Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(2):324-336. doi:10.7150/thno.28201 This issue Cite
Research Paper
β-catenin-activated autocrine PDGF/Src signaling is a therapeutic target in pancreatic cancer
Tzu-Lei Kuo1, Kuang-Hung Cheng2, Yan-Shen Shan3,4, Li-Tzong Chen1,5,6, Wen-Chun Hung1,6 ![]()
1. National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan
2. Institute of Biomedical Sciences, National Sun Yat-Sen University, Kaohsiung 804, Taiwan
3. Institute of Clinical Medicine, National Cheng Kung University, Tainan 704, Taiwan
4. Department of Surgery, National Cheng Kung University Hospital, Tainan 704, Taiwan
5. Division of Hematology/Oncology, Department of Internal Medicine, National Cheng Kung University Hospital, Tainan 704, Taiwan
6. Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 804, Taiwan
Received 2018-6-29; Accepted 2018-12-10; Published 2019-1-1
Abstract

K-ras mutation and p53 loss are the most prevalent genetic alterations in pancreatic cancer. In addition to these two alterations, pancreatic tumors frequently contain a third genetic defect. Mutations in the WNT/ß-catenin signaling molecules occur in 15-20% of pancreatic cancer patients and co-exist with K-ras mutation and p53 loss. However, the contribution of the WNT/ß-catenin pathway in pancreatic tumorigenesis is still unclear.
Methods: We generated Pdx1-CreKrasG12Dp53L/+APCL/+ (KPA) mice and compared their phenotypes with Pdx1-CreKrasG12Dp53L/+ (KPC) mice. The signaling pathways specifically activated in the KPA mice were investigated and the therapeutic effect by targeting the activated pathways was evaluated. We finally validated our findings in human blood and tumor samples.
Results: Survival of the KPA mice was shorter than that of the KPC mice. The KPA cancer cells are highly invasive and exhibit distorted morphology in organoid culture with extensive invadopodia formation and elevated matrix metalloproteinase (MMP) activity. The platelet-derived growth factor (PDGF) pathway is upregulated in the KPA cancer cells, and PDGF production induced by ß-catenin triggers constitutive activation of the Src kinase via the PDGF receptor in the cells. Serum PDGF concentration of the KPA mice is much higher than that of the normal and KPC mice. The Src inhibitor dasatinib effectively inhibits tumor growth and metastasis of the KPA cancer cells. Patient's serum PDGF level is significantly correlated with the expression of PDGF and phosphor-Src in tumors and elevated PDGF/phosphor-Src level in tumors predicts increased recurrence and poor survival. Moreover, mutations of the WNT/ß-catenin signaling molecules are higher in patients with elevated PDGF/phosphor-Src level.
Conclusion: ß-catenin activation, coupled with K-ras mutation and p53 loss, activates an autocrine PDGF/Src signaling in pancreatic cancer and defines a subset of patients who might be sensitive to Src inhibition. In addition, serum PDGF level could be a reliable biomarker for patient selection in clinic.
Keywords: β-catenin, platelet-derived growth factor (PDGF), Src kinase, metastasis, pancreatic cancer
Introduction
Pancreatic cancer is one of the most lethal malignancies, with a 5-year survival rate of less than 8% [1] due to early metastasis and high chemo-/radio-resistance [2]. Global genomic profiling has revealed a number of genetic alterations that might contribute to pancreatic tumorigenesis [3-5]. Specifically, K-ras mutations have been found in more than 90% of patients, and the encoded oncoprotein plays an important role in the initiation and maintenance of pancreatic cancer [6, 7]. P53 inactivated by mutation, deletion, truncation, or gene silencing has been detected in more than 50% of pancreatic cancer patients [8, 9]. It is noteworthy that the deregulation of genes involved in cell cycle control, transforming growth factor beta signaling, and epigenetic modification frequently co-exist with K-ras mutation and p53 inactivation in patients with pancreatic cancer [3-5]. Clinical evidence has also suggested that patients with complex pathway deregulations tend to have poor prognosis [5]. However, the interplay of the third genetic hit with K-ras and p53 in pancreatic tumorigenesis is largely unknown.
The WNT/β-catenin pathway participates in the development of pancreas and might play a role in the tumorigenesis of pancreatic cancer [10]. The transcriptional activity of β-catenin is tightly controlled in cells under physiological circumstances and is deregulated in various pathological conditions, such as cancer. Three potential mechanisms are thought to induce the constitutive activation of β-catenin in cancer cells. First, mutation or deletion of the destruction complex components, including adenomatous polyposis coli (APC), AXIN 1, and AXIN 2; these components mediate the poly-ubiquitination and degradation of β-catenin. Disruption of the destruction complex reduces β-catenin proteolysis and promotes its activation. Second, mutation of the ß-catenin gene may change its protein conformation to hinder its binding to the destruction complex and induce gain-of-function transcriptional activity. Third, dysregulation of WNT ligands or receptors leads to enhancement of β-catenin activation. For example, turnover of the WNT receptor Frizzled is catalyzed by two unique transmembrane E3 ligases, zinc and ring finger 3 (ZNRF3) and ring finger protein 43 (RNF43) [11]. Mutations in ZNRF3 and RNF43, which have been found in a number of cancers, might amplify WNT signaling to promote cancer initiation and progression [12].
Recently, whole-exome sequencing using 109 micro-dissected human pancreatic tumor tissues revealed the dysregulation of multiple signaling pathways in pancreatic cancer [5]. One of the highly upregulated pathways is the WNT/β-catenin axis with loss of RNF43 in ~7% of cancer patients and defect of AXIN1/2 or APC in ~11% of cancer patients. We previously developed a non-K-ras mutation-induced premalignant animal model by deletion of one allele of APC and both alleles of p53 to study the biological function of β-catenin in p53-null mice [13]. Our results showed that heterozygous loss of APC coupled with p53 deficiency induced the formation of mucinous cystic neoplasm (MCN) at an early stage and finally progressed into invasive pancreatic cancer. APC haploinsufficiency promoted β-catenin activation and increased cytokine production and genomic instability. Treatment of the mice with WNT/β-catenin pathway inhibitors suppressed the development of MCN and invasive carcinoma, thereby confirming the tumor-promoting role of β-catenin signaling in pancreatic cancer.
