Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1614-1633. doi:10.7150/thno.30398 This issue Cite
Research Paper
SIRT3-Dependent Mitochondrial Dynamics Remodeling Contributes to Oxidative Stress-Induced Melanocyte Degeneration in Vitiligo
Xiuli Yi*, Weinan Guo*, Qiong Shi*, Yuqi Yang, Weigang Zhang, Xuguang Chen, Pan Kang, Jiaxi Chen, Tingting Cui, Jinyuan Ma, Huina Wang, Sen Guo, Yuqian Chang, Ling Liu, Zhe Jian, Lin Wang, Qian Xiao, Shuli Li ![]() , Tianwen Gao
, Tianwen Gao ![]() , Chunying Li
, Chunying Li ![]()
Department of Dermatology, Xijing hospital, Fourth Military Medical University, Xi'an, Shannxi, China
*These authors contributed equally to this work.
Received 2018-10-3; Accepted 2019-1-22; Published 2019-2-28
Citation:
Yi X, Guo W, Shi Q, Yang Y, Zhang W, Chen X, Kang P, Chen J, Cui T, Ma J, Wang H, Guo S, Chang Y, Liu L, Jian Z, Wang L, Xiao Q, Li S, Gao T, Li C. SIRT3-Dependent Mitochondrial Dynamics Remodeling Contributes to Oxidative Stress-Induced Melanocyte Degeneration in Vitiligo. Theranostics 2019; 9(6):1614-1633. doi:10.7150/thno.30398. https://www.thno.org/v09p1614.htm
Other stylesAbstract

Mitochondrial dysregulation has been implicated in oxidative stress-induced melanocyte destruction in vitiligo. However, the molecular mechanism underlying this process is merely investigated. Given the prominent role of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase Sirtuin3 (SIRT3) in sustaining mitochondrial dynamics and homeostasis and that SIRT3 expression and activity can be influenced by oxidative stress-related signaling, we wondered whether SIRT3 could play an important role in vitiligo melanocyte degeneration by regulating mitochondrial dynamics.
Methods: We initially testified SIRT3 expression and activity in normal and vitiligo melanocytes via PCR, immunoblotting and immunofluorescence assays. Then, cell apoptosis, mitochondrial function and mitochondrial dynamics after SIRT3 intervention were analyzed by flow cytometry, immunoblotting, confocal laser microscopy, transmission electron microscopy and oxphos activity assays. Chromatin immunoprecipitation (ChIP), co-immunoprecipitation (Co-IP), immunoblotting and immunofluorescence assays were performed to clarify the upstream regulatory mechanism of SIRT3. Finally, the effect of honokiol on protecting melanocytes and the underlying mechanism were investigated via flow cytometry and immunoblotting analysis.
Results: We first found that the expression and the activity of SIRT3 were significantly impaired in vitiligo melanocytes both in vitro and in vivo. Then, SIRT3 deficiency led to more melanocyte apoptosis by inducing severe mitochondrial dysfunction and cytochrome c release to cytoplasm, with Optic atrophy 1 (OPA1)-mediated mitochondrial dynamics remodeling involved in. Moreover, potentiated carbonylation and dampened peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) activation accounted for SIRT3 dysregulation in vitiligo melanocytes. Finally, we proved that honokiol could prevent melanocyte apoptosis under oxidative stress by activating SIRT3-OPA1 axis.
Conclusions: Overall, we demonstrate that SIRT3-dependent mitochondrial dynamics remodeling contributes to oxidative stress-induced melanocyte degeneration in vitiligo, and honokiol is promising in preventing oxidative stress-induced vitiligo melanocyte apoptosis.
Keywords: vitiligo, SIRT3, mitochondrial dynamics, melanocyte, apoptosis
Introduction
Vitiligo is an acquired disfiguring skin disease characterized by progressive depigmentation due to the destruction of epidermal melanocytes, and it affects approximately 0.5 % to 1.0 % in the populations worldwide [1-3]. Several pathogenic factors including genetic variation, environmental stimulus, autoimmunity, oxidative stress and mitochondrial dysregulation account for the onset and the development of vitiligo [4-6]. Recently, the critical role of oxidative stress in vitiligo pathogenesis has been well understood [7-9]. Specifically, oxidative stress leads to abnormal immunological response and melanocyte destruction, which is associated with ER stress [9, 10]. However, the administration of anti-oxidative agents alone is not able to reverse the degeneration of melanocytes and induce re-pigmentation of skin in vitiligo patients [11, 12], which is probably due to the occurrence and deposits of peroxidised proteins and lipids, damaged DNA and dysfunctional mitochondria in lesional melanocytes [8, 13-15]. Therefore, further elucidation on the molecular mechanism underlying oxidative stress-induced melanocyte degeneration is important for finding more druggable targets for vitiligo therapy.
Mitochondrion is the crucial cytoplasmic organelle responsible for the generation of energy, and more importantly, for maintaining cellular homeostasis in eukaryotic cells [16]. The dysregulation of mitochondrial function is involved in quite a few diseases, especially oxidative stress-associated tissue and cell damages [17-19]. Previous studies have demonstrated that the polymorphisms of superoxide dismutase (SOD2), a crucial mitochondrial reactive oxygen species (ROS) scavenger, are genetic risk factors for the susceptibility and the progression of vitiligo [20]. Moreover, several lines of evidences have revealed the disordered ultrastructure of mitochondria in vitiligo melanocytes [13, 21], especially the disappearance of cristae, which could impede respiratory chain complex assembly and boost ROS production [22-24], indicating the close linkage between mitochondrial function impairment and melanocyte's susceptibility to oxidative stress. Intriguingly, it is reported that mitochondrial structure is dynamically regulated by two counteracting processes, that are, mitochondrial fusion and fission, with dynamin-related protein 1 (DRP1), fission 1 (Fis1), mitochondrial fission factor (MFF), mitofusin 1 (MFN1), mitofusin 2 (MFN2) and optic atrophy 1 (OPA1) as key regulatory elements [25]. In particular, the deficiency of OPA1-meidated mitochondrial fusion coordinates apoptotic cristae remodeling, cytochrome c release to cytoplasm and attenuated oxidative phosphorylation efficacy under stressful conditions [23, 26, 27]. Therefore, the alteration of mitochondrial dynamics is critical in connecting external stress to cellular homeostasis, while its role has not been clarified in the procedure of melanocyte degeneration in vitiligo.
The processes of mitochondrial fusion and fission are spatiotemporally regulated by multiple post-translational modifications, such as phosphorylation, sumoylation, ubiquitination and acetylation [28-31]. To be specific, protein acetylation, that was evolutionarily conserved, was sensitive to altered intracellular energetic status caused by external stimuli [32], and could be reversibly governed by various acetyltransferases and deacetylases [33]. Sirtuins are nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases comprising of seven members (SIRT1-SIRT7) in humans, among which three (SIRT3, SIRT4 and SIRT5) are exclusively localized to mitochondria [34]. As the most crucial one, SIRT3 has been documented as a versatile molecule in regulating mitochondrial function with the implications in metabolic enzyme activity, oxidative phosphorylation efficacy, anti-oxidant machinery as well as mitochondrial dynamics [35-38]. More importantly, SIRT3 is capable to facilitate mitochondrial fusion via deacetylating OPA1 [29], thus exerting protective effect on cells against detrimental stimuli. Given that excessive oxidative stress could profoundly dampen SIRT3 activity and expression [39, 40], we hypothesized that SIRT3- dependent mitochondrial dynamics remodeling via OPA1 may contribute to melanocyte destruction under oxidative stress.
Honokiol (HKL) [2-(4-hydroxy-3-prop-2-enyl-phenyl)-4-prop-2-enyl-phenol], a natural biphenolic compound derived from the bark of magnolia trees, has been proved as a specific pharmacological activator of SIRT3 and has anti-inflammatory, anti-oxidative, anti-tumor, analgesic and neuroprotective properties [41-44]. In the present study, we finally investigated the therapeutic effect of HKL on protecting melanocytes from oxidative stress and the underlying mechanism.
Results
Impaired SIRT3 expression and activity in vitiligo melanocytes under oxidative stress
To investigate the role of SIRT3 in oxidative stress-driving melanocyte destruction in vitiligo, we first testified the alterations of SIRT3 expression and activity in normal melanocyte cell line PIG1 and vitiligo melanocyte cell line PIG3V after the treatment with 1.0 mM H2O2 (the canonical oxidative stress inducer) for 24 h in vitro that could induce significant melanocyte apoptosis as described in our previous study [7] (Supplementary Figures S1A -C). Notably, the up-regulation of SIRT3 mRNA and protein levels were increased as the concentrations of H2O2 rose in PIG1 cells (Supplementary Figures S1D and E). Moreover, the protein expression level of SIRT3 also increased in a time-dependent manner (Supplementary Figure S1F). As a result, our quantitative real-time PCR (qRT-PCR) and immunoblotting assays showed prominent up-regulation of both SIRT3 mRNA and protein levels in response to H2O2 treatment in PIG1 cells. However, it displayed minimal change of SIRT3 expression in PIG3V cells after H2O2 treatment (Figures 1A and B). Consistent with this, the immunofluorescence analysis displayed that SIRT3 expression was increased in PIG1 cells under oxidative stress, whereas it showed marginal alteration in PIG3V cells (Figure 1C). Aside from this, we discovered that the activity of SIRT3 was profoundly potentiated in PIG1 cells after H2O2 stimulation, but was negligibly changed in PIG3V cells (Figure 1D).
Figure 1
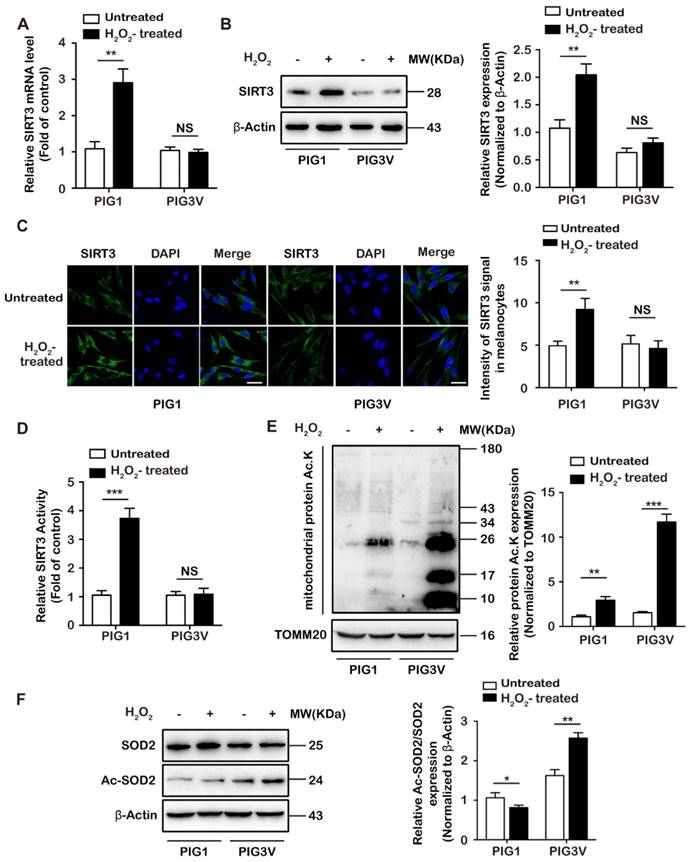
Impaired SIRT3 expression and activity in vitiligo melanocytes under oxidative stress. (A) The relative mRNA level of SIRT3 in PIG1 and PIG3V cells after the treatment of 1.0 mM H2O2 for 24 h. Data represent mean ± SD (n = 3). (B) The protein level of SIRT3 in PIG1 and PIG3V cells after 1.0 mM H2O2 treatment. β-Actin was detected as loading control. Data represent mean ± SD (n = 3). (C) Immunofluorescence staining analysis of SIRT3 expression in PIG1 and PIG3V cells after 1.0 mM H2O2 treatment, Nuclei were counterstained with DAPI (blue). Data are representative of three independently performed experiments. Scale bar = 50 μm (magnification: 600 ×). Intensity of SIRT3 signal in melanocytes was quantified using Image J software. (D) SIRT3 activity in PIG1 and PIG3V cells after 1.0 mM H2O2 treatment. Data represent mean ± SD (n = 3). (E) Acetylation of mitochondiral protein in PIG1 and PIG3V cells after H2O2 treatment. TOMM20 was detected as loading control. Data represent mean ± SD (n = 3). (F) Acetylation of SOD2 in PIG1 and PIG3V cells after 1.0 mM H2O2 treatment for 24 h. β-Actin was detected as loading control. Data represent mean ± SD (n = 3). p value was calculated by two-tailed Student's t-test. *p < 0.05, **p < 0.01, ***p < 0.001, NS, non-significant.
