Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1683-1697. doi:10.7150/thno.30487 This issue Cite
Research Paper
Characterization and Therapeutic Application of Mesenchymal Stem Cells with Neuromesodermal Origin from Human Pluripotent Stem Cells
Huiyan Wang1*, Dairui Li1*, Zhichen Zhai1*, Xiaoran Zhang1, Weijun Huang1, Xiaoyong Chen1, Lihua Huang2, Huanyao Liu1, Jiaqi Sun1, Zhengwei Zou1, Yubao Fan1, Qiong Ke1,3, Xingqiang Lai1, Tao Wang1, Xiaoping Li1, Huiyong Shen4, Andy Peng Xiang1,5 ![]() , Weiqiang Li1,5,6
, Weiqiang Li1,5,6 ![]()
1. Center for Stem Cell Biology and Tissue Engineering, Key Laboratory for Stem Cells and Tissue Engineering, Ministry of Education, Sun Yat-Sen University, Guangzhou, Guangdong, P.R. China.
2. Department of Pediatric Surgery, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou, P.R. China
3. Department of Cell Biology, Zhongshan Medical School, Sun Yat-sen University, Guangzhou, Guangdong, P.R.China.
4. Department of Orthopedics, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, Guangdong, 518033, P.R.China.
5. Department of Biochemistry, Zhongshan Medical School, Sun Yat-sen University, Guangzhou, Guangdong, P.R.China.
6. Guangdong Key Laboratory of Reproductive Medicine, Guangzhou, Guangdong, 510080, China.
*These authors made equal contributions to this work.
Received 2018-10-8; Accepted 2019-2-2; Published 2019-2-28
Abstract

Rationale: Mesenchymal stem cells (MSC) hold great promise in the treatment of various diseases including autoimmune diseases, inflammatory diseases, etc., due to their pleiotropic properties. However, largely incongruent data were obtained from different MSC-based clinical trials, which may be partially due to functional heterogeneity among MSC. Here, we attempt to derive homogeneous mesenchymal stem cells with neuromesodermal origin from human pluripotent stem cells (hPSC) and evaluate their functional properties.
Methods: Growth factors and/or small molecules were used for the differentiation of human pluripotent stem cells (hPSC) into neuromesodermal progenitors (NMP), which were then cultured in animal component-free and serum-free induction medium for the derivation and long-term expansion of MSC. The resulted NMP-MSC were detailed characterized by analyzing their surface marker expression, proliferation, migration, multipotency, immunomodulatory activity and global gene expression profile. Moreover, the in vivo therapeutic potential of NMP-MSC was detected in a mouse model of contact hypersensitivity (CHS).
Results: We demonstrate that NMP-MSC express posterior HOX genes and exhibit characteristics similar to those of bone marrow MSC (BMSC), and NMP-MSC derived from different hPSC lines show high level of similarity in global gene expression profiles. More importantly, NMP-MSC display much stronger immunomodulatory activity than BMSC in vitro and in vivo, as revealed by decreased inflammatory cell infiltration and diminished production of pro-inflammatory cytokines in inflamed tissue of CHS models.
Conclusion: Our results identify NMP as a new source of MSC and suggest that functional and homogeneous NMP-MSC could serve as a candidate for MSC-based therapies.
Keywords: neuromesodermal progenitors, mesenchymal stem cells, human pluripotent stem cells, differentiation, immunomodulatory activity
Introduction
Mesenchymal stem cells (MSC) hold great therapeutic potential for cell-based tissue engineering and regenerative medicine, as they offer the benefits of easy isolation, multipotency, paracrine activity, and immunomodulatory properties [1]. More than 800 clinical trials using MSC to treat various diseases have been registered in the ClinicalTrials.gov database to date; the targeted disorders include bone/cartilage defects, cardiovascular diseases, inflammatory diseases, autoimmune diseases, etc. [2]. In addition, numerous studies have demonstrated that MSC therapy is safe and effective [3]. Nonetheless, perhaps due to variations in donor source, culture protocols, expansion levels, and cryopreservation strategies, researchers have noted that there is functional heterogeneity among MSC [4]. This may explain the largely incongruent data obtained from the various trials of MSC-based therapies [5]. Previous studies showed that bone marrow MSC (BMSC) from healthy donors or different inbred strains of mice possessed different potentials for proliferation and osteogenic differentiation [6-9]. It has also been demonstrated that MSC from different tissues (e.g., bone marrow, adipose, and umbilical cord) have diverse biological characteristics [10], and that intra-population heterogeneity may be found in human and mouse MSC [11, 12]. Therefore, it remains critically challenging to derive and expand homogeneous MSC with specific criteria for therapeutic applications.
Human pluripotent stem cells (hPSC), which include human embryonic stem cells (hESC) and human induced pluripotent stem cells (hiPSC), have the ability to indefinitely proliferate and differentiate to derivatives of all three germ layers. They may therefore represent a potentially ideal source of homogeneous and high-quality MSC [13]. It has been reported that MSC have multiple developmental origins, including the neural crest (neural ectoderm) and mesoderm [14], and can be derived from human pluripotent stem cells through the mesoderm, neural crest, or trophoblast-like intermediate stages [15-17]. Some studies have used fluorescence activated cell sorting (FACS) or serial passage to enrich hiPSC differentiated by certain growth factors for MSC of uncertain developmental origin [18, 19]. However, it remains unclear how similar such cells are in terms of their biology and function, nor do we know how they contribute to the functional heterogeneity of tissue-derived MSC. More importantly, we do not yet know whether MSC of undefined origin exist in the body.
Neuromesodermal progenitors (NMP) co-expressing SOX2 and Brachyury (T) have been found in chick, mouse, and human embryos, where they are localized at the caudal lateral epiblast (CLE) and adjacent node-streak border (NSB) within the primary streak. NMP reportedly have characteristic homeodomain transcription factor (HOX gene) expression profiles (HOX1-13), along with the potential to generate the spinal cord/posterior nervous system and paraxial mesoderm tissues, including spinal bone, cartilage, and muscle cells [20]. This suggests that MSC of NMP origin may exist during fetal development and remain in our bodies throughout life.
Here, we generated highly homogeneous MSC from hPSC via an NMP intermediate, using an efficient and chemically defined method. We demonstrate that NMP-MSC express posterior HOX genes and share characteristics with BMSC, but display stronger immunomodulatory capabilities than BMSC in vitro and in vivo.
Methods
Cell culture
We utilized two human embryonic stem cell (hESC) lines (H1, H9) and one human induced pluripotent stem cell (hiPSC) line that established in our lab as described [21]. The cells were cultured on Matrigel (BD Bioscience, San Diego, CA, USA)-coated plates in mTeSR1 medium (Stemcell Technologies, Vancouver, BC, Canada). The medium was changed daily and the cells were passaged every 4-5 days using the StemPro Accutase Cell Dissociation Reagent (Life Technologies, Carlsbad, CA, USA).
Additional Experimental Procedures
For more detailed and additional information on experimental procedures, please see Supplementary Materials and Methods.
