Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(2):424-435. doi:10.7150/thno.29698 This issue Cite
Research Paper
Low Levels of Sox2 are required for Melanoma Tumor-Repopulating Cell Dormancy
Qiong Jia1*, Fang Yang1*, Wei Huang2, Yao Zhang1, Binghao Bao3, Ke Li1, Fuxiang Wei1, Cunyu Zhang1, Haibo Jia3 ![]()
1. Laboratory for Cellular Biomechanics and Regenerative Medicine, Department of Biomechanical Engineering, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
2. National Engineering Research Center for Nanomedicine, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China
3. Key Laboratory of Molecular Biophysics of Ministry of Education, College of Life Science and Technology, Center for Human Genome Research, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
*These authors contributed equally to the study.
Received 2018-9-4; Accepted 2018-12-3; Published 2019-1-1
Citation:
Jia Q, Yang F, Huang W, Zhang Y, Bao B, Li K, Wei F, Zhang C, Jia H. Low Levels of Sox2 are required for Melanoma Tumor-Repopulating Cell Dormancy. Theranostics 2019; 9(2):424-435. doi:10.7150/thno.29698. https://www.thno.org/v09p0424.htm
Other stylesAbstract

Tumorigenic cells, when facing a hostile environment, may enter a dormant state, leading to long-term tumor survival, relapse, and metastasis. To date, the molecular mechanism of tumor cell dormancy remains poorly understood.
Methods: A soft, 3-dimentional (3D) fibrin gel culture system was used to mechanically select and grow highly malignant and tumorigenic melanoma tumor-repopulating cells (TRCs). We cultured control melanoma TRCs, TRCs with Sox2 knockdown, TRCs with Sox2 knockout, and a 2D control for in vitro and in vivo experiments. Western blotting, immunofluorescence, and flow cytometry analysis were performed to examine TRC dormancy and exit from dormancy.
Results: Under a low-expression condition, we show that Sox2, a stemness molecule participates in dormancy regulation of highly tumorigenic cells that can repopulate a tumor (TRCs). Intriguingly, complete depletion of Sox2 via knockout results in dormancy exit and growth resumption of melanoma TRCs in culture and elevation of melanoma TRC apoptosis. Mice that are injected subcutaneously with Sox2-depleted melanoma TRCs do not form tumors and survive much longer than those injected with melanoma TRCs. We found that complete depletion of Sox2 promotes nuclear translocation of phosphorylated STAT3, where it binds to the p53 gene promoter, thus activating the p53-caspase3 cascade.
Conclusion: These findings provide a novel insight into the role of the Sox2 gene in tumor cell stemness, tumor dormancy, and apoptosis.
Keywords: dormancy, TRCs (tumor-repopulating cells), Sox2 gene, apoptosis, stemness
Introduction
Despite significant progress in cancer therapeutics over the past few decades [1], tumor relapse following long periods of remission after treatment remains a challenging problem. Tumorigenic cells, when facing a hostile environment, may enter a dormant state, leading to long-term tumor survival, relapse, and metastasis. To date, the molecular mechanism of tumor cell dormancy remains poorly understood.
Tumor dormancy is emerging as a key event for tumors escaping intrinsic (immune surveillance) and extrinsic (toxic drugs) attacks [2, 3]. Tumor cell dormancy is defined at cellular levels as a process of induced cell cycle arrest. Tumor cells staying in a dormant state present a key challenge in cancer therapy because of their inhibition of cell proliferation and suppression of cell survival pathways [4, 5]. The dormant tumor cells remain at low numbers after primary tumor resection. These cells are undetectable for long periods and may be the reason for continued asymptomatic residual disease progression and treatment resistance [6-8]. Transmission of cancer from organ transplant recipients has been regarded as an evidence of immunologic tumor dormancy, a dominant type of tumor mass dormancy [9-11]. However, it is still unclear how the immune system induces tumor entry into dormancy and what cellular processes govern these clinical observations. It is also unknown whether the differentiation status of tumorigenic cells plays key roles in the conversion of tumor dormancy and death under immunosurveillance.
Recently, the highly malignant and tumorigenic melanoma tumor-repopulating cells (TRCs) have been screened and grown in our group by culturing single cancer cells in soft fibrin matrices [12]. Remarkably, in addition to being able to generate local primary tumors in wild-type syngeneic mice when injected into tail veins, as few as ten of these cells can generate distant metastatic colonization and grow tumors in the lungs of wild-type non-syngeneic mice [12]. Therefore, we functionally define these soft-fibrin-gel- selected melanoma cells as TRCs based on their high efficiency in repopulating tumors in wild-type syngeneic and non-syngeneic mice when implanted subcutaneously and at secondary sites [12]. These functionallydefined TRCs are distinct from conventional cancer stem cells (CSCs) and from tumor initiating cells (TICs). CSCs are a subset of cancer cells that can self-renew and are highly tumorigenic. CSCs have been identified and sorted using stem cell markers [13], such as CD133, CD44, CD24, and CD90 [14]. However, the approach of identifying cells via their stem cell markers is often unreliable, as subsequent work demonstrates that there is no correlation between surface stem cell markers and tumorigenicity [15]. TICs are heterogeneous and have 3 subtypes: transient, long-term, and delayed-contributing phenotypes [14]. Although these soft-fibrin-gel-selected melanoma TRCs may be also heterogeneous, our previous studies have shown that even as few as about ten TRCs are sufficient to form lung metastasis [12] and the recent finding that 5 TRCs are sufficient to generate subcutaneous tumors [16] suggest that these TRCs are distinct from those TICs that require tens of thousands of cells to generate tumors. Sox2, a stemness molecule that governs the pluripotency of embryonic stem cells [17, 18], is dramatically upregulated in TRCs that grow in soft matrices [19]. TRCs gradually lose Sox2 expression and become differentiated when cultured on 2D rigid plastic dish [19]. When tumor cells are cultured in 3D stiff fibrin matrices, Sox2 expression becomes greatly downregulated and melanoma TRC proliferation substantially decreases [19]. Several other labs have also demonstrated that Sox2 is a regulator of tumor cell self-renewal and tumorigenicity [20, 21]. In 3D stiff matrices, a Cdc42-driven Tet2 epigenetic program drives highly tumorigenic TRCs to enter dormancy [16]. From these published reports, we hypothesize that altering Sox2 expression, independent of the mechanics of 3D matrices, might change the cell cycle state of TRCs to modulate their dormancy propensity.
In this study, we found that low levels of Sox2 in TRCs, via silencing Sox2, induce entry of these tumorigenic cells into dormancy even when these cells are cultured in 3D soft fibrin matrices that should have promoted growth. Low levels of Sox2 in melanoma TRCs induce these cells into dormancy through activating the IDO1-AhR metabolic pathway, leading to upregulation of p27 and p21 and subsequent cell cycle arrests. Intriguingly, complete depletion of Sox2 via knockout results in the exit of melanoma TRCs from dormancy and resumption of proliferation. We found that complete depletion of Sox2 promotes nuclear translocation of activated STAT3 at both tyrosine and serine sites. Activated STAT3 binds to the p53 gene promoter and subsequently activates the p53-caspase3 cascade.