Since SIRT3 is the main deacetylase governing the mitochondrial acetylation network [35], we detected the acetylation modification status of total mitochondrial proteins under oxidative stress. Compared with untreated group, the acetylation of mitochondrial protein in PIG3V cells treated with H2O2 was significantly increased, while in comparison to PIG1 cells, the acetylation of mitochondrial protein was augmented more significantly in PIG3V cells, reflecting the deficient activation of SIRT3 in PIG3V cell line (Figure 1E). To be more specific, we examined the acetylation of superoxide dismutase (SOD2), the crucial mitochondrial ROS scavenger and known as key deacetylase substrate of SIRT3 [37]. Our data revealed that although SOD2 acetylation displayed a slightly decreasing trend in PIG1 cells after H2O2 treatment, it was markedly up-regulated in PIG3V cells, further confirming the deficient SIRT3 activation in vitiligo melanocytes in vitro (Figure 1F). Besides, we examined the expression of SIRT3 in PIG1, PIG3V cell lines and normal human epidermal melanocytes (NHEMs) in the same panel, and found that the SIRT3 expression in PIG3V cells sharply decreased compared to PIG1 cell lines and NHEMs (Supplementary Figure S1G). Importantly, we verified the alterations of SIRT3 expression and activity in NHEMs after treatment with H2O2, and observed a result consistent with that in PIG1 cells, which indicated that SIRT3 expression and activity were both significantly increased in melanocytes under oxidative stress (Supplementary Figure S1H-L).
Figure 2
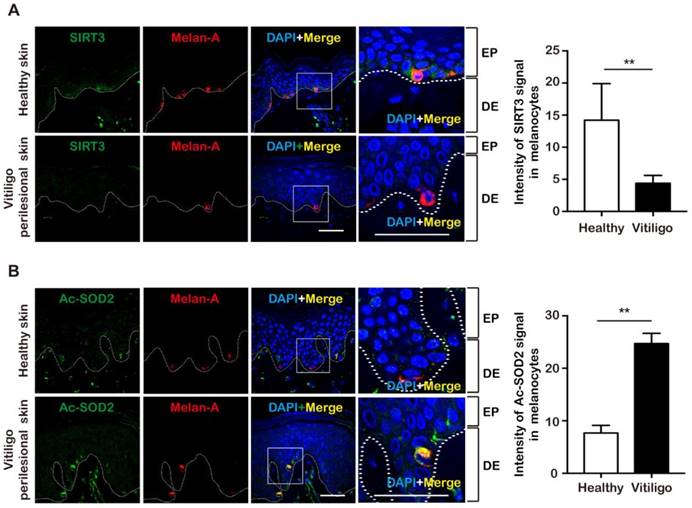
Impaired SIRT3 expression and activity in melanocytes of vitiligo perilesional skin. (A) Representative images of the expression of SIRT3 (green) in healthy skin (n = 8) and vitiligo perilesional skin (n = 8) detected by immunofluorescence. Melanocytes in the epidermis were stained with antibody to Melan-A (red). Nuclei were counterstained with DAPI (blue). Scale bar = 50 μm (Magnification: the left = 600 ×; the right = 1800 ×). EP, epidermis; DE, dermis. Bar graphs represent the mean values of 8 samples and 10 fields of view per sample. Mean ± SD is shown. (B) Representative images of the expression of Ac-SOD2 (green) in healthy skin (n = 8) and vitiligo perilesional skin (n = 8) detected by using immunofluorescence. Melanocytes in the epidermis were stained with antibody to Melan-A (red). Nuclei were counterstained with DAPI (blue). Scale bar = 50 μm (Magnification: the left = 600 ×; the right = 1800 ×). Intensity of SIRT3 signal in melanocytes was quantified using Image J software. Bar graphs represent the mean values of 8 samples and 10 fields of view per sample. Mean ± SD is shown. p value was calculated by two-tailed Student's t-test. **p < 0.01.
To further determine the expression and activity of SIRT3 in vitiligo melanocytes in vivo, we collected perilesional skin samples from vitiligo patients as well as normal skin samples from healthy individuals, and then detected SIRT3 and acetylated superoxide dismutase (Ac-SOD2) expressions in melanocytes by immunofluorescence staining assay. Melan-A was concurrently stained for identifying melanocytes [2]. Compared with normal skin, SIRT3 was weakly stained in melanocytes of vitiligo perilesional skin (Figure 2A). Meanwhile, the difference was significant between eight pairs of vitiligo perilesional skin samples and normal skin samples (Figure 2A). Moreover, we found that Ac-SOD2 expression was significantly higher in melanocytes of vitiligo perilesional skin compared with normal skin (Figure 2B). These findings suggested that both the expression and deacetylase activity of SIRT3 were dampened in vitiligo melanocytes, prompting us to speculate that defect SIRT3 function might be involved in oxidative stress-induced melanocyte degeneration in vitiligo.
SIRT3 deficiency contributes to cell apoptosis and mitochondrial dysfunction in melanocytes
To evaluate the role of SIRT3 in oxidative stress-induced melanocyte destruction in vitiligo, we first knocked down the expression of SIRT3 by (62.17 ± 2.93) % in PIG1 cells and by (64.28 ± 3.6) % in NHEMs, respectively (Supplementary Figures S2A and B). In response to the treatment with different concentrations of H2O2, the knockdown of SIRT3 attenuated cell viability, whereas SIRT3 deficiency alone had marginal impact (Figure 3A and Supplementary Figure S3A). Concurrently, our flow cytometry assay showed that the knockdown of SIRT3 exacerbated oxidative stress-induced cell apoptosis in both PIG1 cells and NEHMs (Figure 3B and Supplementary Figure S3B). In line with this, the immunoblotting analysis showed that under oxidative stress, SIRT3 deficiency promoted the expressions of pro-apoptotic cleaved poly-ADP-ribose polymerase (cleaved-PARP) and B cell lymphoma 2-associated X protein (Bax), and suppressed the expression of anti-apoptotic B cell lymphoma 2 (Bcl2) (Figure 3C and Supplementary Figures S3C and S3D). Notably, the expression of mitochondrial apoptotic pathway mediator cleaved-caspase 9 was also prominently increased, indicating the involvement of mitochondrial dysfunction in initiating cell apoptosis (Figure 3C and Supplementary Figures S3C). Therefore, we analyzed the functional status of mitochondria via the detection of mitochondrial ROS, intracellular ATP level and mitochondrial membrane potential. Not surprisingly, the knockdown of SIRT3 led to more mitochondrial ROS generation (Figure 3D and Supplementary Figure S3D), lessened ATP level (Figure 3E and Supplementary Figure S3E) and dissipative mitochondrial membrane potential (Figure 3F and Supplementary Figure S3F) in both PIG1 cells and NHEMs under oxidative stress.
What's more, we obtained SIRT3 overexpression in PIG3V cells to see whether the rescue of SIRT3 function could ameliorate oxidative stress-induced destruction of vitiligo melanocytes. We first checked the overexpression efficiency of SIRT3 in PIG3V cells. The result showed that SIRT3 expression was up-regulated by 1.132 ± 0.09382 times (Supplementary Figure S4A). As predicted, SIRT3 overexpression suppressed the decline of cell viability (Supplementary Figure S4B) and the increase of cell apoptosis after H2O2 treatment (Supplementary Figure S4C). Consistently, the expressions of pro-apoptotic cleaved-PARP, cleaved-caspase 9 and Bax were reduced, while the expression of anti-apoptotic Bcl2 was increased (Supplementary Figure S4D). In addition, the generation of mitochondrial ROS was significantly reduced (Supplementary Figure S4E), whereas the intracellular ATP level (Supplementary Figure S4F) and mitochondrial membrane potential were increased after SIRT3 overexpression (Supplementary Figure S4G). Collectively, our results demonstrated that SIRT3 deficiency led to the dysfunction of mitochondria and the activation of mitochondrial apoptotic pathway to induce cell death of vitiligo melanocytes.
SIRT3-dependent mitochondrial dynamics remodeling coordinates cytochrome c release and the activities of respiratory complexes under oxidative stress
Since the release of cytochrome c from mitochondria to cytoplasm is the key step for connecting mitochondrial damage to cell apoptosis [45], we wondered whether cytochrome c release was involved in the effect of SIRT3 on cell death under oxidative stress. We conducted the separation of mitochondrial and cytoplasmic compartments to see the distribution of cytochrome c in both PIG1 and PIG3V cell lines. As a result, the knockdown of SIRT3 led to the reduction of mitochondrial cytochrome c and concurrent increase of cytoplasmic cytochrome c in PIG1 cells (Figure 4A). In addition, we obtained the overexpression of SIRT3 in PIG3V cells, and found attenuated translocation of cytochrome c from mitochondria to cytoplasm (Figure 4B). Therefore, SIRT3 was capable to regulate mitochondrial cytochrome c release under oxidative stress. Aside from this, we investigated the alteration of mitochondrial bioenergetic status by examining the activities of respiratory chain complexes, which determines the intracellular ATP level and has critical impact on mitochondrial membrane potential and mitochondrial ROS generation [46]. As was shown, the knockdown of SIRT3 led to a general decline of respiratory chain complex activities in PIG1 cells, whereas the overexpression of SIRT3 exerted promotive role in respiratory capacity in PIG3V cells under oxidative stress (Figure 4C). Collectively, SIRT3 coordinated mitochondrial cytochrome c release and respiratory efficacy to confer protective effect against oxidative stress-induced melanocyte destruction.