Results
Generation of NMP from human pluripotent stem cells by activation of Wnt, FGF and TGFβ signaling
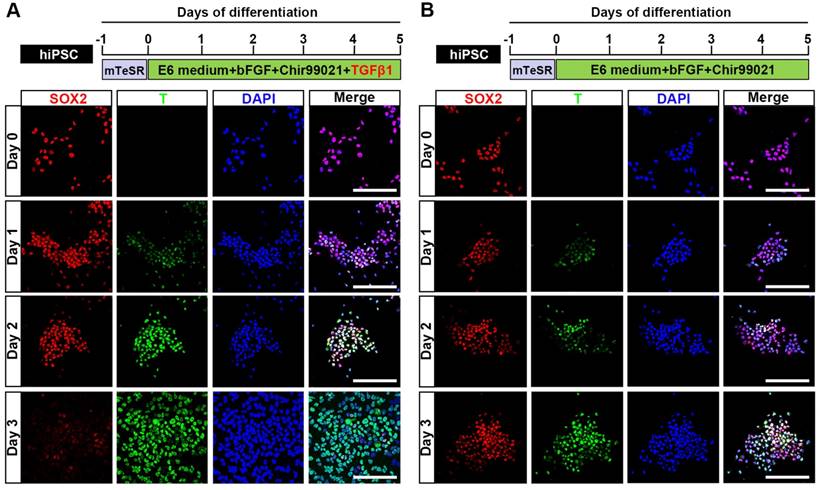
Previous studies demonstrated that Wnt and FGF signaling are involved in the development of NMP [22], and that a Wnt agonist (Chir99021) and bFGF can induce the differentiation of NMP from mouse or human pluripotent stem cells (mPSC or hPSC, respectively) [23]. It was also reported that transforming growth factor β (TGFβ) is a natural inducer of mesoderm during vertebrate development [24], and that supplementation with GDF11 (a TGFβ family member) is necessary for robust activation of lumbosacral HOX genes (HOX10-13) at the later stage (from day 6 to day 9) during the commitment of hPSC to NMP [25]. Here, we examined whether the early addition of TGFβ1 could induce the NMP differentiation of hPSC (hiPSC, H1, and H9) and trigger a similar lumbosacral patterning state. Upon induction by TGFβ1, Chir99021, and bFGF, differentiated hiPSC started to proliferate continuously and the protein expression level of the NMP marker, T, was clearly upregulated in our immunocytochemical analyses. T was first weakly detected 24 hours after induction and then strongly expressed in almost all differentiated hiPSC at 48 hours post-induction. Meanwhile, the expression of SOX2, which is an NMP marker that is intensively expressed by hPSC, was rapidly downregulated during differentiation until only a small population of SOX2-positive cells remained in the differentiation system by 72 hours after NMP commitment was initiated. Uniform co-expression of T and SOX2 (97.17±0.30%) was observed on day 2, as shown by immunofluorescence assays and fluorescence activated cell sorting (FACS) (Fig. 1A; Fig. S1A, S1B; Fig. S2), suggesting that hPSC exposed to TGFβ1, Chir99021, and bFGF for 48 hours represent a population of bipotential neuromesodermal progenitor cells. However, treatment with Chir99021/bFGF for 3 days induced hPSC to differentiate to SOX2+/T+ NMP with a peak efficiency about 86.90±0.62% (Fig. 1B; Fig. S2), which was comparable to that of previous studies [25, 26]. Prolonged treatment with Chir99021/bFGF (4-5 days) rapidly downregulated SOX2 expression, while the expression of T was maintained (data not shown).
Generation of neuromesodermal progenitors from human induced pluripotent stem cells. A. Schematic representation of the differentiation conditions used to generate NMP from hiPSC with TGFβ1 and the expression of T and SOX2 from day 0-day 3 during the NMP differentiation from hiPSC was detected by immunocytochemistry. Scale bar: 100 μm. B. Schematic representation of the differentiation conditions used to generate NMP from hiPSC without TGFβ1 and the expression of T and SOX2 from day 0-day 3 during the NMP differentiation from hiPSC was detected by immunocytochemistry. Scale bar: 100 μm.
Consistent with these immunocytochemical data, qRT-PCR analyses showed that the mRNA levels for NMP-specific markers, including T, NKX1- 2, WNT8A, FGF17, SP5, and CDX1, were significantly upregulated during the differentiation process. These increases in the expression levels of NMP markers were accompanied by downregulation of mRNA encoding pluripotency-specific markers, including OCT4 and NANOG. The mRNA level of SOX2, which is a marker for both PSC and NMP, was remarkably lower in day 2 cells (D2; NMP) than in hPSC, even though a high level of SOX2 protein was detected in NMP (Fig. S3A). These results agreed with those of previous studies showing that SOX2 transcripts are characteristically low in mouse NMP and that the SOX2 protein has a relatively long half-life [23, 27]. Next, we used qRT-PCR to determine whether the differentiated cell population included cells originating from the neural epithelium, neural crest, or endoderm. We found slight increase or decrease in the mRNA levels of SOX1 (neural epithelium), SOX10 (neural crest), and SOX17 (endoderm) (Fig. S3B), but the encoded proteins were barely detectable (data not shown) during NMP induction. qRT-PCR analysis of HOX gene expression revealed that the mRNA expression levels of lumbosacral HOXD10 and HOXA11 were significantly upregulated irrespective of lower (2 ng/ml) or higher (5-10 ng/ml) concentration of TGFβ1, while modest increase in the mRNA transcripts of HOXA10, HOXC10, and HOXC11 could only be detected when treated with higher concentration (5-10 ng/ml) of TGFβ1 in D2 NMP as compared to undifferentiated hPSC (day 0) (Fig. S3C). We also observed that the gene expression of anterior marker, HOXB2, was dramatically increased during the NMP differentiation of hPSC, indicating that anterior NMP might be generated within our differentiation system. The evidence may infer that although TGFβ signaling pathway was necessary for robust activation of lumbosacral markers, it did not significantly inhibit transcription of anterior or rostral HOX genes during development. Collectively, our results indicate that NMP can be efficiently generated from hPSC by activation of Wnt, FGF, and TGFβ signaling.
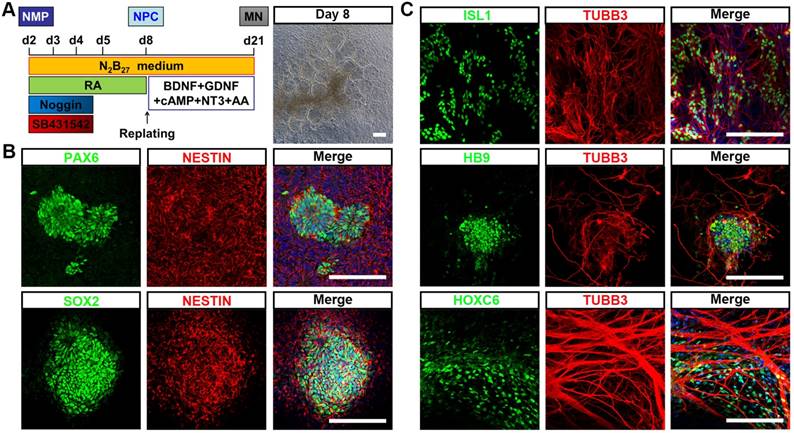
To test whether hPSC-NMP could differentiate into neural progenitor cells (NPC) with spinal cord identity, differentiated cells at D2 were subjected to dual SMAD inhibitor treatment (dSMADi; SB431542 and noggin) together with an RA regime (Fig. 2A), which was previously shown to efficiently promote the anterior neural conversion of hPSC [28]. At 6 days post-treatment, we found that multiple rosette structures resembling primitive neuroepithelium had emerged within the differentiated hPSC-NMP population (Fig. 2A). Immunofluorescence analyses showed that cells in the centers of these neural rosettes robustly expressed the neuroepithelial markers, PAX6, SOX2, and NESTIN (Fig. 2B). We next explored the potential of NPC derived from hPSC-NMP via the dSMADi protocol to give rise to motor neurons (MN) with posterior identity. To induce neuronal differentiation, we cultured hPSC-NMP with BDNF, GDNF, NGF, and db-cAMP for an additional 2-3 weeks. MN differentiation was verified by the positive detection of MN markers, including ISL1, HB9, and TUBB3, and the cervical spinal cord marker, HOXC6 (Fig. 2C). Taken together, these data indicated that exposure of hPSC-NMP to a combination of dSMADi/RA signaling and subsequent neuron-inducing factors could generate spinal cord cells.
Differentiation of neural cells with spinal cord identity from hiPSC-NMP. A. Schematic representation of the differentiation conditions used to generate neuroprogenitors and neurons with spinal cord identity from hiPSC-NMP. Scale bar: 100 μm. B. Immunofluorescence staining for the neuroprogenitor markers, PAX6, SOX2, and NESTIN, in hiPSC-NMP that had been treated with dSMADi/RA for 6 days. Scale bar: 100 μm. C. Motor neuron markers (ISL1, HB9, and TUBB3) and a cervical spinal cord marker (HOXC6) were detected by immunostaining. Scale bar: 100 μm.