Methods
Animals
Four-week-old C57BL/6 mice were purchased from the Center of Medical Experimental Animals of Hubei Province (Wuhan, China). The mice were randomly assigned to be in the control or treated group. All animals received humane care in compliance with the Principles of Laboratory Animal Care Formulated by the National Society of Medical Research and the guide for the US National Institutes of Health. The protocol was approved by the Animal Care and Use Committee of Huazhong University of Science and Technology.
Cell culture
Murine melanoma cell line B16-F1 and human melanoma cell line A375 were purchased from China Center for Type Culture Collection (CCTCC, Wuhan, China). Cells were cultured with minimum essential medium (MEM) and Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA]), supplemented with 10% fetal bovine serum (Invitrogen). The mESC line W4 was obtained from New England Biolabs (Ipswich, MA, USA). These undifferentiated mESCs were cultured and maintained in high-glucoseDMEM (Invitrogen) supplemented with 15% ES-qualified fetal bovine serum (Invitrogen), 2 mM L-glutamine (Invitrogen), 1 mM sodium pyruvate, 0.1 mM nonessential amino acids (Invitrogen), 1% penicillin-streptomycin, 0.1 mM beta-mercaptoethanol (Sigma, St. Louis, MO, USA), and 1,000 U/mL recombinant LIF (ESGRO; Millipore, Billerica, MA, USA) at 37 oC in 5% CO2.
3D fibrin gel preparation
Salmon fibrinogen and thrombin were purchased from Reagent Proteins (San Diego, CA, USA). Three-dimensional fibrin gels were prepared as described previously [12, 19]. Briefly, 250 μL of 1 mg/mL (~90 Pa) fibrinogen and single-cell solution mixture were seeded into each well of a 24-well plate and mixed well with 5 μL pre-added thrombin (100 U/mL). The cell culture plate was then incubated in a 37 °C cell culture incubator for 25 min. Finally, 1 mL of MEM containing 10% fetal bovine serum and antibiotics was added.
qPCR analysis
Total mRNA was isolated from the cells using the Trizol reagent according to the supplier's instructions (Invitrogen, Carlsbad, CA, USA). Reverse transcription (RT) was performed using the TransScript First-strand cDNA Synthesis Super Mix (TransGen, Beijing, China). qPCR was performed using GoTaq qPCR Master Mix (Promega, Madison, WI, USA). The primer sequences are shown in Table S1.
Cell cycle and apoptosis analysis
Cells were incubated with 50 mM BrdU (BD Bioscience, Franklin Lakes, NJ, USA) for 2 h, and cell cycle analysis was performed using BD Pharmingen APC-BrdU Flow Kits according to the manufacturer's protocol (BD Bioscience, Franklin Lakes, NJ, USA). The samples were analyzed by flow cytometry on a BD Accuri C6 Flow Cytometer (BD Bioscience). Cell apoptosis was detected using the Annexin V-FITC Apoptosis Kit (Biolegend, San Diego, CA) according to the manufacturer's protocol. The cells were cultured for 48 h with normal medium and then treated with or without drugs for 24 h. Cells were collected and re-suspended in 1× binding buffer at a concentration of 1 × 106 cells per mL. Then, the cells (1 × 105) were incubated with 5 μL of Annexin V-FITC and 5 μL of PI for 15 min at 24 °C in the dark, followed by the addition of another 400 μL of 1× binding buffer. Samples were analyzed by flow cytometry. The data were analyzed using FlowJo software (San Francisco, CA, USA).
Immunofluorescence
Cells were fixed with 4% formaldehyde (BioSharp, Hefei, China) for 10 min at room temperature. Cells were then permeabilized with 0.5% Triton X-100 (BioSharp, Hefei, China) for 2 min and treated with a blocking serum (Solarbio, Beijing, China) for more than 5 h. Primary antibody, anti-COUP TF1 (sc-74560, 1:50, Santa Cruz, TX, USA), and anti-Ki67 (ab15580, 1:200, Abcam, Cambridge, UK) were incubated overnight for immunofluorescence. After being washed to remove unbound primary antibodies, cells were incubated with the secondary antibody Dnk pAb to Rb IgG (Alexa Fluor® 488) (ab150074, 1:1000, Abcam, UK), donkey anti-mouse IgG H&L (Alexa Fluor 594) (ab15010B, 1:1000, Abcam, UK), and DAPI (Biosharp, Hefei, China) for 2 h under dark conditions. Images were acquired with a Leica SP8 (Wetzlar, Germany) confocal microscope.
Western blotting assay
Cells and tumor tissues were lysed with RIPA Lysis buffer (Beyotime, Shanghai, China), and protein concentrations were determined by the BCA kit (Beyotime, Shanghai, China). Each sample was separated by 8-12% SDS-PAGE, blocked with 5% BSA (Albumin from bovine serum) for 2 h at room temperature, and incubated with primary antibodies to β-tubulin, Sox2 (#14962, 1:100, CST, MA, USA; sc-365964, 1:100, Santa Cruz, TX, USA; ab97959, 1:1000, Abcam, Cambridge, UK), AhR (ab2769, 1:500, Abcam, Cambridge, UK), STAT3 (#12640, 1:1000, CST, MA, USA), Y-STAT3 (ab76315, 1:2000, Abcam, Cambridge, UK), S-STAT3 (ab86430, 1:250, Abcam, Cambridge, UK), PCNA (#2586, 1:2000, CST, MA, USA), IDO1 (#51851, 1:500, CST, MA, USA), p53 (#2524, 1:1000, CST, MA, USA), p27 (#3686, 1:1000, CST, MA, USA), and p21 (sc-397, 1:200, Santa Cruz, TX, USA) overnight at 4 oC. Primary antibodies were detected with a goat anti-rabbit IgG-HRP (sc-2004, 1:2000, Santa Cruz, TX, USA) or anti-mouse IgG-HRP (sc-2005, 1:2000, Santa Cruz, TX, USA). The blots were developed using Super Signal West Pico chemiluminescent substrate (Millipore, Billerica, MA, USA).
Glucose consumption
Cells were collected and lysed for 10 min at 24 oC and then treated for 30 min at 37 oC using the Glucose Oxidase Method Kit (ATI, Beijing, China). Absorbance values were determined at 550 nm. Glucose consumption was calculated using the standard curve.
SA-β-gal activity assay
B16 TRCs were fixed for 15 min at room temperature and then stained overnight at 37 oC using the β-Galactosidase Staining Kit (CST, Danvers, MA, USA). SA-β-gal-positive and negative cells were then counted under the microscope.
Chromatin immunoprecipitation assay
We performed a chromatin immunoprecipitation (ChIP) assay following the manufacturer's instructions (EZ-ChIP kit, Millipore). Briefly, cells were subjected to cross-linking with 1% formaldehyde in medium for 10 min at 37 °C and then lysed on ice. Chromatin was sonicated to shear DNA to an average length of 0.2-1.0 kb. ChIP was performed using a control rabbit IgG or antibody against S-STAT3 or Y-STAT3 (Abcam, UK). The DNA fragments in the precipitates were purified for real-time qPCR analysis. Primer sets corresponded to p53 and p27 promoter regions. Primer sequences are shown in Table S1.
