Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and Discussions
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results and Discussions
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1572-1579. doi:10.7150/thno.31986 This issue Cite
Research Paper
Polymerase Chain Reaction using “V” Shape Thermal Cycling Program
Rong Chen1,2, Xue Lu1, Mei Li1, Gangyi Chen1, Yun Deng3, Feng Du1, Juan Dong1, Xin Huang1, Xin Cui1, Zhuo Tang1 ![]()
1. Natural Products Research Center, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, P. R. China
2. Ethnomedicine College, Chengdu University of Traditional Chinese Medicine; Chengdu 611137, P. R. China
3. College of pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, P. R. China
Received 2018-12-3; Accepted 2019-1-14; Published 2019-2-28
Citation:
Chen R, Lu X, Li M, Chen G, Deng Y, Du F, Dong J, Huang X, Cui X, Tang Z. Polymerase Chain Reaction using “V” Shape Thermal Cycling Program. Theranostics 2019; 9(6):1572-1579. doi:10.7150/thno.31986. https://www.thno.org/v09p1572.htm
Other stylesAbstract

Polymerase chain reaction (PCR) is the most commonly used technique in molecular biology and diagnostics. To achieve faster PCR reaction time, two strategies were employed by previous studies. That includes improving the thermal ramp rate by developing novel devices to reduce the time wasted on temperature transitions and cutting the holding time in every step, which could even lead to compromise in amplification efficiency. Hence the need to further improve the technique.
Methods: A different way to achieve fast DNA amplification is developed by using the previously thought wasted time spent on heating and cooling the samples to finish the amplification. That means the holding time of the three procedures are omitted and this could be carried out on the ordinary PCR thermal cyclers.
Results: 2/3 of the amplification time is easily saved, compared to the conventionally used method. Additionally, the reaction time could be further reduced by using longer primers with higher melting temperature (Tm). The record time of the “V” shape Polymerase chain reaction (VPCR) conducted on ordinary PCR machine for amplification of a 98 bp fragment is 8 min. Furthermore, VPCR still retains the merits of traditional PCR technique, including specificity, sensitivity, generality, and compatibility with quantitative detection.
Conclusion: It is confirmed that the three procedures of PCR could be completed during the dynamic heating and cooling process when the cyclers are run at a moderate thermal ramp rate. As VPCR described here is based on the current PCR system, it could be implemented in any biological Lab immediately and provide great convenience to the people working in the field of life science and human health.
Keywords: VPCR, Ordinary PCR thermal cyclers, DNA amplification finished within 8 min
Introduction
Since the invention of polymerase chain reaction (PCR) in 1985 by Karry Mullis, it has become a well- established method for amplifying trace amounts of DNA as well as quantifying specific DNA or RNA. The robustness and exquisite sensitivity have made PCR the most powerful tool for clinical diagnostics [1], forensic identification [2], food and drug authentication [3], environmental monitoring [4] and so on. A typical PCR process consists of three discrete, multiple repeated steps: denaturation of the double-stranded DNA at 94 °C for 30-60 s, binding of primers to their target site around the melting temperature (Tm) for 20 s-30 s and extension of primers with a Taq DNA polymerase at 72 °C for 30 s-60 s (Figure 1A Mode 1). In the early days, the PCR technique was laborious and slow, requiring manual transferring between water baths at different temperatures. The closed DNA thermal cyclers were introduced based on thermoelectric element technology, achieving simple and reliable PCR amplification that are widely used today. With the nucleic acid testing entering clinical practice, rapid amplification of nucleic acid is highly desirable especially for the timely diagnosis of pathogens and acute diseases [5]. To achieve faster PCR assay, two strategies were involved in previous studies. That includes improving the thermal ramp rate by developing novel devices to reduce the time wasted on temperature transitions and cutting the holding time in every temperature, which could even lead to compromising in amplification efficiency [2, 6-13].