Efficient derivation of MSC from hPSC-NMP
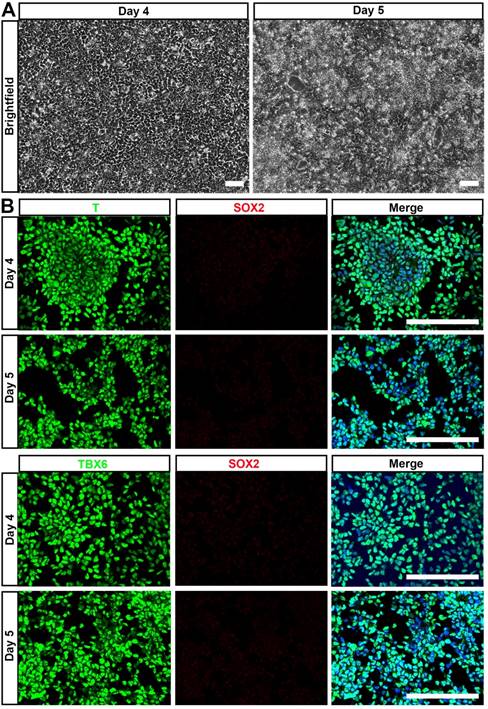
NMP is a common precursor of the spinal cord and paraxial mesoderm, the latter of which initially forms somites and later during vertebrate development gives rise to all skeletal muscles of the body, the axial skeleton, and part of the dermis [29]. Thus, we hypothesized that NMP could be a major source of MSC. Accordingly, we examined the mesenchymal lineage differentiation potential of hPSC-NMP. To avoid spontaneous neural commitment, we treated hPSC with bFGF, TGFβ1, and Chir99021 for more than 3 days, with the goal of completely inactivating SOX2 expression. Indeed, cells treated for 4-5 days proliferated quickly and became confluent by day 4 (Fig. 3A). Immunostaining failed to reveal any SOX2-positive cell and most of the differentiated cells expressed the paraxial mesoderm markers, T and TBX6 (Fig. 3B). These results indicated that hPSC were successfully induced to differentiate into paraxial mesoderm cells (hPSC-NMP-PM).
Generation of paraxial mesoderm from hiPSC by extended treatment with bFGF, TGFβ1, and Chir99021. A. hiPSC were treated with bFGF, TGFβ1, and Chir99021 for 4-5 days and observed under phase-contrast microscopy, which showed that cells proliferated quickly and became confluent by day 4. Scale bar: 100 μm. B. Immunostaining was used to detect paraxial mesoderm markers (T and TBX6) and SOX2. Scale bar: 100 μm.
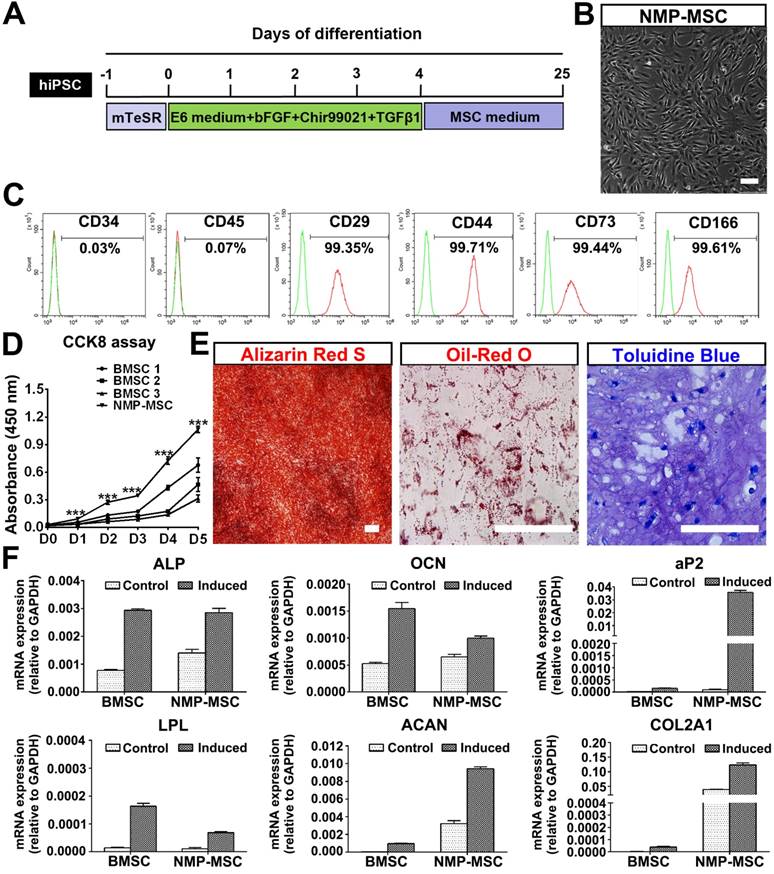
To investigate whether hPSC-NMP-PM could undergo MSC differentiation, the cells were exposed to MesenCult™-ACF Plus Medium (animal component-free and serum-free medium) for 2-3 weeks (Fig. 4A). We observed significant changes in the morphology of hPSC-NMP-PM, along with the emergence and active proliferation of spindle-shaped cells that exhibited a parallel or spiral arrangement (NMP-MSC) (Fig. 4B). To examine whether these cells possessed MSC characteristics, we analyzed their surface marker expression, proliferation ability, and multilineage differentiation potential. FACS analysis (the gating strategy was shown as Fig. S4A) showed that most of the NMP-MSC (>98%) expressed surface markers typical of human MSC, including CD29, CD44, CD73, and CD166, but nearly all were negative for CD34 or CD45 (Fig. 4C; Fig. S4B). CCK8 assays indicated that NMP-MSC cultured in MesenCult™- ACF Plus Medium expanded more quickly than control BMSC cultured in the same medium (p<0.001; Fig. 4D). The in vitro migration ability of NMP-MSC was assessed by time-lapse analysis, transwell assays, and wound-healing assays, in which we failed to observe any significant difference between NMP-MSC and BMSC (data not shown). Moreover, NMP-MSC cultured under specific conditions were able to differentiate into osteoblasts, adipocytes, and chondrocytes, respectively, as confirmed by Alizarin Red S staining, oil red O staining, and toluidine blue staining, respectively (Fig. 4E; Fig. S4C). qRT-PCR results also confirmed the multilineage differentiation ability of NMP-MSC (Fig. 4F). We further demonstrated that NMP-MSC from all three hPSC lines could be maintained in serum-free MesenCult™-ACF Plus Medium for over 20 passages without losing their surface marker expression, mitotic activity, or tri-lineage differentiation ability (data not shown). These results demonstrate that NMP-MSC resemble human BMSC in terms of their marker expression, self-renewal, and multipotency.
Derivation and characterization of NMP-MSC from hiPSC. A. Strategy for deriving MSC from hiPSC-NMP. B. Cells were observed under phase-contrast microscope following exposure of hiPSC-NMP-PM to serum-free MSC inducing medium for about 21 days. Scale bar: 100 μm. C. FACS analysis for detection of typical MSC surface markers in NMP-MSC derived from hiPSC. D. The CCK8 assay was used to detect the proliferation of NMP-MSC derived from hiPSC and control BMSC. The data represent mean ± SEM of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant. E. The osteogenic, adipogenic, and chondrogenic differentiation potentials of NMP-MSC were verified by Alizarin Red S staining, oil red O staining, and toluidine blue staining, respectively. Scale bar: 100 μm. F. qRT-PCR analysis was used to detect osteogenic (ALP and OCN), adipogenic (aP2 and LPL), and chondrogenic (ACAN and COL2A1) markers. The data represent mean ± SEM of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant.
