Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1599-1613. doi:10.7150/thno.30372 This issue Cite
Research Paper
Identification of miR-29c and its Target FBXO31 as a Key Regulatory Mechanism in Esophageal Cancer Chemoresistance: Functional Validation and Clinical Significance
Bin Li1,2,3, Pan Hong3, Can-Can Zheng3, Wei Dai4, Wen-You Chen5, Qing-Sheng Yang5, Liang Han2, Sai Wah Tsao2, Kin Tak Chan6, Nikki Pui Yue Lee6, Simon Law6, Li Yan Xu7, En Min Li7, Kwok Wah Chan1,8, Yan Ru Qin9, Xin Yuan Guan4, Maria Li Lung4, Qing-Yu He3, Wen Wen Xu10,1,2 ![]() , Annie LM Cheung1,2,
, Annie LM Cheung1,2, ![]()
1. The University of Hong Kong-Shenzhen Institute of Research and Innovation (HKU-SIRI), China
2. School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China
3. Key Laboratory of Functional Protein Research of Guangdong Higher Education Institutes, Institute of Life and Health Engineering, College of Life Science and Technology, Jinan University, Guangzhou 510632, China
4. Department of Clinical oncology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China
5. Department of Thoracic Surgery, First Affiliated Hospital, Jinan University, Guangzhou 510632, China
6. Department of Surgery, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China
7. The Key Laboratory of Molecular Biology for High Cancer Incidence Coastal Chaoshan Area, Shantou University Medical College, Shantou, Guangdong, China
8. Department of Pathology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China
9. Department of Clinical Oncology, First Affiliated Hospital, Zhengzhou University, Zhengzhou, China
10. Institute of Biomedicine, Guangdong Provincial Key Laboratory of Bioengineering Medicine, National Engineering Research Center of Genetic Medicine, Jinan University, Guangzhou, China.
Received 2018-10-2; Accepted 2019-1-18; Published 2019-2-28
Citation:
Li B, Hong P, Zheng CC, Dai W, Chen WY, Yang QS, Han L, Tsao SW, Chan KT, Lee NPY, Law S, Xu LY, Li EM, Chan KW, Qin YR, Guan XY, Lung ML, He QY, Xu WW, Cheung ALM. Identification of miR-29c and its Target FBXO31 as a Key Regulatory Mechanism in Esophageal Cancer Chemoresistance: Functional Validation and Clinical Significance. Theranostics 2019; 9(6):1599-1613. doi:10.7150/thno.30372. https://www.thno.org/v09p1599.htm
Other stylesAbstract

Rationale: Dysregulated microRNA (miRNA) expressions in cancer can contribute to chemoresistance. This study aims to identify miRNAs that are associated with fluorouracil (5-FU) chemoresistance in esophageal squamous cell carcinoma (ESCC). The potential of miR-29c as a novel diagnostic, prognostic and treatment-predictive marker in ESCC, and its mechanisms and therapeutic implication in overcoming 5-FU chemoresistance were explored.
Methods: The miRNA profiles of an ESCC cell model with acquired chemoresistance to 5-FU were analyzed using a Taqman miRNA microarray to identify novel miRNAs associated with 5-FU chemoresistance. Quantitative real-time PCR was used to determine miR-29c expression in tissue and serum samples of patients. Bioinformatics, gain- and loss-of-function experiments, and luciferase reporter assay were performed to validate F-box only protein 31 (FBXO31) as a direct target of miR-29c, and to identify potential transcription factor binding events that control miR-29c expression. The potential of systemic miR-29c oligonucleotide-based therapy in overcoming 5-FU chemoresistance was evaluated in tumor xenograft model.
Results: MiR-29c, under the regulatory control of STAT5A, was frequently downregulated in tumor and serum samples of patients with ESCC, and the expression level was correlated with overall survival. Functional studies showed that miR-29c could override 5-FU chemoresistance in vitro and in vivo by directly interacting with the 3'UTR of FBXO31, leading to repression of FBXO31 expression and downstream activation of p38 MAPK. Systemically administered miR-29c dramatically improved response of 5-FU chemoresistant ESCC xenografts in vivo.
Conclusions: MiR-29c modulates chemoresistance by interacting with FBXO31, and is a promising non-invasive biomarker and therapeutic target in ESCC.
Keywords: diagnosis and prognosis, chemoresistance, p38 signaling, STAT5A, microRNA therapy
Introduction
Cisplatin (DDP) and 5-FU-based chemotherapy regimens constitute an important component of multimodal cancer treatment, but acquisition of resistance to these standard chemotherapeutic agents is a major challenge in the management of cancer [1, 2], including esophageal cancer which is the sixth most common cause of death worldwide with a 5-year survival rate of ~14% [3]. Therefore, comprehensive elucidation of the mechanisms governing chemoresistance and development of strategies to overcome chemoresistance are urgently needed.
MicroRNAs (miRNAs) are non-coding small RNAs of 19-22 nucleotides that negatively modulate protein expression [4, 5]. Their potential as diagnostic and prognostic biomarkers in cancer and other diseases has been documented [6-10]. Increasing evidence shows that miRNAs significantly contribute to different malignant phenotypes including chemoresistance [11-18]. However, there are few studies that identify and experimentally validate chemoresistance-related miRNAs in esophageal squamous cell carcinoma (ESCC). In our previous study, we established ESCC cell line models of acquired chemoresistance to fluorouracil (5-FU) (FR cells) [19]. By profiling the miRNA expression between parental cells and resistant cells in the present study, miR-29c-3p (referred to as miR-29c) was found to be one of the differentially expressed miRNAs in the FR cells, suggesting its potential role in modulating chemoresistance. However, the role of miR-29c in 5-FU chemoresistance has not been reported previously.
Here, we identified F-box only protein 31 (FBXO31), a novel F Box protein with prognostic significance in ESCC [20], as a protein that was upregulated in FR cells and a potential target of miR-29c. Our previous studies demonstrated that FBXO31 deactivates MAPK p38 and JNK signaling in ESCC cells subjected to genotoxic stresses, and that this mechanism contributes to DDP chemoresistance [20, 21], but the functional role of FBXO31 in 5-FU chemoresistance and its regulatory mechanisms have not been reported. In this study, the potential of miR-29c as a non-invasive biomarker and its mechanism in 5-FU chemoresistance of ESCC were investigated. Moreover, the efficacy of systemic delivery of miR-29c as cancer therapeutics was evaluated.
Methods
Cell culture and drugs
The 5-FU-resistant human ESCC sublines KYSE150FR and KYSE410FR, established through continuous treatment of KYSE150 and KYSE410 (DSMZ, Braunschweig, Germany) [22] with increasing concentrations of 5-FU [19], were maintained in RPMI 1640 (Sigma, St. Louis, MO,USA) supplemented with 10% fetal bovine serum (Invitrogen, Gaithersburg, MD, USA) at 37ºC in 5% CO2. Fluorouracil was purchased from Calbiochem (Darmstadt, Germany) and dissolved in dimethyl sulfoxide (DMSO). Cisplatin was purchased from Merck (Kenilworth, NJ, USA).
ESCC tumor, serum samples and tissue microarray
All clinical samples were collected with informed consent following approval by the Queen Mary Hospital (Hong Kong), Zhengzhou University (Zhengzhou, China) and Shantou University (Shantou, China). Primary tumors and the corresponding adjacent non-tumorous esophageal tissues were collected from a cohort of 50 patients who received frontline surgery without any prior chemoradiotherapy between 2011 and 2012 at the Queen Mary Hospital, and at the First Affiliated Hospital, Zhengzhou University from 2008 to 2010. Another independent cohort of 32 patients with survival data who underwent surgical resection without pre- operative chemoradiotherapy at Queen Mary Hospital was also included in this study. All tissues were frozen immediately in liquid nitrogen for subsequent protein or RNA extraction. In addition, serum samples from 50 healthy controls and 114 preoperative serum samples from patients who subsequently received 5-FU- and DDP-based neoadjuvant chemoradiotherapy were obtained from Shantou University Medical College and Queen Mary Hospital respectively. A tissue microarray containing 104 cases of ESCC and 75 cases of non-tumor tissue (Shanghai Outdo Biotech, Shanghai, China) was used to determine expression of STAT5A.
Taqman miRNA profiling
Total RNA from cells was extracted using Trizol reagent according to the manufacturer's protocol (Invitrogen), and the quality of total RNA was assessed with the Agilent 2100 Bioanalyzer Platform (Agilent Technologies, Santa Clara, CA, USA). Reverse transcription was performed with Megaplex RT primers (Applied Biosystems, Carlsbad, CA, USA), and the miRNA expression profiles were determined with TaqMan human MicroRNA Low- Density Array Set (Applied Biosystems). Expression levels were normalized against endogenous U6 small nuclear RNA.