The earliest study of fast PCR can date back to 1990's. Wittwer [14] developed a thermal cycler which uses hot air for heat transferring and reduces the total amplification time from 2 h to 10-30 min. In the last decades, many researchers have also tried to improve thermal ramp rate by taking advantage of various heating methods, such as using hot and cold water [8], infrared lamp [15], photonic heat converter [12] for heat transferring, or microfluidic devices which avoid repeated heating and cooling by making use of a space conversion [16], etc. It seems that the development of fast PCR becomes a “who can run faster” competition. The highest recorded heating rate of 175 °C/s and cooling rate of 125 °C/s have been achieved in a silicon-based micro machine system [17].
Figure 1
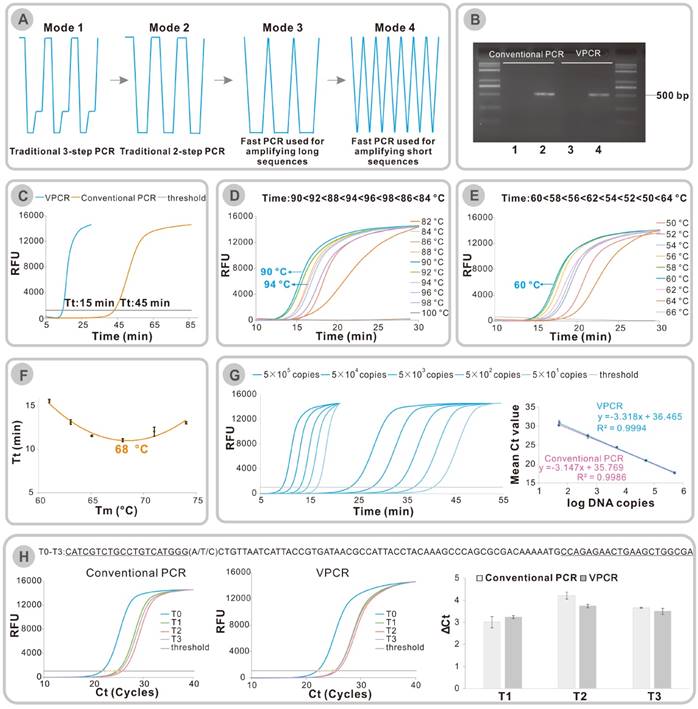
(A) Development of temperature modes from traditional 3-step PCR to the reported fast PCR techniques. (B) Amplification of a 500 bp segment of λ DNA using conventional PCR or VPCR. Lane 1 and lane 2 were the amplification results of conventional PCR; Lane 3 and lane 4 were the amplification results of VPCR. Lane 1-4: 1. No template control, 2. λ DNA (final concentration:10 pg/μl), 3. No template control, 4. λ DNA (final concentration:10 pg/μl). (C) Fluorescence vs time for 40 cycles of VPCR (blue) and conventional PCR (orange). (D) Effect of high temperature (TH) on the amplification time of a 98 bp template. (E) Effect of low temperature (TL) on the amplification time of a 98 bp template. (F) Threshold time (Tt) vs Tm of primers in VPCR. (G) Real-time Quantitative assay. Left: Real-time monitoring of VPCR and conventional PCR with SYBR Green I. Right: Ct values Vs log (DNA copies). (H) The comparison of ΔCt values generated by conventional PCR and VPCR: ΔCt(T1/T2/T3) =Ct(T1/T2/T3)-Ct(T0), error bars were standard deviations of three parallel assays.
On the other hand, in seeking the shortest cycling time, the classic three-step PCR protocol was simplified to two steps by combining the annealing and extension steps (Figure 1A Mode 2). The time spent on every step was further reduced, it was proved that the time required for denaturation could be as short as 3 s [6] or it even can be finished instantaneously [8, 12] while the time in the combined annealing and extension step should be adjusted according to the length of the amplification sequences [11] (Figure 1A Mode 3 or Mode 4). In some extreme cases, when the amplification lengths are very short (40-100 bp), the extension time required for amplification is usually very short (less than 1 s) [11, 12]. It looks like the holding time in the two temperatures are omitted (Figure 1A Mode 4). According to these studies, we come up with another idea that the three procedures of PCR may be completed during the dynamic heating and cooling process. That is to say, that the previously thought wasted time in heating and cooling the samples probably could be used to finish the amplification rather than being wasted. The holding time in the two temperatures can both be omitted. Therefore, if the hypothesis is true, we can conduct a fast DNA amplification only on the ordinary PCR thermal cycler. As the temperature-time curve of the newly proposed rapid DNA amplification method forms a repeated “V” curve, we name it “VPCR”.