In this study, we introduced a K-ras mutation into our mouse model to generate Pdx1-CreKrasG12Dp53L/+APCL/+ (KPA) mice and compared their phenotypes with Pdx1-CreKrasG12Dp53L/+ (KPC) mice to identify potential therapeutic targets for pancreatic cancer with three genetic alterations. We found a significant elevation of PDGF production and constitutive activation of Src kinase via autocrine PDGF receptor stimulation in the KPA tumors; these tumors were highly sensitive to Src inhibitors. Thus, by detecting serum levels of PDGF or via genetic screening of K-ras, p53, and WNT/β-catenin signaling molecules, it is possible to identify a group of pancreatic cancer patients who might benefit from dasatinib treatment.
Methods
Genetically modified mice and primary culture of mouse pancreatic cancer cells
Pdx-1Cre, LSLKrasG12D, p53Loxp/Loxp, and APCCKO/CKO mice, obtained from the Mouse Models of Human Cancers Consortium (MMHCC) under material transfer agreements, were generously made available by Drs. Andrew M. Lowy, Tyler Jacks, Anton Berns and Raju Kucherlapati, respectively [14-16]. The mice were genotyped as described by the MMHCC PCR protocols for strains 01XL5, 01XJ6, 01XC2 and 01XAA. Animal studies were approved by the Institutional Animal Care and Use Committee of the National Health Research Institutes.
Data extraction for the analysis of genetic alterations
To explore the presence of mutations, deletions or amplifications in the identified genes (K-RAS, p53, APC, CTNNB1, AXIN1/2, RNF43, and TCF4), we used databases published in cBioportal (www.cbioportal.org) for analysis [17].
Organoid culture
The organoid culture was performed according to the procedure described previously [18].
Immunohistochemistry (IHC) and immunofluorescence (IF)
Periodic Acid-Schiff stain (PAS) and Alcian blue staining kits were purchased from Scy-Tek Laboratories (Logan, Utah, USA) and performed according to the manufacturer's protocols. The images of the IHC-stained slides were captured using a Carl Zeiss Axioskop 2 plus microscope (Carl Zeiss, Thornwood, NY, USA). IF images were studied using the ECLIPSE TE2000U laser scanning confocal microscope (Nikon, Melville, NY, USA) and analyzed using the EZ-C1 software (Nikon, Melville, NY, USA). Standard procedures for IHC and IF analysis were described in detail previously [13], and antibodies used in these studies are listed in Table S1.
Primary culture of mouse pancreatic cancer cells
The KPA or KPC tumor tissues were minced and digested overnight with 0.012% (w/v) collagenase XI and 0.012% (w/v) dispase in DMEM media containing 1% fetal bovine serum (FBS). The isolation of mouse pancreatic cancer cells was performed as described previously [13]. Tumorigenicity of the isolated primary mouse pancreatic cancer cells was tested by injecting the cells into severe combined immunodeficient (SCID) mice and the tumors were histopathologically characterized by a pathological examination. Primary cancer cells of less than 6 passages were used for microarray expression profile analyses.
Cell proliferation assay, Western blot analysis, real-time quantitative PCR analysis and microarray analysis
The procedures for these assays were described in our previous study [13]. The primers used are listed in Table S2.
RNA extraction and microarray assay
Cells scraped from the plates were collected by centrifugation, and total RNA was isolated by RNeasy Mini Kit (QIAGEN, P/N 74104, Germantown, MD, USA). RNA quantity and purity were assessed using a Nanodrop (ND-1000; Labtech. International, Vienna, Austria). Microarray analysis was performed using the Affymetrix GeneChip MoGene 1.0 ST array (Phalanx Biotech Group, Hsinchu, Taiwan). All samples were analyzed twice, and the intensities were normalized for each microarray. The significant lists were uploaded from a Microsoft Excel spreadsheet onto the Metacore 6.13 software (GeneGo pathways analysis) (http://www.genego.com). GeneGo recognizes the Affymetrix identifiers and maps the tissues to the MetaCore™ data analysis suite to describe common pathways or molecular connections between pancreatic tissues on the list. Graphical representations of the molecular relationships between genes were generated using the GeneGo pathway analysis, based on processes showing significant association. The Ingenuity Pathway Analysis (IPA) (www.ingenuity.com/products/ipa) software was also performed to predict upstream regulators with their activation score and p-value.
PDGF ELISA assay
Cell culture supernatant (500 μL) was collected and centrifuged at 4,000 rpm for 5 min to remove particulates before being stored at -80 °C. Blood samples were obtained from the facial vein of the mice by using a lancet. Mouse PDGF-AA and PDGF-BB were quantified using Quantikine® ELISA kits according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA) on a PowerWave340 microplate spectrophotometer (BioTek, Winooski, VT, USA). The standards and the samples were run in duplicates. The serum PDGF-BB level of pancreatic cancer patients was quantified similarly by using Human PDGF-BB Quantikine ELISA kit (R&D Systems).
Detection of PDGFR phosphorylation by immunoprecipitation/Western blotting
To measure levels of endogenous phosphorylation of PDGFR-β, cells were harvested in lysis buffer (50 mM Tris-HCl pH 7.5, 1% Tween-20, 200 mM NaCl, 0.2% NP-40, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and protease inhibitors). Anti-PDGFR-β antibody was used to immunoprecipitate the receptor protein from a whole cell lysate containing 2 mg of cellular proteins. Immunoprecipitates were washed, subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with an anti-phosphor-tyrosine antibody to study the phosphorylation status of PDGFR-β.