Tumor-specific CTL killing assay
Briefly, splenocytes were harvested from mice preimmunized with B16 TRCs 20 days after tumor implantation, and single-cell suspensions were prepared by homogenization using frosted glass slides. Splenocytes were cultured with tumor cells. Several days later, the cells were harvested and used as CTL effector cells in a standard lactate dehydrogenase cytotoxicity assay.
Transfection and stable cell line generation
Cells were transfected with gRNA-Cas9, shRNA, or complementary DNA using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. Sox2 shRNAs and scramble shRNA sequences are shown in Table S2. The Sox2 cDNA was obtained from OriGene (MG204615, Rockville, MD, USA). IFNγ was purchased from PeproTech (Rocky Hill, NJ, USA). Sox2 shRNA-transfected cells were purified with Puromycin (Sigma, St. Louis, MO, USA) for 7 days. CRISPR/Cas9 was applied to generate frameshift mutations in the Sox2 coding sequence. gRNA-Cas9 plasmids were purchased from Cyagen Biosciences (Guangzhou, China). gRNA sequences are shown in Table S3. The GFP+ populations were purified from cell lines harboring each individual guide RNA from a single cell, and then the targeted loci were analyzed by Sanger sequencing. TIDE analysis (https://tide.deskgen.com/) was used to calculate the indel mutation efficiency at the targeted loci.
Colony number assay
By changing the focal planes along the z axis (the direction of gel depth), the colony number was counted view by view. At least three wells of colonies were counted per condition per day.
Statistical analysis
All statistical analyses were performed using a two-tailed Student's t-test with an unequal variance, except for analyzing data from mice experiments which was performed using the Fisher's exact test and Welch's unpaired t-test.
Results
Sox2 is required for TRC stemness and growth
We have reported that Sox2 is a critical and highly expressed gene in melanoma TRCs, and knocking down Sox2 substantially inhibits tumor cell proliferation and tumorigenic capacity [12, 19]. To further explore Sox2 function in TRCs, we knocked out the Sox2 gene in the B16-F1 cell line using the CRISPR-Cas9 genome editing technique. We designed two different gRNAs and chose gRNA2 since it has a higher mutation rate (Figure S1A). Sox2 mutant cell lines #1 and #2 were selected. The cells transfected with scrambled plasmids grew in a manner similar to control cells (Figure S1B-C ). Then, we cultured negative control cells (transfected with scrambled shRNA), Sox2 knockdown cells (Sox2 shRNA, silenced with short-hairpin RNA of Sox2), Sox2 overexpressed cells, control cells, and Sox2 knockout cells (Sox2 KO) in the soft 90-Pa 3D fibrin gels for 5 days, and calculated their colony numbers and sizes. Knocking down Sox2 dramatically decreased colony sizes (Figure 1A-B ), but decreased colony numbers (an index of how many colonies could form for a given cultured tumor cell population) only by ~20% (Figure 1C). These cells expressed low levels of Sox2 as a result of Sox2 silencing (Figure 1D-E) and almost stopped growing (Figure 1A-B ). Transfection efficiency of Sox2 shRNA and overexpression plasmid were measured by western blotting and qPCR (Figure S1D-E). Comparing brightfield images of colonies from Sox2 siRNA and Sox2 shRNA TRCs (Figure S2A), we found that the colonies in the Sox2 shRNA group were more round and uniform in size than those of the Sox2 siRNA group and there were many spread colonies in the Sox2 siRNA group, suggesting that many cells in the Sox2 siRNA group began to differentiate, consistent with the previously published report [12]. In contrast, overexpressing Sox2 significantly increased colony sizes and numbers (Figure 1A-C). Intriguingly, complete depletion of Sox2 via knockout (Figure 1D-E) only decreased colony sizes and numbers by ~50% and moderate proliferation capability was maintained (Figure 1A-C).
Low levels of Sox2 induce TRC dormancy in culture
Sox2 knockdown colonies exhibited typical growth arrests, which is consistent with our previous report [19]. To determine if these cells could resume proliferation at longer times, we extended the observation time to 10 days. The colony size did not change on day 10 when compared with that on day 5 (Figure 2A, left). Overexpressing Sox2 in these Sox2 shRNA transfected, day-10 colonies reactivated colony growth (Figure 2A, right), suggesting that these Sox2-silenced tumor cells were not apoptotic. Apoptotic analyses confirmed that shRNA Sox2 did not cause apoptosis in these melanoma cells (Figure S3). Moreover, cell cycle analysis showed that knocking down Sox2 significantly increased the G0/G1 ratio (Figure 2B-C), suggesting that low levels of Sox2 may induce TRCs to undergo cell cycle arrest. Tumor dormancy can be functionally defined as G0/G1 arrest and cell re-growth once the inducers of dormancy are removed. We hypothesized that knocking down Sox2 might induce TRCs to enter dormancy. Among the genes for the dormancy signature, COUP-TF1+ Ki67- has been identified as a dominating dormancy marker [22-24]. Using immunofluorescence assays, we found that the percentage of COUP-TF1+Ki67- cells was significantly increased and glucose consumption was decreased after Sox2 was silenced (Figure 2D-E). When compared to the positive control Etoposide- (a DNA re-ligation inhibitor, 50 μM)treated group [25], the Sox2 shRNA-treated colony did not undergo senescence as shown by β-galactosidase activity (Figure 2F). It is known that dormant tumor cells increase their resistance to chemotherapeutic drugs [26]. Two anti-tumor retinoid drugs, tazarotene [27] and ATRA [28], were used in control and Sox2 knockdown TRCs. The colony size and apoptotic ratio results show that the drug resistance was also increased in the knockdown group (Figure S3A-B). Silencing Sox2 using siRNA also induced cell cycle arrest and elevated the percentage of COUP-TF1+Ki67- cells, suggesting cell dormancy (Figure S2B-C). However, the Sox2 shRNA cells appeared to exhibit higher G0/G1 ratios and a higher COUP-TF1+Ki67- percentage (compare Figure S2B, D with Figure 2B-D). Together, these data suggest that low levels of Sox2 are required for TRC dormancy.
Recently, it was reported that pleiotropic cytokine IFNγ may induce TRC entry into immunologicallyinduced dormancy through the IDO-AhR metabolic pathway [29]. Consistent with the published results, we found that IFNγ treatment also decreased Sox2 expression (Figure 2G). Silencing Sox2, independent of IFNγ, significantly increased IDO1 and AhR expression and AhR nuclear translocation (Figure 2G and Figure S4A-B).
To further test whether silencing Sox2 induces cellular dormancy in other cells types, we used human melanoma cell line A375 and mouse embryonic stem cell line W4. For the cell lines cultured in 3D soft fibrin matrices, colony growth was substantially arrested after Sox2 was silenced (Figure S5A-B ) and IDO1 expression was increased (Figure S5C). These data suggest that Sox2 knockdown (Figure S5D) colonies from different cell lines all undergo dormancy as B16 TRCs do.