Figure 3
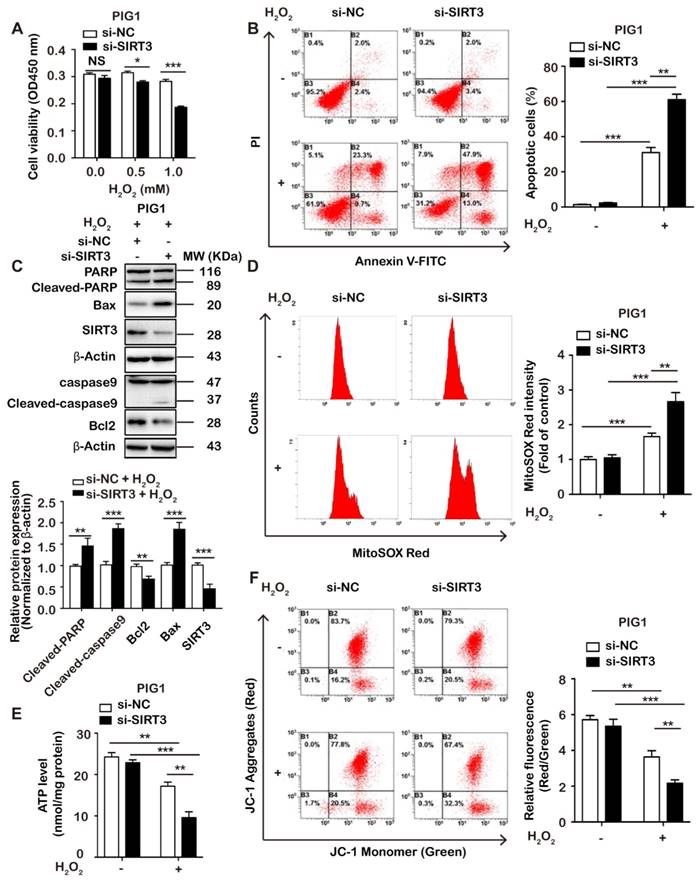
SIRT3 deficiency contributes to cell apoptosis and mitochondrial dysfunction in melanocytes. (A) PIG1 cells transfected with si-NC or si-SIRT3 were treated by different concentrations of H2O2 (0, 0.5, 1.0 mM), and the cell viability was determined by CCK-8. si-NC refers to negative control small interfering RNA, and si-SIRT3 refers to small interfering RNA against SIRT3. Data represent mean ± SD (n = 3). (B) The apoptotic level of PIG1 cells transfected with si-NC or si-SIRT3 after H2O2 treatment for 24 h was examined by flow cytometry. Bar graphs represent mean ± SD (n = 3). (C) The level of apoptosis-related proteins in PIG1 cells was detected by immunoblotting. Data are representative of three independently experiments. Quantitative analysis on the apoptosis-related proteins of interest as indicated (mean ± SD, n = 3). (D) The mitochondrial ROS level of PIG1 cells was examined by MitoSOX™ Red mitochondrial superoxide indicator staining. Bar graphs represent mean ± SD (n = 3). (E) Assessment of ATP level in PIG1 cells with treatment as indicated. Data represent mean ± SD of triplicates. (F) The mitochondrial membrane potential level of PIG1 cells was examined by JC-1 staining. The scatter plot of the flow cytometry analysis shows the distribution of JC-1 aggregates (Red) and JC-1 monomer (Green) cell population. Histogram calculated the relative ratio of Red against Green fluorescence (mean ± SD, n=3). p value was calculated by two-tailed Student's t-test. *p < 0.05, **p < 0.01, ***p < 0.001, NS, non-significant.
Figure 4
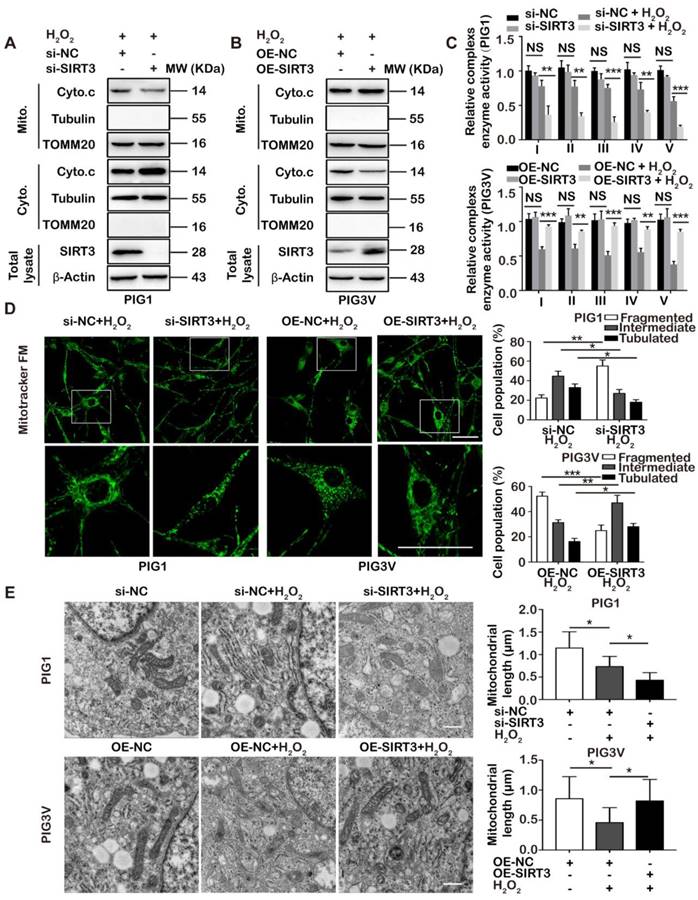
SIRT3-dependent mitochondrial dynamics remodeling coordinates cytochrome c release and respiratory complex activities under oxidative stress. (A, B) PIG1 cells were transfected with si-NC or si-SIRT3 following the treatment of 1.0 mM H2O2 for 24 h. si-NC refers to negative control small interfering RNA, and si-SIRT3 refers to small interfering RNA against SIRT3. PIG3V cells were transfected with plasmid OE-NC or OE-SIRT3 following treatment with 1.0 mM H2O2 for 24 h. OE-NC refers to transfection with control plasmids, and OE-SIRT3 refers to transfection with SIRT3 plasmids. The expression of cytochrome c in mitochondria or cytoplasm was detected. TOMM20 was detected as loading control for mitochondrial fraction. Tubulin was detected as loading control for cytosolic fraction. β-Actin was detected as loading control for the whole cell lysate. Data are representative of three independent experiments. (C) Relative enzyme activity of respiratory complexes I, II, III, IV and V were measured in PIG1 and PIG3V cells with treatment as indicated. Data represent mean ± SD of triplicates. (D) Representative confocal microscope images of the mitochondrial network in PIG1 and PIG3V cells with treatment as indicated. Scale bar = 50 μm (Magnification: the upper = 600 ×; the under = 1800 ×). The proportion of PIG1 and PIG3V cells (n = 100 cells for each sample) with tubulated, intermediate and fragmented mitochondria was quantified. Data are representative of three independent experiments. (E) Representative transmission electron microscopy images of the mitochondrial network in PIG1 and PIG3V cells. Scale bar = 500 nm. Mitochondrial length was quantified using Image J software in PIG1 and PIG3V cells. One hundred mitochondria were randomly selected in each treatment group. Data are representative of three independent experiments. Mean ± SD is shown. p value was calculated by two-tailed Student's t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
It has been well documented that mitochondrial cristae contain the bulk of cytochrome c. The cristae remodeling caused by dynamics alteration could lead to cytochrome c mobilization and the activation of apoptotic pathway under stressful conditions [27, 47]. Moreover, mitochondrial dynamics-mediated cristae shape could influence respiratory chain complex assembly and activity [23]. Given these, we wondered whether SIRT3 deficiency-induced cytochrome c release and suppressed complex activities were associated with mitochondrial dynamics remodeling. We first employed immunofluorescence staining of mitochondrial dye MitoTracker Green and found that in response to H2O2 treatment, the knockdown of SIRT3 led to dramatic mitochondrial fission in PIG1 cells, as indicated by a significant increase in the percentage of cells with mitochondria fragmentation. In parallel, the overexpression of SIRT3 promoted the elongation of mitochondria in PIG3V cells, with increased cells with mitochondrial tubulation (Figure 4D). Moreover, we used transmission electron microscope assay to see the change of mitochondrial ultrastructure within the cells. As was shown, the knockdown of SIRT3 resulted in prominent fragmentation along with the disappearance of normal cristae shape in PIG1 cells under oxidative stress. Consistent with this, the overexpression of SIRT3 led to mitochondrial elongation and the recovery of cristae shape in PIG3V cells (Figure 4E). Therefore, SIRT3 was greatly implicated in the regulation of mitochondrial dynamics and subsequent cristae formation in melanocytes under oxidative stress.
Mitochondrial dynamics is governed by a series of regulators, including DRP1, Fis1, MFN1, MFN2 and OPA1 [25]. To investigate how SIRT3 impacted mitochondrial dynamics in melanocytes under oxidative stress, we first examined the expressions of DRP1, Fis1, MFN1, MFN2 and OPA1, but discovered that the knockdown of SIRT3 showed marginal influence (Supplementary Figure S5). It has been well recognized that post-translational modifications of these regulators are greatly involved in regulating mitochondrial fusion and fission [28-31]. More importantly, OPA1 was a substrate of SIRT3 that could be directly deacetylated and activated to promote mitochondrial fusion [29]. Therefore, we performed co-immunoprecipitation analysis and found that the knockdown of SIRT3 increased the acetylation of OPA1 under oxidative stress in melanocytes, indicating that OPA1 could mediate the effect of SIRT3 on mitochondrial dynamics (Figure 5A). Then, we overexpressed OPA1 after the knockdown of SIRT3, and our immunofluorescence staining analysis displayed that OPA1 overexpression reversed SIRT3 deficiency-induced mitochondrial fragmentation (Figure 5B). Intriguingly, the overexpression of OPA1 reversed SIRT3 deficiency-induced cytochrome c release (Figure 5C) and the impairment of respiratory chain complex activities under oxidetive stress (Figure 5D). Therefore, SIRT3 regulated mitochondrial dynamics remodeling via OPA1 to coordinate cytochrome c release and respiratory complex activities, which was greatly implicated in oxidative stress-induced cell apoptosis and mitochondrial dysfunction in vitiligo melanocytes.
SIRT3 deficiency contributes to cell apoptosis and mitochondrial dysfunction via OPA1
Thereafter, we went on to investigate whether the protective role of SIRT3 against oxidative stress-induced melanocyte apoptosis relied on OPA1. As was shown, OPA1 overexpression prominently repressed SIRT3 deficiency-induced melanocyte apoptosis under oxidative stress (Figure 5E and Supplementary Figure S6A). Furthermore, the expressions of pro-apoptotic cleaved-caspase 9, cleaved- PARP and Bax were significantly decreased, while the expression of anti-apoptotic Bcl2 was increased (Figure 5F and Supplementary Figure S6B). Moreover, we found that OPA1 overexpression remarkably inhibited the generation of mitochondrial ROS (Figure 5G and Supplementary Figure S6C) and the dissipation of mitochondrial membrane potential (Figure 5H and Supplementary Figure S6D) induced by the knockdown of SIRT3 after H2O2 treatment. Meanwhile, the decline of intracellular ATP level was significantly reversed (Figure 5I). Taken together, SIRT3 deficiency contributed to cell apoptosis and mitochondrial dysfunction in an OPA1-dependent manner.
A recent study shows that SIRT4 interacts with OPA1 to regulate mitochondrial quality and mitophagy [48]. In order to see whether OPA1-mediated mitochondrial dynamics remodeling was associated with SIRT4 after SIRT3 intervention under oxidative stress, we examined the expression of SIRT4 in SIRT3-silenced PIG1 cells with or without H2O2 treatment. Our results showed that there was no obvious alteration of SIRT4 expression after the intervention of SIRT3 under oxidative stress (Supplementary Figure S6E). Therefore, the alterations of the function of OPA1 and mitochondrial dynamics were not attributed to SIRT4 after SIRT3 intervention.