Materials and Methods
Materials
Primers used in this study were all PAGE-purified by Sangon Biotech Co., Ltd. (Shanghai, China). EasyTaq DNA polymerase, Taq buffer, and dNTPs were purchased from TransGen Biotech (Beijing, China). KAPA2G Robust DNA Polymerase was purchased from Kapa Biosystems. Ex Taq DNA Polymerase was purchased from Takara Biomedical Technology (Beijing) Co., Ltd. Q5 High-Fidelity DNA Polymerase was purchased from New England Biolabs. Taq DNA polymerase was purchased from TIANGEN Biotech (BEIJING) Co., Ltd. The λ-DNA was purchased from Takara Biomedical Technology (Beijing) Co., Ltd. The DNA isolation kit was purchased from Foregene Co., Ltd. (Chengdu, China). SYBR Green I was purchased from probes.invitrogen. com. HBV Quantitative Real Time PCR Kit was purchased from Liferiver Bio-Tech (United States) Corp.
Taq DNA polymerase PCR system
Both the conventional PCR and VPCR were performed in a final volume of 10 μl (until otherwise noted) mixture containing 1x taq buffer (20 mM Tris-HCl (pH 8.4), 20 mM KCl, 10 mM (NH4)2SO4 and 2 mM MgSO4), 0.2 mM dNTPs, 0.1 U/μL of EasyTaq DNA polymerase, 0.4 x SYBR green dyes (only in Real-time PCR or Real-time VPCR), appropriate concentration of each primer and a certain amount of template. VPCR amplifications were usually conducted under the following cycling conditions: 94 °C (until otherwise noted) for 0 s, 50-78 °C (according to different primers) for 0 s, 30 cycles (End-point VPCR) or 40-50 cycles (Real-time VPCR). Conventional PCR amplifications usually consist of 30 (End-point PCR) or 40 (Real-time PCR) cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s.
KAPA2G Robust DNA Polymerase PCR system
Both the conventional PCR and VPCR were carried out in a final volume of 10 μl mixture containing 1x KAPA2G Buffer A, additional 1 mM MgCl2, 0.2 mM dNTPs, 0.5 units of KAPA2G Robust DNA Polymerase, 0.4 x SYBR green dyes (only in Real-time VPCR), appropriate concentration of each primer and a certain amount of template. VPCR amplifications were usually conducted under the following cycling conditions: 94 °C (until otherwise noted) for 0 s, 50-78 °C (according to different primers) for 0 s, 30 cycles (End-point VPCR) or 40-50 cycles (Real-time VPCR). Conventional PCR amplifications usually consist of 30 (End-point PCR) or 40 (Real-time PCR) cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s.
The fastest VPCR
The fastest VPCR was carried out in a final volume of 5 μl mixture containing 1x taq Buffer (20 mM Tris-HCl (pH 8.4), 20 mM KCl, 10 mM (NH4)2SO4 and 2 mM MgSO4), additional 3 mM MgCl2, 0.2 mM dNTPs, 0.25 units of KAPA2G Robust DNA Polymerase, 0.5 μmol/L of each primer (LG/LRG), 0.1 ng/μl of λDNA. The amplification was conducted under the following cycling conditions: 89 °C for 0 s (temperature decreases 0.1 °C per cycle), 77 °C for 0 s (temperature increases 0.1 °C per cycle), 25 cycles.
Results and Discussions
Hypothesis validation
In order to prove that the three procedures of PCR can be finished during the dynamic heating and cooling process, a DNA amplification was conducted on ordinary PCR equipment using either conventional PCR protocol or VPCR protocol. A fragment (500 bp in length) of λ DNA was chosen as the target sequence, and two primers were designed accordingly. The amplification was first performed on an ordinary PCR cycler with a classic PCR procedure (30 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s), which took approximately 66 min. After amplification, the PCR products with expected length were detected on the agarose gel (Figure 1B, lane 1-2). When the same reaction had been carried out using VPCR protocol (94 °C for 0 s, 60 °C for 0 s), it took only 16 min 51 s to complete 30 thermal cycles, which saved more than 2/3 of the amplification time compared to the traditional method. Surprisingly, VPCR efficiently amplified the same DNA sample, affording the identical PCR products (Figure 1B, lane 3-4).