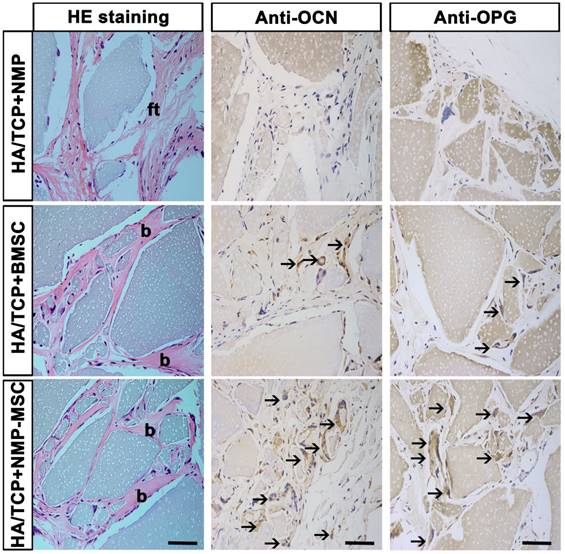
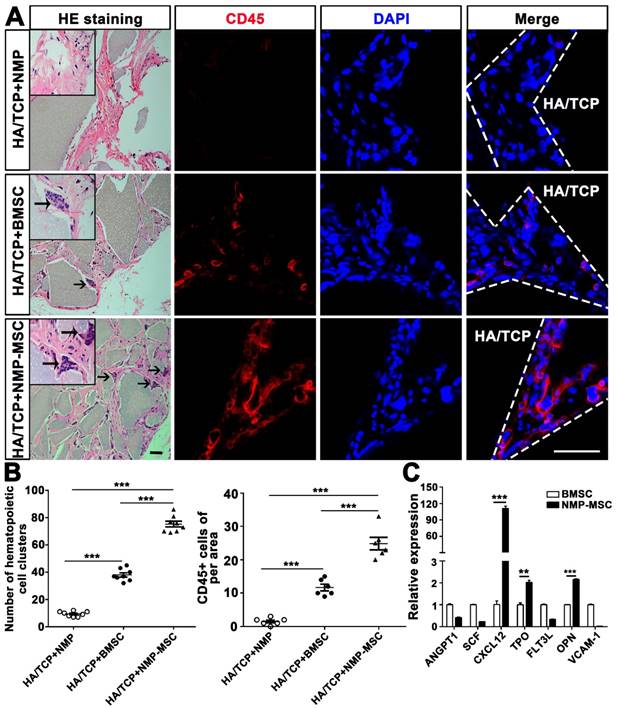
To examine the in vivo bone formation ability of NMP-MSC, we performed heterotopic transplantation into immunocompromised mice. NMP-MSC were allowed to adhere to scaffolds, the hydroxyl-apatite/ tricalcium phosphate ceramic powder (HA/TCP), and the generated cell-scaffold complexes were subjected to osteogenic differentiation for 3 days and then transplanted subcutaneously into nude mice. NMP was served as control cells. Eight weeks later, immunohistochemistry showed that there were more osteocalcin (OCN)- and osteoprotegerin (OPG)-positive osteoblasts in the BMSC and NMP-MSC groups than in the NMP control group (Fig. 5). HE staining revealed that NMP control group failed to form either bone or hematopoietic marrow but rather fibrous tissue at the transplantation site, and that NMP-MSC-I njected mice showed enhanced bone formation (Fig. 5), more hematopoietic cell clusters (9.38±0.68 for NMP group; 38±1.56 for BMSC group; 75.25±2.12 for NMP-MSC group) and CD45+ cells (pan-leukocyte marker; 1.5±0.43/field for NMP group; 11.67±0.99/field for BMSC group; 24.83±1.85/field for NMP-MSC group) when compared with the BMSC group (Fig. 6A, 6B). We then analyzed the expression of genes that regulate hematopoietic supporting activity and qRT-PCR indicated that the expression of CXCL12 was over 100-fold higher, and the expression of TPO and OPN was about 2-fold higher in NMP-MSC than BMSC (Fig. 6C). These results suggest that NMP-MSC can reconstitute the hematopoietic microenvironment in vivo, which may be due to higher expression of hematopoiesis supporting genes (CXCL12, TPO, and OPN) in NMP-MSC. We also transplanted NMP-MSC subcutaneously into 8-week-old NOG mice and assessed the tumorigenicity of these cells. Eight weeks later, we failed to find any sign of tumor formation in mice of the NMP-MSC group, whereas control mice injected with undifferentiated hiPSC exhibited efficient formation of teratomas (100%) (Fig. S5).
In vivo bone formation of NMP-MSC derived from hiPSC. The samples of in vivo bone formation were analyzed by hematoxylin and eosin (H&E) staining, and osteocalcin (OCN)- and osteoprotegerin (OPG)-expressing osteocytes were detected by immunohistochemistry. b, bone; ft, fibrous tissue; black arrows showed the location of OCN+ or OPG+ cells. Scale bar: 50 μm.
Hematopoietic clusters could be found in the samples of in vivo bone formation. A. HE staining was used to observe the hematopoietic clusters and immunostaining with anti-CD45 antibody was applied for the detection of the nucleated cells of hematopoietic origin in in vivo transplants. More hematopoietic clusters and CD45+ cells were detected in the NMP-MSC group compared to the BMSC and control groups. Black arrows showed the location of hematopoietic cell clusters. HA/TCP, hydroxyl-apatite/tricalcium phosphate ceramic powder. Scale bar: 50 μm. B. Quantification of hematopoietic clusters (n=8) and CD45+ cells (n=6) were performed in different group. The data are expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant. C. The expression of hematopoietic supporting genes in cultured NMP-MSC and BMSC were detected by qRT-PCR. The data represent mean ± SEM of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant.
Transcriptome profiling of NMP-MSC by RNA sequencing
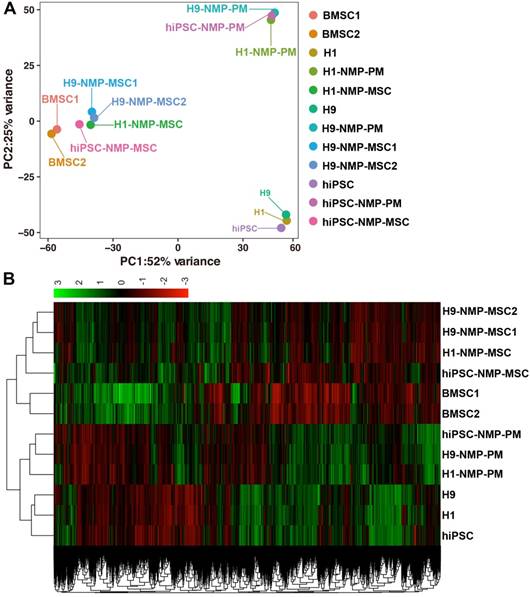
RNA sequencing (RNA-Seq) was performed to examine changes in the global gene expression profile during the NMP-MSC differentiation of hPSC (GSE120420). We established global lineage relationships by identifying genes that were differentially expressed among all populations. Principal component analysis (PCA) of all mapped genes showed that undifferentiated pluripotent cells and the NMP- derived paraxial mesoderm cells in the intermediate differentiation stage each grouped together, while NMP-MSC of the different cell lines together with control BMSC exhibited distinct profiles (Fig. 7A). We also compared the TPM of all genes in each sample and used them to generate a two-way clustering heat map of whole-gene expression profiles. This analysis showed that the gene expression patterns were differed between the groups of cells (Fig. 7B), which was consistent with the results of our PCA. To assess the global relatedness of the gene expression profiles between NMP-MSC and BMSC, we calculated Pearson correlations of pair-wise comparisons for all expressed genes. The results suggested that NMP- MSC and BMSC shared relatively similar gene expression patterns (R2>0.80). Moreover, NMP-MSC derived from hiPSC exhibited a high correlation with NMP-MSC derived from the human embryonic stem cell lines, H1 and H9 (R2>0.84). Notably, we observed an extremely high level of similarity between H1-NMP-MSC and H9-NMP-MSC (R2 > 0.95) (Fig. S6), suggesting that our protocol could generate highly homogeneous MSC populations from human ESC.
Principal component analysis and heatmap of transcriptome profiles from hPSC, hPSC-NMP-PM, and NMP-MSC. A. Principal component analysis (PCA) showed that undifferentiated hPSC and hPSC-NMP-PM grouped together, whereas NMP-MSC derived from the various cell lines or control BMSC each represented distinct expression profiles. B. Heat map of global gene expression profiles showed that the cell groups exhibited different gene expression patterns consistent with the results of our PCA.
The RNA-Seq data further demonstrated that the transcript profile of undifferentiated hPSC was enriched for genes related to pluripotency (OCT4, SOX2, NANOG, and others), while hPSC-NMP-PM strongly expressed paraxial mesoderm-specific transcription factors (e.g., CDX1, CDX2, TBX6, and others). More importantly, all the NMP-MSC samples were highly enriched in transcripts associated with MSC-specific molecules as described [15, 30, 31], including cell adhesion/CD epitope encoding genes (such as CD29/ ITGB1, CD44, CD73/ NT5E, CD166/ ALCAM, and others), metalloproteinases (such as MMP14), tissue inhibitors of metalloproteinases (TIMPs; such as TIMP2), and BMP pathway molecules (such as BMP1, BMPR1B) (Fig. S7). HOX genes were also enriched in hPSC-NMP-PM, hPSC- NMP-MSC and human BMSC as well. In contrast, undifferentiated pluripotent stem cells did not express any of these HOX genes (Fig. S8A). The results were further confirmed by qRT-PCR in hiPSC (Fig. S8B). These findings support our earlier contention that hPSC-NMP-MSC maintain the NMP- specific HOX gene expression pattern and have a gene expression profile similar to that of BMSC.