Matrix degradation assay
Alexa Fluor 488-conjugated gelatin was prepared according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). Cells were plated on glass coverslips coated with 20 ng/mL Alexa Fluor 488-conjugated gelatin. After various times, the cells were fixed and stained for F-actin and nuclei. Coverslips were viewed using a laser-scanning confocal microscope image system (Nikon C1-Si, Melville, NY, USA) to study the co-localization of F-actin-positive protrusions and the fluorescent-negative digested area. The areas in which Alexa Fluor 488-conjugated gelatin were degraded were measured using the ImageJ software (National Institutes of Health, USA) and a total of 10 random fields of view, equivalent to 2 mm2, were measured.
Orthotopic animal model
Six-week-old, pathogen-free SCID mice were purchased from BioLASCO Taiwan Co., Ltd. (Taipei, Taiwan). Luciferase-tagged KPC and KPA cancer cells (1×106 cells suspended in 50 μL Hank's balanced salt saline) were injected into the pancreas of mice. After 1 week, PBS or dasatinib (10 mg/kg) was given to the mice by an oral gavage daily for 2 weeks, and tumor growth was monitored by the IVIS imaging system (PerkinElmer, Melville, NY, USA). For in vivo imaging, mice were intraperitoneally injected with d-luciferin (150 mg/kg in saline) immediately after administering anesthesia with isoflurane. After the administration of d-luciferin for 2 min, the anesthetized mice were placed onto the light-tight camera box with continuous exposure to isoflurane. The IVIS camera system was used to visualize the tumors, and photon measurement was defined around the tumor area. The signal was quantified as total number of photons by using the Living Image software (Xenogen, Corp, Alameda, CA, USA). Blood samples were obtained from the mice at sacrifice and blood chemistry parameters were analyzed by Fuji Dri-Chem 4000i autoanalyzer of the Taiwan Mouse Clinic (Taipei, Taiwan).
Patients and clinical samples
A total of 77 pancreatic cancer patients who underwent tumor resection in the National Cheng Kung University Hospital (NCKUH) were included in this study. Anonymously archived samples, including malignant tissues and serum of the patients, were obtained from the Human Biobank of the NCKUH. Tumor sections were examined by pathologists and graded histologically. Blood samples obtained from 20 patients before surgery were frozen at -80 °C until use. Informed consent was obtained from all patients under a protocol approved by the Institutional Review Board (IRB) of the NCKUH.
Statistical analysis
Statistical analysis was performed by one- or two-way ANOVA using the Prism 5.0 software (GraphPad, San Diego, CA, USA). The median survival was estimated using the Kaplan-Meier method. The association between the studied variables was evaluated using the Spearman correlation coefficient test in SPSS 17.0 (IBM, Armonk, NY, USA). Data measured on a continuous scale was analyzed using Student's t-test and categorical data were subjected to Chi-square test. A p-value less than 0.05 was considered significant.
Results
Genetic alterations in the WNT/ß-catenin pathway co-existed with K-ras and p53 mutations in pancreatic cancer patients
There are four public pancreatic cancer databases at The Cancer Genome Atlas (TCGA) website (https://cancergenome.nih.gov). We performed our analyses using two databases containing information on both mutation and copy number alterations. In the TCGA provisional dataset, genetic alterations, including amplification, deletion, and mutations in WNT/β-catenin signaling mediators, co-existed with K-ras mutations and p53 defects in 11.4% (21/185) of pancreatic tumors (Figure S1A). Analysis of the UT Southwestern dataset, which includes whole-exome sequencing information on 109 micro-dissected human pancreatic tumor tissues, revealed the co-existence of genetic alterations in the WNT/β-catenin pathway with K-ras and p53 mutations in 31 cases (28%) (Figure S1B). These results prompted us to study the functional interplay between these genes in pancreatic tumorigenesis.
KPA mice developed poorly differentiated and highly metastatic tumors
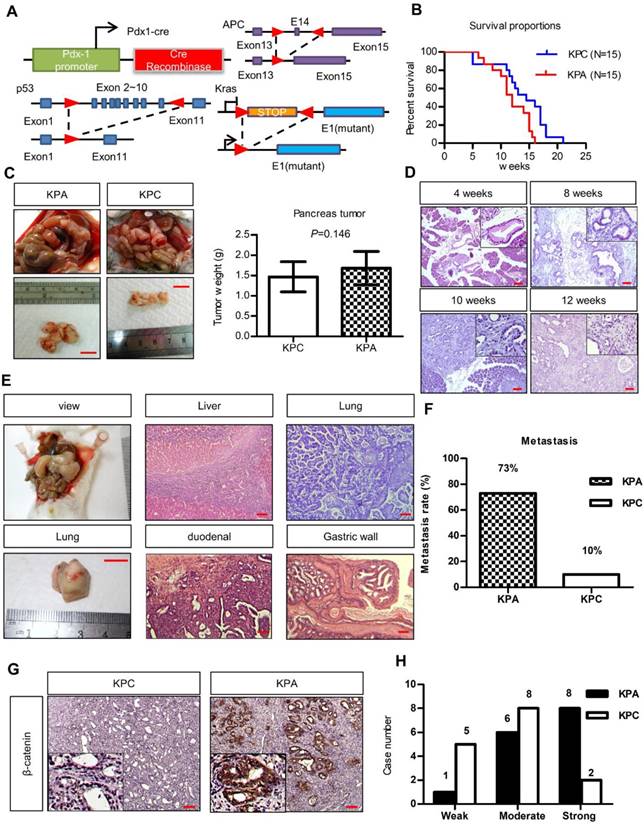
To study the interplay between ß-catenin, K-ras, and p53, we generated Pdx1-CreKrasG12Dp53L/+ (KPC) and Pdx1-CreKrasG12Dp53L/+APCL/+ (KPA) mice (Figure 1A). Survival of the KPA mice (n = 15) was shorter than that of the KPC mice (n=15, p=0.023). The average tumor size of the KPA mice was also larger, but the difference in tumor weight was not statistically significant (n=15, p=0.146; Figure 1B). Pathological analysis of the pancreas of the KPA mice demonstrated the appearance of low-grade pancreatic intraepithelial neoplasia (PanIN) at 4 weeks, and high-grade PanIN and carcinoma at about 8-10 weeks (Figure 1C). The percentage of 12-week-old KPC and KPA mice with distant metastasis were 10% and 73%, respectively (Figure 1D and Table S3). The preferential metastatic sites of the KPA mice were intestine (73%), liver (40%), lung (33%), and stomach (33%), and 50% of the mice developed ascites (Table S3). Compared to the KPC tumors, more than 90% of the KPA tumors showed moderate-to-strong β-catenin staining, indicating that β-catenin activation was caused by APC deletion (Figure 1E). In addition, the KPA tumors had reduced cytokeratin-19 and E-cadherin levels and increased α-smooth muscle actin expression (Figure S2), suggesting that these tumors were poorly differentiated and exhibited strong stromal activation.