Previously it has been reported that a combined annealing/extension step of 3 s is required for the amplification of a 500 bp fragment in the fast PCR system in which the temperature change rate and reagents concentrations are relatively high [11]. Here we successfully prove that the amplification can also be finished during the heating and cooling process when the cycler is run at a moderate thermal ramp rate. It is worth mentioning that a fast DNA polymerase named KAPA2G Robust which was employed in the previous fast PCR study [11] was used in the hypothesis validation experiment described above. We have also tried the conventional used Taq DNA polymerase in this section. Figure S1 demonstrates that VPCR can also work in the Taq DNA polymerase system. The effect of different polymerase on VPCR will further be discussed in the generality part.
Since the holding time set for denaturation, annealing and extension are no longer in existence, a systematic investigation of VPCR process should be conducted and all the parameters need to be recharacterized technically. Because the targets used in most of the DNA identification cases are around 100 bp, a 98 nt fragment of λ DNA which is used in a reported fast PCR study [12] is chosen as our target in the following investigations. Ordinary Taq DNA polymerase was used unless otherwise noted.
Real-time monitoring of VPCR
Considering that the end-point detection of PCR product on gel analysis only reveals the final outcome of DNA amplification, a Real-time assay with the commonly used SYBR green dyes was conducted to monitor the whole process of VPCR. It is found that the threshold cycle (Ct) value of VPCR is similar to that of conventional PCR (Figure S2, △Ct=1), implying no apparent decrease in amplification efficiency of each VPCR cycle. If the fluorescence of PCR reaction has been recorded in a time-resolved way, the Tt (threshold time which is an analogy to the Ct in Real-time PCR analysis) value of VPCR is 15 min, while that of conventional PCR is 45 min (Figure 1C). These results indicate that the amplification process of VPCR is also completed by cycles, which are composed of repeated DNA melting and enzymatic replication of the DNA amplicons. As there is no pause time in the VPCR protocol, the steps of denaturation, annealing and extension must be fulfilled during the dynamic heating and cooling process, in which the reaction temperature changes constantly.
Temperature Settings
Furthermore, we studied the two temperatures in the newly developed VPCR protocol. As shown in Figure 1D, the High temperature (TH) of the Real-time VPCR amplifications were investigated from 82 °C to 100 °C while the Low temperature (TL) was set at 60 °C arbitrarily. It turns out that the amplification could be achieved when the TH was set from 84 °C to 98 °C, and there was no significant difference in the time spent to reach the threshold when it ranges from 88 °C to 96 °C (Figure 1D). On the other side, the TL was studied by changing the temperature from 50 °C to 66 °C, while the TH was set at 94 °C. As a result, VPCR fails to amplify the template when the TL is higher than 64 °C. The best result of Real-time VPCR in terms of time is obtained when the TL is set at 60 °C, which is equal to the calculated Tm value of the primers (Figure 1E). Different primer pairs with distinct Tm values have been applied in VPCR, and we arrived at similar results that the optimal TL is around the Tm value of corresponding primers (Figure S3). It is worth mentioning that several empirical equations can be used to calculate the Tm of a primer. The Tm used here is calculated by an equation considering the exact sequence, primer concentrations and metallic ion concentrations, which is available in a public web [18].
Primer Optimization
As the amplification time can be typically reduced when the low temperature (TL) is set around the Tm of the primers, it is reasonable to expect that the use of longer primers with higher melting temperature could further shorten the amplification time. Thus, a series of primers with the length from17 to 46 and melting temperature (Tm) ranged from 61 °C to 80 °C were designed. Then the amplification time of each primer pair is compared under their own optimal VPCR condition (Figure S4). As shown in Figure 1F, the reaction time drastically reduced when the Tm increases from 60 °C to 68 °C, but it could not be further shortened when the Tm is higher than 68 °C. To amplify 0.5 pg/μl λDNA, the corresponding Tt is 11 min for the optimum primers (Tm=68 °C) while it is 15 min for the commonly used primers (Tm=60 °C). This result indicates that the primer should have appropriate length, instead of the longer, the better.