Figure 5
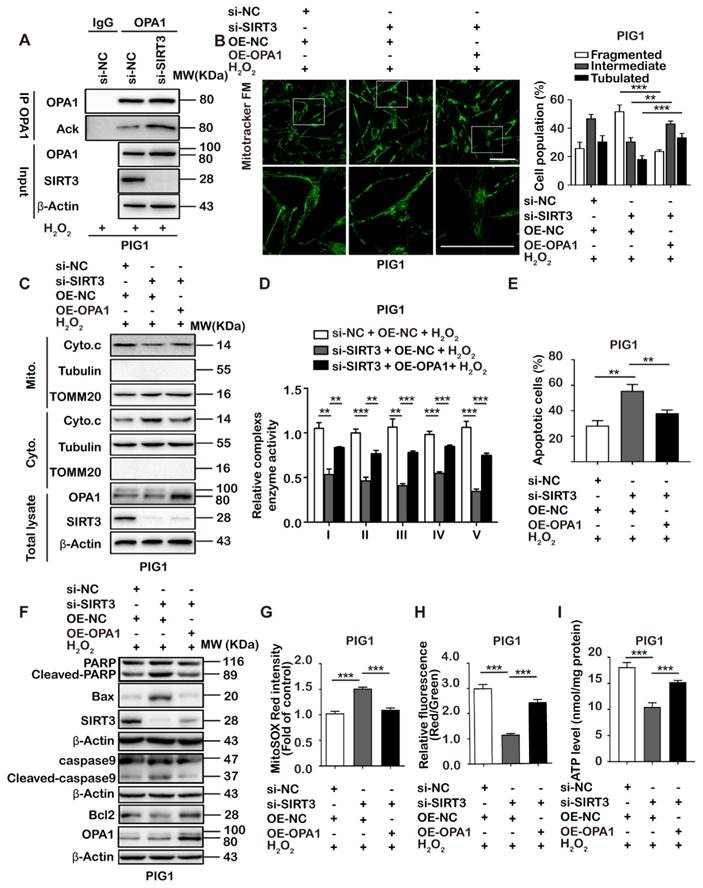
SIRT3 deficiency contributes to cell apoptosis and mitochondrial dysfunction via OPA1. (A) The acetylation level of OPA1 was determined in PIG1 cells transfected with si-NC or si-SIRT3 following the treatment with H2O2 by co-immunoprecipitation analysis. Data are representative of three independent experiments. (B) PIG1 cells were co-transfected with si-SIRT3 and OPA1 plasmids (OE-OPA1) or control plasmids (OE-NC), and then were treated with 1.0 mM H2O2 for 24 h. si-NC refers to negative control small interfering RNA, and si-SIRT3 refers to small interfering RNA against SIRT3. Representative confocal microscope images of the mitochondrial network in PIG1 cells were shown. Scale bar = 50 μm (magnification: the upper = 600 ×; the lower = 1800 ×).The proportion of PIG1 and PIG3V cells (n = 100 cells for each sample) with tubulated, intermediate and fragmented mitochondria was quantified. Data are representative of three independent experiments. (C) The expression of cytochrome c in mitochondrial or cytoplasmic compartments in PIG1 cells with indicated treatment. TOMM 20 was detected as loading control for mitochondrial fraction. Tubulin was detected as loading control for cytosolic fraction. β-Actin was detected as loading control for the whole cell lysate. (D) Relative enzyme activity of respiratory complexes I, II, III, IV and V were measured in PIG1 cells with indicated treatment. Data represent mean ± SD of triplicates. (E) The apoptotic level of PIG1 cells was examined by annexin V-FITC/PI staining (mean ± SD, n = 3). (F) The level of apoptosis-related proteins in PIG1 cells with indicated treatment. (G) The mitochondrial ROS level of PIG1 cells with indicated treatment was examined by MitoSOX™ Red mitochondrial superoxide indicator staining. Bar graphs represent mean ± SD (n = 3). (H) The mitochondrial membrane potential level of PIG1 cells was analyzed by JC-1 staining. The scatter plot of the flow cytometry analysis shows the distribution of JC-1 aggregates (Red) and JC-1 monomer (Green) cell population. Histogram calculated the relative ratio of Red against Green fluorescence (mean ± SD, n = 3). (I) Assessment of ATP level in PIG1 cells with treatment as indicated. Data represent mean ± SD of triplicates. p value was calculated by two-tailed Student's t-test. **p < 0.01, ***p < 0.001.
Oxidative stress simultaneously impairs SIRT3 activity and transcription
We have proved that SIRT3 deficiency contributed to melanocyte destruction under oxidative stress by regulating mitochondrial dynamics. Thereafter, we wondered how oxidative stress resulted in impaired SIRT3 activity and expression in vitiligo melanocytes. It has been proved that H2O2 treatment leads to excessive carbonyl stress in vitiligo melanocytes [8], and SIRT3 activity could be prominently dampened after carbonylation modification [39], the most frequent type of irreversible protein modification in response to oxidative stress. Hence, we speculated that oxidative stress might impair SIRT3 activity via carbonylation in vitiligo melanocytes. We first examined the levels of lipid peroxidation products 4-Hydroxynoenal (4-HNE) and Malondialdehyde (MDA). The results showed that H2O2 treatment led to more prominent increase of 4-HNE and MDA in PIG3V cells compared with PIG1 cells (Supplementary Figures S7A and B). In parallel, the protein carbonylation was also significantly increased, indicating more oxidative toxicity in vitiligo melanocytes (Supplementary Figure S7C). To further confirm our findings in vivo, we detected the level of 8-hydroxy-2 deoxyguanosine (8-OHdG), the oxidative DNA adducts that reflect the occurrence of oxidative damage [49], by using immunofluorescence staining in vitiligo perilesional skin samples and healthy skin samples. As was shown, the intensity of 8-OHdG co-localized to Melan-A was higher in vitiligo perilesional skin than that in healthy controls, demonstrating more severe oxidative stress in vitiligo melanocytes in vivo, which was consistent with previous studies (Figure 6A). To be more specific, we performed immunoprecipitation assay to see the carbonylation of SIRT3. It showed that in response to H2O2 treatment, PIG3V cells displayed prominent increase of SIRT3 carbonylation, whereas PIG1 cells showed slight reduction of SIRT3 carbonylation (Figure 6B). Therefore, the impaired activity of SIRT3 in vitiligo melanocytes could be partially resulted from potentiated carbonylation. Aside from the enzymatic activity, the expression of SIRT3 was also markedly dampened in vitiligo melanocytes under oxidative stress. Of note, we proved that this alteration occurred at the transcriptional level (Figure 1A). It has been documented that mitochondrial master regulator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) is among the most critical transcriptional factors of SIRT3 [50]. Herein, we obtained the knockdown of PGC1α expression, which expectedly led to significant reduction of SIRT3 expression in melanocytes (Supplementary Figures S7D and E). Subsequently, we turned to see whether PGC1α was involved in the impeded expression of SIRT3 in H2O2-treated vitiligo melanocytes. Through qRT-PCR and immunoblotting analysis, we found that the expression of PGC1α was drastically increased in PIG1 cells, but exerted marginal change in PIG3V cells after H2O2 stimulation (Figures 6C and D). More importantly, our chromatin immunoprecipitation assay showed impaired recruitment of PGC1α to the promoter of SIRT3 in vitiligo melanocytes under oxidative stress (Figure 6E). Moreover, we performed immunofluorescence staining analysis and discovered that compared with normal skin, the expression of PGC1α in melanocytes was decreased in perilesional skin from vitiligo patients (Figure 6F). Forwardly to see the relationship between oxidative stress and PGC1α-SIRT3 axis in vivo, we conducted correlation analysis of the immunofluorescence signal intensities. As a result, the oxidative stress indicator 8-OHdG was highly associated with SIRT3 and PGC1α expressions in vitiligo melanocytes (Supplementary Figure S7F). In addition, SIRT3 expression was prominently correlated with PGC1α expression in vitiligo melanocytes (Supplementary Figure S7F). Therefore, it was the suppression of PGC1α expression and transcriptional function that accounted for the impeded SIRT3 expression in vitiligo melanocytes. Collectively, oxidative stress was capable to simultaneously impair SIRT3 activity and transcription by regulating protein carbonylation and PGC1α transcriptional function.
HKL protects vitiligo melanocytes against oxidative stress by activating SIRT3-OPA1 axis
HKL, a natural biphenolic compound derived from the bark of magnolia trees, has been reported as a specific pharmacological activator of SIRT3 [51]. Intriguingly, it can enhance SIRT3 expression by nearly two-fold and bind to SIRT3 to further increase its activity [51]. Given that the dysregulation of SIRT3 contributed to melanocyte degeneration under oxidative stress, we wondered whether the specific activation of SIRT3 by HKL could be useful therapeutic approach for treating vitiligo. To this end, we treated vitiligo PIG3V cells with different concentrations of HKL after the exposure to H2O2. Our CCK8 assay revealed that the impaired cell viability of PIG3V cells induced by H2O2 was significantly recovered by the pre-treatment with HKL (Supplementary Figure S8A). Importantly, HKL treatment markedly increased the expression of SIRT3 at both mRNA and protein levels (Figures 7A and B). In addition, HKL was capable to enhance SIRT3 activity and reduce the acetylation of SOD2 (Figures 7C and D), confirming that HKL treatment promoted SIRT3 expression and activity in melanocytes concurrently. We previously showed that PGC1α was involved in the impeded expression of SIRT3 in H2O2-treated vitiligo melanocytes.
Figure 6
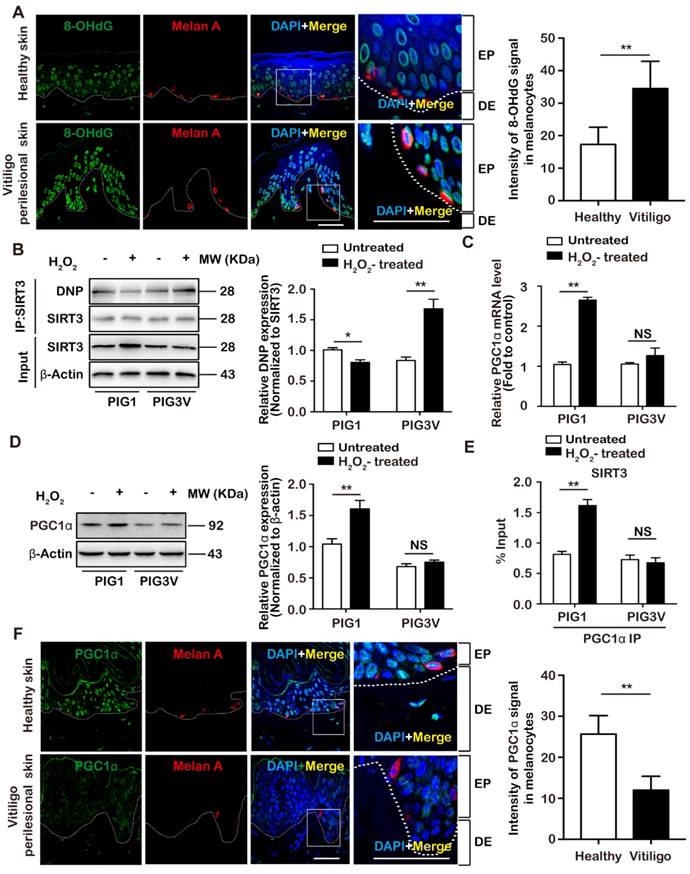
Oxidative stress simultaneously impairs SIRT3 activity and transcription. (A) Representative images of the expression of 8-OHdG (green) in healthy skin (n = 8) and vitiligo perilesional skin (n = 8) detected by using immunofluorescence. Melanocytes were stained with antibody to Melan-A (red). Nuclei were counterstained with DAPI (blue). Scale bar = 50 μm (Magnification: the left = 600 ×; the right = 1800 ×). Intensity of 8-OHdG signal in melanocytes was quantified using Image J software. Bar graphs represent mean values of 8 samples and 10 fields of view per sample. Mean ± SD is shown. (B) PIG1 and PIG3V cells were treated with 1.0 mM H2O2 for 24 h. Cell extracts were subjected to testify the carbnylation of SIRT3 via examing the interation between SIRT3 and DNPH. Data represent mean ± SD (n = 3). (C, D) PIG1 and PIG3V cells were treated with 1.0 mM H2O2 for 24 h. The mRNA and the protein levels of PGC1α in PIG1 and PIG3V cells were detected. Data represent mean ± SD (n = 3). (E) Enrichment of PGC1α to the promoter of SIRT3 after indicated treatment. Data are presented as mean ± SD. (F) Representative images of the expression of PGC1α (green) in healthy skin (n = 8) and vitiligo perilesional skin (n = 8) detected by using immunofluorescence. Melanocytes were stainied with antibody to Melan-A (red). Nuclei were counterstained with DAPI (blue). Scale bar = 50 μm (Magnification: the left = 600 ×; the right = 1800 ×). Intensity of PGC1α signal in melanocytes was quantified using Image J software. Bar graphs represent mean values of 8 samples and 10 fields of view per sample. Mean ± SD is shown. p value was calculated by two-tailed Student's t-test. *p < 0.05, **p < 0.01, ***p < 0.001, NS, non-significant.
