wei P, Li T. Effect of hyperthermic CO2-treated dendritic cell-derived exosomes on the human gastric cancer AGS cell line. Oncol Lett. 2015;10:71-76
14. Guo F, Chang CK, Fan HH, Nie XX, Ren YN, Liu YY. et al. Anti-tumour effects of exosomes in combination with cyclophosphamide and polyinosinic-polycytidylic acid. J Int Med Res. 2008;36:1342-1353
15. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G. et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883-891
16. Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P. et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002;195:1303-1316
17. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453-461
18. Yang M, Chen J, Su F, Yu B, Su F, Lin L. et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer. 2011;10:117
19. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014;6:1670-1690
20. Mills CD, Lenz LL, Harris RA. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016;76:513-516
21. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309-322
22. Zheng P, Luo Q, Wang W, Li J, Wang T, Wang P. et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein E. Cell Death Dis. 2018;9:434
23. Zheng P, Chen L, Yuan X, Luo Q, Liu Y, Xie G. et al. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J Exp Clin Cancer Res. 2017;36:53
24. Reales-Calderon JA, Vaz C, Monteoliva L, Molero G, Gil C. Candida albicans Modifies the Protein Composition and Size Distribution of THP-1 Macrophage-Derived Extracellular Vesicles. J Proteome Res. 2017;16:87-105
25. McDonald MK, Tian Y, Qureshi RA, Gormley M, Ertel A, Gao R. et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain. 2014;155:1527-1539
26. Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K. et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood. 2013;121:984-995
27. Morishita M, Takahashi Y, Matsumoto A, Nishikawa M, Takakura Y. Exosome-based tumor antigens-adjuvant co-delivery utilizing genetically engineered tumor cell-derived exosomes with immunostimulatory CpG DNA. Biomaterials. 2016;111:55-65
28. Kooijmans SA, Vader P, van Dommelen SM, van Solinge WW, Schiffelers RM. Exosome mimetics: a novel class of drug delivery systems. Int J Nanomedicine. 2012;7:1525-1541
29. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N. et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21:185-191
30. Ha D, Yang N, Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B. 2016;6:287-296
31. Turturici G, Tinnirello R, Sconzo G, Geraci F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Physiol Cell Physiol. 2014;306:C621-C633
32. Chen G, Zhu JY, Zhang ZL, Zhang W, Ren JG, Wu M. et al. Transformation of cell-derived microparticles into quantum-dot-labeled nanovectors for antitumor siRNA delivery. Angew Chem Int Ed Engl. 2015;54:1036-1040
33. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123-134
34. Zhang W, Yu ZL, Wu M, Ren JG, Xia HF, Sa GL. et al. Magnetic and Folate Functionalization Enables Rapid Isolation and Enhanced Tumor-Targeting of Cell-Derived Microvesicles. ACS Nano. 2017;11:277-290
35. Escudier B, Dorval T, Chaput N, Andre F, Caby MP, Novault S. et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005;3:10
36. Cheng L, Wang Y, Huang L. Exosomes from M1-Polarized Macrophages Potentiate the Cancer Vaccine by Creating a Pro-inflammatory Microenvironment in the Lymph Node. Mol Ther. 2017;25:1665-1675
37. Eitan E, Zhang S, Witwer KW, Mattson MP. Extracellular vesicle-depleted fetal bovine and human sera have reduced capacity to support cell growth. J Extracell Vesicles. 2015;4:26373
38. Yu G, Jung H, Kang YY, Mok H. Comparative evaluation of cell- and serum-derived exosomes to deliver immune stimulators to lymph nodes. Biomaterials. 2018;162:71-81
39. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569-579
40. Du W, Zhang K, Zhang S, Wang R, Nie Y, Tao H. et al. Enhanced proangiogenic potential of mesenchymal stem cell-derived exosomes stimulated by a nitric oxide releasing polymer. Biomaterials. 2017;133:70-81
41. Tian T, Zhang HX, He CP, Fan S, Zhu YL, Qi C. et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137-149
42. Kim MS, Haney MJ, Zhao Y, Yuan D, Deygen I, Klyachko NL. et al. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: in vitro and in vivo evaluations. Nanomedicine. 2018;14:195-204
43. Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL. et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine. 2016;12:655-64
44. Jiang L, Li L, He X, Yi Q, He B, Cao J. et al. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials. 2015;52:126-139
45. Luo LM, Huang Y, Zhao BX, Zhao X, Duan Y, Du R. et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34:1102-1114
46. Munagala R, Aqil F, Jeyabalan J, Gupta RC. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016;371:48-61
47. Wan Y, Wang L, Zhu C, Zheng Q, Wang G, Tong J. et al. Aptamer-Conjugated Extracellular Nanovesicles for Targeted Drug Delivery. Cancer Res. 2018;78:798-808
48. Agrawal AK, Aqil F, Jeyabalan J, Spencer WA, Beck J, Gachuki BW. et al. Milk-derived exosomes for oral delivery of paclitaxel. Nanomedicine. 2017;13:1627-1636
49. Costa Verdera H, Gitz-Francois JJ, Schiffelers RM, Vader P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J Control Release. 2017;266:100-108
50. Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z. et al. Exosomes as drug delivery vehicles for Parkinson's disease therapy. J Control Release. 2015;207:18-30
51. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383-2390
52. Sun J, Jiang L, Lin Y, Gerhard EM, Jiang X, Li L. et al. Enhanced anticancer efficacy of paclitaxel through multistage tumor-targeting liposomes modified with RGD and KLA peptides. Int J Nanomedicine. 2017;12:1517-1537
53. Choo YW, Kang M, Kim HY, Han J, Kang S, Lee JR. et al. M1 Macrophage-Derived Nanovesicles Potentiate the Anticancer Efficacy of Immune Checkpoint Inhibitors. ACS Nano. 2018;12:8977-8993
54. Cai L, Xu G, Shi C, Guo D, Wang X, Luo J. Telodendrimer nanocarrier for co-delivery of paclitaxel and cisplatin: A synergistic combination nanotherapy for ovarian cancer treatment. Biomaterials. 2015;37:456-468
55. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D. et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998;4:594-600
56. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM. et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3:9
57. Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D. et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology. 2016;5:e1071008
58. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F. et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425-6440
59. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986-994
60. Daldrup-Link HE, Golovko D, Ruffell B, Denardo DG, Castaneda R, Ansari C. et al. MRI of tumor-associated macrophages with clinically applicable iron oxide nanoparticles. Clin Cancer Res. 2011;17:5695-5704
61. Zhao G, Rodriguez BL. Molecular targeting of liposomal nanoparticles to tumor microenvironment. Int J Nanomedicine. 2013;8:61-71
62. Cao Q, Yan X, Chen K, Huang Q, Melancon MP, Lopez G. et al. Macrophages as a potential tumor-microenvironment target for noninvasive imaging of early response to anticancer therapy. Biomaterials. 2018;152:63-76
63. Zhu S, Niu M, O'Mary H, Cui Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol Pharm. 2013;10:3525-3230
64. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462-472
65. Duluc D, Corvaisier M, Blanchard S, Catala L, Descamps P, Gamelin E. et al. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer. 2009;125:367-373
66. Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739-1742
67. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166-6173
68. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677-686
69. Narayanan R, Huang CC, Ravindran S. Hijacking the Cellular Mail: Exosome Mediated Differentiation of Mesenchymal Stem Cells. Stem Cells Int. 2016;2016:3808674
70. Eloy JO, Petrilli R, Topan JF, Antonio HMR, Barcellos JPA, Chesca DL. et al. Co-loaded paclitaxel/rapamycin liposomes: Development, characterization and in vitro and in vivo evaluation for breast cancer therapy. Colloids Surf B Biointerfaces. 2016;141:74-82
Author contact
![]() Corresponding authors: Lei Wang, e-mail: wangleiedu.cn and Weidong Chen, e-mail: wdchenedu.cn
Corresponding authors: Lei Wang, e-mail: wangleiedu.cn and Weidong Chen, e-mail: wdchenedu.cn