Plasmids, transfection and infection
The transient expressing plasmids of miR-29c (pcDNA6.2-GW/EmGFP-miR-29c) and the scrambled miRNA control (pcDNA6.2-GW/EmGFP-miR-CON) were constructed using the Block-iT Pol II miR RNAi Expression Vector kit (Invitrogen). The pLenti- CMV-miR-29c and pLenti-CMV-miR-CON constructs used for stable expression were established using the Gateway approach (Invitrogen). The plasmids expressing miR-zip-29c and miR-zip-CON were purchased from System Biosciences (Palo Alto, CA, USA). The miR-29c mimic, miR-29c inhibitor and the negative controls were purchased from Ambion Inc (Austin, TX, USA). The FBXO31-overexpressing and -knockdown plasmids were prepared as described in our previous paper [21]. The 3'UTR of FBXO31 was introduced into pGL3-control to establish the pGL3-FBXO31-3'UTR plasmid. The pGL3-basic plasmid containing the promoter of miR-29c, pGL3- miR29c-pro-WT, was kindly provided by Professor Carlos Lo´pez-Otı´n (Universidad de Oviedo, Spain). Full-length STAT5A was obtained by PCR amplification and cloned into the pcDNA3 vector (Invitrogen). The siRNAs against STAT5A (si-STAT5A) were purchased from TranSheep Bio (Shanghai, China) and the target sequences were 5'-gcaacctgtggaacctgaaaccatt-3' for si-STAT5A#1 and 5'-atggatatgtgaaaccaca-3' for si-STAT5A#2. The STAT5A-knockdown plasmids were purchased from TranSheep Bio. Transfection was peformed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. For infection, 293T cells were transfected with lentiviral plasmids and packaging mix (Invitrogen) using Lipofectamine 2000, and the virus-containing culture media were collected and used to infect target cells, and the stable cell lines were selected with puromycin. The cells with stable overexpression of miR-29c were established and maintained at least two weeks before experiments.
Reverse transcription and quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer's protocol as previously described [23]. MicroRNA was converted to cDNA using the miScript II RT Kit (Qiagen, Hilden, Germany), and qPCR was performed using the miScript SYBR® Green PCR Kit (Qiagen). The forward primers for the 12 shortlisted miRNAs are listed in Table S1. Taqman MicroRNA Reverse Transcription kit and Taqman miRNA assays (Applied Biosystems) were also used to determine the expression of miR-29c and that of U6 as control [23]. All the experiments were performed on MyIQTM2 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The expression level of miR-29c was categorized into “high” and “low” using the median value as cut-off point.
Chromatin immunoprecipitation-quantitative PCR (ChIP)
The chromatin immunoprecipitation (ChIP) assay was performed by using simple ChIP enzymatic chromatin IP kit (Cell Signaling, Beverly, MA, USA) according to the manufacturer's manual as described previously [23]. In brief, in vivo protein and DNA crosslinking was performed using 37% formaldehyde, followed by sonication and chromatin digestion. The protein-DNA complexes were immunoprecipitated using STAT5A antibody or negative control IgG antibody. The purified DNA was subjected to SYBR Green PCR analysis (Bio-Rad). Relative mRNA expression was calculated using the comparative Ct method after normalization to GAPDH control.
Western blot
Western blot was performed as previously described [24]. The primary antibodies used included FBXO31 antibody from Abcam (Cambridge, UK), caspase-3 and cleaved caspase-3 from Cell Signaling Technology, phosphorylated p38 (p-p38), p38 from BD Biosciences (San Diego, CA, USA), STAT5A from Proteintech (Rosemont, IL, USA) and actin from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
MTT assay
Cell viability was determined as described previously [25]. Briefly, cells were plated in 96-well plates at a density of 1000 cells per well, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added at the end of experiment. The absorbance was measured at a wavelength of 570 nm on an automated microplate spectrophotometer (BioTek Instruments, Winooski, VT, USA).
Colony formation assay
Cells were seeded onto 6-well plates at a density of 500 cells per well and cultured for 14 days, and then fixed in 75% ethanol and stained with 0.2% crystal violet. The number of colonies was counted [26].
Flow cytometry analysis
Apoptosis was evaluated using an annexin V‐fluoroisothiocyanate apoptosis detection kit according to the manufacturer's instructions (KeyGen Biotech, Jiangsu, China). In brief, cells were pelleted by centrifugation and then resuspended in 100 μL of annexin V binding buffer. After incubation, cells were washed and subjected to flow cytometer analysis. The data were analyzed by FlowJo software (FlowJo LLC, Ashland, OR, USA).
Site-directed mutagenesis and luciferase assay
QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) was used to generate constructs that expressed mutated 3'UTR of FBXO31 and mutation of pGL3-miR29c-pro-WT. The primers used are listed in Table S2. The luciferase activity was measured by using dual-luciferase reporter assay (Promega, Madison, WI, USA) as previously described [19].
Tumor xenograft experiment
All the animal experiments were approved by the Committee on the Use of Live Animals in Teaching and Research, the University of Hong Kong, or Jinan University. Female BALB/c nude mice aged 6-8 weeks were used. Tumor xenografts were established as described previously [27]. When the subcutaneous tumors reached ~5 mm diameter, the mice were intraperitoneally injected twice weekly with 5-FU (20 mg/kg) or vehicle. In the experiment involving systemic miR-29c treatment, KYSE410FR cells were subcutaneously injected into mice and the mice were randomized into four groups when the tumors reached ~5 mm in diameter. The miR-29c oligonucleotide or miR-CON (GenePharma, Shanghai, China) was formulated with a polymer-based agent (in vivo-jetPEITM; Polyplus, Illkirch, France) as described previously [28] and injected intravenously into mice with or without 5-FU treatment. Tumor size was monitored with caliper every three days, and tumor volume was calculated using the equation V= (length x width2)/2. All measured tumor sizes were normalized to the average tumor volume of the corresponding DMSO-treated group. Tumors were collected for immunohistochemistry and Western blot analyses.
Immunohistochemistry
Immunohistochemistry analysis was performed on the paraffin-embedded tumor sections as previously described [29]. After antigen retrieval and blocking with normal serum, the slides were incubated with Ki-67 antibody (Dako, Mississauga, ON, Canada) or STAT5A antibody (Proteintech) overnight at 4 ºC, washed with phosphate buffered saline, followed by biotinylated secondary antibody and peroxidase-conjugated avidin-biotin complex (Dako). Immunostaining was visualized using 3,3'-diaminobenzidine (Dako), which served as a chromogen, and the sections were counterstained with hematoxylin. Ki-67 proliferation index and staining scores for STAT5A were determined as described previously [25, 30]. For STAT5A, specimens assigned scores of 0~1 were categorized as low expression, whereas scores 2~3 were determined as high expression.
Hematologic analyses
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in mouse serum were determined using commercial kits (HuiLi Biotech Ltd., Changchun, China). Hemoglobin (HGB) and blood cells from mice including lymphocytes, neutrophils, platelets (PLT), red blood cells (RBC) and white blood cells (WBC) were analyzed by a fully automatic hematology analyzer (BC-2800Vet, Mondrary, Shenzhen, China).
Bioinformatics study
Two transcription factor binding site prediction software programs ConTra V2 [31] and TRRD [32] were used to identify the potential STAT5A binding sites in the promoter region of miR-29c (chr1 207995872-208000037). Two software programs including TargetScan (http://www.targetscan.org/vert_ 50/) and miRanda (http://www.microrna.org/microrna/getExpr Form.do) were used to predict the biological targets of miR-29c, as previously described [23].
Public datasets analysis
The expression values of miR-29c and clinical data were retrieved from the public datasets and analyzed as previously described [29]. The expression values of miR-29c in different cancer types in TCGA datasets were analyzed via OncomiR website (http:// www.oncomir.org/oncomir/search_miR_tumor.html). The values were divided as low and high using the 50% value as cut-off point.
Statistical analysis
The data were expressed as the mean ± SD and compared using ANOVA. The association between the expression levels of miR-29c and FBXO31 was determined using Pearson rank correlation coefficient. The expression level of miR-29c in tumor and serum samples was compared with that in non-tumor tissues and healthy control serum, respectively, using paired or unpaired t-test. The correlation between miR-29c and clinicopathological parameters was assessed using Fisher exact test. Survival analysis was performed by Kaplan-Meier method with the log-rank test. P values < 0.05 were deemed significant. All in vitro experiments were repeated at least three times.