Figure 7
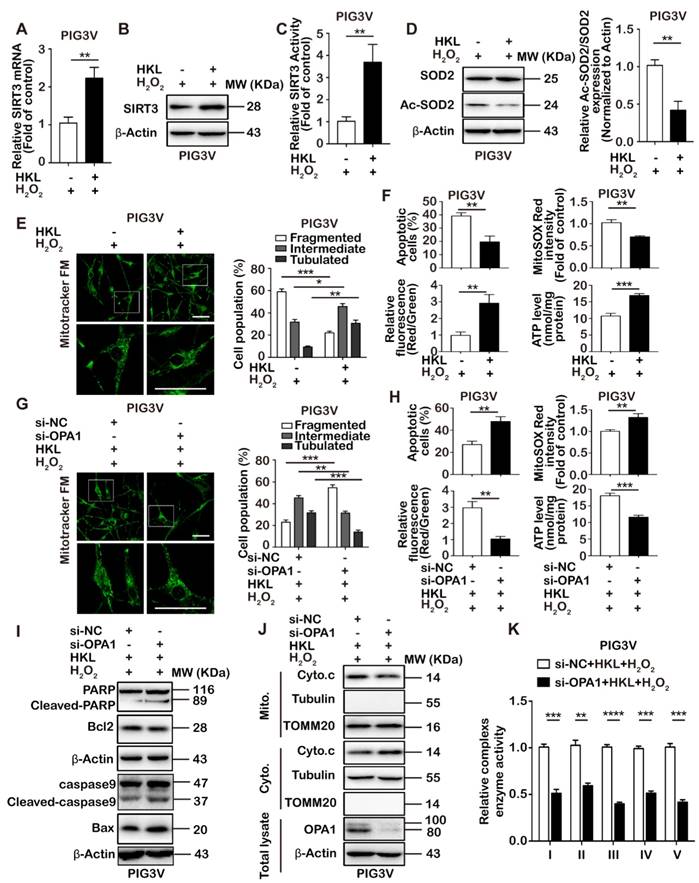
HKL protects vitiligo melanocytes against oxidative stress by activating SIRT3-OPA1 axis. (A-B) PIG3V cells were pre-treated with 5 μM HKL for 24 h and then treated with 1.0 mM H2O2 for 24 h. The mRNA and the protein levels of SIRT3 were detected by qRT-PCR and immunoblotting. (C) SIRT3 activity of PIG3V cells with treatment as indicated was measured based on an enzymatic reaction using a SIRT3 activity assay kit. Data represent mean ± SD (n = 3). (D) The protein level of SOD2 and Ac-SOD2 in PIG3V cells with treatment as indicated were detected. Mean ± SD is shown (n = 3). (E) Representative confocal microscope images of the mitochondrial network in PIG3V cells with treatment as indicated. Scale bar = 50 μm (Magnification: the upper = 600 ×; the under = 1800 ×). The proportion of PIG3V cells (n=100 cells for each sample) with tubulated, intermediate and fragmented mitochondria was quantified. (F) The apoptosis level of PIG3V cells with treatment as indicated was examined by annexin V-FITC/PI staining. The mitochondrial ROS level of PIG3V cells with treatment as indicated was examined by MitoSOX™ Red mitochondrial superoxide indicator staining. The mitochondrial membrane potential level of PIG3V cells with treatment as indicated was analyzed by JC-1 staining. Assessment of ATP level in PIG3V cells with treatment as indicated. Data represent mean ± SD of triplicates. (G) Representative confocal microscope images of the mitochondrial network in PIG3V cells with treatment as indicated. si-NC refers to negative control small interfering RNA, si-OPA1 refers to small interfering RNA against OPA1. Scale bar = 50 μm (Magnification: the upper = 600 ×; the under = 1800 ×). The proportion of PIG3V cells (n=100 cells for each sample) with tubulated, intermediate and fragmented mitochondria was quantified. (H) The apoptosis level of PIG3V cells with treatment as indicated was examined by annexin V-FITC/PI staining. The mitochondrial ROS level of PIG3V cells with treatment as indicated was examined by MitoSOX™ Red mitochondrial superoxide indicator staining. The mitochondrial membrane potential level of PIG3V cells with treatment as indicated was analyzed by JC-1 staining. Assessment of ATP level in PIG3V cells with treatment as indicated. Data represent mean ± SD of triplicates.(I) The level of apoptosis-related proteins in PIG3V cells with treatment as indicated was detected by immunoblotting. (J) The level of cytochrome c in mitochondria or cytoplasm in PIG3V cells with treatment as indicated was detected by western blotting. TOMM20 was detected as loading control for mitochondrial fraction. Tubulin was detected as loading control for cytosolic fraction. β-Actin was detected as loading control for the whole cell lysate. (K) Relative enzyme activity of respiratory complexes I, II, III, IV and V were measured in PIG3V cells with treatment as indicated. Data represent mean ± SD of triplicates. p value was calculated by two-tailed Student's t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
Importantly, HKL treatment could increase the expression of PGC1α at both mRNA and protein levels (Supplementary Figure S8B and C). Furthermore, our chromatin immunoprecipitation assay showed more recruitment of PGC1α to the promoter of SIRT3 in vitiligo melanocytes under oxidative stress after HKL treatment (Supplementary Figure S8D), indicating that HKL-induced increased expression of SIRT3 was highly associated with potentiated PGC1α expression and transcriptional function. Not surprisingly, HKL treatment led to significant mitochondrial fusion under oxidative stress (Figure 7E). Moreover, oxidative stress-induced cell apoptosis was markedly inhibited (Figure 7F and Supplementary Figure S8E). In parallel, the generation of mitochondrial ROS, the dissipation of mitochondrial membrane potential and the reduced intracellular ATP level were markedly reversed (Figure 7F and Supplementary Figures S8F and G), demonstrating that HKL activated SIRT3 to prevent cell death and mitochondrial dysfunction under oxidative stress. Furthermore, we obtained the knockdown of OPA1 in PIG3V cells to see whether HKL exerted its protective function via OPA1. As was shown, OPA1 deficiency abrogated the facilitative role of HKL in mitochondrial fusion under oxidative stress (Figure 7G). In line with this, the knockdown of OPA1 expression led to increased cell apoptosis, potentiated mitochondrial ROS, reduced mitochondrial membrane potential and lessened ATP level in PIG3V cells after HKL treatment under oxidative stress (Figure 7H and Supplementary Figures S8H-J). Through the immunoblotting analysis, we found that OPA1 deficiency promoted the expressions of pro-apoptotic cleaved-caspase 9, cleaved-PARP and Bax, but suppressed the expression of anti-apoptotic Bcl2 (Figure 7I and Supplementary Figure S8K). What's more, the release of cytochrome c from mitochondria to cytoplasm was increased (Figure 7J). Importantly, we also found that OPA1 deficiency led to a general decline of respiratory chain complex activities in case of HKL treatment under oxidative stress (Figure 7K), indicating that OPA1-mediated cytochrome c translocation and respiratory efficacy was involved in the role of HKL in preventing cell apoptosis and mitochondrial dysfunction in vitiligo melanocytes. Altogether, our results demonstrated that HKL protected vitiligo melanocytes against oxidative stress by activating SIRT3-OPA1 axis.
Discussion
In the present study, we first revealed the impairment of SIRT3 expression and activity in vitiligo melanocytes in response to oxidative stress. In addition, we proved that SIRT3 deficiency contributed to oxidative stress-induced melanocyte apoptosis via the exacerbation of mitochondrial damage and the release of cytochrome c to cytoplasm for activating apoptotic pathway. Subsequently, we showed that SIRT3 deacetylated OPA1 to regulated mitochondrial dynamics, thus impacting cytochrome c translocation and mitochondrial respiratory efficacy. Our data further revealed that oxidative stress dampened SIRT3 activity and transcription by potentiating its carbonylation and inhibited PGC1α transcriptional function. Finally, we proved that the specific activation of SIRT3 by HKL was capable to protect vitiligo melanocytes against oxidative stress via the regulation of OPA1-dependent mitochondrial dynamics. Collectively, our study demonstrates that SIRT3-dependent mitochondrial dynamics remodeling contributes to oxidative stress-induced melanocyte degeneration in vitiligo, and more importantly, the specific activation of SIRT3 in melanocyte could be a promising therapeutic approach for treating vitiligo (Figure 8).
Oxidative stress plays a key role in vitiligo pathogenesis because it is capable to directly induce melanocyte apoptosis and potentiate the autoimmune response that subsequently causes melanocyte destruction [7-9]. Previously, we have revealed that impaired anti-oxidant capacity and dysregulated miRNA expression profile may account for oxidative stress-induced cell death of melanocytes in vitiligo [2, 8]. However, our findings fail to fully explain the dysregulation of melanocyte biology in vitiligo, especially the dysregulated mitochondrial function of melanocytes [13, 14]. Dell'Anna et al. have observed increased mitochondria-derived ROS production and impaired activity of electron transport chain complex in vitiligo melanocytes [21]. Consistent with their findings, we also detected increased mitochondrial ROS generation, decreased mitochondrial membrane potential, lessened intracellular ATP level and impaired respiratory chain complex activities in melanocytes after H2O2 stimulation, further confirming the involvement of damaged mitochondria in inducing melanocyte destruction in vitiligo. Recent studies have documented that mitochondrial morphology is dynamically shaped under the control of opposing processes of fission and fusion, which remarkably influences mitochondrial function [25]. Distinct from the notion that reduced cardiolipin may be responsible for mitochondrial defects of vitiligo melanocytes [13], we for the first time showed the occurrence of excessive mitochondrial fragmentation induced more cytochrome c release and inhibited respiratory chain function that made melanocytes more vulnerable to oxidative stress. Therefore, restoring mitochondrial fusion could be novel therapeutic approach for preventing H2O2-induced melanocyte destruction in vitiligo.
Figure 8
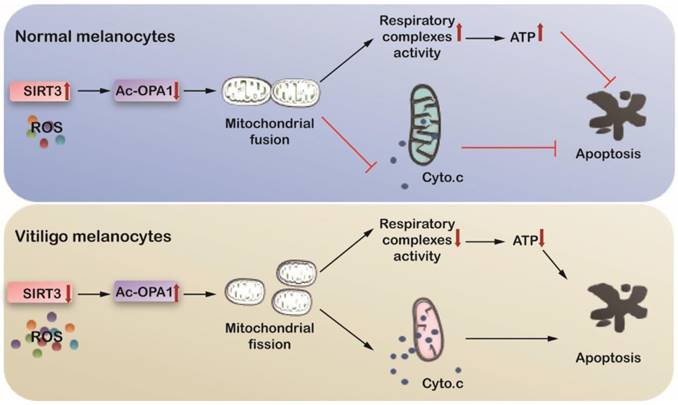
SIRT3-OPA1 pathway regulates mitochondrial dynamics and cell apoptosis in melanocytes under oxidative stress. Oxidative stress potentiates SIRT3 expression and activity in normal human melanocytes, but suppresses both the expression and the activity of SIRT3 in vitiligo melanocytes. SIRT3 deficiency in vililigo melanoncytes contributes to mitochondiral fission via inducing hyperacetylation of OPA1, leading to cytochrome c release from mitochondria to cytoplasm and deficient respiratory complex activities, which ultimately activates apoptotic pathway and results in melanocyte destruction in vitiligo.