Figure 1
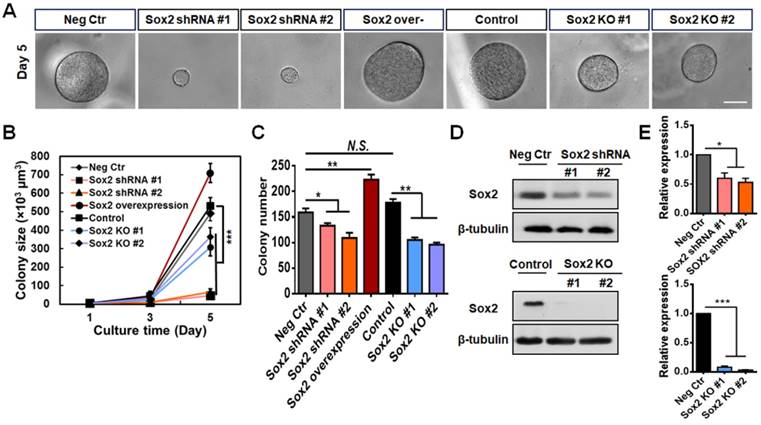
Silencing but not knocking out Sox2 in TRCs inhibits colony growth of TRCs. (A) Single individual B16-F1 melanoma cells were cultured in 90-Pa fibrin gels after Sox2 was silenced (Sox2 shRNA), overexpressed, or knocked out (Sox2 KO). Representative images of colonies on day 5 (all colonies started as single cells at day 0). Scale bar: 50 μm. (B, C) Colony size and colony number were quantified (mean ± SEM; n = 15 samples; three separate experiments). (D) Representative images of western blotting of Sox2 expression after Sox2 knockdown or knockout. (E) Quantified data of western blotting (mean ± SEM; n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 2
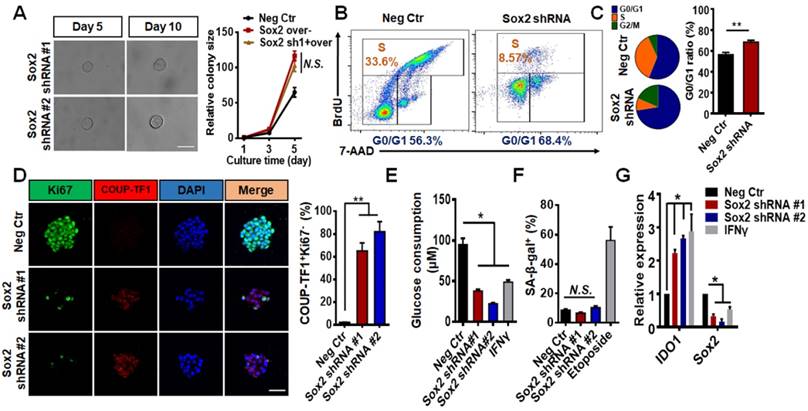
Silencing Sox2 induces entry of cultured TRCs into dormancy. (A) B16 cells, transfected with Sox2 shRNA#1 or shRNA#2, were cultured as single individual cells in 90-Pa fibrin gels for 10 days. Left: bright field images were taken on days 5 and 10; right: B16 cells transfected with negative control, Sox2 overexpression plasmids, or Sox2 shRNA#1 (day 10) plus Sox2 overexpression plasmids. Colony sizes were quantified (mean ± SEM; n = 15 samples; three independent experiments. N.S.: not significantly different). (B) Cell cycles were analyzed by flow cytometry after seeding cells in 90-Pa fibrin gels for 5 days. (C) Quantification of cell cycle analysis (mean ± SEM; n = 3 independent experiments) **P < 0.01. (D) Representative images (left panels) and quantification (right panel) of double immunostaining of COUP-TF1 and Ki67 under various conditions. Scale bar: 50 μm. The values represent mean ± SEM from n = 10 randomly chosen fields of view from two different experiments. (E) Glucose consumption was assayed in B16 TRCs transfected with shRNA or treated with IFN-γ (100 ng/ml) for 3 days. (F) SA-β-gal staining was quantified. B16 TRCs were transfected with shRNA or treated with 50 μM Etoposide for 3 days. N.S.: not significantly different. (G) B16 cells transfected with negative control, Sox2 shRNA #1, Sox2 shRNA #2, or treated with IFN-γ (100 ng/ml) were cultured in 90-Pa fibrin gels for 5 days and total mRNAs were extracted for quantitative analysis of IDO1 or Sox2 expression by real-time PCR (mean ± SEM; three independent experiments). *P < 0.05, **P < 0.01.
Complete depletion of Sox2 triggers TRC exit from dormancy
In sharp contrast, Sox2 KO TRCs in soft fibrin matrices exhibited proliferation but not dormancy. Cell cycle analyses show that the G0/G1 ratio was only slightly increased in Sox2 knockout TRCs (Figure 3A). The expression of PCNA (proliferating cell nuclear antigen) protein, a well-accepted cell proliferation marker [30], was decreased by ~15% (Figure 3B) and the dormancy marker COUP-TF1+ Ki67- did not increase in Sox2 KO TRCs (Figure S6). However, we observed that the expression of cancer cell stemness marker CD133 [31-33] was decreased in Sox2 KO TRCs (Figure 3B), implying that these Sox2 KO colonies may lose some of their CD133-mediated stemness. In contrast, CD133 levels were increased in Sox2 shRNA TRCs (Figure S7), suggesting that CD133-mediated stemness increased, although we do not know the underlying mechanism. Since Sox2-shRNA cells enter dormancy and are not tumorigenic, our result suggests that CD133 plays a different role from Sox2 in tumorigenicity. Our data are consistent with findings from the Sean Morrison group [15] that CD133 is not a reliable stemness and tumorigenic marker in highly tumorigenic melanoma cells. To test the capacity of drug resistance of Sox2 KO TRCs, we used Tazarotene, ATRA, Temozolomide, Cisplatin, and the dormancy inducer IFNγ to treat Sox2 KO and control TRCs, respectively. Sox2 KO TRCs exhibited a much lower resistance to apoptosis, as the colony size was decreased much more than that of control TRCs (Figure 3C and Figure S8E). The apoptotic ratio was still high even when treated with IFNγ (Figure 3D), which has been reported to induce TRC dormancy [28]. Furthermore, western blot data showed that in Sox2 KO TRCs, the expression of IDO1 was not increased even after IFNγ treatment (Figure 3Eand Figure S4C). TRC Sox2-null cells were further compared with 2D control B16 cells. Western blotting of PCNA protein levels (Figure S8A) and cell cycle analysis (Figure S8B) results demonstrated that their proliferation levels are similar. However, the CD133 expression (Figure S8C-D) and drug resistance of Sox2 KO TRCs are lower than those in 2D control cells. These data suggested that knockout of Sox2 triggers TRCs to lose part of their stemness and exit from dormancy. To eliminate the concern regarding the off-target effects of Sox2 knockdown, we knocked down Sox2 in the Sox2 knockout cells. Both colony number and size were not affected after Sox2 silencing (Figure S9A-B). The expression of IDO1 was not increased after Sox2 silencing in Sox2 KO cells (Figure S9C) Together, these results suggest that the Sox2 silencing-induced TRC dormancy is Sox2 deficiencyspecific, and not a result of off-target effects.