Results
miR-29c is downregulated in 5-FU chemoresistant ESCC cells
In order to screen and identify the miRNAs which regulate 5-FU chemoresistance, a miRNA microarray was used to profile the miRNAs differentially expressed between FR cells (KYSE150-FR) and parental KYSE150 cells. A literature search was conducted for the top 100 downregulated miRNAs (Table S3) to narrow the candidates. Sixty-seven miRNAs were discarded because they are frequently upregulated in a wide variety of cancer and therefore less likely to have tumor-suppressive function in ESCC (Figure S1A). Of the remaining 33 miRNAs, we further excluded 21 miRNAs which had already been reported to be related to chemoresistance, leaving 12 novel ones for further validation (Table S1). Our qRT-PCR data showed that, among the 12 shortlisted candidates, miR-29c was the most consistently and significantly decreased miRNA in two FR sublines, compared with corresponding parental cells (Figure S1B-C), suggesting its potential role in regulating chemoresistance.
Figure 1
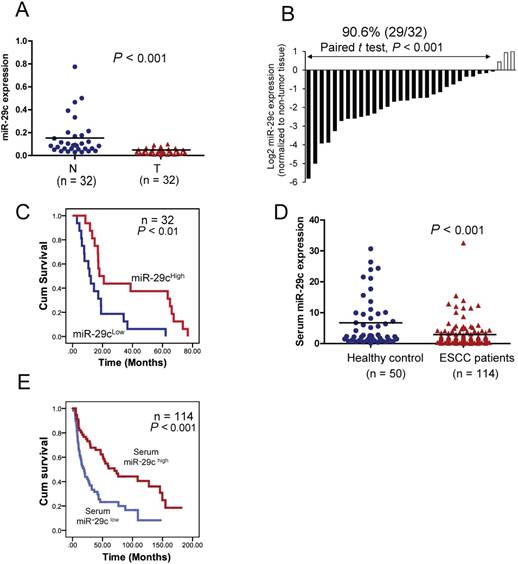
Clinical significance of miR-29c in ESCC. (A) Mean miR-29c expression in primary esophageal cancer (T) and the paired non-tumor tissues (N) was compared using Taqman qRT-PCR. The horizontal bars represent the mean values. (B) miR- 29c expression in 32 primary ESCC tumor samples relative to the corresponding non-tumor tissues. Lower expression of miR- 29c was observed in the majority of tumors examined (29/32, 90.6%), compared with their corresponding non-tumor tissues.(C) Kaplan-Meier analysis based on the 32 tumor tissues showing that miR-29c expression was positively associated with patient survival. (D) Serum miR-29c level in 114 cancer patients and 50 healthy control subjects. The horizontal bars represent mean values. (E) Kaplan-Meier analysis of overall survival of 114 patients with ESCC stratified according to admission serum miR-29c level.
miR-29c is aberrantly downregulated and correlated with poor survival outcome in patients with several types of cancer
To investigate the clinical significance of miR-29c in ESCC, the expression of miR-29c was determined in a cohort including 32 pairs of tumors and matched adjacent non-tumor tissues by qRT-PCR. The results showed that miR-29c expression was significantly lower in tumor tissues than adjacent non-tumor tissues (P < 0.001) (Figure 1A), and that downregulation was detected in 90.6% (29/32) of ESCC cases (Figure 1B). As shown in Figure 1A, the mean miR-29c expression in the tumor tissues was about 3.9-fold lower than that in the paired non-tumor tissues. More importantly, our results showed that miR-29c was significantly associated with overall survival of ESCC patients, with the mean survival time dropping from 36.8 months in the patients with high miR-29c expression to 17.0 months in the patients with low miR-29c expression (Figure 1C). Taken together, these data suggest that miR- 29c could be a prognostic marker to predict the outcome of ESCC patients. By analyzing the Gene Omnibus Express (GEO) and TCGA datasets, we found frequent downregulation of miR-29c in tumor tissues of multiple cancer types, compared with non- tumor tissues (Figures S2A and S3A). In the public databases, low miR-29c expression was also associated with poor prognosis in several other cancer types (Figures S2B and S3B).
High expression level of miR-29c in serum is associated with better prognosis in patients with ESCC
We further explored the clinical relevance of miR-29c as a non-invasive marker in ESCC by comparing the expression level of miR-29c in admission serum samples of 114 ESCC patients with that in serum samples of 50 healthy individuals. The expression level of serum miR-29c was significantly downregulated in the ESCC patients (Figure 1D). Moreover, Kaplan- Meier survival analysis showed that the ESCC patients with high serum miR-29c expression had significantly prolonged overall survival time (Figure 1E). Collectively, these data indicated that serum miR-29c level could predict the response of ESCC patients to chemoradiotherapy.
Figure 2
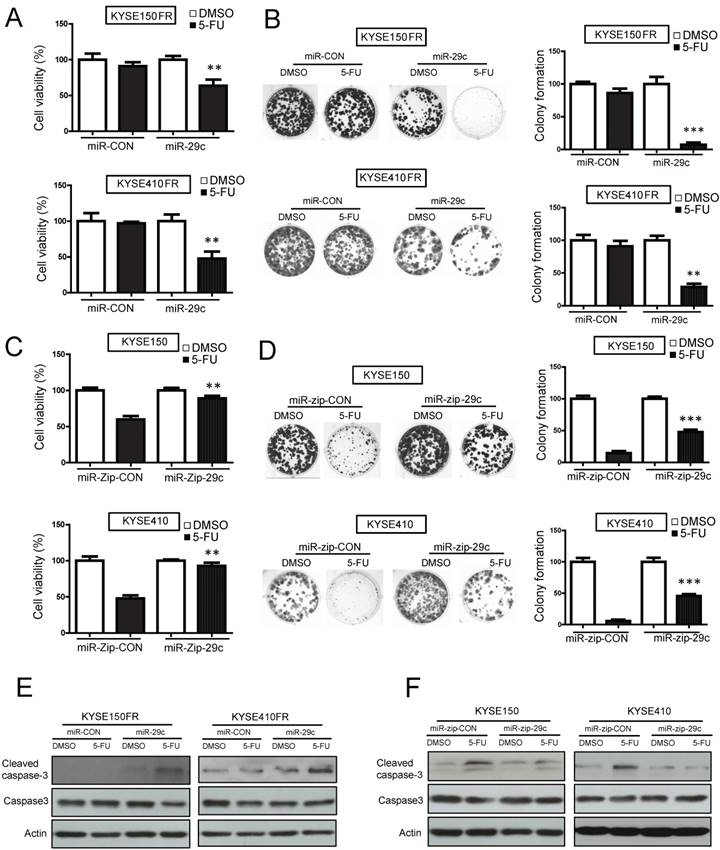
miR-29c expression sensitizes ESCC cells to 5-FU treatment. (A, B) FR ESCC cells KYSE150FR and KYSE410FR which stably overexpressed miR-29c or the scrambled miRNA control were treated with 5-FU (10 μM) or DMSO for 48 hours, and then subjected to MTT assay (A) and colony formation assay (B). (C) ESCC cells with miR-29c-knockdown and the control cells expressing miR-zip-CON were exposed to 5-FU (10 μM) for 48 hours, and cell viability was determined by MTT assay. (D) Colony formation assay showing the effect of miR-29c knockdown on the sensitivity of ESCC cells to 5-FU. (E) Expression levels of cleaved caspase-3 and caspase-3 were determined in the miR-29c-overexpressing cells and control cells upon 5-FU treatment by Western blot. Actin was included as loading control. (F) Western blot data showing that 5-FU could not induce expression of cleaved caspase-3 when miR-29c expression was inhibited in KYSE150 and KYSE410 cells. Bars, SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with corresponding 5-FU treated control cells.
Ectopic expression of miR-29c overrides 5-FU chemoresistance in ESCC cells
To study the role of miR-29c in ESCC chemoresistance, miR-29c was stably overexpressed in two FR sublines (KYSE150FR-miR-29c and KYSE410FR- miR-29c) (Figure S4A), and then the cells were treated with 5-FU in functional assays. The results from MTT and colony-formation assays showed that ectopic expression of miR-29c overcame the resistance of KYSE150FR and KYSE410FR cells to 5-FU treatment (Figure 2A-B). Conversely, knockdown of miR-29c by miR-zip-29c (Figure S4A) markedly conferred 5-FU chemoresistance to ESCC cells (Figure 2C-D). Moreover, we found that miR-29c promoted apoptosis of the 5-FU-treated cells, indicated by the increased 5-FU-induced cleaved caspase-3 expression in miR-29c-overexpressing FR cells and the decreased 5-FU-induced cleaved caspase-3 expression in miR-29c-knockdown cells (Figure 2E-F). Flow cytometry confirmed the effects of miR-29c on apoptosis (Figure S4B). Taken together, these data demonstrated that miR-29c could enhance the sensitivity of ESCC cells to 5-FU.