Real-time Quantitative detection
With all the basic technical characters well studied, we were very curious about the quantitative performance of the VPCR. Therefore, serially diluted synthetic DNA was amplified on Real-time PCR instrument using the developed VPCR protocol to investigate the quantitative performance of VPCR. SYBR Green dyes were used for fluorescence monitoring. According to the standard curve and liner equation in Figure 1G, the amplification efficiency of VPCR is 100.16% in the four-log dynamic range, similar to that of the conventional PCR. R2 value of 0.9994 compared to 0.9986 of conventional PCR successfully demonstrates a good liner relationship, which is suitable for DNA quantification. Additionally, the detection limit of VPCR is the same as the conventional PCR. Both are as low as 50 copies. The total time for the 40-cycle amplification is reduced from 54 min 36 s (two-step PCR) to about 20 min 17 s. In Figure 1G, the error bars in standard curves represent the variation of triplicate experiments, which are too short to be seen, successfully demonstrating a good reproducibility of the developed VPCR system. What's more, the cleavage-based TaqMan Real-time VPCR was also tested by similar experiment and proved to be viable (Figure S5). The corresponding amplification efficiency is 91.53% in VPCR compared with 90.70% in conventional PCR. Likewise, 40 cycles of VPCR requires 25 min 32 s, whereas ordinary PCR is completed in 64 min 50 s.
Specificity
Specificity is also an important character for a DNA amplification method. Four different templates with one mutation at the 3 end of the upstream primer were prepared to investigate the specificity of VPCR. As shown in Figure 1H, the original template is G, and the other three are mutated to A, T, and C, respectively. The four analogous templates were amplified by VPCR or the conventional PCR, respecttively. Figure 1H illustrates that the delta Ct values (ΔCt(T1/T2/T3) =Ct(T1/T2/T3)-Ct(T0) generated by VPCR are very close to, or even greater than that of conventional PCR, indicating that the specificity of the VPCR is similar to or sometimes superior to conventional PCR, which is consistent with the previous rapid PCR studies [2].
Generality
Methodological validation of PCR usually involves different DNA templates, DNA polymerase, reaction volumes, and thermal cyclers. In this research, DNA from different sources (including plasmid DNA of pEGFP-N1, genomic DNA of bacteriophage λ c|857, Staphylococcus aureus, Carthamus tinctorius, and Sus scrofa) were isolated by routinely used DNA extraction method and amplified by the developed VPCR. In Figure S6, the clear bands on agarose gel successfully demonstrated that VPCR can work with DNA templates from different sources. Moreover, a GC-rich template (GC content: 75%) was amplified either by conventional PCR or VPCR. Figure S7 shows that the templates in the two systems can both be amplified with similar efficiency, indicating that VPCR also works for the amplification of GC-rich templates. In addition, different DNA polymerase from several mainstream suppliers were also tested. It turns out that all the polymerase could work well in the VPCR system and give the expected amplification products on the agarose gel (Figure S8). What's more, the effects of amplification volumes on the developed VPCR approach were studied by amplifying 5, 10, 25 and 50 μl of samples using ordinary thermal cyclers. Figure S9 shows that different volumes of samples can all be amplified with equivalent yield, indicating a wider application of the VPCR technology. In addition, the performance of VPCR on instruments from different manufacturers was also investigated by analyzing serially diluted λDNA with SYBR green dyes. Good linear relationships and amplification efficiencies in Figure S10 prove that the developed amplification system is suitable for quantification. Despite the instrumental choice, the total time for the 40 cycles of amplification is all reduced from nearly 1 h to about 20 min. At last, a TaqMan based Hepatitis B Virus quant assay (serum samples) and sequencing assisted Single Nucleotide Polymorphism genotyping of a multi-drug resistance gene (saliva samples) were performed to test the applicability of the developed VPCR to the clinically-relevant assay. Figure S11- Figure S13 indicate that the results obtained by VPCR are the same as those obtained by conventional PCR, demonstrating the practicability of VPCR in clinical assays.