The KPA mice develop highly aggressive and poorly differentiated tumors. (A)Pdx-1Cre mediated deletion of APC in the pancreas of Pdx1-CreLSLkrasG12Dp53L/+ (KPC) mice. Structure of the APC floxed allele: Loxp sites were inserted into the introns surrounding exons 13 and 15. (B) The survival proportions of KPA mice were compared to that of KPC mice (n=15). (C) A representative picture showed the tumor obtained from the mice at sacrifice. Scale bars represent 1 cm. The tumors (n=15) of KPA mice were larger than that of KPC mice, but the difference was not statistically significant. Error bars are Means ± SEM. (D) The development of PanIN and carcinoma in the pancreas of KPA mice at different ages. (E) Gross images of distant metastases sites; lung. Scale bars represent 1 cm. H&E histological examination revealing a KPA mouse with distant liver, lung, duodenal and gastric wall metastasis. (F) The percentage of metastasis of pancreatic cancer to various organs in KPC and KPA mice (n=15). (G) The expression of ß-catenin in KPC and KPA tumors was checked by immunohistochemical staining and a representative picture was shown. (H) The signal intensity of tumors (n=15) was summarized. Scale bars in D, E, and G represent 100 μm.
KPA cancer cells are highly invasive and tumorigenic
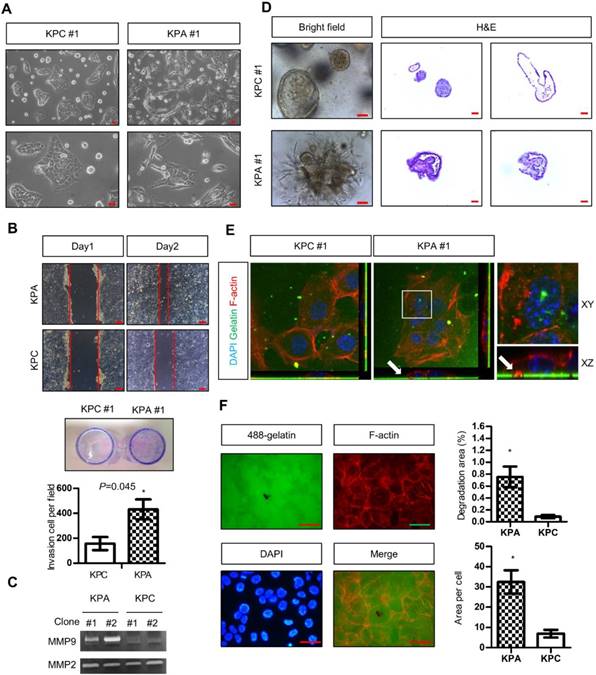
Two independent KPC (#1 and #2) and KPA (#1 and #2) cancer cell clones were isolated from different tumors. The KPC cancer cells exhibited a flat and epithelial morphology in two-dimensional (2D) cell culture, whereas the KPA cancer cells were spindle-shaped (a typical image of clone #1, Figure 2A). Colony formation of the KPA cancer cells was higher than that of the KPC cancer cells (Figure S3A). The migratory and invasive abilities were also higher in the KPA cancer cells (Figure 2B and Figure S3B). Gelatin enzymography demonstrated that KPA cancer cells exhibited a much higher matrix metalloproteinase-9 (MMP-9) activity than did KPC cancer cells (Figure 2C). The 3D organoids formed by the KPC cancer cells had a sphere structure with a smooth surface and a hollow central lumen (a typical image of clone #1, Figure 2D and Figure S4A). Conversely, KPA cancer cell organoids exhibited irregular morphology with an extensive invadopodia-like structure (a typical image of clone #1, Figure 2D and Figure S4A). E-cadherin was located at the cell-cell junction and F-actin appeared at the lumen part of the KPC organoids (Figure S4B), as previously described [19]. These two proteins were randomly distributed in different areas of the KPA organoids, indicating a disrupted structure (Figure S4B). The proliferative activity demonstrated by Ki-67 staining was higher in the KPA organoids (Figure S4B). Two approaches were used to study invadopodia formation. First, the KPA cancer cells were seeded on a fluorescent dye-conjugated gelatin, and the digested sites were studied by confocal microscopy. As shown in Figure 2E, the digested sites were co-localized with invadopodia-related puncta, confirming the invasion by actin-positive protrusions extending from the cancer cells. Second, we quantified the total digested area and digested area per cell, and observed a significant increase in matrix digestive activity in the KPA cancer cells (Figure 2F).