In addition, we determined whether miR-29c dyregulation may contribute to DDP chemoresistance. ESCC cells with miR-29c-knockdown showed enhanced viability and colony formation ability, compared with the control cells expressing miR-zip-CON, when exposed to DDP, which suggests that miR-29c can sensitize ESCC cells to DDP treatment (Figure S5A-B).
FBXO31 is a direct target of miR-29c
We next investigated the mechanism by which miR-29c regulates 5-FU resistance in ESCC cells. In silico analysis using TargetScan and MiRanda target prediction algorithms was performed to search for potential targets of miR-29c (Figure 3A). FBXO31, which we previously reported to have prognostic significance and regulatory function on DDP chemoresistance in ESCC [20, 21], was identified as a potential target of miR-29c. The binding site between miR-29c and FBXO31 3'UTR is illustrated in Figure 3B. Interestingly, FBXO31 was found to be significantly upregulated in the FR cells, compared with the parental cells (Figure 3C). A decrease in FBXO31 expression and an increase in p-p38 expression were observed in the FR cell lines with stable overexpression of miR-29c (Figure 3D). Conversely, expression level of FBXO31 was upregulated, whereas p-p38 expression was downregulated, in the ESCC cells with stable knockdown of miR-29c (Figure 3E). The inhibitory effect of miR-29c on FBXO31 expression was confirmed by transient transfection of miR- 29c mimic or anti-miR-29c inhibitor (Figure S6A-B). To further study the significance of interaction between FBXO31 and miR-29c, dual-luciferase reporter assay was performed, plasmids expressing wild type FBXO31 3'UTR and miR-29c, respectively, were co-transfected into KYSE150 cells, and the results showed that miR-29c overexpression led to a significant decrease in luciferase activity of FBXO31 3'UTR in a dose-dependent manner (Figure 3F). However, the inhibitory effect of miR-29c on luciferase activity of FBXO31 3'UTR was abolished when the binding site of miR-29c on FBXO31 3'UTR was mutated (Figure 3G), confirming that miR-29c can directly bind to and decrease FBXO31 expression. Furthermore, the expression levels of miR-29c and FBXO31 were determined in 50 pairs of ESCC tumor and matched non-tumor tissues (Figure S7). Markedly higher expression of FBXO31 (Figures 3H) and lower expression of miR-29c (Figure 3I) were found in ESCC tissues compared with non-tumor esophageal tissues, and a significant inverse correlation between miR-29c and FBXO31 was observed (Figure 3J). These data make it evident that miR-29c affects FBXO31 expression by directly binding to its 3'UTR region, thus suggesting that aberrant downregulation of miR-29c may enhance FBXO31-mediated chemoresistance in ESCC.
FBXO31 mediates the effect of miR-29c on 5-FU chemoresistance in vitro and in vivo
The effect of FBXO31 in 5-FU chemoresistance has not been reported previously. We found that knockdown of FBXO31 by stable transfection with shRNA FBXO31 (Figure S8A) significantly reduced 5-FU chemoresistance in KYSE150FR and KYSE410FR cells, as determined using MTT and colony formation assays (Figure S8B-C). To study whether FBXO31 mediates the effect of miR-29c on 5-FU chemoresistance, the endogenous FBXO31 expression in the FR ESCC cells was first suppressed by overexpressing miR-29c, which resulted in increased phosphorylation/activation of p38 (Figure 4A). Western blot analysis confirmed that these effects were abolished by co-expressing FBXO31 in the FR cells (Figure 4A). The FR cells co-overexpressing miR-29c and FBXO31 (KYSE150FR-miR-29c-FBXO31, KYSE410FR-miR-29c-FBXO31), or miR-29c alone (KYSE150FR-miR-29c-CON, KYSE410FR-miR-29c-CON), and the control cells (KYSE150-FR-miR-CON-CON, KYSE410FR-miR-CON-CON) were then treated with 10 μM 5-FU. The results from MTT assay showed that overexpression of miR-29c led to a significant increased sensitivity to 5-FU, and that this effect was significantly diminished when FBXO31 expression was restored (Figure 4B). Consistent results were obtained at a higher concentration of 5-FU (i.e. 20 μM) (Figure S9). Western blot data confirmed that miR-29c enhanced 5-FU-induced expression of cleaved caspase-3 in KYSE150FR and KYSE410 cells, whereas ectopic expression of FBXO31 significantly abrogated these effects (Figure 4C). On the contrary, inhibition of FBXO31 expression level with lentiviral shRNA in the miR-29c-knockdown ESCC cells (KYSE150-miR-zip- 29c-shFBXO31 and KYSE410-miR-zip-29c-shFBXO31) markedly counteracted the 5-FU chemoresistance induced by miR-29c knockdown, as indicated by Western blot and MTT assays (Figure 4D-F). We next performed flow cytometry and the results showed that FBXO31-knockdown significantly increased 5-FU-induced apoptosis (Figure S10).
Figure 3
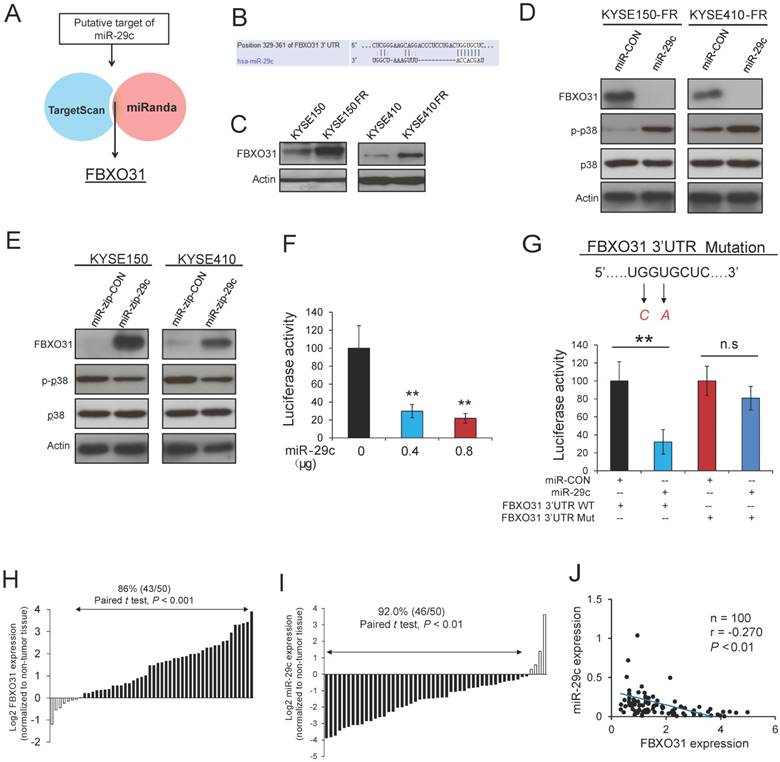
FBXO31 is a target of miR-29c. (A) Diagram showing the approach used to identify the putative targets of miR-29c by in silico software prediction. (B) Alignment of miR-29c and its corresponding complementary binding sequence in FBXO31 3'UTR. (C) FBXO31 expression was determined in 5-FU-resistant ESCC cells and parental cells by Western blot. (D) Western blot analysis showing the effects of stable miR-29c overexpression on FBXO31, p-p38 and total p38 expressions in KYSE150FR and KYSE410FR cells. (E) Effects of stable knockdown of miR-29c on FBXO31, p-p38, p38 expressions in ESCC cells, as determined by Western blot. (F) Ectopic miR-29c expression caused dose-dependent decrease in the luciferase activity of FBXO31 3'-UTR. (G) Luciferase activity in KYSE150 cells co-transfected with the indicated plasmids. Mutations introduced into the FBXO31 3'UTR by site-directed mutagenesis to generate the mutant FBXO31 3'UTR are shown above the graph. (H) Expression level of FBXO31 was determined in 50 pairs of ESCC (T) and matched non-tumor (N) tissues by Western blot. Graph showing the FBXO31 expression in the primary tumors which was quantified relative to that of corresponding non-tumor tissue samples. (I) Expression level of miR-29c was determined in the 50 pairs of ESCC tissues and non-tumor tissues by Taqman qRT-PCR. In the majority of the cases (46/50, 92.0%), miR-29c expression was lower in tumor tissues relative to the corresponding non-tumor tissues. (J) Inverse correlation between miR-29c and FBXO31 in clinical ESCC tissues. Bars, SD; n.s., no significance;**, P < 0.01.