STAT3/p53 activation mediates TRC exit from dormancy
We aimed to elucidate the molecular mechanism through which Sox2 knockdown induces TRC dormancy while Sox2 knockout induces an exit from dormancy. In addition, we tested key cell cycle inhibitors, p27 [34, 35]and p21 [36, 37], and apoptosis- associated marker p53 in control, Sox2 shRNA and Sox2 KO TRCs, IFNγ (dormancy positive control), and Etoposide- (a senescence positive control)treated groups (Figure 4A-B). We found that, similar to the IFNγ-treated group, the Sox2 shRNA group showed p27high p21high expression. The Etoposide group also showed p21highp53high expression. However, the Sox2 KO group exhibited p21low p53high expression, which is different from the dormancy or senescence gene expression patterns. Low levels of p21 may contribute to proliferation of Sox2 KO colonies. However, a high level of p53 may be the key factor for the Sox2 KO cells' low resistance to apoptosis and exit from dormancy. Moreover, the apoptosis analysis showed that p53 inhibition significantly rescues the Sox2 KO cells from apoptosis under Tazarotene treatment (Figure 4C).
It is reported that STAT3 phosphorylation is the key event that switches TRCs from dormancy to apoptosis by regulating p53 and p27 expression [38]. Serine-, but not tyrosine-, phosphorylated STAT3 mediates TRC dormancy by binding to the promoter of p27, while serine- and tyrosine-phosphorylated STAT3 together induce TRC apoptosis by binding to the promoter of p53, but not that of p27. Consistently, we found that S-STAT3 (serine-phosphorylated STAT3) was increased in both Sox2 shRNA and Sox2 KO groups, and Y-STAT3 (tyrosine-phosphorylated STAT3) was significantly decreased in the Sox2 shRNA group (Figure 4D). We further performed a ChIP assay to test p53 and p27 promoters (Figure 4E-F). For the Sox2 shRNA group (S-STAT3high Y-STAT3low), STAT3 activated p27 expression by binding to the promoter of p27, and subsequently induced dormancy. However, for the Sox2 KO group (S-STAT3high Y-STAT3high), STAT3 activated p53 expression by binding to the promoter of p53, subsequently switching TRCs from dormancy to pre-apoptosis. As for the cells on 2D rigid dishes, we examined their STAT3/p53-caspase3 pathway activation levels by western blotting (Figure S10). p53 and Y-STAT3, but not S-STAT3, were slightly increased in 2D Sox2 KO cells, suggesting that Sox2 knockout on 2D rigid dishes partially activates the STAT3/p53 pathway.
Figure 3
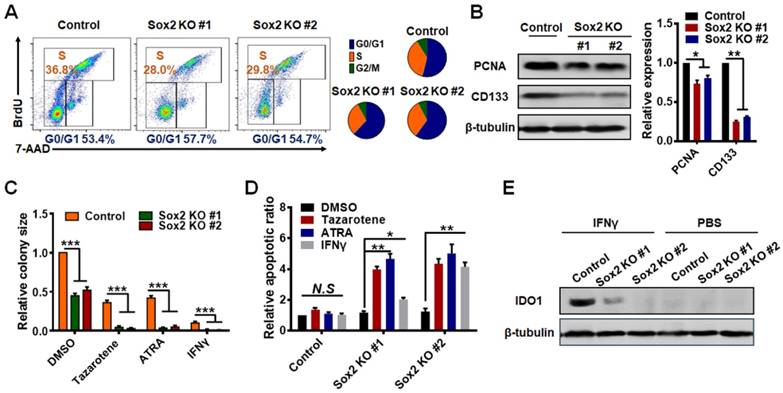
Knocking out Sox2 triggers TRC exit from dormancy. (A) Cell cycles of Sox2-knocked out (KO) cells were analyzed by flow cytometry after seeding in 90-Pa fibrin gels for 5 days. (B) Cells were cultured in 90-Pa fibrin gels for 5 days and total proteins were extracted for analyses of PCNA or CD133 expression by western blotting. Left: representative blotting images; right: quantitative data (mean ± SEM; n = 3 independent experiments). (C) Both control cells and Sox2 knockout cells were treated with 0.1% DMSO, Tazarotene (100 μM), or all-trans retinoic acid (ATRA) (100 μM) for 3 days in 90-Pa fibrin gels. Similarly, cells were cultured in 900 Pa fibrin gels for 2 days and then treated with IFN-γ (100 ng/ml) for 24 h. Colony sizes were measured on day 5 (mean ± SEM; n = 3 independent experiments). (D) Cell apoptotic ratio was measured by flow cytometry ( mean ± SEM; n = 3 independent experiments). (E) Representative westernblotting images of IDO1 expression in control B16 cells or Sox2-knocked out cells, either treated with IFN-γ or with PBS. Quantification results are shown in Figure S4C.*P < 0.05, **P < 0.01, ***P < 0.001.
Figure 4
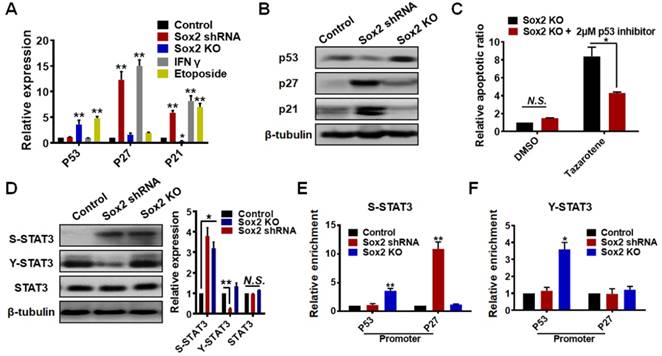
Dormancy exit is mediated via STAT3 and p53 activation. (A) p53, p27, or p21 expression was quantified by real-time PCR. Total mRNAs of control B16 cells, B16 cells transfected with Sox2 shRNA, Sox2-KO cells, or cells treated with IFN-γ (100 ng/ml) or treated with Etoposide (a DNA re-ligation inhibitor, 50 μM) were extracted. The values represent mean ± SEM from three independent experiments. (B) Western blotting analysis of p53, p27, or p21 protein level in control B16 cells, B16 cells transfected with Sox2 shRNA, or Sox2 KO cells from three independent experiments. (C) Sox2 knockout cells, with or without the p53 inhibitor, Pifithrin (2 μM), were treated with 0.1% DMSO or Tazarotene (100 μM) for 3 days in 90-Pa fibrin gels. The cell apoptotic ratio was measured by flow cytometry. The values represent mean ± SEM from three independent experiments. (D) Representative images and quantitative analysis of S-STAT3 (phospho-S727), Y-STAT3 (phospho-Y705), or total STAT3 expression in control B16 cells, B16 cells transfected with Sox2 shRNA, or Sox2-KO cells by western blotting. The values represent mean ± SEM from three independent experiments. N.S.: not statistically significant. (E, F) ChIP (chromatin immunoprecipitation) assays were performed using normal rabbit IgG (negative control), S-STAT3, or Y-STAT3 antibody on control Sox2 shRNA and Sox2-KO cell lysates. Two sets of primers were used for p27 and p53 promoter regions. Relative enrichment was determined by real time PCR. The values represent mean ± SEM ( n = 3). *P < 0.05, **P < 0.01.