Mitochondrial fusion and fission are spatiotemporally regulated by multiple post-translational modifications, including phosphorylation, ubiquitination and acetylation [28, 30, 31]. To be specific, SIRT3 is the most crucial mitochondria- localized deacetylase that regulates mitochondrial fusion via the direct deacetylation and activation of OPA1 [29]. In normal human melanocytes, SIRT3 was physiologically activated to confer protective effect against oxidative stress. However, we observed impaired activity and expression of SIRT3 in vitiligo melanocytes after H2O2 treatment in vitro. In addition, SIRT3 expression was decreased whereas acetylated- SOD2 expression was markedly increased in melanocytes in vitiligo lesion, indicating impaired SIRT3 expression and activity in vivo. Notably, the deficiency of SIRT3 prominently increased the vulnerability of melanocytes to oxidative stress, paralleled with more fragmented mitochondria, more cytochrome c release and suppressed mitochondrial respiratory efficacy. Therefore, dysregulated SIRT3- induced disordered mitochondrial dynamics was responsible for mitochondrial dysfunction and melanocyte destruction in vitiligo. However, we found that the expressions of mitochondrial dynamics regulators MFN1, MFN2, OPA1, Fis1 and DRP1 were not impacted by SIRT3, while the acetylation of OPA1 was significantly increased in case of SIRT3 deficiency instead. More importantly, we proved that the protective effect of SIRT3 on melanocytes against oxidative stress was in an OPA1-dependent manner. As is reported, OPA1 deficiency-induced mitochondrial network fragmentation and cristae remodeling with widening of cristae junctions are required for the release of cytochrome c, which could execute cell demise under stressful conditions [26, 27, 47]. Moreover, OPA1-mediated cristae shape could influence respiratory chain complex assembly and activity [23]. In line with this, the re-introduction of OPA1 expression suppressed the translocation of cytochrome c from mitochondria to cytoplasm, the impaired respiratory chain complex activities, and the induction of cell apoptosis in SIRT3-deficient melanocytes. Thus, our findings demonstrate that SIRT3-OPA1 axis could connect mitochondrial network to the execution of cell death in vitiligo melanocytes, and the intervention of mitochondrial dynamics could uncouple stressful stimuli with apoptotic signaling pathways.
Previous studies have proved that vitiligo melanocytes have higher basal level of oxidative stress compared with normal melanocytes, which is attributed to excessive generation of ROS and deficient activity of anti-oxidant molecules like Nrf2 in vitiligo melanocytes [7, 8, 52]. More importantly, due to the impaired scavenging capacity, the accumulation of ROS tends to be more significant in vitiligo melanocytes than normal melanocytes under oxidative stress, which has been confirmed by our previous studies [8]. Accumulative evidences have revealed that the expression and the activity of SIRT3 are regulated by multiple transcriptional and post-translational modifications [50, 53-55]. However, the linkage between oxidative stress and SIRT3 function is merely elucidated. A previous study has reported that lipid peroxidation product 4-HNE could allosterically inhibit SIRT3 activity via carbonylation [39]. In addition, we have previously found that oxidative stress could lead to more accumulation of lipid peroxidation products such as 4-HNE and MDA in vitiligo melanocytes compared with normal melanocytes [8]. Herein, we confirmed that the generation of 4-HNE and MDA was more prominent in vitiligo melanocytes after H2O2 treatment, and more importantly, the carbonylation of SIRT3 was consistently increased, which could partially account for the impairment of SIRT3 activity in vitiligo melanocytes. In addition to this, we observed that the up-regulation of PGC1α expression was diminished in vitiligo melanocytes after H2O2 treatment, which resulted in impaired PGC1α-mediated transcriptional activation of SIRT3. Therefore, the excessive carbonylation and dampened PGC1α transcriptional function depicted the crosstalk between oxidative stress and dysregulated SIRT3 function in vitiligo. It is reported that PGC1α and SIRT3 could form a positive feedback loop. On one hand, PGC1α could activate SIRT3 gene transcription via co-activation of the estrogen-related receptor-α (ERRα), which binds to the upstream element of SIRT3 gene promoter [50, 56]. On the other hand, SIRT3 has a facilitative effect on PGC1α expression via impacting AMPK and CREB phosphorylation [57]. Therefore, the down-regulation of PGC1α in vitiligo could be resulted from impaired function of SIRT3, which was caused by its augmented carbonylation under excessive oxidative stress. The impaired SIRT3 function and defect PGC1α expression form a negative feedback loop in vitiligo melanocytes to induce mitochondrial dysregulation under oxidative stress. Previously, Mansuri et al. reported the possible downregulation of SIRT1 through miR-1 in vitiligo skin, indicating the plausible role of miR-1 and SIRT1 in melanocyte destruction under oxidative stress and inflammatory micro-environment in vitiligo lesion [58]. In this respect, in addition to be regulated by PGC1α at the transcriptional level and by carbonylation modification at post-translational level, defective SIRT3 function in vitiligo might also be relevant to the change of miRNAs. In our previous study, we performed a miRNA microarray screening using serum samples from vitiligo patients and healthy controls. The assay identified 48 aberrantly expressed miRNAs (two fold changes, p<0.01), among which 24 miRNAs (e.g., let-7b) were upregulated, whereas another 24 ones (e.g., miR-1914) were downregulated [2]. However, all previously reported miRNAs that could regulate SIRT3 expression had no change in our miRNA microarray study [59-65]. According to our present research, it was the excessive carbonylation of SIRT3 and dampened activation of PGC1α transcriptional function contributing to defective expression and function of SIRT3 in vitiligo [56]. Since miRNAs play a critical role in the pathogenesis of vitiligo, we will analyze more miRNAs that may regulate SIRT3 expression in further study.
HKL has been proved as a specific pharmacological activator of SIRT3 via the simultaneous enhancement of its expression and activity [51]. Since dysregulated SIRT3 function was greatly implicated in oxidative stress-induced melanocyte destruction in vitiligo, activating SIRT3 could be promising in treating vitiligo. We proved that HKL regulated mitochondrial network in an OPA1-dependent manner, thus suppressing cytochrome c release, increasing respiratory chain complex activities and preventing cell apoptosis. Previous studies have revealed that HKL is valuable in ameliorating tissue or cell damage in oxidative stress-related diseases [66-68]. Gupta et al. have observed that HKL injection can block the cardiac hypertrophic response in vivo [51]. Our study expands the therapeutic spectrum of HKL by proving that HKL is efficient in reducing vitiligo melanocyte apoptosis under oxidative stress. Moreover, different from other systematic oxidative stress-related diseases, the application of HKL to the treatment of vitiligo could be designed as topical ointment rather than systemic administration [51, 68]. It is noteworthy of investigating the effect of topical HKL ointment in treating vitiligo through clinical trials in the future.
Increasing evidences demonstrate that vitiligo is a tissue-specific autoimmune disease of the skin caused by cytotoxic T cells-mediated destruction of melanocytes [69-71]. Previous studies have showed that downregulation of pro-survival genes like SIRT3 and the increase of ROS lead to the release of pro-inflammatory cytokines and chemokines, which result in cell senescence, inflammation and the development of autoimmune disorder [72-74]. Moreover, SIRT3 is a key effector of neutrophils- and lymphocytes-mediated immune response after exercise [75, 76]. Therefore, it is possible that SIRT3 is also involved in regulating dysregulated autoimmunity in vitiligo, which needs to be further investigated.
In conclusion, our study demonstrates that SIRT3-dependent mitochondrial dynamics remodeling contributes to oxidative stress-induced melanocyte degeneration in vitiligo. More importantly, the specific activation of SIRT3 could be promising therapeutic approach for suppressing melanocyte destruction in vitiligo. Additional studies using vitiligo mice or human skin xenografts are needed to confirm the therapeutic potential of SIRT3 activator HKL for vitiligo.
Materials and Methods
Patient specimens
Skin specimens for immunofluorescence staining analysis were taken from eight vitiligo patients (from the perilesional skin) and eight matched healthy volunteers. The patients and controls were selected as described in our previous study [9]. All the clinical specimens were obtained from patients given a diagnosis of vitiligo according to clinical and histologic manifestation in Department of Dermatology, Xijing Hospital, Fourth Military Medical University from 2016 to 2017. All subjects enrolled in the study displayed initiation of depigmentation within 3 months and had not received any systemic or topical therapy for at least 3 months before sample collection. The healthy volunteers were matched to the cases by age and sex, and informed consent was obtained from all the patients and control subjects. The detailed clinical information of patients and healthy controls were shown in Supplementary Table S1 and Table S2. The research protocol was approved by the ethics review board of Fourth Military Medical University, according to the Declaration of Helsinki Principles.
Cell culture
Normal human epidermal melanocyte (NHEM) was a gift from Professor David Schrama (University Hospital Würzburg, Germany) and cultured in HAM's F10 medium (Sigma-Aldrich, St Louis, MO, USA) supplemented with 20% fetal bovine serum (Invitrogen, San Diego, CA, USA), 200 nM cholera toxin (Sigma-Aldrich), 100 μM 3-Isobutyl-1-methylxanthine (IBMX, Sigma-Aldrich), 5μM Phorbol 12-myristate 13-acetate (TPA, Sigma-Aldrich) and ITS (containing 1.0 mg/ml recombinant human insulin, 0.55 mg/ml human transferrin and 0.5 μg/ml sodium selenite at the 100 × concentration, Sigma-Aldrich) respectively. The immortalized normal human melanocyte cell line PIG1 and vitiligo melanocyte cell line PIG3V (both gifted by Dr. Caroline Le Poole, Loyola University Chicago, Maywood, IL, USA) were grown in Medium 254 (Gibco, Grand Island, NY, USA) containing Human Melanocyte Growth Supplement (Gibco) and 5% fetal bovine serum (Gibco). Cells were grown in 5% CO2 humidified atmosphere at 37 °C. H2O2 (Sigma-Aldrich) was used at concentration of 1.0 mM. Honokiol (HKL, Sigma-Aldrich) treatment was performed 24 h prior to H2O2 treatment. HKL was first dissolved in dimethylsulfoxide (DMSO, Sigma-Aldrich) to make a stock solution of 100 mM, which was then diluted in medium and made into different final concentrations.
Immunoblotting analysis
Cells were washed with phosphate buffered saline for two times, and lysed with RIPA buffer (Beyotime biotechnology, Beijing, China) supplemented with a protease inhibitor phenylmethanesulfonyl fluoride (Sigma-Aldrich). Lysates were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). The primary antibodies in this study were as follows: anti-SIRT3 Antibody (#2627, 1:1000, Cell Signaling Technology, Beverly, MA), anti-SOD2 Antibody (ab13533,1:2000, Abcam, Cambridge, MA), rabbit polyclonal anti-SOD2 (acetyl K68) antibody (ab137037, 1:2000, Abcam), anti-OPA1 (D7C1A) Antibody (#67589, 1:1000, Cell Signaling Technology), anti-cytochrome c Antibody (ab13575, 1:1000, Abcam), anti-PGC1 alpha Antibody (ab54481, 1:1000, Abcam), anti-acetyl Lysine Antibody (ab22550, 1:1000, Abcam), anti-Bax Antibody (#2772, 1:1000, Cell Signaling Technology), anti-Bcl-2 Antibody (#2872, 1:1000, Cell Signaling Technology), anti-PARP (46D11) Antibody (#9532, 1:1000, Cell Signaling Technology), anti-Caspase-9 Antibody (#9502, 1:1000, Cell Signaling Technology), anti-Mitofusin 1 Antibody (ab129154, 1:1000, Abcam), anti-Mitofusin 2 Antibody (ab124773, 1:1000, Abcam), anti-DRP1 Antibody (ab184247, 1:1000, Abcam), anti-Fis1 Antibody (10956-1-AP, 1:8000, Proteintech Group, Rosemont, USA), anti-TOMM20 Antibody (ab56783, 1:1000, Abcam) and anti-β-Actin Antibody (#3700, 1:5000, Cell Signaling Technology). PVDF membranes were incubated with primary antibody overnight at 4 °C, and followed by incubating with peroxidase-conjugated anti-rabbit (111-035-003, 1:5000, Jackson ImmunoResearch, West Grove, PA) and anti-mouse secondary antibodies (115-035-003, 1:5000, Jackson ImmunoResearch) for 1 h at room temperature. Signals were detected using Western ECL Substrate (Thermo Scientific, Waltham, MA, USA).