Thermal Ramp Rate
Here we have proved that the VPCR protocol can reduce two-thirds of the amplification time by only using ordinary PCR machines. Nevertheless, there is another parameter that we haven't discussed: thermal ramp rate, which is often unchanged in conventional PCR machines. The fastest temperature ramp rates of mainstream PCR thermal cyclers are all claimed to be as fast as 6 °C /s or even 8 °C/s. However, the average temperature change rates in practical operation are slower than 2 °C/s according to a simple calculation. Under this condition, the effects of the temperature ramp rate on the developed rapid PCR method were also examined. As is shown in Figure S14A-B, for the 98 bp template, increased temperature ramp rates can result in decrease time to arrive at the plateau, but the optimal temperature ramp rate is unavailable due to the limited cooling and heating speed of the current instrument.
However, as the denaturation, annealing and extension are finished during the heating and cooling process and the extension rate of Taq DNA polymerase is limited, usually 35-100 nucleotides/s [15], we reasoned that the time required for elongation should vary with the length of the amplification products. Therefore, a series of primers were designed to amplify fragments with length from 200 bp to 500 bp.
For the 200 bp template (Figure S14C-D), the optimal cooling and heating speed is still unavailable. But for other longer fragments, it is found that the optimal temperature ramp rate is inversely related to the product length. The best ramp rate decreases from 1.6 °C/s for 250 bp to 0.7 °C/s for 500 bp fragment (Figure 2A and Figure S14E-L). Over-rapid temperature changes will lead to a decrease in extension time which will result in incomplete extension and unexpected amplification band on agarose gel, especially for long sequences (Figure S14H and Figure S14L). Fortunately, most of the targets, especially in TaqMan-based technology used in the rapid diagnostics are about 100 bp. Therefore, conventional used Taq DNA polymerase can meet most needs of the rapid VPCR cycling. Due to the limit of current instrumentation, there is no need to consider the thermal ramp rate when the target is within 200 bp.
Figure 2
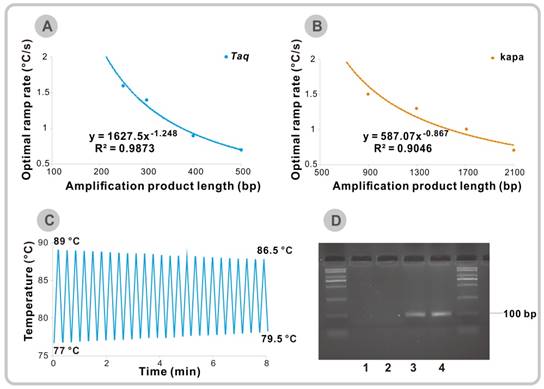
(A) Optimal observed temperature ramp rates for efficient amplification of 250 bp to 500 bp fragments using ordinary Taq DNA polymerase. (B) Optimal observed temperature ramp rates for efficient amplification of 900 bp to 2100 bp fragments using KAPA2G Robust DNA Polymerase. (C) Thermal cycler settings for 25 cycles of touch-down and touch-up VPCR. (D) Agarose gel electrophoresis after 25 cycles of the touch-down and touch-up VPCR. Lane 1-2: No template control, Lane 3-4: λ DNA (final concentration: 0.1 ng/μl).
Besides, the extension rate varies among different kinds of polymerase. The KAPA2G Robust DNA Polymerase which is commonly used in rapid PCR cycling has the ability to extend 155 nucleotides/s [9]. We also tried to apply this KAPA2G Robust DNA Polymerase to amplify DNA fragments with different length. It turns out that the optimal temperature change speed will also decrease as the product size increases (Figure 2B and Figure S15). According to the approximate equation in Figure 2A and 2B, we can expect that the optimal ramp rates are 5.2 °C/s for 100 bp and 12.3 °C/s for 50 bp amplicons when Taq DNA polymerase is used for amplification. Although the predicted best temperature change rates of KAPA2G Robust DNA Polymerase are much higher than that of Taq DNA polymerase (10.8 °C/s for 100 bp and 19.8 °C/s for 50 bp), we can still conclude that the thermal ramp rate is not the faster, the better.