Low levels of Sox2 induce TRC dormancy in vivo
To further examine the role of Sox2 in vivo, we cultured control melanoma TRCs, TRCs with Sox2 knockdown, TRCs with Sox2 knockout, and melanoma cells grown on 2D rigid dishes (2D control) for 5 days and injected 3 × 104 cells per mouse into mice subcutaneously. The tumor formation rates of control TRCs, knockdown TRCs, and 2D control cells were similar (Table 1). Silencing Sox2 in TRCs substantially suppressed B16 melanoma growth and prolonged the survival of mice (Figure 5A-C ). The growth of the tumor mass was confirmed by immunohistochemistry and H&E staining. On day 28 after injection, the control melanoma cells were more compact than on day 14. On day 28, silencing Sox2 in TRCs did not alter tumor cell density or numbers compared with those on day 14, suggesting that Sox2 knockdown induces growth arrest of melanoma TRCs in vivo (Figure 5B). These findings are consistent with the in vitro results and confirm that low levels of Sox2 induce TRC dormancy. Intriguingly, the tumor formation rate of the Sox2 KO group was zero (0 out of 8 mice) (Table 1). None of the mice injected with Sox2 KO cells had succumbed to the tumor on day 50, when all the mice from all the other groups had died. Hence, Sox KO greatly extended the survival time of the mice (Figure 5C). To elucidate how Sox2 KO TRCs were eliminated in vivo, we performed a tumor-specific cytotoxic T lymphocytes (CTLs) killing assay to test their specific lysis [39]. Cytolytic analysis indicated that splenic T cells from mice immunized with B16 TRCs were much more potent in killing Sox2 KO cells than control or Sox2 shRNA cells (Figure 5D). For a further comparative study using western blotting and immunohistochemistry assays, in order to obtain the tumor tissue from all the mice, we tried several numbers of Sox2 KO TRCs (5 × 105, 1 × 106, and 2 × 106). We chose to utilize 5 × 105 cells (Figure 5E and Figure 6C) since the number of Sox2 KO B16 cells is enough to form a sizeable tumor in mice. Too many cells may cause skin tissue ulceration in mice. Melanoma tissues of control and Sox2 KO mice at the injection sites were extracted for western blotting analysis. p53 and Y-STAT3 in Sox2 KO TRCs were increased (Figure 5E and Figure S11). Expression of cleaved (active) caspase 3 was significantly increased in Sox2 KO melanoma cells (Figure 5E and Figure S11). These results suggest that Sox2 knockout melanoma cells became apoptotic via upregulating the p53-caspase3 cascade. Single-cell analysis was performed to correlate Sox2 expression with TRC dormancy and exit from dormancy. Sox2 expressions under different conditions were further tested by immunofluorescence and flow cytometry, both in 2D cells (Figure S12) and in 3D TRCs (Figure 6A-B ). Additional immunohistochemistry analysis of Ki67 and COUP-TF1 from different tumor tissues further demonstrates that Sox2 partial depletion is closely associated with TRC dormancy and Sox2 knockout is closely associated with exit from dormancy (Figure 6C).
Discussion
Cancer dormancy is governed by a combination of external and internal cues [40]. It is critical to increase our understanding of the key factors and various pathways involved in cellular dormancy. In this study, we show that Sox2 participates in melanoma TRC dormancy under a low-expression condition. Low levels of Sox2 induce the entry of melanoma TRCs into dormancy, mediated by activating the expression of the IDO1/AhR pathway, which then induces the expression of p27 and p21, contributing to TRC growth arrest and dormancy. With the knockdown manipulation, Sox2 expression was maintained at a low level in the soft matrix of the mechanical microenvironment. Previously, it was reported that the 3D soft fibrin matrix promotes melanoma TRCs to grow and proliferate rapidly [12] and that the 3D stiff fibrin matrix inhibits TRC growth [19] and induces TRCs to enter dormancy [16]. Our current finding of low levels of Sox2 inducing TRCs to enter dormancy in 3D soft fibrin matrix reveals the role of Sox2 in regulating tumor cell dormancy, independent of matrix stiffness. On the contrary, complete depletion of Sox2 in TRCs induces exit of dormancy. High expression levels of p53 and low expression levels of p21 mediate high colony proliferation and moderate apoptosis, while activation of the p53-caspase3 cascade triggers TRC apoptosis and tumor elimination in vivo. For differentiated melanoma cells on 2D rigid dishes, Sox2 KO does not activate caspase 3 but activates p53 and Y-STAT3 (Figure S10), suggesting that triggering of those differentiated melanoma cells to apoptosis is substantially reduced.
Figure 5
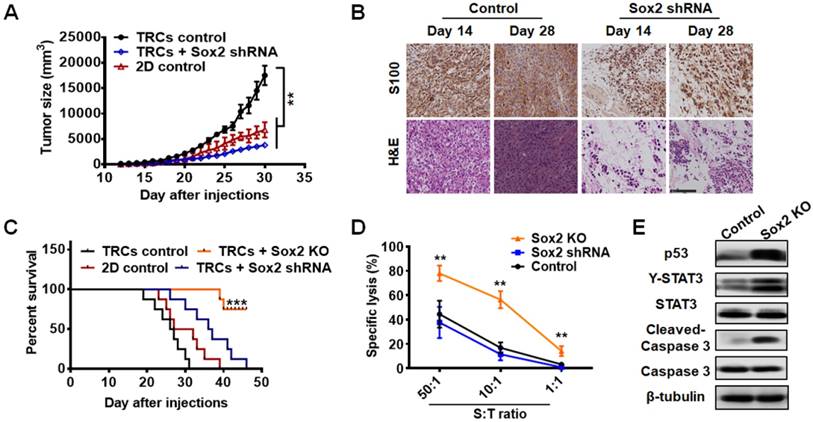
Knocking down of Sox2 in TRCs induces melanoma dormancy in vivo. B16 cells (3 × 104) of 2D control (cells were plated on rigid plastic), TRCs control (cells were plated in soft 90-Pa fibrin gels), TRCs + Sox2 shRNA (TRCs transfected with Sox2 shRNA), or TRCs+ Sox2 KO (Sox2 was knocked out with CRISPR) TRCs were subcutaneously injected into C57BL/6 mice (n = 8 mice per group). (A) Tumor size of mice were measured (n = 8 mice per group). Note that in the TRCs + Sox2 KO group, since no tumors were detected, the tumor size was always 0. **P < 0.01. (B) On day 14 and 28, tumors were isolated and analyzed by immunostaining against S100 or H&E staining (400×). Upper: immunohistochemical analysis of melanoma tissues using S100 (a specific melanoma cell marker). S100 signal is shown as brown. DAPI (blue) staining for the nucleus. Lower: H&E staining for cells in the tumor tissues. Nucleus (Blue), cytoplasm (Light purple). Twenty-eight days after injection, control melanoma cells were more compact than those on day 14, while Sox2 knockdown TRCs almost maintained the original tumor cell density and numbers as observed on day 14. Scale bar: 100 μm. (C) Long-term survival were measured (n = 8 mice per group), no mice in the TRCs + Sox2 KO group succumbed to the tumor on day 50, when the observation was stopped because all mice from the other groups had died. Two of eight mice in the TRCs + Sox2 KO group died only because of accidental tearing damage during long-term feeding. ***P < 0.001. (D) Splenocytes were harvested from mice preimmunized with B16 TRCs 20 days after tumor implantation, and then cultured with 2D B16 control cells. 5 days later, the cells were harvested and used as CTL effector cells. Co-cultures of the control B16 TRCs, B16 TRCs with Sox2 shRNA, or Sox2 KO TRCs with CTL effector cells at different ratio (S: T ratio) for 24h. Specific lysis were analyzed by standard lactate dehydrogenase cytotoxicity assay. The values represent mean ± SEM from three independent experiments. **P < 0.01. (E) Representative western blotting images from tumor tissues. To harvest tumor tissue proteins, 5 × 105 control TRCs and Sox2 knockout TRCs were subcutaneously injected into C57BL/6 mice for 7 days (at least 5 mm × 5 mm size of melanoma was collected for each group). Melanoma tissues were collected and lysed for protein analyses. These are representative images of p53, Y-STAT3, STAT3, Cleaved-Caspase3 (activated Caspase 3), or Caspase3 expression. Quantified results are shown in Figure S11.