The KPA cancer cells are highly invasive and tumorigenic. (A) Morphology of the KPA and KPC cancer cells, observed under a light microscope. Scale bars represent 50 μm. (B)The migration and invasion of the KPC and KPA cancer cells were assessed by wound healing (left panel) and transwell (right panel) assays. The data represent mean ± SEM (n=3). *p<0.05. Scale bars represent 100 μm. (C) Morphology of the organoids established from the KPC and KPA cancer cells. Scale bars represent 75 μm. The organoids were processed for cryosection and H&E staining. Scale bars represent 50 μm. (D) Activity of MMP-2 and MMP-9 in the conditioned medium of two independent cancer cell clones isolated from different KPC and KPA tumors was assessed by gelatin zymography. (E) Confocal microscope images showing the result of the matrix digestion assay; the white arrow indicates the co-localization of F-actin-positive cell protrusion and gelatin digested area (without fluorescence). The z stack images were obtained and reconstituted by confocal microscopy. The XY and XZ sections of the boxed areas are shown. (F) The total digested area and the digested area per cell of Alexa Fluor 488-conjugated gelatin by the KPC and KPA cancer cells are shown. The experiments were repeated three times and the data represents mean ± SEM (n=3). *p<0.05. Scale bars represent 20 μm.
PDGF expression is significantly increased in KPA cancer cells
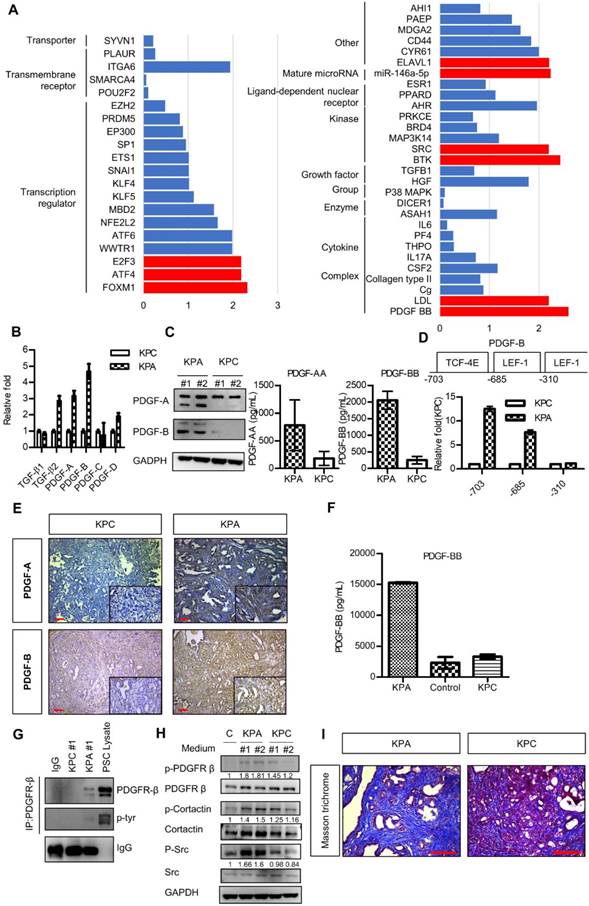
Next, we compared the gene expression profile of the KPA and KPC cancer cells by a complementary DNA microarray analysis, and found upregulation of the WNT, PDGF, vascular endothelial growth factor, and TGF-β signaling pathways in the KPA cancer cells (Figure S5). Ingenuity pathway analysis showed that PDGF-B was the most highly activated gene in the KPA cancer cells (Figure 3A). Indeed, compared to KPC cells, KPA cancer cells had a higher PDGF-A, PDGF-B, and PDGF-D mRNA expression (Figure 3B) and secreted more PDGF-A and PDGF-B than did KPC cancer cells (Figure 3C). To confirm that the PDGF-B gene is a direct transcriptional target of β-catenin, we searched the consensus TCF-4 and LEF-1 sequences in the human PDGF-B gene promoter and identified three potential binding sites within the -703/-310 region upstream of the transcription start site (Figure 3D). Our Chromatin immunoprecipitation assay demonstrated a strong binding of β-catenin to the TCF-4 and LEF-1 sites located around -703 and -685 regions of the PDGF-B gene promoter in the KPA cancer cells, while no significant binding was found in the KPC cancer cells (Figure 3D). In addition to isolated cancer cells, our IHC data also confirmed that the KPA tumors expressed much higher PDGF-A and PDGF-B than did the KPC tumors (Figure 3E). Because PDGF-B was the most highly upregulated protein of the PDGF family, we focused on this growth factor in our subsequent experiments. PDGF-B serum concentration in 7-week-old KPA mice (>15000 pg/mL) was much higher than that in the KPC mice (3314 ± 366 pg/mL) and normal mice (2313 ± 953 pg/mL) (Figure 3F). Both KPA and KPC cancer cells expressed PDGFR-α and PDGFR-β mRNA (Figure S6A); however, their protein levels were low, in accordance with findings from a previous study [20]. Therefore, we immunoprecipitated the PDGFR-β receptor, and confirmed its activation by detecting phosphorylated tyrosine (p-Tyr) levels. Serum-cultured pancreatic stellate cells (PSCs) containing endogenous p-PDGFR-β were used as a positive control. As shown in Figure 3G, PDGFR-β was constitutively activated in KPA cancer cells, whereas its activity was undetectable in KPC cancer cells. PDGF released by cancer cells might also stimulate PDGFR-β of PSCs via paracrine mechanisms. We found that the conditioned medium of KPA cancer cells stimulated PDGFR-β phosphorylation in serum-starved PSCs more significantly than did the conditioned medium of KPC cancer cells (Figure 3H). In addition, the phosphorylation of PDGFR downstream mediators Src and cortactin was increased in the PSCs. PDGF has been shown to trigger PSC activation and collagen production in vivo [21, 22]. By using Masson's trichrome staining, we demonstrated that collagen production and tissue fibrosis (blue staining) were increased in KPA tumors compared to KPC tumors, supporting the fact that PDGF derived from KPA cancer cells induces PSC activation and promotes desmoplasia formation (Figure 3I).