The immunomodulatory capacity of NMP-MSC in vitro and in vivo
Human BMSC reportedly possess immunomodulatory properties toward both innate and adaptive immune cells, and BMSC transplantation has been used to treat a variety of inflammatory and autoimmune diseases in preclinical and clinical studies [1, 32]. To investigate the immunoregulatory abilities of hPSC-NMP- MSC, we examined the potential regulatory effects of NMP-MSC on the proliferation and pro-inflammatory cytokine production of CD3+ T cells. Our CFSE assay demonstrated that both NMP-MSC (P5) and BMSC (P5) could efficiently inhibit the proliferation of CD3+ T cells (79.73±2.53% for control; 30.44±2.85% for BMSC1; 15.67±2.46% for BMSC2; 24.99±2.17% for BMSC3; 4.78±0.59% for hiPSC-NMP-MSC) and suppress the percentages of CD3+ T cells that produced tumor necrosis factor α (TNF-α) (50.86±0.99% for control; 26.59±1.24% for BMSC1; 24.97±0.23% for BMSC2; 31.27±1.94% for BMSC3; 5.20±0.18% for hiPSC-NMP-MSC) and interferon γ (IFN-γ) (19.64±1.81% for control; 10.60±1.03% for BMSC1; 11.93±2.35% for BMSC2; 10.47±1.42% for BMSC3; 1.95±0.50% for hiPSC- NMP-MSC) (Fig. 7A). Notably, NMP-MSC derived from hiPSC, H1, and H9 all exhibited markedly higher immunomodulatory activities than the tested BMSC (Fig. 8A; Fig. S9). Most strikingly, although NMP- MSC showed a slight decrease in their immunoregulatory ability as the in vitro culture was prolonged, the immunomodulatory capability of NMP-MSC at P20 was greater than that of BMSC at P5 (Fig. S10).
Immunoregulatory effect of NMP-MSC derived from hiPSC on CD3+ T cells in vitro. A. T cells were cultured with or without NMP-MSC or BMSC for 3 days, and their proliferation and productions of TNF-α and IFN-γ were examined by flow cytometry. The data represent mean ± SEM of four independent experiments. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant. B. mRNA transcripts of proinflammatory cytokines (IL-6, IL-8, and CCL2) and anti-inflammatory mediators (PDL1, TSG6, and IDO) were analyzed by qRT-PCR in NMP-MSC and control BMSC that had been exposed to an inflammatory environment. The data represent mean ± SEM of three independent experiments. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant.
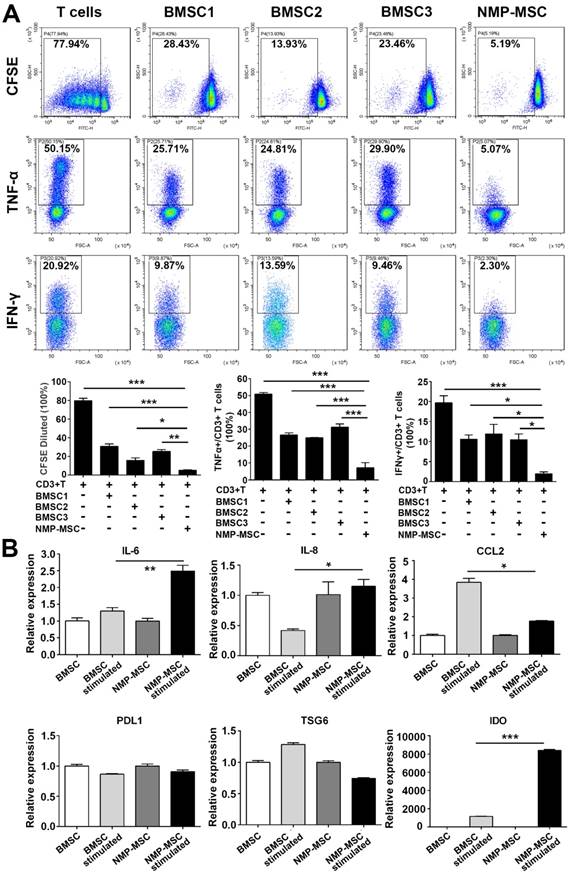
We next studied a potential underlying cellular mechanism for the robust immunoregulatory ability of NMP-MSC by exposing cells to proinflammatory factors (modelling an inflammatory environment) [17] and examining changes in the mRNA expression levels of proinflammatory cytokines (IL-6 and CCL2) and anti-inflammatory mediators (PDL1, TSG6, and IDO). qRT-PCR revealed that IFN-γ stimulation induced the expression of the proinflammatory cytokines, IL-6 in NMP-MSC (about 2-fold higher than untreated cells) and CCL2 in BMSC (about 4-fold higher than untreated cells) (Fig. 8B). With respect to the anti-inflammatory genes (PDL1, TSG6, and IDO), the gene expression of IDO was greatly upregulated by IFN-γ in both BMSC and NMP-MSC, with a level in NMP-MSC reaching 6-fold that in BMSC (p<0.001); in contrast, the mRNA expression levels of PDL1 and TSG6 were not influenced by IFN-γ treatment (Fig. 8B). Moreover, IDO specific inhibitor 1-MT significantly antagonized the immunoregulatory ability of NMP-MSC (Fig. S11). These preliminary results indicate that the enhanced immunomodulatory capability of NMP-MSC may be partially mediated via IDO pathway.
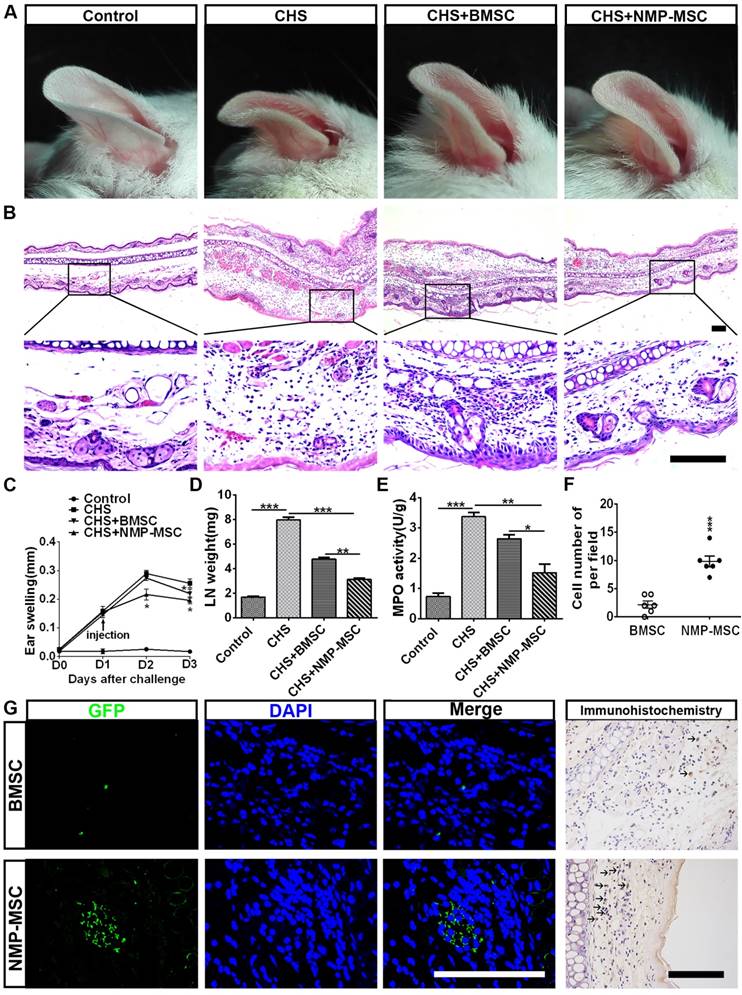
We further questioned whether NMP-MSC could modulate inflammation and contribute to tissue regeneration through their potent immunomodulatory effects. Toward this end, we transplanted cells into a mouse model of contact hypersensitivity (CHS). We injected cells on day 1 post-challenge and measured swelling/inflammation (in terms of external ear thickness) every 24 hours for the next 3 days. The results revealed that NMP-MSC had better treatment efficacy than BMSC, as characterized by the ability to decrease ear thickness (Fig. 9A) and leukocyte infiltration (Fig. 9B). NMP-MSC significantly attenuated CHS as early as 24 hours post-injection (CHS+ NMP-MSC versus CHS, p<0.05; CHS+NMP-MSC versus CHS+BMSC, p<0.05; CHS+BMSC versus CHS, p>0.05) and had even greater effects 48 hours post-injection (CHS+NMP-MSC versus CHS, p<0.05; CHS+NMP-MSC versus CHS+BMSC, p<0.05; CHS+ BMSC versus CHS, p<0.05) (Fig. 9C). The gross weight of regional draining lymph nodes (dLNs) from CHS mice 24 hr post cell transplantation showed a markedly reduction in NMP-MSC group (3.13±0.10mg) and BMSC group (4.79±0.13mg) than that in CHS group (7.96±0.24mg) (Fig. 9D). We used qRT-PCR and ELISA to examine the mRNA and protein levels of pro-inflammatory mediators (IFN-γ, TNF-α, IL-17, and IL-6) in the inflamed ears respectively and observed significant downregulation of these pro-inflammatory cytokines in the NMP-MSC group, whereas relatively lower fold changes were seen in the BMSC treatment group versus the CHS group (Fig. S12A, S12B). As an index for evaluating the severity of CHS [32], the MPO activity in ear homogenates obtained from CHS mice 24 hr post- injection was examined. We found that MPO activity was notably decreased in the NMP-MSC group (1.52±0.29) and differed slightly in the BMSC group (2.64±0.13), when compared with the CHS group (3.37±0.13) (Fig. 9E). We also investigated whether NMP-MSC could migrate toward inflamed ears following intravenous infusion in vivo. The inflamed ears were collected 24 hours post-injection and subjected to in situ anti-EGFP immunofluorescence staining and EGFP-positive cells were quantified per microscopic field of ear cryosections (Fig. 9F, 9G). Our results revealed that small amount of BMSC could be detected (2.17±0.65 cells of per field), whereas a relatively higher number of NMP-MSC (9.83±0.95 cells of per field) was observed in the inflamed ears of CHS mice. These results demonstrate that NMP-MSC have better therapeutic ability for improving the symptoms of CHS than BMSC, possibly due to their enhanced ability to suppress pro-inflammatory cytokines and undergo targeted migration to inflamed ears in vivo.