Figure 6
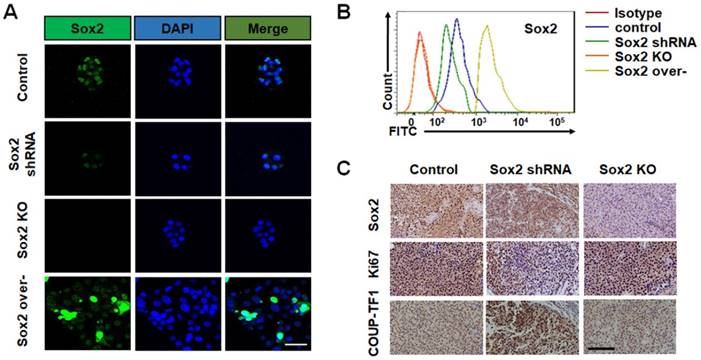
Sox2 expression is closely associated with TRC dormancy by single-cell analysis. (A) Representative images of immunostaining of Sox2 for B16 control cells, cells transfected with Sox2 shRNA, Sox2 KO cells, or cells transfected with Sox2 overexpression plasmid after being cultured in 90-Pa fibrin gels for 5 days (n = 10 randomly chosen fields of view from two different experiments). Scale bar: 50 μm. (B) The expression of Sox2 was analyzed by flow cytometry. Data are representative of three independent experiments (C) Tumors were isolated and analyzed by immunohistochemistry analysis against Sox2, Ki67, or COUP-TF1 staining (400×). Signals are shown in brown, with DAPI (blue) staining for the nucleus. Scale bar: 100 μm.
Table 1
Tumorigenicity of B16-F1 cells in C57BL/6 mice.
| Tumor cells | Tumor formation |
|---|---|
| TRCs control | 8/8 |
| TRCs + Sox2 shRNA | 7/8 |
| TRCs + Sox2 KO | 0/8* |
| 2D control | 8/8 |
3 × 104 B16-F1 cells were subcutaneously injected into C57BL/6 mice (n=8 mice per group). Number of tumor formation per group was measured in 30 days. Note that no tumor was formed in the TRCs + Sox2 KO group. *P<0.05 (Fisher's exact test).
Sox2 is critical in pluripotency maintenance of embryonic stem cells and fate determination of adult stem cells [17, 18]. Our previous studies have shown that Sox2 is essential for proliferation and self-renewal of melanoma TRCs [12, 19]. Understanding the intersection between cancer dormancy and the cancer stem cell model, which is still poorly characterized, may offer insight into the underlying biology in both realms. Our 3D fibrin gel culture system presents a useful model for research on cancer stem-like cells and cancer dormancy. Recently, several reports have shown that IFNγ and IFNβ treatment and matrix stiffness induce the dormancy of tumor-repopulating cells [16, 28, 38]. Although cancer dormancy signature has not been welldocumented, it has been wellaccepted that indolamine 2, 3-dioxygenase/aryl hydrocarbon receptor (IDO1/AhR) metabolic pathway activation and/or cell cycle inhibiting regulators p27/p21 represent dormancy of TRCs [16, 28, 38]. We have shown that Sox2 expression is decreased in IFNγ-induced dormant TRCs, suggesting that Sox2 may be a key factor downstream of IFNγ and upstream of the IDO1/AhR dormancy-related pathway. There are still questions that need to be answered to determine how IFNγ affects the stemness marker gene Sox2 expression and the underlying pathological significance during the process of tumor formation and progression. Sox2 is known to be highly expressed in lung and esophageal carcinomas [41]. In the present study, we have only demonstrated that low levels of Sox2 are required for melanoma TRC dormancy. In the future, it should be determined if low levels of Sox2 are required for entry into dormancy of other types of cancer.
In this study, we show two different cell fates of TRCs by using two different approaches for Sox2 gene modification. These two widely used approaches are RNA interference (shRNA), which destabilizes a targeted transcript and induces partial loss-of-function, and CRISPR-Cas9 mutagenesis, which utilizes the nuclease Cas9 to induce frameshift mutations at a targeted locus and induces complete depletion. Several groups have reported that gene knockdown and gene knockout result in very different phenotypes [42, 43]. For future applications of knockdown and knockout approaches, it is important to consider the molecular causes of the phenotypic discrepancies. Our findings suggest the possibility that complete depletion of Sox2 might be an effective tumor elimination therapy for melanoma using the CRISPR strategy. However, it remains to be seen how to effectively introduce CRISPR to human melanoma in vivo. Nevertheless, a deep understanding of Sox2 function in tumorigenic cells may provide a novel insight for improved cancer therapy.
Abbreviations
BSA: Albumin from bovine serum; ChIP: chromatin immunoprecipitation; CSCs: cancer stem cells; CTLs: cytotoxic T lymphocytes; DMEM: dulbecco's modified Eagle's medium; DMSO: Dimethyl sulfoxide; H&E: hematoxylin-eosin staining; IDO1/ AhR: indolamine 2, 3-dioxygenase/aryl hydrocarbon receptor; IFNγ: Interferon γ; MEM: minimum essential medium; Neg Ctr: negative control; PBS: phosphate buffered saline; PCNA: proliferating cell nuclear antigen; RT: Reverse transcription; shRNA: short-hairpin RNA; siRNA: Small interfering RNA; Sox2 KO: Sox2 knockout; TICs: tumor initiating cells; TRCs: tumor-repopulating cells.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We greatly appreciate the guidance and leadership of Professor Ning Wang throughout the project. The current work is a follow-up study of the 2012 Nature Materials paper (J Liu et al.) on 3D soft matrix-selected tumor-repopulating cells (TRCs), the 2014 Nature Communications paper (Y Tan et al.) on H3K9 demethylation-mediated Sox2 expression in TRCs, and the 2018 Cancer Research paper (Y Liu et al.) on 3D stiff matrix-induced tumor dormancy. We thank Professors Ning Wang and Bo Huang for their valuable comments on the manuscript and helpful discussions. We also thank the technical support from the central facilities at School of Life Science and Technology, Huazhong University of Science and Technology. This work was supported by the Ministry of Science and Technology of China grant [2016YFA0101100], the National Natural Science Foundation of China [81770102 & 81874149], and a China Postdoctoral Science Foundation grant [0106170082].