We further performed in vivo experiment to validate these findings, and the results showed that ectopic miR-29c expression could override the resistance of FR tumor xenografts to 5-FU treatment in nude mice, and that overexpression of FBXO31 significantly attenuated this effect (Figure 4G-H). Collectively, these results demonstrated that miR-29c could sensitize ESCC cells to 5-FU treatment through downregulation of FBXO31 expression.
STAT5A regulates the expression of miR-29c at transcriptional level
To obtain a better understanding of why miR-29c is underexpressed in ESCC, in silico prediction algorithm was applied to determine the upstream regulatory mechanism of miR-29c. Three potential binding sites for STAT5A were identified in the promoter region of miR-29c, suggesting that STAT5A may participate in the regulation of miR-29c. Gain- and loss-of-function experiments showed that STAT5A expression negatively regulated the expression of miR-29c (Figure 5A and Figure S11A). We also compared the expression of STAT5A in parental cells and corresponding FR cells, and noted that STAT5A did increase in the FR cells (Figure S11B). Chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) assay was carried out to investigate whether there is physical interaction between STAT5A protein and the promoter region of miR-29c. We found that, of the three potential STAT5A binding sites in the promoter region of miR-29c (designated as Site 1, Site 2 and Site 3), only the Site 3 fragment was enriched in the STAT5A immunoprecipitates (Figure 5B). Site-specific mutations and luciferase assay further confirmed that only Site 3, but not the other two fragments, functions as the STAT5A-responsive element (Figure 5C).
Figure 4
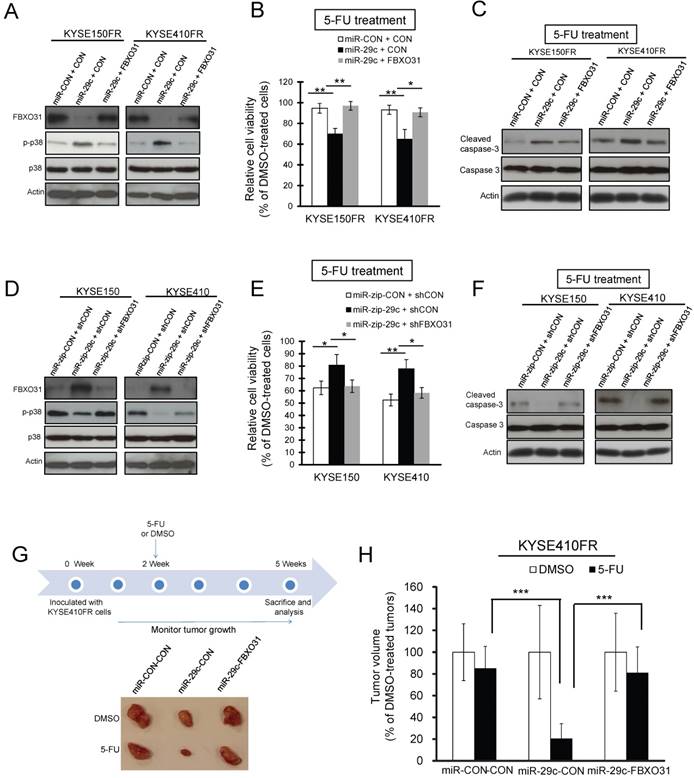
FBXO31 mediates the effect of miR-29c on 5-FU chemoresistance in vitro and in vivo. (A) Western blot analyses showing that restoration of FBXO31 expression abolished p38 activation in miR-29c-overexpressing KYSE150FR and KYSE410FR cells. (B, C) KYSE150FR and KYSE410FR cells with stable overexpression of miR-29c, co-overexpression of miR-29c and FBXO31, or control plasmids, were treated with 5-FU (10 μM) or DMSO for 48 hours and then subjected to MTT assay (B) and Western blot analyses for detection of cleaved caspase-3 and caspase-3 (C). (D) Western blots showing that knockdown of FBXO31 expression reactivated p38 in KYSE150 and KYSE410 cells with stable expression of miR-zip-29c. (E,F) Knockdown of FBXO31 abrogated the effect of miR-29c inhibition on enhancing the chemoresistance of ESCC cells to 5-FU treatment, as shown in MTT assay (E) and Western blot analysis of apoptotic markers (F). (G) Experimental scheme of tumor xenograft model established with the indicated cells, and photograph of representative tumors from the six groups (n = 6 per group). (H) Comparison of KYSE410FR-miR-CON-CON, KYSE410FR-miR-29c-CON, and KYSE410FR-miR-29c-FBXO31 tumor xenografts in nude mice for 5-FU chemosensitivity. Bars, SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Figure 5
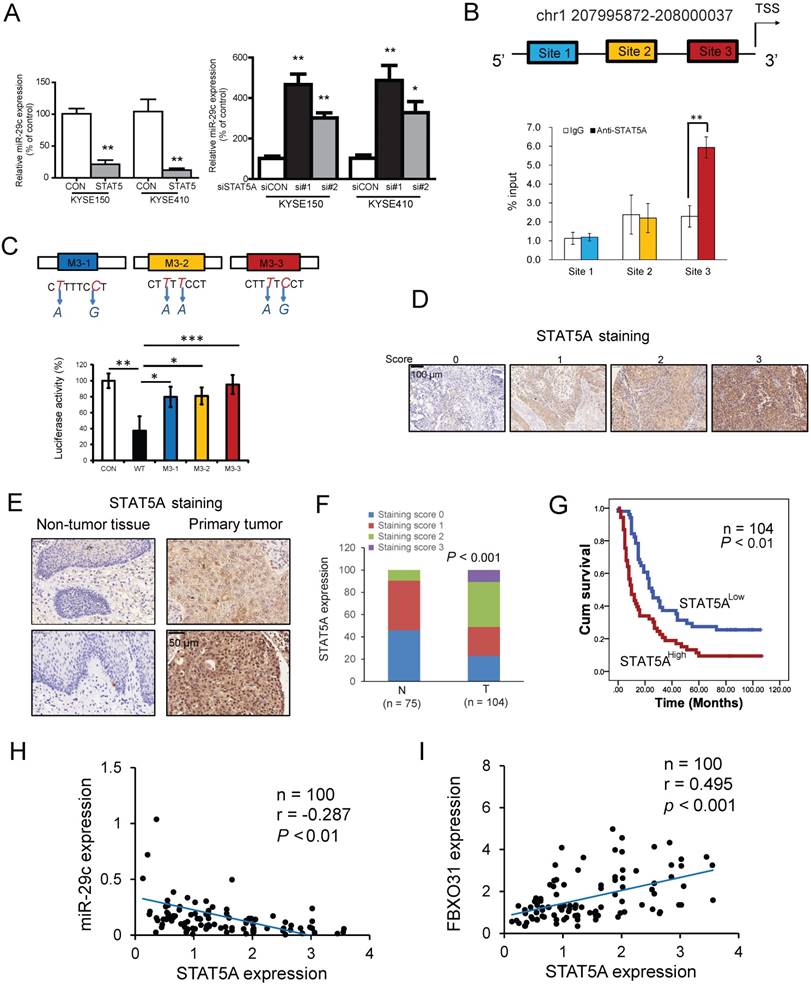
STAT5A regulates miR-29c and is inversely correlated with miR-29c expression in ESCC tissues. (A) Taqman miRNA assay showing the changes in miR-29c expression upon overexpression and knockdown of STAT5A. (B) Diagram illustrating the putative STAT5A binding sites in the promoter of miR-29c by in silico prediction. The enrichment of STAT5A in miR-29c promoter region was determined by ChIP-qPCR. (C) Upper panel shows the site-specific mutations introduced in the reporter plasmid containing miR-29c promoter (Upper panel). Lower panel shows the luciferase activity in ESCC cells co-transfected with STAT5A-expressing plasmid and the plasmid expressing wide type or mutated miR-29c promoter. (D) Representative images of ESCC with immunohistochemical staining scores of 0-3 for STAT5A. (E) Two examples of primary ESCC with high immunohistochemical expression of STAT5A, compared with matched normal epithelium. (F) Stacked graph showing the expression of STAT5A in 104 cases primary tumor tissue and 75 cases non-tumor tissues. (G) Kaplan-Meier analysis based on the 104 tumor tissues showing that STAT5A expression was negatively associated with survival in patients with ESCC (Log Rank = 7.86, P < 0.01). (H) Inverse expression correlation between miR-29c and STAT5A in clinical ESCC tissues. (I) Inverse expression correlation between FBXO31 and STAT5A in clinical ESCC tissues. Bars, SD; *, P < 0.05; **, P < 0.01; ***, P <0.001.