Injection of NMP-MSC derived from hiPSC attenuates CHS in vivo. A. Representative photos of ear samples from mice of each group. B. Representative HE-stained ear samples from mice of each group. Forty-eight hours post-injection, ear tissues were collected, paraffin-embedded, sectioned, and stained. Scale bar: 100 μm. C. The degree of swelling was calculated as the thickness of the right ear (challenged ear) minus the baseline thickness of the left ear (unchallenged ear) (n = 6 /group). The data are expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant. D. The gross weight of regional draining lymph nodes (dLNs) from CHS mice 24 hours post cell transplantation (n=3). The data are expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant. E. MPO activity was examined by colorimetric assay of ear tissue homogenates obtained from each group during DNFB-induced CHS (24 hours post-injection) (n=4).The data are expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant. F. Number of EGFP+ cells in inflamed ears from different group were counted (n=6). The data are expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001, and n.s. is non-significant. G. The targeted migration of NMP-MSC toward inflamed ears was evaluated by immunofluorescence and immunocytochemistry assay. Scale bar: 100 μm.
Discussion
We herein describe a detailed protocol for deriving mesenchymal stem cells via a neuromesoderm intermediate stage. We demonstrate that the hPSC-derived NMP-MSC have typical HOX gene expression profiles and resemble human BMSC in their surface marker expression pattern and the ability to differentiate into multiple mesenchymal lineages. Most importantly, NMP-MSC displayed immunomodulatory effects superior to those of BMSC, as assessed by measuring their ability to suppress the proliferation and pro-inflammatory cytokine production of T cells in vitro and in vivo.
The transplantation of MSC, which exhibit immunoregulatory ability and tissue regeneration potential, has emerged as a promising candidate for cell therapy. Previous studies showed that MSC can be derived from neural crest [14], trophoblast [17], and lateral plate mesoderm [14, 33], but it was unclear whether there are MSC of other developmental origins within the human body. The bipotent NMP that give rise to the spinal cord and paraxial mesoderm were first discovered in mouse embryos and later identified later in human embryos [20]. Their ability to generate paraxial mesoderm tissues (e.g., bone, cartilage, and muscle cells) prompted us to hypothesize that NMP may represent a major source of MSC, and that NMP-MSC may play essential roles in paraxial mesoderm formation during human body development. However, the cellular and molecular mechanisms of NMP-MSC development and their biological features were previously unknown. Moreover, no previous study had reported the isolation of primary NMP or NMP-MSC from human tissues, largely because access to gastrula-stage human embryos and/or adult spine tissues is invasive and limited by numerous ethical and legal questions. In this study, we demonstrated that NMP-MSC could be efficiently derived from hPSC through the intermediate stage of NMP for the first time. Thus, the NMP-MSC could provide a valuable tool for understanding paraxial mesoderm development, modeling human paraxial mesoderm-related diseases, and developing NMP-MSC related cell therapy.
As mentioned above, scientists have successfully derived functional MSC from hPSC via different intermediate stages, including neural crest [16, 21], trophoblast [17], and mesoderm [15]. Various other studies have used certain growth factors or small molecules to trigger the directed differentiation of MSC from hPSC without a distinct intermediate stage, but this could yield a heterogeneous mixed population of MSC with different origins [18, 19, 34, 35]. Different methods have been used to generate MSC from hPSC, but most have proven inefficient. For example, human ESC directly co-cultured with mouse OP9 stromal cells differentiated towards a mesoderm fate, and were then induced with FGF2 and PDGFBB for the generation of MSC [15]. Although the resulting cells exhibited chondro-, osteo- and adipogenic differentiation potentials, however, the contaminated mouse feeder cells limited the clinical application. Other studies reported monolayer [18] or embryoid body (EB)-based [19] protocols for obtaining MSC from hPSC, but these methods were required to purify the MSC using FACS via specific cell surface marker, such as CD73, CD90, or CD105. More importantly, most of these studies used medium containing fetal bovine serum [16-19, 34, 35], which has an undefined composition and may exhibit between-batch differences and/or functional heterogeneity.
Here, we sought to generate NMP-MSC with NMP origin from human pluripotent stem cells (e.g., hESC and hiPSC). We observed that treatment with Chir99021/bFGF for 3 days induced hPSC to differentiate to SOX2+/T+ NMP with similar efficiency to that of previous studies [25, 26]. Adding TGFβ1 to the induction medium at the beginning of the differentiation process could yield 95% NMP (i.e., those expressing typical NMP markers and posterior HOX gene mRNA expression) within 2 days, as shown by anti-SOX2/T immunostaining, RNA-Seq and qRT-PCR analysis. These data indicate that supplementation of TGFβ1 at the early differentiation stage may accelerate the specification of NMP from hPSC without any loss of the lumbosacral patterning effect. Next, we tested whether our system could be used to efficiently induce MSC from hPSC-NMP under the serum-free culture conditions. We treated hPSC-NMP cells with bFGF, TGFβ1, and Chir99021 for 4-5 days (whereupon they expressed paraxial mesoderm-specific transcription factors) and then switched the medium to defined MesenCult™-ACF Plus Medium for 2-3 weeks. This strategy readily generated NMP-MSC without the need for additional FACS enrichment. As expected, the NMP-MSC had a surface marker expression pattern and multi-differentiation potential similar to those of human BMSC. Furthermore, the RNA-Seq data illustrated that NMP-MSC derived from different hPSC exhibited highly similar global gene expression patterns, suggesting that our protocol can reliably generate homogeneous MSC, which could be further verified by Single-cell mRNA sequencing.