SIRT3 deacetylase activity assay
SIRT3 deacetylase activity was examined using a SIRT3 Fluorimetric Activity Assay Kit (#ab156067, Abcam) following the manufacturer's protocol. In brief, the mitochondrial extract was incubated with the SIRT3 assay buffer, and then co-incubated with Fluoro-Substrate Peptide, NAD and Developer at 37 °C for 1 h. Fluorescent intensity was measured using a Fluoroskan Ascent® microplate fluorometer (Thermo Electron Corp., Milford, MA, USA) at 350 nm/450 nm [77].
Flow cytometry assays
The level of cell apoptosis, mitochondrial membrane potential and mitochondrial ROS were analyzed by flow cytometry. For detection of cell apoptosis, cells were collected after experimental treatment and detected using a FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA) according to the manufacturer's instructions. For detection of mitochondrial membrane potential, cells were harvested and washed twice in PBS, followed by staining with JC-1 (10 μg/ml, Invitrogen, Rockford, IL, USA) for 10-20 min at 37 °C. Stained cells were analyzed by flow cytometry. For detection of mitochondrial ROS, cells were harvested and washed twice in PBS, followed by staining with Mito SoxRed (5μM, Invitrogen) for 10-20 min at 37 °C. Stained cells were analyzed by flow cytometry. All the analyzed samples were identified in flow cytometer (Beckman Coulter, Miami, USA) within an hour. The data analysis was done with Expo32 software (Beckman Coulter). All experiments were performed in triplicate.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the EZ ChIPTM chromatin immunoprecipitation kit (Millipore catalog number 17-371) according to the manufacturer's instruction as described before [78]. In brief, immunoprecipitation was performed with PGC1α antibody (#20658-1-AP, Proteintech Group, Rosemont, IL, USA). Rabbit IgG (ab46540, Abcam) was used as an isotype control antibody. The immunoprecipitated DNA was treated with RNase A and proteinase K and purified using phenol- chloroform extraction and ethanol precipitation. Input DNA starting from aliquots of cell lysates was purified using phenol-chloroform extraction and ethanol precipitation. The purified DNA and input genomic DNA were analyzed by real time PCR. Primer sequences of SIRT3 promoter for amplifying purified DNA in this study are as follows: Forward primer: 5'- GCCTACTCAAGGAGGTCG-3', Reverse primer: 5'- TGTTTATGCCTGGTGCTG-3'. % Input was calculated using 100% × 2(-△CT), where △CT = Ct (Ip) - Ct (Input).
Isolation of cytosolic and mitochondrial fractions
In order to obtain mitochondrial extracts, cells with indicated treatments were collected and processed to the isolation of cytosolic and mitochondrial fraction using Mitochondria Isolation Kit for Cultured Cells (Thermo Scientific, Rockford, IL, USA) following the manufacturer's protocol. In brief, 2 × 107 cells were pelleted by centrifuging harvested cell suspension, and then mitochondria Isolation Reagent was added into the cell pellets. The cell resuspension was centrifuged at 700 × g for 10 min at 4 °C, and then the supernatant was transferred to a new 2.0 ml tube and centrifuged at 12,000 × g for 15 min at 4 °C. The supernatant (cytosolic fraction) was transferred to a new tube, and the pellet contained the isolated mitochondria. Then, the integrity of isolated mitochondria was examined by electron microscopy. Moreover, the purity of the mitochondria was determined by detecting the expression of tubulin that was the loading control of cytosolic protein.
Mitochondrial network imaging by transmission electron microscopy and confocal microscopy
For transmission electron microscopy analysis, cells were collected and washed twice with PBS and fixed with ice-cold glutaraldehyde for 24 h at 4 °C. Then the preparation of section was performed as described before [79]. Ultrathin section analysis was visualized using a Tecnai™ G2 Spirit Bio Twin (FEI, Hillsboro, OR). Confocal immunofluorescent analysis with MitoTracker green FM Molecular Probes (Invitrogen) was performed as described previously [22]. For morphometric analysis, the length of mitochondria was measured using Image J software (NIH, Bethesda, MD, USA).
Co-immunoprecipitation
Cells after treatments were collected, and co-immunoprecipitation was performed using Pierce co-immunoprecipitation (Co-IP) kit (Thermo Scientific) according to manufacturer's instructions. In brief, 20 μg anti-OPA1 (D7C1A) antibody (#67589, Cell Signaling Technology) was added directly to the resin in the spin column. The column was capped and incubated at room temperature for 90-120 min using a rotating body or mixer. After the antibody was immobilized, the protein extracts (500 μg) were added to the resin, followed by being evenly turned over at 4 °C overnight. Following the elution of Co-IP samples, SDS-PAGE samples were prepared and further immunoblotted with anti-acetyl Lysine antibody (#ab22550,1:1000, Abcam).
Total protein and SIRT3 carbonylation assay
Following treatment, the cells were collected, and carbonylation assays were performed using Oxidized Protein Western Blot Kit (#ab178020, Abcam) following the manufacturer's protocol. In brief, the cell pellets were solubilized at 2 × 107/ml in 1 × Extraction Buffer, and then incubated on ice for 20 min. Following being centrifuged, the supernatant samples were transferred into new tubes. Then the protein samples were treated with derivatization reaction by thawing 1 × DNPH Solution, and the reaction products were used for western blot by using anti-DNP primary antibody. Besides, the detection of the SIRT3 carbonylation was performed as described before with some modifications [80]. SIRT3 was firstly immunoprecipitated using whole-cell extracts and anti-SIRT3 antibody. To determine the carbonylation of SIRT3, the products of immunoprecipitation were processed to derivatization reaction by thawing 1 × DNPH Solution, and the reaction products were processed to western blotting by using anti-DNP primary antibody.
Measurement of mitochondrial respiratory chain complex activity
The measurement of mitochondrial respiratory chain complex activity was performed by the MitoTox Complete OXPHOS Activity Assay Panel (# ab110419, Abcam) following the manufacturer's protocol [22]. In brief, each of the complexes was obtained from isolated mitochondria of the treated cells using highly specific monoclonal antibodies attached to 96-well microplates. Complex activity was determined by measuring the decrease in optical density per min as described in the manufacturer's protocol. Specified wavelengths (340 nm for complexes I and V, 550 nm for complexes III and IV, and 600 nm for complex II) were used to measure the absorbance.
Statistical analyses
All statistical analyses were performed with GraphPad Prism software (7.0; GraphPad software, San Diego, Calif). Dual comparisons were made with the unpaired two-tailed Student t test. *p < 0.05, **p < 0.01 and ***p < 0.001 were considered to be statistically significant. Data represent mean ± S.D. for at least 3 independent experiments.
Abbreviations
NAD: nicotinamide adenine dinucleotide; SIRT3: Sirtuin3; OPA1: Optic atrophy 1; HKL: honokiol; PGC1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; H2O2: hydrogen peroxide; ER: Endoplasmic reticulum; ROS: reactive oxygen species; SOD2: superoxide dismutase; qRT-PCR: real-time quantitative reverse transcription-PCR; Fis1: fission 1; MFF: mitochondrial fission factor; MFN1: mitofusion-1; MFN2: mitofusion-2; DRP1: Dynamin- related protein 1; Ac-SOD2: acetylated superoxide dismutase; cleaved-PARP: cleaved poly-ADP-ribose polymerase; Bax: B cell lymphoma 2-associated X protein; Bcl-2: B cell lymphoma 2; Co-IP: co-immunoprecipitation; 4-HNE: 4-Hydroxynoenal; MDA: Malondialdehyde; 8-OHdG: 8-hydroxy-2 deoxyguanosine; Cyto.c: Cytochorome.c; Nrf2: nuclear factor-E2- related factor 2; CRT: Calreticulin; TNFα: Tumor necrosis factor α.
Acknowledgements
The authors thank the doctors and the patients who participated in our study. This work was supported by National Natural Science Foundation of China (no. 91742201, no. 81472863, no. 81803112, no. 81602764, no. 81602750).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xie H, Zhou F, Liu L. et al. Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity? J Dermatol Sci. 2016;81:3-9
2. Shi Q, Zhang W, Guo S. et al. Oxidative stress-induced overexpression of miR-25: the mechanism underlying the degeneration of melanocytes in vitiligo. Cell Death Differ. 2016;23:496-508
3. Ezzedine K, Lim HW, Suzuki T. et al. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25:E1-13
4. Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond). 2011;120:99-120
5. Laddha NC, Dwivedi M, Mansuri MS. et al. Vitiligo: interplay between oxidative stress and immune system. Exp Dermatol. 2013;22:245-50
6. Rodrigues M, Ezzedine K, Hamzavi I. et al. New discoveries in the pathogenesis and classification of vitiligo. J Am Acad Dermatol. 2017;77:1-13
7. Jian Z, Li K, Liu L. et al. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J Invest Dermatol. 2011;131:1420-7
8. Jian Z, Li K, Song P. et al. Impaired activation of the Nrf2-ARE signaling pathway undermines H2O2-induced oxidative stress response: a possible mechanism for melanocyte degeneration in vitiligo. J Invest Dermatol. 2014;134:2221-30
9. Li S, Zhu G, Yang Y. et al. Oxidative stress drives CD8(+) T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J Allergy Clin Immunol. 2017;140:177-89 e9
10. Mansuri MS, Singh M, Jadeja SD. et al. Could ER Stress Be A Major Link Between Oxidative Stress And Autoimmunity In Vitiligo? J Pigmentary Disorders. 2014;1:1000123
11. Patel DC, Evans AV, Hawk JL. Topical pseudocatalase mousse and narrowband UVB phototherapy is not effective for vitiligo: an open, single-centre study. Clin Exp Dermatol. 2002;27:641-4
12. Yuksel EP, Aydin F, Senturk N. et al. Comparison of the efficacy of narrow band ultraviolet B and narrow band ultraviolet B plus topical catalase-superoxide dismutase treatment in vitiligo patients. Eur J Dermatol. 2009;19:341-4
13. Dell'Anna ML, Ottaviani M, Albanesi V. et al. Membrane lipid alterations as a possible basis for melanocyte degeneration in vitiligo. J Invest Dermatol. 2007;127:1226-33
14. Liu B, Jian Z, Li Q. et al. Baicalein protects human melanocytes from H(2)O(2)-induced apoptosis via inhibiting mitochondria-dependent caspase activation and the p38 MAPK pathway. Free Radic Biol Med. 2012;53:183-93
15. Mitra S, De Sarkar S, Pradhan A. et al. Levels of oxidative damage and proinflammatory cytokines are enhanced in patients with active vitiligo. Free Radic Res. 2017;51:986-94
16. Hroudova J, Singh N, Fisar Z. Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer's disease. Biomed Res Int. 2014;2014:175062
17. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524-37
18. Kornfeld OS, Hwang S, Disatnik MH. et al. Mitochondrial reactive oxygen species at the heart of the matter: new therapeutic approaches for cardiovascular diseases. Circ Res. 2015;116:1783-99
19. Manucha W. Mitochondria and oxidative stress participation in renal inflammatory process. Medicina (B Aires). 2014;74:254-8
20. Laddha NC, Dwivedi M, Gani AR. et al. Involvement of superoxide dismutase isoenzymes and their genetic variants in progression of and higher susceptibility to vitiligo. Free Radic Biol Med. 2013;65:1110-25