Western blot was undertaken to determine the effect of manipulation of STAT5A expression on FBXO31, and the results indicated a positive regulation as shown in Figure S12A. To study the significance of STAT5A in chemoresistance, shRNA against STAT5A was stably transfected into the KYSE150FR and KYSE410FR cells. The data from MTT assay and colony formation assays showed that knockdown of STAT5A significantly enhanced 5-FU sensitivity (Figure S12B-C).
The clinical relevance of STAT5A in ESCC is largely unknown. We made use of a tissue microarray consisting of 104 cases of primary ESCC tissues and 75 cases of non-tumor tissues and performed immunohistochemistry for STAT5A (Figure 5D). Fifty-one percent of the ESCC tumors received high immunohistochemical scores of 2 to 3, whereas 90.5% of non-tumor esophageal tissue samples showed no or weak staining (scores 0 to 1) (χ2 test, P < 0.001) (Figure 5E-F). Survival analysis showed that STAT5A expression was negatively correlated with overall survival. Patients with low STAT5A expression had a longer survival (mean survival = 40.35 months) than the patients with high STAT5A expression (mean survival = 22.56 months) (Figure 5G). In addition, there is a significant inverse correlation between STAT5A expression and pathological N-stage (Table 1). Furthermore, the expression of STAT5A was analyzed in the same 50 pairs of ESCC tumor and matched non-tumor tissues mentioned in Figure 3. The data showed a markedly higher expression of STAT5A in ESCC tissues compared with non-tumor esophageal tissues (Figure S13), and a significant inverse correlation between miR-29c and STAT5A (r = -0.287, P < 0.01) (Figure 5H). Moreover, there was a positive correlation between STAT5A and FBXO31 (r = 0.495, P < 0.001) (Figure 5I). We also selected 12 pairs of ESCC and matched non-tumor samples to determine the expression of p-p38, and found that p-p38 had an inverse correlation with FBXO31 and STAT5A (Figure S14A-C).
Systemic delivery of miR-29c increases chemosensitivity of ESCC cells to 5-FU in vivo
We next assessed the therapeutic potential of systemically delivered miR-29c in treating 5-FU chemoresistant ESCC. The 5-FU-resistant ESCC tumor xenografts were established by subcutaneously injecting KYSE410FR cells into the flanks of nude mice. We found that administration of a combination of miR-29c oligonucleotide and 5-FU resulted in a significant decrease in tumor volume compared with 5-FU treatment alone (Figure 6A-B). Furthermore, systemic miR-29c treatment facilitated 5-FU-induced apoptosis and suppressed proliferation of tumor cells, as indicated by increased expression of cleaved caspase-3 and p-p38 (Figure 6C) and decreased percentage of Ki-67-positive tumor cells (Figure 6D), respectively, in the tumor xenografts of mice treated with both 5-FU and miR-29c, compared with the groups treated with 5-FU or miR-29c alone. Notably, the administration of miR-29c did not cause toxicity to the mice, as evidenced by the lack of significant changes in body weight (Figure 6E), and blood biochemical and hematological parameters (Figure 6F).
Table 1
Correlation between STAT5A expression levels and clinicopathological parameters in 104 cases of esophageal cancer.
| Variable | n | Low STAT5A | High STAT5A | P value |
|---|---|---|---|---|
| Age ( years) | ||||
| ≤55 | 20 | 11 | 9 | |
| >55 | 84 | 40 | 44 | 0.552 |
| Gender | ||||
| Female | 28 | 17 | 11 | |
| Male | 76 | 34 | 42 | 0.160 |
| T-Stage | ||||
| 1/2 | 16 | 9 | 7 | |
| 3/4 | 86 | 42 | 44 | 0.586 |
| N-Stage | ||||
| N0 | 47 | 29 | 18 | |
| N1 | 56 | 22 | 34 | 0.023* |
| Pathologic stage | ||||
| Stages I & II | 76 | 37 | 39 | |
| Stages III & IV | 28 | 14 | 14 | 0.905 |
Discussion
Resistance to chemotherapy drugs is a major cause of tumor recurrence. A relapse phenotype may be due to pre-existing resistance of the original tumor to therapy (intrinsic resistance), or therapy-dependent selection of resistant cancer cells (acquired resistance) [2]. A number of miRNAs and their target genes are known to play a role in chemoresistance. The miR34a/GOLPH3 axis, for example, was reported to abrogate urothelial bladder cancer chemoresistance via reduced cancer stemness [18]. In another study, Lai et al. showed that miRNAs, namely miR-205-5p and miR-342-3p, can cooperate to repress E2F1 and thereby decrease tumor chemoresistance [33]. MiRNA arrays are frequently used to profile differentially expressed miRNAs in matched tumor tissues and adjacent non-tumor tissue [34, 35], but studies that make use of miRNA array to profile chemoresistance- related miRNAs are rare. This is the first study that used miRNA array to compare matched parental versus 5-FU chemoresistant cells to identify resistance-related miRNA signatures. The advantages of using matched parental versus resistant cells are obvious: 1) this cell model mimics the process of acquired chemoresistance, and this comparative approach allows us to capture the genetic alterations that occur during this process; 2) the use of matched cell lines with same genetic background can circumvent the confounding factor of massive genetic heterogeneity (associated with tumor tissues) which may create unnecessary interference in profiling resistance-related miRNAs. Using this approach, we have provided the first evidence of miR-29c downregulation in 5-FU resistant cancer cells, and shown that restoration of miR-29c can sensitize ESCC cells to 5-FU treatment in vitro and in vivo (Figures 2 and 4).
Figure 6
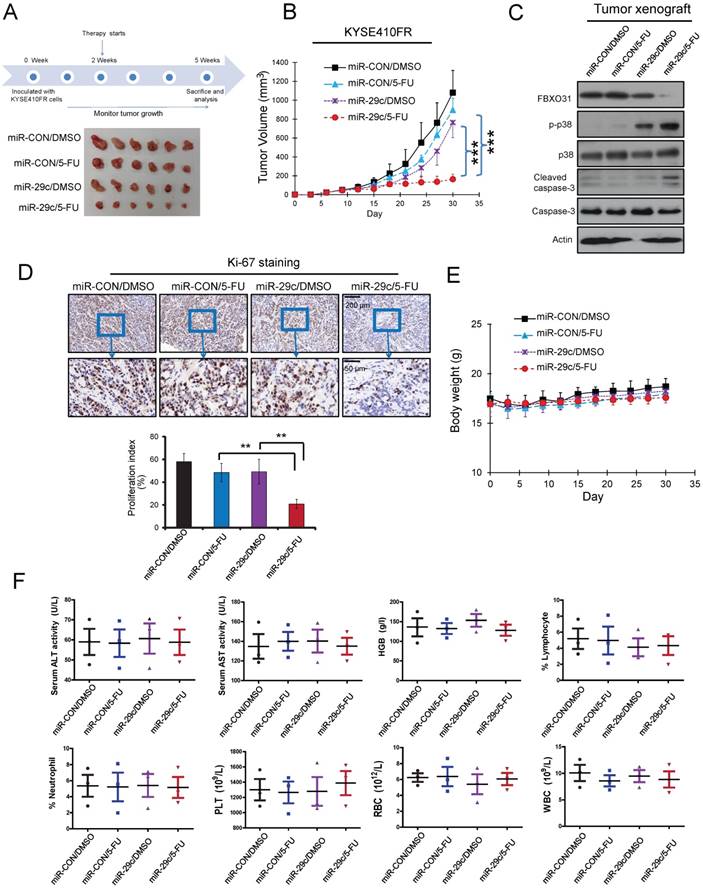
Systemic delivery of miR-29c oligonucleotide overrides 5-FU chemoresistance of FR cells in vivo. (A) Schematic diagram of the miR-29c oligonucleotide treatment experiment and photograph of tumors excised from the control and treatment groups. (B) Tumor growth curves showing the chemosensitizing effect of miR-29c oligonucleotide on KYSE410FR-derived tumor xenografts to 5-FU treatment (n = 6). (C) Western blot analyses comparing the expressions of cleaved caspase-3 and caspase-3 in the tumors of the four groups of nude mice. (D) Quantification of Ki-67 proliferation index in the tumors. (E) Body weight of the nude mice in the treatment and control groups as indicated was monitored. (F) No significant differences were detected in the blood chemistry and blood cell counts among the groups; ALT (alanine aminotransferase), AST (aspartate aminotransferase), lymphocytes, neutrophils, HGB (hemoglobin), PLT (platelets), RBC (red blood cells), WBC (white blood cells).