The regenerative potential, bone marrow niche function and immunomodulation capability are essential properties for the clinical translation of hPSC-MSC and were intensively discussed in many studies [17, 36-43]. Kang R et al. reported that hPSC-MSC in vitro possessed osteogenic and chondrogenic efficacy similar to that of BM-MSCs, but dispayed much lower adipogenic differentiation ability than those from BM-MSCs [42]. However, Chijimatsu R et al. found that tissue-engineered constructs formed by MSC derived from hiPSC via neural crest development failed to repair rat osteochondral defects in vivo, although these cells exhibited good chondrogenic potential in vitro under appropriate conditions, while transplantation with BMSC derived tissue-engineered constructs showed active cartilaginous tissue and subchondral bone formation [36]. It was shown that hPSC-MSC and BM-MSC had comparable supportive nature on the expansion of CD34+ hematopoietic stem/progenitor cells in vitro [43]. While in our study, NMP-MSC that transplantated into immunocompromised mice showed enhanced bone formation, increased osteocalcin (OCN)- and osteoprotegerin (OPG)-positive osteoblasts, and more hematopoietic clusters when compared with the BMSC group, although we did not observe significantly difference between these cells lines in in vitro assay. These results indicate that hPSC-NMP-MSC may be suitable for the treatment of bone defects.
Previous research also revealed that hPSC-derived MSC had active immunoregulatory abilities. Kimbrel EA et al. reported that hPSC-MSC could suppress T-cell proliferation, inhibit CD83 up-regulation and IL-12p70 secretion from dendritic cells, and help to prolong the lifespan of lupus-prone mice and result in a reduction of symptoms in an autoimmune model of uveitis in vivo [37]. Roux C et al. demonstrated that hPSC-MSC significantly reduced the T cell activation and increased the generation of functional CD4+ FoxP3+ regulatory T (Treg) in vitro and in vivo [38]. These studies, however, did not compare the immunoregulatory activity of hPSC-MSC with that of BMSC. Other studies demonstrated that hPSC-MSC had similar immunomodulation effect with those of BMSC [17, 39, 40, 43]. Notably, Wang X et al. proved that the experimental autoimmune encephalitis (EAE) disease-modifying effect of hESC-MSC was significantly greater than that of human bone-marrow-derived MSCs (BM-MSCs) [41]. Here, we evaluated the immunomodulatory potential of NMP-MSC using a CHS mouse model, which was characterized by T cell-mediated antigen-specific skin inflammation induced by skin exposure of sensitized mice to haptens [32]. The generated NMP-MSC displayed much stronger suppression of the proliferation and expression of the proinflammatory cytokines in T cells in vitro and in vivo than the three tested BMSC lines (at P5), which may at least partly reflect the dramatically higher expression of IDO by NMP-MSC (compared to BMSC) upon exposure to an inflammatory environment. More strikingly, NMP-MSC from all three hPSC cell lines exhibited robust proliferation ability in MesenCult™-ACF Plus Medium and could be propagated for at least 20 passages without losing their MSC characteristics or immunomodulatory ability. Due to the potential clinical application of NMP-MSC in the future, it is of great importance to generate a sufficient number of cells for transplantation, which may represent one of the key challenges due to the limited number of MSC isolated from donated tissues [39, 44]. Here we found that hPSC-derived MSC had a higher proliferation index and shorter population doubling time when cultured in serum-free, chemical defined medium than bone marrow-derived MSC, which was consistent with most of reported hPSC-MSC studies [15, 18, 34, 42], and no obvious cell death of hPSC-MSC was detected during in-vitro culture in this study. Some studies reported that the expansion potential of hiPSC-MSC could not be expanded beyond 14 or 17 passages, in regardless of using serum-containing medium or xeno-free and Good Manufacturing Practice-compatible media [34, 45]. Significantly, other studies revealed that clonally derived hiPSC-MSC were highly expandable up to 40 passages or could generate up to 1022 total cells without losing characteristics of MSC [15, 18]. Recently, Yan L et al. also described a 3D protocol for scalable production of hESC-MSC (up to 1012~1013 cells at Passage 6) by directly converting hPSC spheres to MSC spheres [39]. We then evaluate whether our method can be used for large-scale production of NMP-MSC, and we found that we could approximately generate 1.2×107 NMP-MSC (Passage 0; about 6 T75 flasks) from 1×106 undifferentiated hPSC (1 well of six-well plates) within 3-4 weeks, and will steadily get 4.18×1016 NMP-MSC at Passage 20 (e.g., 1:3 split ratio; reaching confluence within 3-4 days for each passage) from different hPSC lines (H1, H9, and hiPSC). This suggests that our protocol could be scaled up for the clinical translation of NMP-MSC for treating inflammatory and autoimmune diseases.
Conclusions
We herein describe a simple, efficient, and chemically defined protocol for the derivation of high-quality and homogeneous NMP-MSC. These cells may serve as a powerful tool for studying the pathogenesis of NMP-MSC-related disorders and supply a potentially unlimited cell source for therapeutic application.
Abbreviations
MSC: Mesenchymal stem cells; BMSC: Bone marrow mesenchymal stem cells; hPSC: Human pluripotent stem cells; NMP: Neuromesodermal progenitors; CHS: Contact hypersensitivity; T: Brachyury; CLE: Caudal lateral epiblast; NSB: Node-streak border; hESC: Human embryonic stem cells; hiPSC: Human induced pluripotent stem cells; mPSC: Mouse pluripotent stem cells; HOX: Homeodomain transcription factor; TGFβ: Transforming growth factor β; qRT-PCR: Quantitative reverse transcription-polymerase chain reaction; FACS: Fluorescence activated cell sorting; NPC: Neural progenitor cells; dSMADi: dual SMAD inhibitor; MN: Motor neurons; BDNF: Brain-derived neurotrophic factor; GDNF: Glial cell line-derived neurotrophic factor; NGF: Nerve growth factor; NT3: Neurotrophin -3; AA: Ascorbic acid; db-Camp: dibutyryl-cAMP; PM: Paraxial mesoderm; OCN: Osteocalcin; OPG: Osteoprotegerin; RNA-Seq: RNA sequencing; PCA: Principal component analysis; TIMP: Tissue inhibitors of metalloproteinases; CFSE: Carboxyfluorescein succinimidyl ester; CCK8: Cell Counting Kit-8; TNF-α: Tumor necrosis factor α; IFN-γ: Interferon γ.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The National Key Research and Development Program of China (2017YFA0103802, 2018YFA0107200, 2017YFA0103403); the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16020701, XDA16010102); The National Natural Science Foundation of China (U1501245, 81501901, 81570487, 81730005, 31771616, 81570161); Key Scientific and Technological Program of Guangzhou City (201704020223, 201604020010, 201604020008, 201604020189); Frontier and Innovation of Key Technology Project in Science and Technology Department of Guangdong Province (2016B030229002, 2015B020228001, 2017B020231001, 2016B030230001); The Natural Science Foundation of Guangdong Province (2016A030310158); The Fundamental Research Funds for the Central Universities (17ykzd07); China Postdoctoral Science Foundation (2015M572407).