Src is a major downstream kinase differentially activated in KPA cancer cells
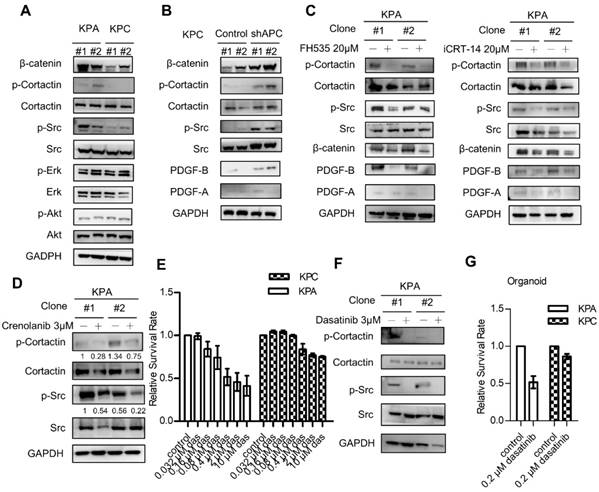
Next, we next studied PDGFR downstream signaling molecules in the KPC and KPA cancer cells. Interestingly, the activity of extracellular signal-regulated kinase (ERK) and AKT kinase was similar in both cells (Figure 4A). However, KPA cancer cells exhibited a much higher Src activity, as demonstrated by the increase of p-Src and p-cortactin. When APC was depleted by short hairpin RNA in the KPC cancer cells, activation of β-catenin, an increase of PDGF-B production, and the phosphorylation of Src and cortactin were observed (Figure 4B). Treatment with the β-catenin inhibitor FH535 or iCRT-14 reduced PDGF-B, p-Src, and p-cortactin in the KPA cancer cells in two independent cell clones (Figure 4C and Figure S6B). These data are consistent with our data (Figure 2D) and a previous finding that the PDGF-B gene is a transcriptional target of β-catenin [23]. To validate that the activation of Src was induced by PDGFR, we treated the KPA cancer cells with crenolanib, a potent and selective PDGFR-α/PDGFR-β inhibitor. Our data showed that the phosphorylation of Src and cortactin was significantly suppressed, indicating that Src activity was mainly activated via the PDGFR (Figure 4D). Depletion of PDGFR-β by siRNA also reduced the phosphorylation of Src and cortactin in the KPA cancer cells (Figure S7). Selective activation of Src in the KPA cancer cells led us to test whether these cells were more sensitive to Src inhibition. Dasatinib, a potent Src inhibitor, induced more than 50% of growth inhibition at a concentration of 10 μM in the KPA cancer cells, whereas only 20% of inhibition was found in the KPC cancer cells (Figure 4E). In addition, this concentration of dasatinib completely suppressed Src activity in the KPA cancer cells (Figure 4F), and significantly inhibited growth of the KPA organoids but not of the KPC organoids (Figure 4G).
The PDGF signaling pathway is significantly upregulated in the KPA tumors. (A) The differentially activated pathways were analyzed by Ingenuity Pathway Analysis and the genes with activation Z-score >2 were highlighted by red color. (B) Expression of the PDGF family genes in the KPC and KPA cancer cells was investigated by real-time RT-PCR. The experiments were repeated three times and the data represent mean ± SEM (n=3). (C) Intracellular protein level of PDGF-A and PDGF-B was investigated by Western blotting and secreted PDGF-A and PDGF-B was determined by ELISA assay. Error bars are Means ± SEM. (n=3), *p<0.05. (D) The potential TCF-4 and LEF-1 consensus sequences in the human PDGF gene promoter were identified by bioinformatics prediction and the locations of these sites were indicated. Chromatin immunoprecipitation assays were done to study the interaction of ß-catenin with these sites. Quantitative PCR was performed to compare the amount of ß-catenin binding to the three sites in the KPC and KPA cells. (E) Expression of PDGF-A and PDGF-B in the KPC and KPA tumor tissues were examined by IHC staining. The images are representative images of examined mouse tissues (n=5). Scale bars represent 100 μm. (F) Serum were obtained from the facial vein of normal, KPC and KPA mice and the PDGF-B concentration was determined using ELISA assay. Error bars are Means ± SEM (n=3), *p<0.05. (G) Cellular proteins (2 mg) were used for immunoprecipitation by anti-PDGFR-β and the phosphorylation status of PDGFR-β was studied by an anti-phosphor-tyrosine antibody. Cell lysate of serum-stimulated PSCs was used as a positive control. (H) Serum-depleted PSCs were cultured overnight in a regular culture medium (control, C) or conditioned medium obtained from two independent KPC or KPA cancer cell clones. Phosphorylation of PDGFR-β and its downstream mediators was investigated by Western blotting. (I) Tissue fibrosis was studied by Masson's trichrome staining and the blue color indicates the stained collagen fiber in the tissues. The images are representative images of examined mouse tissues (n=5). Scale bars represent 100 μm.