Biomarkers that can facilitate early diagnosis and predict treatment response are instrumental in optimizing therapy and improving the outcome for patients with ESCC. The clinical significance of miR-29c dysregulation was evaluated in the present study (Figure 1). We found that the expression levels of miR-29c is significantly lower in primary ESCC tumor tissue compared with matched non-tumor tissues, and that high tumor miR-29c expression level predicts better survival. Our results also showed that serum miR-29c level has diagnostic significance, as reported previously by Xu et al, thus affirming its significance as a non-invasive marker for ESCC screening [36]. More importantly, the data from our larger cohort of patients demonstrate that serum miR-29c level has the potential to be a predictive marker for response to chemoradiotherapy.
The biogenesis of miRNAs is under tight control at various levels. A previous study showed that miR-29 is repressed by NF-kB through Yin Yang 1 (YY1) and the Polycomb group, and that disruption of this regulatory circuit contributes to rhabdomyosarcoma [37]. Another study demonstrated that miR-29 is under negative regulation by TGF-beta-Smad3 signaling via dual mechanisms of both inhibiting MyoD binding and enhancing YY1-recruited Polycomb association [38]. In our previous study, we showed that IGF2 regulates miR-29c at transcriptional level in a p53-dependent manner [23]. In this study, by using in silico prediction, followed by ChIP and gain- and loss-of-function study, we have uncovered another novel mechanism whereby STAT5A represses the transcription of miR-29c by directly binding to its promoter (Figure 5). The function of this mechanism in regulating FBXO31 expression and chemoresistance is represented schematically in Figure 7.
Figure 7
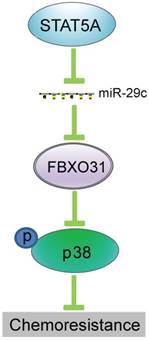
Schematic illustration of how transcriptional regulation of miR-29c by STAT5A can regulate FBXO31 expression and chemoresistance.
F-box proteins are well recognized as the substrate-recognition components of the SKP1/ CUL1/F-box protein (SCF) class of E3 ubiquitin ligases [39]. Our recent report showed that higher FBXO31 expression level is significantly correlated with histological grade, clinical stage and post-operative survival in patients with ESCC [20]. Another of our previous studies also showed that FBXO31 binds to MKK6 and decreases its protein stability, thereby negatively regulating the activation of p38 in cancer cells upon genotoxic stresses [21]. The molecular mechanism on upstream regulation of FBXO31 was unknown. Here, our results show for the first time that miR-29c directly binds to and represses the expression level of FBXO31. We further confirmed an inverse correlation between miR-29c and FBXO31 in 50 pairs of ESCC tissues (Figure 3). Our results showing that enforced expression of FBXO31 abolished the 5-FU-chemosensitizing effect of miR-29c in ESCC cells (Figure 4) revealed for the first time the key role of FBXO31 in mediating miR-29c-regulated 5-FU chemoresistance. Nevertheless, we do not rule out the possibility that other targets of miR-29c such as cyclin E may also mediate the chemosensitizing effects of miR-29c [40]. We have focused on FBXO31 in this study because its biological role of FBXO31 in cancer was rarely studied, and its potential function in 5-FU chemoresistance had not been reported.
In conclusion, this study establishes that miR-29c can sensitize ESCC cells to 5-FU treatment through regulation of FBXO31-p38 signaling, and that the expression levels of miR-29c in the tumor and serum of ESCC patients have diagnostic and prognostic significance. Our preclinical data, which proved that systemic delivery of miR-29c oligonucleotide could significantly alleviate the 5-FU chemoresistance of ESCC tumors, are strong evidence supporting the use of miR-29c as adjuvant therapy in the management of ESCC. Since a considerable percentage of ESCC overexpress STAT5A and STAT5A can suppress miR-29c, targeting STAT5A may be an additional strategy that is worth considering.
Abbreviations
ALT: Alanine aminotransferase; AST: aspartate aminotransferase; ChIP: Chromatin immunoprecipitation; DDP: cisplatin; DMSO: dimethyl sulfoxide; ESCC: esophageal squamous cell carcinoma; FBS: fetal bovine serum; FBXO31: F-box only protein 31; 5-FU: fluorouracil; GEO: Gene Omnibus Express; HGB: Hemoglobin; miRNA: microRNA; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PLT: platelets; RBC: red blood cells; SCF: SKP1/ CUL1/F-box protein; Taqman qRT-PCR: Taqman quantitative real time polymerase chain reaction; WBC: white blood cells.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Project Nos. 81472790, 81672953); Guangzhou Science and Technology Project (201707010260); Research Grants Council of the Hong Kong SAR, China (GRF Project Nos. 17103814 and 17111917). We thank Professor Yutaka Shimada (University of Toyama, Toyama, Japan) and DSMZ for the KYSE cell lines; Hua Zhen Tan, Shan Shan Li and Yi Wei Xu (Shantou University Medical College, China) for their help in collecting human serum samples. We acknowledge the Centre for Genomics Sciences, The University of Hong Kong, for their services in miRNA profiling.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-26
2. Lippert TH, Ruoff HJ, Volm M. Intrinsic and acquired drug resistance in malignant tumors. The main reason for therapeutic failure. Arzneimittel-Forschung. 2008;58:261-4
3. Enzinger PC, Mayer RJ. Esophageal cancer. N Eng J Med. 2003;349:2241-52
4. Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390-4
5. Srinivasan S, Selvan ST, Archunan G, Gulyas B, Padmanabhan P. MicroRNAs - the next generation therapeutic targets in human diseases. Theranostics. 2013;3:930-42
6. Kleivi Sahlberg K, Bottai G, Naume B, Burwinkel B, Calin GA, Borresen-Dale AL. et al. A serum microRNA signature predicts tumor relapse and survival in triple-negative breast cancer patients. Clin Cancer Res. 2015;21:1207-14
7. de Jong MC, Ten Hoeve JJ, Grenman R, Wessels LF, Kerkhoven R, Te Riele H. et al. Pretreatment microRNA expression impacting on epithelial-to-mesenchymal transition predicts intrinsic radiosensitivity in head and neck cancer cell lines and patients. Clin Cancer Res. 2015;21:5630-8
8. Roberts BS, Hardigan AA, Moore DE, Ramaker RC, Jones AL, Fitz-Gerald MB. et al. Discovery and validation of circulating biomarkers of colorectal adenoma by high-depth small RNA sequencing. Clin Cancer Res. 2018;24:2092-9
9. Berger F, Reiser MF. Micro-RNAs as potential new molecular biomarkers in oncology: have they reached relevance for the clinical imaging sciences? Theranostics. 2013;3:943-52
10. Bertoli G, Cava C, Castiglioni I. MicroRNAs: new biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics. 2015;5:1122-43
11. Zhang JX, Chen ZH, Xu Y, Chen JW, Weng HW, Yun M. et al. Downregulation of microRNA-644a promotes esophageal squamous cell carcinoma aggressiveness and stem cell-like phenotype via dysregulation of PITX2. Clin Cancer Res. 2017;23:298-310
12. Qian D, Chen K, Deng H, Rao H, Huang H, Liao Y. et al. MicroRNA-374b suppresses proliferation and promotes apoptosis in T-cell lymphoblastic lymphoma by repressing AKT1 and Wnt-16. Clin Cancer Res. 2015;21:4881-91
13. Fang T, Lv H, Lv G, Li T, Wang C, Han Q. et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9:191
14. Yao J, Liang L, Huang S, Ding J, Tan N, Zhao Y. et al. MicroRNA-30d promotes tumor invasion and metastasis by targeting Galphai2 in hepatocellular carcinoma. Hepatology. 2010;51:846-56