Author Contributions
The conception and design of the study, or acquisition of data, or analysis and interpretation of data: Huiyan Wang, Dairui Li, Zhichen Zhai, Xiaoran Zhang, Weijun Huang, Xiaoyong Chen, Lihua Huang, Huanyao Liu, Jiaqi Sun, Zhengwei Zou, Yubao Fan, Qiong Ke, Xingqiang Lai, Tao Wang, Xiaoping Li, Huiyong Shen, Andy Peng Xiang, Weiqiang Li. Drafting the article or revising it critically for important intellectual content: Huiyan Wang, Dairui Li, Zhichen Zhai, Andy Peng Xiang, Weiqiang Li. Final approval of the version to be submitted: Weiqiang Li, Andy Peng Xiang.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Peng Y, Chen X, Liu Q, Zhang X, Huang K, Liu L. et al. Mesenchymal stromal cells infusions improve refractory chronic graft versus host disease through an increase of CD5+ regulatory B cells producing interleukin 10. Leukemia. 2015;29:636-46
2. Trounson A, McDonald C. Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell. 2015;17:11-22
3. Squillaro T, Peluso G, Galderisi U. Clinical Trials With Mesenchymal Stem Cells: An Update. Cell Transplant. 2016;25:829-48
4. Phinney DG. Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. J Cell Biochem. 2012;113:2806-12
5. Galipeau J. The mesenchymal stromal cells dilemma-does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy. 2013;15:2-8
6. Phinney DG, Kopen G, Righter W, Webster S, Tremain N, Prockop DJ. Donor variation in the growth properties and osteogenic potential of human marrow stromal cells. J Cell Biochem. 1999;75:424-36
7. Phinney DG, Kopen G, Isaacson RL, Prockop DJ. Plastic adherent stromal cells from the bone marrow of commonly used strains of inbred mice: variations in yield, growth, and differentiation. J Cell Biochem. 1999;72:570-85
8. Zhou S, Greenberger JS, Epperly MW, Goff JP, Adler C, Leboff MS. et al. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell. 2008;7:335-43
9. Wagner W, Ho AD. Mesenchymal stem cell preparations-comparing apples and oranges. Stem Cell Rev. 2007;3:239-48
10. Hass R, Kasper C, Bohm S, Jacobs R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal. 2011;9:12
11. Muraglia A, Cancedda R, Quarto R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J Cell Sci. 2000;113( Pt 7):1161-6
12. Lei J, Hui D, Huang W, Liao Y, Yang L, Liu L. et al. Heterogeneity of the biological properties and gene expression profiles of murine bone marrow stromal cells. Int J Biochem Cell Biol. 2013;45:2431-43
13. Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295-305
14. Isern J, Garcia-Garcia A, Martin AM, Arranz L, Martin-Perez D, Torroja C. et al. The neural crest is a source of mesenchymal stem cells with specialized hematopoietic stem cell niche function. Elife. 2014;3:e03696
15. Vodyanik MA, Yu J, Zhang X, Tian S, Stewart R, Thomson JA. et al. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell. 2010;7:718-29
16. Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T. et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol. 2007;25:1468-75
17. Wang X, Lazorchak AS, Song L, Li E, Zhang Z, Jiang B. et al. Immune modulatory mesenchymal stem cells derived from human embryonic stem cells through a trophoblast-like stage. Stem Cells. 2016;34:380-91
18. Lian Q, Zhang Y, Zhang J, Zhang HK, Wu X, Zhang Y. et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation. 2010;121:1113-23
19. Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J. et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348:1160-3
20. Henrique D, Abranches E, Verrier L, Storey KG. Neuromesodermal progenitors and the making of the spinal cord. Development. 2015;142:2864-75
21. Li W, Huang L, Zeng J, Lin W, Li K, Sun J. et al. Characterization and transplantation of enteric neural crest cells from human induced pluripotent stem cells. Mol Psychiatry. 2018;23:499-508
22. Wilson V, Olivera-Martinez I, Storey KG. Stem cells, signals and vertebrate body axis extension. Development. 2009;136:1591-604
23. Gouti M, Tsakiridis A, Wymeersch FJ, Huang Y, Kleinjung J, Wilson V. et al. In vitro generation of neuromesodermal progenitors reveals distinct roles for wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biol. 2014;12:e1001937
24. Kimelman D, Kirschner M. Synergistic induction of mesoderm by FGF and TGF-beta and the identification of an mRNA coding for FGF in the early Xenopus embryo. Cell. 1987;51:869-77
25. Lippmann ES, Williams CE, Ruhl DA, Estevez-Silva MC, Chapman ER, Coon JJ. et al. Deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem Cell Reports. 2015;4:632-44
26. Verrier L, Davidson L, Gierlinski M, Dady A, Storey KG. Neural differentiation, selection and transcriptomic profiling of human neuromesodermal progenitor-like cells in vitro. Development. 2018:145
27. Buckley SM, Aranda-Orgilles B, Strikoudis A, Apostolou E, Loizou E, Moran-Crusio K. et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell. 2012;11:783-98
28. Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275-80
29. Pourquie O. Vertebrate somitogenesis. Annu Rev Cell Dev Biol. 2001;17:311-50
30. Pontikoglou C, Deschaseaux F, Sensebe L, Papadaki HA. Bone marrow mesenchymal stem cells: biological properties and their role in hematopoiesis and hematopoietic stem cell transplantation. Stem Cell Rev. 2011;7:569-89
31. Pelekanos RA, Li J, Gongora M, Chandrakanthan V, Scown J, Suhaimi N. et al. Comprehensive transcriptome and immunophenotype analysis of renal and cardiac MSC-like populations supports strong congruence with bone marrow MSC despite maintenance of distinct identities. Stem Cell Res. 2012;8:58-73
32. Zhang X, Huang W, Chen X, Lian Y, Wang J, Cai C. et al. CXCR5-Overexpressing Mesenchymal Stromal Cells Exhibit Enhanced Homing and Can Decrease Contact Hypersensitivity. Mol Ther. 2017;25:1434-47
33. Sheng G. The developmental basis of mesenchymal stem/stromal cells (MSCs). BMC Dev Biol. 2015;15:44
34. Zhao Q, Gregory CA, Lee RH, Reger RL, Qin L, Hai B. et al. MSCs derived from iPSCs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow MSCs. Proc Natl Acad Sci U S A. 2015;112:530-5
35. Lee EJ, Hwang I, Lee JY, Park JN, Kim KC, Kim GH. et al. Hepatocyte Growth Factor Improves the Therapeutic Efficacy of Human Bone Marrow Mesenchymal Stem Cells via RAD51. Mol Ther. 2018;26:845-59
36. Chijimatsu R, Ikeya M, Yasui Y, Ikeda Y, Ebina K, Moriguchi Y. et al. Characterization of Mesenchymal Stem Cell-Like Cells Derived From Human iPSCs via Neural Crest Development and Their Application for Osteochondral Repair. Stem Cells Int. 2017;2017:1960965
37. Kimbrel EA, Kouris NA, Yavanian GJ, Chu J, Qin Y, Chan A. et al. Mesenchymal stem cell population derived from human pluripotent stem cells displays potent immunomodulatory and therapeutic properties. Stem Cells Dev. 2014;23:1611-24
38. Roux C, Saviane G, Pini J, Belaid N, Dhib G, Voha C. et al. Immunosuppressive Mesenchymal Stromal Cells Derived from Human-Induced Pluripotent Stem Cells Induce Human Regulatory T Cells In Vitro and In Vivo. Front Immunol. 2017;8:1991
39. Yan L, Jiang B, Li E, Wang X, Ling Q, Zheng D. et al. Scalable Generation of Mesenchymal Stem Cells from Human Embryonic Stem Cells in 3D. Int J Biol Sci. 2018;14:1196-210
40. Yun YI, Park SY, Lee HJ, Ko JH, Kim MK, Wee WR. et al. Comparison of the anti-inflammatory effects of induced pluripotent stem cell-derived and bone marrow-derived mesenchymal stromal cells in a murine model of corneal injury. Cytotherapy. 2017;19:28-35
41. Wang X, Kimbrel EA, Ijichi K, Paul D, Lazorchak AS, Chu J. et al. Human ESC-derived MSCs outperform bone marrow MSCs in the treatment of an EAE model of multiple sclerosis. Stem Cell Reports. 2014;3:115-30
42. Kang R, Zhou Y, Tan S, Zhou G, Aagaard L, Xie L. et al. Mesenchymal stem cells derived from human induced pluripotent stem cells retain adequate osteogenicity and chondrogenicity but less adipogenicity. Stem Cell Res Ther. 2015;6:144
43. Moslem M, Eberle I, Weber I, Henschler R, Cantz T. Mesenchymal Stem/Stromal Cells Derived from Induced Pluripotent Stem Cells Support CD34(pos) Hematopoietic Stem Cell Propagation and Suppress Inflammatory Reaction. Stem Cells Int. 2015;2015:843058
44. Jiang B, Li Y, Wang X, Li E, Murphy K, Vaccaro K. et al. Mesenchymal stem cells derived from human pluripotent cells, an unlimited and quality-controllable source, for therapeutic applications. Stem Cells. 2018
45. McGrath M, Tam E, Sladkova M, AlManaie A, Zimmer M, de Peppo GM. GMP-compatible and xeno-free cultivation of mesenchymal progenitors derived from human-induced pluripotent stem cells. Stem Cell Res Ther. 2019;10:11
Author contact
![]() Corresponding authors: Weiqiang Li, E-mail: liweiq6sysu.edu.cn; Tel: +8620-87335982; Fax: +8620-87335858 and Andy Peng Xiang, E-mail: xiangpsysu.edu.cn; Tel: +8620-87335822; Fax: +8620-87335858
Corresponding authors: Weiqiang Li, E-mail: liweiq6sysu.edu.cn; Tel: +8620-87335982; Fax: +8620-87335858 and Andy Peng Xiang, E-mail: xiangpsysu.edu.cn; Tel: +8620-87335822; Fax: +8620-87335858