21. Dell'Anna ML, Ottaviani M, Kovacs D. et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci Rep. 2017;7:13663
22. Li J, Huang Q, Long X. et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene. 2017;36:4901-12
23. Cogliati S, Frezza C, Soriano ME. et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160-71
24. Plecita-Hlavata L, Jezek P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int J Biochem Cell Biol. 2016;80:31-50
25. Eisner V, Picard M, Hajnoczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018;20:755-65
26. Frezza C, Cipolat S, Martins de Brito O. et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177-89
27. Cipolat S, Rudka T, Hartmann D. et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163-75
28. Kashatus JA, Nascimento A, Myers LJ. et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57:537-51
29. Samant SA, Zhang HJ, Hong Z. et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34:807-19
30. Guerra de Souza AC, Prediger RD, Cimarosti H. SUMO-regulated mitochondrial function in Parkinson's disease. J Neurochem. 2016;137:673-86
31. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013:5
32. Marino G, Pietrocola F, Eisenberg T. et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53:710-25
33. Sabari BR, Zhang D, Allis CD. et al. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 2017;18:90-101
34. Hallows WC, Smith BC, Lee S. et al. Ure(k)a! Sirtuins Regulate Mitochondria. Cell. 2009;137:404-6
35. Yang W, Nagasawa K, Munch C. et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell. 2016;167:985-1000 e21
36. Ozden O, Park SH, Wagner BA. et al. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic Biol Med. 2014;76:163-72
37. Qiu X, Brown K, Hirschey MD. et al. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662-7
38. Morigi M, Perico L, Rota C. et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest. 2015;125:715-26
39. Fritz KS, Galligan JJ, Smathers RL. et al. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem Res Toxicol. 2011;24:651-62
40. Song Y, Li S, Geng W. et al. Sirtuin 3-dependent mitochondrial redox homeostasis protects against AGEs-induced intervertebral disc degeneration. Redox Biol. 2018;19:339-53
41. Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid Redox Signal. 2009;11:1139-48
42. Bai X, Cerimele F, Ushio-Fukai M. et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501-7
43. Chen XR, Lu R, Dan HX. et al. Honokiol: a promising small molecular weight natural agent for the growth inhibition of oral squamous cell carcinoma cells. Int J Oral Sci. 2011;3:34-42
44. Woodbury A, Yu SP, Wei L. et al. Neuro-modulating effects of honokiol: a review. Front Neurol. 2013;4:130
45. Chen H, Wang Y, Yao Y. et al. Sequential Delivery of Cyclopeptide RA-V and Doxorubicin for Combination Therapy on Resistant Tumor and In Situ Monitoring of Cytochrome c Release. Theranostics. 2017;7:3781-93
46. Milenkovic D, Blaza JN, Larsson NG. et al. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017;25:765-76
47. Varanita T, Soriano ME, Romanello V. et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834-44
48. Lang A, Anand R, Altinoluk-Hambuchen S. et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging (Albany NY). 2017;9:2163-89
49. Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2' -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:120-39
50. Giralt A, Hondares E, Villena JA. et al. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J Biol Chem. 2011;286:16958-66
51. Pillai VB, Samant S, Sundaresan NR. et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun. 2015;6:6656
52. Zhang Y, Liu L, Jin L. et al. Oxidative stress-induced calreticulin expression and translocation: new insights into the destruction of melanocytes. J Invest Dermatol. 2014;134:183-91
53. Satterstrom FK, Swindell WR, Laurent G. et al. Nuclear respiratory factor 2 induces SIRT3 expression. Aging Cell. 2015;14:818-25
54. Liu R, Fan M, Candas D. et al. CDK1-Mediated SIRT3 Activation Enhances Mitochondrial Function and Tumor Radioresistance. Mol Cancer Ther. 2015;14:2090-102
55. Fang Y, Wang J, Xu L. et al. Autophagy maintains ubiquitination-proteasomal degradation of Sirt3 to limit oxidative stress in K562 leukemia cells. Oncotarget. 2016;7:35692-702
56. Wang Q, Li L, Li CY. et al. SIRT3 protects cells from hypoxia via PGC-1alpha- and MnSOD-dependent pathways. Neuroscience. 2015;286:109-21
57. Palacios OM, Carmona JJ, Michan S. et al. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging (Albany NY). 2009;1:771-83
58. Mansuri MS, Singh M, Begum R. miRNA signatures and transcriptional regulation of their target genes in vitiligo. J Dermatol Sci. 2016;84:50-58
59. Cai H, Li Y, Li H. et al. Identification and characterization of human ovary-derived circular RNAs and their potential roles in ovarian aging. Aging (Albany NY). 2018;10:2511-34
60. Zhang X, Ji R, Liao X. et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation. 2018;137:2052-67
61. Shiozawa K, Shuting J, Yoshioka Y. et al. Extracellular vesicle-encapsulated microRNA-761 enhances pazopanib resistance in synovial sarcoma. Biochem Biophys Res Commun. 2018;495:1322-27
62. Sun W, Zhao L, Song X. et al. MicroRNA-210 Modulates the Cellular Energy Metabolism Shift During H2O2-Induced Oxidative Stress by Repressing ISCU in H9c2 Cardiomyocytes. Cell Physiol Biochem. 2017;43:383-94
63. Katta S, Karnewar S, Panuganti D. et al. Mitochondria-targeted esculetin inhibits PAI-1 levels by modulating STAT3 activation and miR-19b via SIRT3: Role in acute coronary artery syndrome. J Cell Physiol. 2018;233:214-25
64. Cheng Y, Mai J, Hou T. et al. MicroRNA-421 induces hepatic mitochondrial dysfunction in non-alcoholic fatty liver disease mice by inhibiting sirtuin 3. Biochem Biophys Res Commun. 2016;474:57-63
65. Poulsen RC, Knowles HJ, Carr AJ. et al. Cell differentiation versus cell death: extracellular glucose is a key determinant of cell fate following oxidative stress exposure. Cell Death Dis. 2014;5:e1074
66. Matsui N, Takahashi K, Takeichi M. et al. Magnolol and honokiol prevent learning and memory impairment and cholinergic deficit in SAMP8 mice. Brain Res. 2009;1305:108-17
67. Tang P, Gu JM, Xie ZA. et al. Honokiol alleviates the degeneration of intervertebral disc via suppressing the activation of TXNIP-NLRP3 inflammasome signal pathway. Free Radic Biol Med. 2018;120:368-79
68. Chen HH, Chang PC, Chen C. et al. Protective and therapeutic activity of honokiol in reversing motor deficits and neuronal degeneration in the mouse model of Parkinson's disease. Pharmacol Rep. 2018;70:668-76
69. van den Boorn JG, Konijnenberg D, Dellemijn TA. et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. 2009;129:2220-32
70. Schallreuter KU, Bahadoran P, Picardo M. et al. Vitiligo pathogenesis: autoimmune disease, genetic defect, excessive reactive oxygen species, calcium imbalance, or what else? Exp Dermatol. 2008;17:139-40 discussion 41-60
71. Spritz RA. Six decades of vitiligo genetics: genome-wide studies provide insights into autoimmune pathogenesis. J Invest Dermatol. 2012;132:268-73
72. Benigni A, Corna D, Zoja C. et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524-30
73. Capettini LS, Montecucco F, Mach F. et al. Role of renin-angiotensin system in inflammation, immunity and aging. Curr Pharm Des. 2012;18:963-70
74. Alhazzazi TY, Kamarajan P, Verdin E. et al. Sirtuin-3 (SIRT3) and the Hallmarks of Cancer. Genes Cancer. 2013;4:164-71
75. Ferrer MD, Tauler P, Sureda A. et al. Antioxidant regulatory mechanisms in neutrophils and lymphocytes after intense exercise. J Sports Sci. 2009;27:49-58
76. Mestre-Alfaro A, Ferrer MD, Banquells M. et al. Body temperature modulates the antioxidant and acute immune responses to exercise. Free Radic Res. 2012;46:799-808
77. Pi H, Xu S, Reiter RJ. et al. SIRT3-SOD2-mROS-dependent autophagy in cadmium-induced hepatotoxicity and salvage by melatonin. Autophagy. 2015;11:1037-51
78. Yang Y, Guo W, Ma J. et al. Downregulated TRPV1 Expression Contributes to Melanoma Growth via the Calcineurin-ATF3-p53 Pathway. J Invest Dermatol. 2018;138:2205-15
79. He Y, Li S, Zhang W. et al. Dysregulated autophagy increased melanocyte sensitivity to H2O2-induced oxidative stress in vitiligo. Sci Rep. 2017;7:42394
80. Gu C, Xing Y, Jiang L. et al. Impaired cardiac SIRT1 activity by carbonyl stress contributes to aging-related ischemic intolerance. PLoS One. 2013;8:e74050
Author contact
![]() Corresponding authors: S Li, Department of Dermatology, Xijing hospital, Fourth Military Medical University, 127# Changle xi Road, Xi'an, Shannxi 710032, China. Tel: +86 29 8477 5406; Fax: +86 29 8477 5401; E-mail: xjpf10edu.cn or T Gao, Department of Dermatology, Xijing hospital, Fourth Military Medical University, 127# Changle xi Road, Xi'an, Shannxi 710032, China. Tel: +86 29 8477 5406; Fax: +86 29 8477 5401; E-mail: gaotwedu.cn or C Li, Department of Dermatology, Xijing hospital, Fourth Military Medical University, 127# Changle xi Road, Xi'an, Shannxi 710032, China. Tel: +86 29 8477 5406; Fax: +86 29 8477 5401; E-mail: lichyingedu.cn
Corresponding authors: S Li, Department of Dermatology, Xijing hospital, Fourth Military Medical University, 127# Changle xi Road, Xi'an, Shannxi 710032, China. Tel: +86 29 8477 5406; Fax: +86 29 8477 5401; E-mail: xjpf10edu.cn or T Gao, Department of Dermatology, Xijing hospital, Fourth Military Medical University, 127# Changle xi Road, Xi'an, Shannxi 710032, China. Tel: +86 29 8477 5406; Fax: +86 29 8477 5401; E-mail: gaotwedu.cn or C Li, Department of Dermatology, Xijing hospital, Fourth Military Medical University, 127# Changle xi Road, Xi'an, Shannxi 710032, China. Tel: +86 29 8477 5406; Fax: +86 29 8477 5401; E-mail: lichyingedu.cn
Citation styles
APA
Yi, X., Guo, W., Shi, Q., Yang, Y., Zhang, W., Chen, X., Kang, P., Chen, J., Cui, T., Ma, J., Wang, H., Guo, S., Chang, Y., Liu, L., Jian, Z., Wang, L., Xiao, Q., Li, S., Gao, T., Li, C. (2019). SIRT3-Dependent Mitochondrial Dynamics Remodeling Contributes to Oxidative Stress-Induced Melanocyte Degeneration in Vitiligo. Theranostics, 9(6), 1614-1633. https://doi.org/10.7150/thno.30398.
ACS
Yi, X.; Guo, W.; Shi, Q.; Yang, Y.; Zhang, W.; Chen, X.; Kang, P.; Chen, J.; Cui, T.; Ma, J.; Wang, H.; Guo, S.; Chang, Y.; Liu, L.; Jian, Z.; Wang, L.; Xiao, Q.; Li, S.; Gao, T.; Li, C. SIRT3-Dependent Mitochondrial Dynamics Remodeling Contributes to Oxidative Stress-Induced Melanocyte Degeneration in Vitiligo. Theranostics 2019, 9 (6), 1614-1633. DOI: 10.7150/thno.30398.
NLM
Yi X, Guo W, Shi Q, Yang Y, Zhang W, Chen X, Kang P, Chen J, Cui T, Ma J, Wang H, Guo S, Chang Y, Liu L, Jian Z, Wang L, Xiao Q, Li S, Gao T, Li C. SIRT3-Dependent Mitochondrial Dynamics Remodeling Contributes to Oxidative Stress-Induced Melanocyte Degeneration in Vitiligo. Theranostics 2019; 9(6):1614-1633. doi:10.7150/thno.30398. https://www.thno.org/v09p1614.htm
CSE
Yi X, Guo W, Shi Q, Yang Y, Zhang W, Chen X, Kang P, Chen J, Cui T, Ma J, Wang H, Guo S, Chang Y, Liu L, Jian Z, Wang L, Xiao Q, Li S, Gao T, Li C. 2019. SIRT3-Dependent Mitochondrial Dynamics Remodeling Contributes to Oxidative Stress-Induced Melanocyte Degeneration in Vitiligo. Theranostics. 9(6):1614-1633.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.