Minimum VPCR Record time using ordinary PCR equipment
Finally, we tried to set a record time for our newly developed VPCR technique by only using the ordinary PCR thermal cycler. The KAPA2G Robust DNA Polymerase with a faster extension rate is applied in this part of experiment to save more amplification time. All the amplification parameters for amplifying the previously mentioned 98 bp target were fully optimized on the basis of the KAPA2G Robust DNA Polymerase system (Figure S16A- D). It turns out that the biggest time-saver in this system is primer pair LG/LRG and the corresponding TL and TH is 78 °C and 88 °C, respectively. As the TH required for denaturation of PCR products is lower than that of the genomic DNA and the accumulation of target sequences can increase the possibility of primer binding, the reaction time could be further shortened by gradually decreasing the TH and increasing the TL over the amplification cycles. Figure 2C shows the corresponding temperature-time profile during the PCR process. Finally, we successfully obtained the 98 bp PCR products in 8 min by using the touch-down and touch-up VPCR program (Figure 2D). The shrinkage in temperature brings an advance in amplification time and shows a new direction to cut the time for VPCR. Meanwhile, the fastest record for amplification of the same 98 bp fragment is 5 min by using a novel photonic heat converter, which is unavailable in most labs [12].
Conclusions
In this study, we proved that the three procedures of PCR can be finished during the dynamic heating and cooling process and hence the holding time for denaturation, annealing, and extension in conventional PCR can be omitted. According to this finding, only based on current PCR systems, we developed a new kind of DNA amplification method which can easily save 2/3 of the reaction time compared to the conventional used method. What's more, according to a systematic investigation of the amplification process, it is found that the reaction time could be further reduced by using longer primers with higher melting temperature. Because no compromise in amplification efficiency, the developed VPCR can also be used to perform quantitative Real-time PCR. Both the nonspecific fluorescent dyes based and specific Taqman probe based quantitative Real-time PCR show high amplification efficiencies and good linear relationships. A good compatibility with different instruments, polymerase and templates are also confirmed.
Last but not least, this work helps us to take a new look at the PCR process and provides another way to realize fast DNA amplification. As most of the previously reported fast PCR devices have yet to reach the commercial market, they are not available for common users. In contrast, VPCR described here is based on the current equipment and PCR reaction mixture, so it could be tried or implemented in any biological Lab immediately. For the amplification of short sequences, the shortest cycling time using VPCR technique is still unavailable due to the limit of the current instrument, but there is no need to blindly improve thermal ramp rate if we want to develop novel PCR equipment, as over-rapid temperature changes will lead to compromise in efficiency and yield. What's more, as PCR technique is the basis of other important technologies like RT-PCR, digital PCR, SNP assay and even sequencing, the application and development of VPCR will completely change the understanding and using habits of those researchers who are using or will use PCR technology. It will also provide great convenience to the people working in the field of life science and human health.
Abbreviations
PCR: Polymerase Chain Reaction; VPCR: “V” Shape Polymerase Chain Reaction; TL: Low temperature; Tm: Melting temperature; TH: High temperature.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Sciences Foundation of China [grant numbers 81403185, 21402189, 21572222]; the Innovative Team of Sichuan Province [grant number 2017TD0021], Chengdu Municipal Bureau of Science and Technology [grant number 2015-HM02-00099-SF].