β-catenin activation induces constitutive activation of Src which could be repressed by the Src kinase inhibitor dasatinib. (A) The activity of various downstream mediators of the PDGF signaling pathway was investigated by Western blotting. Two independent cell clones (#1 and #2) were used in the study to confirm the specific activation of Src in the KPA cancer cells. (B) Two independent clones of the KPC cancer cells were transfected with control or APC shRNA for 48 h. Expression of β-catenin and activation of Src were investigated. (C) Two independent clones of the KPA cancer cells were treated overnight with β-catenin inhibitor FH535 or iCRT-14. Intracellular PDGF level and Src activity were studied by Western blotting. (D) Two independent clones of the KPA cancer cells were treated overnight with crenolanib, a potent and selective PDGFR-α/-β inhibitor. The phosphorylation of Src and cortactin was investigated by Western blotting. (E) The KPC and KPA cancer cells were treated with different concentrations of dasatinib for 24 h and cell viability was studied by MTT assay. The efficacy of dasatinib on the viability of the KPC and KPA cancer cells was compared. The experiments were repeated three times and the data represents mean ± SEM (n=3). *p<0.05. (F) Two independent KPA cancer cell clones were treated with 10 μM of dasatinib for 24 h and the phosphorylation of Src and cortactin was studied by Western blotting. (G) The KPC and KPA cancer cells treated without or with dasatinib (0.2 μM) overnight and the cells were subjected to organoid culture. The number of organoids was assessed one week after cell seeding. Error bars are Means ± SEM (n=3), *p<0.05.
Growth of KPA cancer cells in mice is suppressed by dasatinib
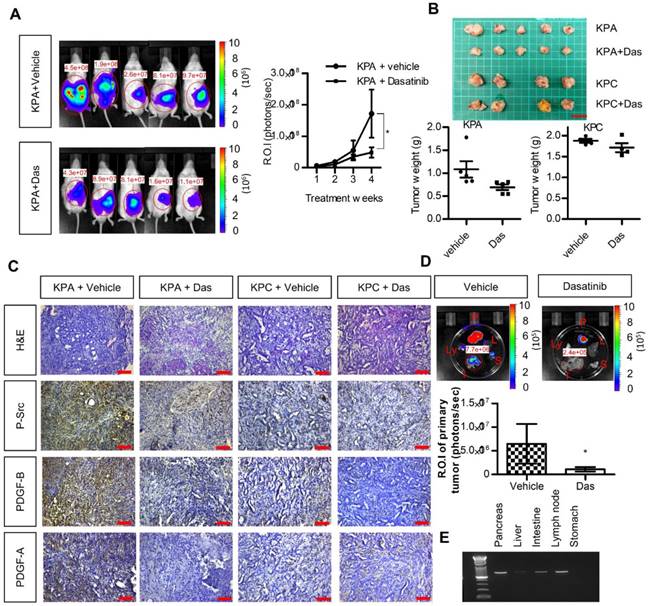
We next investigated the anti-cancer effects of dasatinib in vivo using an orthotopic animal model. Luciferase-tagged KPA or KPC cancer cells were injected into the pancreases of mice, and tumor growth was monitored by the IVIS imaging system. As shown in Figure 5A-B, dasatinib inhibited the growth of the KPA tumors but not of the KPC tumors. In addition, Src activity was much higher in the KPA tumors, and was significantly inhibited by the dasatinib treatment (Figure 5C). Interestingly, we also observed decreased PDGF-A and PDGF-B staining in dasatinib-treated KPA tumors, suggesting that PDGF-induced Src activation may in turn promote the production of PDGF to elicit a positive feedback loop. The emitted photon signal of the primary tumors was decreased by 90% in dasatinib-treated mice (Figure 5D). In addition, vehicle-treated KPA orthotopic mice showed extensive metastasis to various organs, which could be reduced by dasatinib (Figure 5D). The dissemination of KPA cancer cells to the liver, intestine, and lymph nodes was further confirmed by PCR amplification of the luciferase gene (Figure 5E). During treatment, dasatinib did not cause body weight loss or significant serum biochemistry changes (Figure S8).
Co-expression of PDGF/p-Src in human pancreatic tumors is associated with a higher mutation rate of WNT/ß-catenin signaling molecules, elevated serum PDGF, and poor survival
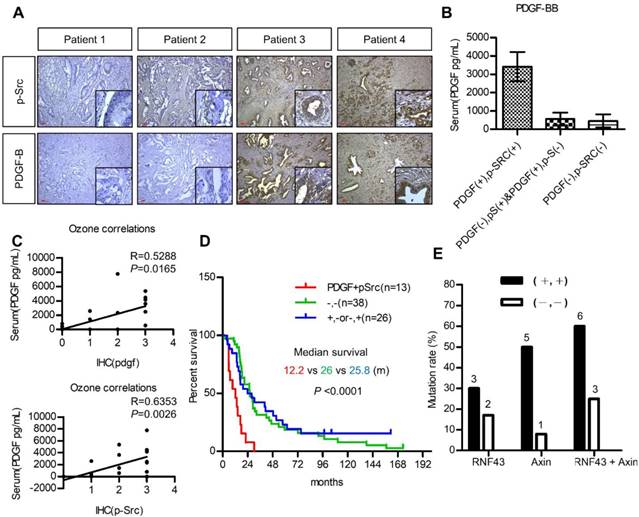
The utilization of PDGF as a biomarker for patient selection and the importance of PDGF/Src signaling in the prognosis of pancreatic cancer were further validated. We first investigated the association between serum PDGF level and the expression of PDGF and p-Src in 20 pancreatic cancer patients. Figure 6A shows pancreatic tumors with various expression levels of PDGF and p-Src. Our data demonstrated that serum PDGF concentration in patients with a strong tumor PDGF/p-Src expression was much higher than that in patients with a low PDGF/p-Src expression (Figure 6B). More importantly, serum PDGF level was positively associated with tumor PDGF (p = 0.0165) and p-Src (p = 0.0026) expression, suggesting that serum PDGF level may be a useful biomarker for the selection of patients with high tumor expression of PDGF/p-Src (Figure 6C). Next, we used another large cohort (n = 77) of pancreatic tumor tissues to study prognostic significance. We found that high PDGF/p-Src expression was associated with large tumor size, lymph node invasion, high carcinoembryonic antigen level, and increased tumor recurrence (Table S4). In addition, patients with high PDGF/p-Src expression had the worst survival (Figure 6D). Finally, we determined whether tumors with high PDGF/p-Src expression indeed had more mutations in WNT/β-catenin signaling molecules. We chose 10 and 12 tumor tissues from groups with high and low PDGF/p-Src expression, respectively, and sequenced the RNF43 and AXIN1 genes, the most highly mutated genes in the WNT/β-catenin pathway. Our data showed that RNF4 was mutated in 30% and 17% of patients in the high and low PDGF/p-Src groups, respectively (Figure 6E and Table S5), and the mutation rates of AXIN1 were 50% and 8%, respectively. Combined together, we detected mutations in RNF43 and AXIN1 in 60% of high-expressing PDGF/p-Src tumors, whereas only 25% of low-expressing PDGF/p-Src tumors contained mutations in these two genes. Together, these data show that activation of the PDGF/Src signaling axis in human pancreatic tumors correlates with a higher mutation rate of WNT/ β-catenin pathway components and is associated with poor clinical outcome.