Contributions
HJ conceived the initial idea with Professor Ning Wang. QJ, FY, and HJ designed the experiments. QJ, FY, WH, YZ, BB, FW, and CZ carried out the experiments and analyzed the data. QJ, HJ, and FY wrote the manuscript with input from all other authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
2. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611-22
3. Goss PE, Chambers AF. Does tumour dormancy offer a therapeutic target? Nat Rev Cancer. 2010;10:871-7
4. Adav AS, Pandey PR, Butti R. et al. The biology and therapeutic implications of tumor dormancy and reactivation. Frontiers in Oncology. 2018;8:72-8
5. Ross JS, Slodkowska EA. Circulating and disseminated tumor cells in the management of breast cancer. Am J Clin Pathol. 2009;132:237-45
6. Páez D, Labonte MJ, Bohanes P. et al. Cancer dormancy: a model of early dissemination and late cancer recurrence. Clin Cancer Res. 2012;18:645-53
7. Gay LJ, Malanchi I. The sleeping ugly: tumour microenvironment's act to make or break the spell of dormancy. Biochim Biophys Acta. 2017;1868:231-8
8. Dasgupta A, Lim AR, Ghajar CM. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol Oncol. 2017;11:40-61
9. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137-48
10. Elder GJ, Hersey P, Branley P. Remission of transplanted melanoma-clinical course and tumour cell characterisation. Clinical transplantation. 1997;11:565-8
11. Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes & development. 2011;25:2559-72
12. Liu J, Tan Y, Zhang H. et al. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat. Mater. 2012;11:734-41
13. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994Feb17;367(6464):645-8
14. Dieter SM, Ball CR, Hoffmann CM. et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell. 2011;9:357-65
15. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593-8
16. Liu Y, Lv J, Liang X. et al. Fibrin Stiffness Mediates Dormancy of Tumor-Repopulating Cells via a Cdc42-Driven Tet2 Epigenetic Program. Cancer Res. 2018;78:3926-37
17. Masui S, Nakatake Y, Toyooka Y. et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol. 2007;9:625-35
18. Sarkar A. & Hochedlinger, K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15-30
19. Tan Y, Tajik A, Chen J. et al. Matrix softness regulates plasticity of tumour repopulating cells via H3K9 demethylation and Sox2 expression. Nat. Commun. 2014;5:4619
20. Santini R, Pietrobono S, Pandolfi S, Montagnani V, D'Amico M, Penachioni JY, Vinci MC, Borgognoni L, Stecca B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene. 2014Sep18;33(38):4697-708
21. Wen Y, Hou Y, Huang Z, Cai J, Wang Z. SOX2 is required to maintain cancer stem cells in ovarian cancer. Cancer Sci. 2017Apr;108(4):719-731
22. Kim RS, Avivarvalderas A, Estrada Y. et al. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PloS one. 2012;7:e35569
23. Sosa MS, Parikh F, Maia AG. et al. NR2F1 controls tumor cell dormancy via SOX9 and RARβdriven quiescence programs. Nat commun. 2015;6:61-70
24. Fluegen G, Avivar-Valderas A, Wang Y. et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19:120-32
25. Dolega A. Cytotoxic mechanism and antineoplastic action of etoposide. Postepy Hig Med Dosw. 1998;52:67-87
26. Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer. 2015;15:238-47
27. Brigger D, Schläfli AM, Garattini E, Tschan MP. Activation of RARα induces autophagy in SKBR3 breast cancer cells and depletion of key autophagy genes enhances ATRA toxicity. Cell Death Dis. 2015;6:e1861
28. Liu Y, Liang X, Yin X. et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. 2017;8:15207
29. Chandraratna RA. Disubstituted acetylenes bearing heteroaromatic and heterobicyclic groups having retinoid like activity. US patent. 1995;117:1249
30. Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051-60
31. Monzani E, Facchetti F, Galmozzi E. et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935-46
32. Bresdcia P, Ortensi B, Fornasari L, Levi D, Broggi G, Pelicci G. CD133 is esential for glioblastoma stem cell maintennance. Stem cells. 2013;31:857-69
33. Ren F, Sheng WQ, Du X. CD133: a cancer stem cells marker, is used in colorectal cancers. World J Gastroenterol. 2013;19:2603-11
34. Dai L, Liu Y, Liu J. et al. A novel cyclinE/cyclinA-CDK inhibitor targets p27 (Kip1) degradation, cell cycle progression and cell survival: implications in cancer therapy. Cancer Lett. 2013;333:103-12
35. Ou L, Waddell MB, Kriwacki RW. Mechanism of cell cycle entry mediated by the intrinsically disordered portein p27 (Kip1). ACS Chem Biol. 2012;7:678-82
36. Yoon MK, Mitrea DM, Ou L, Kriwacki RW. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem Soc Trans. 2012;40:981-8
37. Al-Maghribi J, Al-Ahwal M, Buhmeida A. et al. Expression of cell cycle regulators p21 and p27 as predictors of disease outcome in colorectal carcinoma. J Gastrointest Cancer. 2012;43:279-87
38. Liu Y, Lv J, Liang X. et al. STAT3/p53 pathway activation disrupts IFN-β-induced dormancy in tumor-repopulating cells. J Clin Invest. 2018;128:1057-73
39. Abe R, Peng T, Sailors J, Bucala R, Metz CN. Regulation of the CTL response by macrophage migration inhibitory factor. J Immunol. 2001;166:747-53
40. Yeh AC, Ramaswamy S. Mechanisms of Cancer Cell Dormancy-Another Hallmark of Cancer? Cancer Res. 2015;75:5014-22
41. Bass AJ, Watanabe H, Mermel CH. et al. Sox2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009;41:1238-42
42. Lin Ann Giuliano, Christopher J Sayles et al. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. eLife. 2017:6
43. Lawson ND. Reverse Genetics in Zebrafish: Mutants, Morphants, and Moving Forward. Trends Cell Biol. 2016;26:77-9
Author contact
![]() Corresponding author: Dr. Haibo Jia, Phone/fax: (+86) 2787792072; Email: haibo.jiaedu.cn
Corresponding author: Dr. Haibo Jia, Phone/fax: (+86) 2787792072; Email: haibo.jiaedu.cn
Citation styles
APA
Jia, Q., Yang, F., Huang, W., Zhang, Y., Bao, B., Li, K., Wei, F., Zhang, C., Jia, H. (2019). Low Levels of Sox2 are required for Melanoma Tumor-Repopulating Cell Dormancy. Theranostics, 9(2), 424-435. https://doi.org/10.7150/thno.29698.
ACS
Jia, Q.; Yang, F.; Huang, W.; Zhang, Y.; Bao, B.; Li, K.; Wei, F.; Zhang, C.; Jia, H. Low Levels of Sox2 are required for Melanoma Tumor-Repopulating Cell Dormancy. Theranostics 2019, 9 (2), 424-435. DOI: 10.7150/thno.29698.
NLM
Jia Q, Yang F, Huang W, Zhang Y, Bao B, Li K, Wei F, Zhang C, Jia H. Low Levels of Sox2 are required for Melanoma Tumor-Repopulating Cell Dormancy. Theranostics 2019; 9(2):424-435. doi:10.7150/thno.29698. https://www.thno.org/v09p0424.htm
CSE
Jia Q, Yang F, Huang W, Zhang Y, Bao B, Li K, Wei F, Zhang C, Jia H. 2019. Low Levels of Sox2 are required for Melanoma Tumor-Repopulating Cell Dormancy. Theranostics. 9(2):424-435.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.