15. Liang L, Wong CM, Ying Q, Fan DN, Huang S, Ding J. et al. MicroRNA-125b suppressesed human liver cancer cell proliferation and metastasis by directly targeting oncogene LIN28B2. Hepatology. 2010;52:1731-40
16. Ye G, Huang K, Yu J, Zhao L, Zhu X, Yang Q. et al. MicroRNA-647 targets SRF-MYH9 axis to suppress invasion and metastasis of gastric cancer. Theranostics. 2017;7:3338-53
17. Wu DW, Wang YC, Wang L, Chen CY, Lee H. A low microRNA-630 expression confers resistance to tyrosine kinase inhibitors in EGFR-mutated lung adenocarcinomas via miR-630/YAP1/ERK feedback loop. Theranostics. 2018;8:1256-69
18. Zhang Q, Zhuang J, Deng Y, Yang L, Cao W, Chen W. et al. miR34a/GOLPH3 axis abrogates urothelial bladder cancer chemoresistance via reduced cancer stemness. Theranostics. 2017;7:4777-90
19. Li B, Xu WW, Guan XY, Qin YR, Law S, Lee NP. et al. Competitive binding between Id1 and E2F1 to Cdc20 regulates E2F1 degradation and thymidylate synthase expression to promote esophageal cancer chemoresistance. Clin Cancer Res. 2016;22:1243-55
20. Liu J, Lv L, Gong J, Tan Y, Zhu Y, Dai Y. et al. Overexpression of F-box only protein 31 predicts poor prognosis and deregulates p38α- and JNK-mediated apoptosis in esophageal squamous cell carcinoma. Int J Cancer. 2018;142:145-55
21. Liu J, Han L, Li B, Yang J, Huen MS, Pan X. et al. F-box only protein 31 (FBXO31) negatively regulates p38 mitogen-activated protein kinase (MAPK) signaling by mediating lysine 48-linked ubiquitination and degradation of mitogen-activated protein kinase kinase 6 (MKK6). J Biol Chem. 2014;289:21508-18
22. Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69:277-84
23. Xu WW, Li B, Guan XY, Chung SK, Wang Y, Yip YL. et al. Cancer cell-secreted IGF2 instigates fibroblasts and bone marrow-derived vascular progenitor cells to promote cancer progression. Nat Commun. 2017;8:14399
24. Li B, Tsao SW, Li YY, Wang X, Ling MT, Wong YC. et al. Id-1 promotes tumorigenicity and metastasis of human esophageal cancer cells through activation of PI3K/AKT signaling pathway. Int J Cancer. 2009;125:2576-85
25. Li B, Tsao SW, Chan KW, Ludwig DL, Novosyadlyy R, Li YY. et al. Id1-induced IGF-II and its autocrine/endocrine promotion of esophageal cancer progression and chemoresistance-implications for IGF-II and IGF-IR-targeted therapy. Clin Cancer Res. 2014;20:2651-62
26. Li B, Li J, Xu WW, Guan XY, Qin YR, Zhang LY. et al. Suppression of esophageal tumor growth and chemoresistance by directly targeting the PI3K/AKT pathway. Oncotarget. 2014;5:11576-87
27. Xu WW, Li B, Lam AK, Tsao SW, Law SY, Chan KW. et al. Targeting VEGFR1- and VEGFR2-expressing non-tumor cells is essential for esophageal cancer therapy. Oncotarget. 2015;6:1790-805
28. Li B, Xu WW, Han L, Chan KT, Tsao SW, Lee NPY. et al. MicroRNA-377 suppresses initiation and progression of esophageal cancer by inhibiting CD133 and VEGF. Oncogene. 2017;36:3986-4000
29. Xu WW, Li B, Zhao JF, Yang JG, Li JQ, Tsao SW. et al. IGF2 induces CD133 expression in esophageal cancer cells to promote cancer stemness. Cancer Lett. 2018;425:88-100
30. Zhong Y, Yang J, Xu WW, Wang Y, Zheng CC, Li B. et al. KCTD12 promotes tumorigenesis by facilitating CDC25B/CDK1/Aurora A-dependent G2/M transition. Oncogene. 2017;36:6177-89
31. Broos S, Hulpiau P, Galle J, Hooghe B, Van Roy F, De Bleser P. ConTra v2: a tool to identify transcription factor binding sites across species, update 2011. Nucleic Acids Res. 2011;39:W74-8
32. Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV. et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362-7
33. Lai X, Gupta SK, Schmitz U, Marquardt S, Knoll S, Spitschak A. et al. MiR-205-5p and miR-342-3p cooperate in the repression of the E2F1 transcription factor in the context of anticancer chemotherapy resistance. Theranostics. 2018;8:1106-20
34. Guo Y, Chen Z, Zhang L, Zhou F, Shi S, Feng X. et al. Distinctive microRNA profiles relating to patient survival in esophageal squamous cell carcinoma. Cancer Res. 2008;68:26-33
35. Mathe EA, Nguyen GH, Bowman ED, Zhao Y, Budhu A, Schetter AJ. et al. MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clin Cancer Res. 2009;15:6192-200
36. Xu H, Yao Y, Meng F, Qian X, Jiang X, Li X. et al. Predictive value of serum miR-10b, miR-29c, and miR-205 as promising biomarkers in esophageal squamous cell carcinoma screening. Medicine. 2015;94:e1558
37. Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J. et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer cell. 2008;14:369-81
38. Zhou L, Wang L, Lu L, Jiang P, Sun H, Wang H. Inhibition of miR-29 by TGF-beta-Smad3 signaling through dual mechanisms promotes transdifferentiation of mouse myoblasts into myofibroblasts. PloS one. 2012;7:e33766
39. Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369-81
40. Ding DP, Chen ZL, Zhao XH, Wang JW, Sun J, Wang Z. et al. miR-29c induces cell cycle arrest in esophageal squamous cell carcinoma by modulating cyclin E expression. Carcinogenesis. 2011;32:1025-32
Author contact
![]() Corresponding authors: Dr. Annie L. M. Cheung, School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong SAR, China. Phone: (852) 39179293; Fax: (852) 28170857; Email: lmcheunghk and Dr. Wen Wen Xu, Institute of Biomedicine, Guangdong Provincial Key Laboratory of Bioengineering Medicine, National Engineering Research Center of Genetic Medicine, Jinan University, Guangzhou, China. Phone: (86)-20-85221062; Fax: (86)-20-85221062; Email: xuwen6966com
Corresponding authors: Dr. Annie L. M. Cheung, School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong SAR, China. Phone: (852) 39179293; Fax: (852) 28170857; Email: lmcheunghk and Dr. Wen Wen Xu, Institute of Biomedicine, Guangdong Provincial Key Laboratory of Bioengineering Medicine, National Engineering Research Center of Genetic Medicine, Jinan University, Guangzhou, China. Phone: (86)-20-85221062; Fax: (86)-20-85221062; Email: xuwen6966com
Citation styles
APA
Li, B., Hong, P., Zheng, C.C., Dai, W., Chen, W.Y., Yang, Q.S., Han, L., Tsao, S.W., Chan, K.T., Lee, N.P.Y., Law, S., Xu, L.Y., Li, E.M., Chan, K.W., Qin, Y.R., Guan, X.Y., Lung, M.L., He, Q.Y., Xu, W.W., Cheung, A.LM. (2019). Identification of miR-29c and its Target FBXO31 as a Key Regulatory Mechanism in Esophageal Cancer Chemoresistance: Functional Validation and Clinical Significance. Theranostics, 9(6), 1599-1613. https://doi.org/10.7150/thno.30372.
ACS
Li, B.; Hong, P.; Zheng, C.C.; Dai, W.; Chen, W.Y.; Yang, Q.S.; Han, L.; Tsao, S.W.; Chan, K.T.; Lee, N.P.Y.; Law, S.; Xu, L.Y.; Li, E.M.; Chan, K.W.; Qin, Y.R.; Guan, X.Y.; Lung, M.L.; He, Q.Y.; Xu, W.W.; Cheung, A.LM. Identification of miR-29c and its Target FBXO31 as a Key Regulatory Mechanism in Esophageal Cancer Chemoresistance: Functional Validation and Clinical Significance. Theranostics 2019, 9 (6), 1599-1613. DOI: 10.7150/thno.30372.
NLM
Li B, Hong P, Zheng CC, Dai W, Chen WY, Yang QS, Han L, Tsao SW, Chan KT, Lee NPY, Law S, Xu LY, Li EM, Chan KW, Qin YR, Guan XY, Lung ML, He QY, Xu WW, Cheung ALM. Identification of miR-29c and its Target FBXO31 as a Key Regulatory Mechanism in Esophageal Cancer Chemoresistance: Functional Validation and Clinical Significance. Theranostics 2019; 9(6):1599-1613. doi:10.7150/thno.30372. https://www.thno.org/v09p1599.htm
CSE
Li B, Hong P, Zheng CC, Dai W, Chen WY, Yang QS, Han L, Tsao SW, Chan KT, Lee NPY, Law S, Xu LY, Li EM, Chan KW, Qin YR, Guan XY, Lung ML, He QY, Xu WW, Cheung ALM. 2019. Identification of miR-29c and its Target FBXO31 as a Key Regulatory Mechanism in Esophageal Cancer Chemoresistance: Functional Validation and Clinical Significance. Theranostics. 9(6):1599-1613.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.