Author Contributions
Z.T. and Y.D. conceived and designed the research. R. C., X.L, M.L., GY.C, F.D., J.D. and X.H., X.C. performed experiments. R. C. and Z.T. analyzed the data and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ. et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10:1003-5
2. Le Roux D, Root BE, Reedy CR, Hickey JA, Scott ON, Bienvenue JM. et al. DNA Analysis Using an Integrated Microchip for Multiplex PCR Amplification and Electrophoresis for Reference Samples. Anal Chem. 2014;86:8192-9
3. Mishra P, Kumar A, Nagireddy A, Mani DN, Shukla AK, Tiwari R. et al. DNA barcoding: an efficient tool to overcome authentication challenges in the herbal market. Plant Biotechnol J. 2016;14:8-21
4. Shannon KE, Lee DY, Trevors JT, Beaudette LA. Application of real-time quantitative PCR for the detection of selected bacterial pathogens during municipal wastewater treatment. Sci Total Environ. 2007;382:121-9
5. Niemz A, Ferguson TM, Boyle DS. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011;29:240-50
6. Zhang C, Xing D. Miniaturized PCR chips for nucleic acid amplification and analysis: latest advances and future trends. Nucleic Acids Res. 2007;35:4223-37
7. Kopf-Sill AR, Portola Valley C. Inefficient Fast PCR. United States: Caliper Technologies Corp. 2001
8. Wheeler EK, Hara CA, Frank J, Deotte J, Hall SB, Benett W. et al. Under-three minute PCR: Probing the limits of fast amplification. Analyst. 2011;136:3707-12
9. Fuchiwaki Y, Nagai H, Saito M, Tamiya E. Ultra-rapid flow-through polymerase chain reaction microfluidics using vapor pressure. Biosens Bioelectron. 2011;27:88-94
10. Lee SH, Kim SW, Lee S, Kim E, Kim DJ, Park S. et al. Rapid detection of Mycobacterium tuberculosis using a novel ultrafast chip-type real-time polymerase chain reaction system. Chest. 2014;146:1319-26
11. Farrar JS, Wittwer CT. Extreme PCR: Efficient and Specific DNA Amplification in 15-60 Seconds. Clin Chem. 2015;61:145-53
12. Son JH, Cho B, Hong S, Lee SH, Hoxha O, Haack AJ. et al. Ultrafast photonic PCR. Light-Science & Applications. 2015;4:e280
13. Song H-O, Kim J-H, Ryu H-S, Lee D-H, Kim S-J, Kim D-J. et al. Polymeric LabChip Real-Time PCR as a Point-of-Care-Potential Diagnostic Tool for Rapid Detection of Influenza A/H1N1 Virus in Human Clinical Specimens. Plos One. 2012;7:e53325
14. Wittwer CT, Fillmore GC, Garling DJ. Minimizing the Time Required for DNA Amplification by Efficient Heat Transfer to Small Samples. Anal Biochem. 1990;186:328-31
15. Huhmer AFR, Landers JP. Noncontact infrared-mediated thermocycling for effective polymerase chain reaction amplification of DNA in nanoliter volumes. Anal Chem. 2000;72:5507-12
16. Kopp MU, Mello AJ, Manz A. Chemical amplification: continuous-flow PCR on a chip. Science. 1998;280:1046-8
17. Neuzil P, Zhang C, Pipper J, Oh S, Zhuo L. Ultra fast miniaturized real-time PCR: 40 cycles in less than six minutes. Nucleic Acids Res. 2006;34:e77
18. Promega Corporation. 2018. http://www.promega.com/a/apps/
Author contact
![]() Corresponding author: tangzhuoac.cn, Phone/Fax: +86-28-85243250
Corresponding author: tangzhuoac.cn, Phone/Fax: +86-28-85243250
Citation styles
APA
Chen, R., Lu, X., Li, M., Chen, G., Deng, Y., Du, F., Dong, J., Huang, X., Cui, X., Tang, Z. (2019). Polymerase Chain Reaction using “V” Shape Thermal Cycling Program. Theranostics, 9(6), 1572-1579. https://doi.org/10.7150/thno.31986.
ACS
Chen, R.; Lu, X.; Li, M.; Chen, G.; Deng, Y.; Du, F.; Dong, J.; Huang, X.; Cui, X.; Tang, Z. Polymerase Chain Reaction using “V” Shape Thermal Cycling Program. Theranostics 2019, 9 (6), 1572-1579. DOI: 10.7150/thno.31986.
NLM
Chen R, Lu X, Li M, Chen G, Deng Y, Du F, Dong J, Huang X, Cui X, Tang Z. Polymerase Chain Reaction using “V” Shape Thermal Cycling Program. Theranostics 2019; 9(6):1572-1579. doi:10.7150/thno.31986. https://www.thno.org/v09p1572.htm
CSE
Chen R, Lu X, Li M, Chen G, Deng Y, Du F, Dong J, Huang X, Cui X, Tang Z. 2019. Polymerase Chain Reaction using “V” Shape Thermal Cycling Program. Theranostics. 9(6):1572-1579.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.