Src kinase inhibitor dasatinib inhibits tumor growth and metastasis of the KPA cancer cells in the orthotopic model. (A)The luciferase-tagged KPA or KPC cancer cells were injected into the pancreas of mice. One week after cancer cell inoculation, dasatinib was given by an oral gavage at the dose of 10 mg/kg/day for consecutive two weeks and tumor growth was monitored by the IVIS imaging system. Error bars are Means ± SEM (n=5); *p<0.05. (B) The mice (n=5 for the KPA group and n=4 for the KPC group) were sacrificed one week after the end of dasatinib treatment and tumor weight of the control and dasatinib groups was compared. (C) Tumor tissues of the control (vehicle) and dasatinib (Das)-treated mice were subjected to immunohistochemical staining to detect the expression of PDGF-A, PDGF-B and p-Src. The images are representative images of examined mouse tissues (n=5). Scale bars represent 100 μm. (D) Organs including pancreas, liver, stomach, intestine and mesenteric lymph nodes were collected from the vehicle- or dasatinb-treated mice at sacrifice. The luciferase activities of the primary tumors at the pancreas were detected by the IVIS imaging system and the emitted photon signal of the pancreas tumors (n=5 for the KPA group and n=4 for the KPC group) was compared. Error bars are Means ± SEM. *p<0.05. (E) Metastasis of luciferase-tagged cancer cells to different organs was confirmed by PCR amplification of luciferase gene in the organs.
Activation of PDGF/Src signaling predicts poor clinical outcome in pancreatic cancer patients. (A) Representative images show various expressions of PDGF-B and p-Src in pancreatic tumor tissues. Scale bars represent 50 μm. (B) Serum PDGF level (Mean ± SD.) of 20 pancreatic cancer patients with high co-expression of tumor PDGF/p-Src (+/+) was determined by ELISA assay and was compared to the other patients without co-expression (-/+, +/- or -/-). (C) The associations between serum PDGF level with the expression of PDGF and p-Src in tumor tissues were tested. Each dot represents an individual patient. (D) Overall survival of a cohort of pancreatic cancer patients (n=77) with high tumor PDGF/p-Src (+/+, median survival=12.2 months) was compared to the patients with low tumor PDGF/p-Src (-/-, median survival=25.8 months) or with high PDGF or high p-Src alone (+/- or -/+, median survival=26 months). (E) Ten and twelve tumor tissues of the high or low PDGF/p-Src (+/+ and -/-) groups were subjected to genomic DNA isolation and sequencing. The mutation rates of RNF43 and AXIN1 genes in these two groups were shown separately. The total mutation rates were also included.
Discussion
To study the interplay of genetic alterations in the WNT/ β-catenin pathway with K-ras and p53 mutations in pancreatic tumorigenesis, we generated KPA mice and unexpectedly found a constitutive activation of Src in KPA tumors due to autocrine PDGF/PDGFR stimulation. In addition, we showed that dasatinib significantly suppressed growth of the KPA tumors. Dasatinib is a multiple kinase inhibitor with potent inhibitory effects on Bcr-Abl and Src kinases and has been approved for first-line use in patients with chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia [24]. Results from two recent clinical trials showed that the combination of dasatinib and gemcitabine did not improve the overall survival of patients with advanced pancreatic cancer compared to those receiving gemcitabine alone [25, 26]. One possible explanation for the negative results of these studies is the lack of patient selection. In these trials, Src activity in tumor biopsy samples was not checked, and no biomarker was used for patient selection. Our results highlight the potential ability of concurrent genetic changes in K-ras and p53 and WNT signaling molecules, and/or overexpression of p-Src in tumor tissues, to serve as biomarkers to identify patients most likely to benefit from Src-inhibitor treatment. Our findings of significantly elevated serum levels of PDGF in KPA mice and in patients with high PDGF/p-Src expression suggest that screening for serum PDGF levels may be a novel, alternative strategy for selecting pancreatic cancer patients for dasatinib therapy.
Our mechanistic study suggests that Src activity can only be elevated by stimulation with constitutive production of high levels of PDGF in vivo. As shown in Figure 4A, the activities of ERK and AKT were similar in both KPC and KPA cancer cells. Conversely, Src was significantly activated in the KPA cancer cells that constitutively produced high levels of PDGF. Our immunohistochemical staining also confirmed that only KPA tumors showed a strong p-Src staining (Figure 5C). Thus, the activity of various downstream mediators is fine-tuned by growth factors in time- and dose-dependent manners. Because Src controls cytoskeleton remodeling and MMP activation, we also found that KPA cancer cells exhibited strong invasive activity in 2D and organoid culture. This explains why dasatinib not only significantly decreased tumor growth but also reduced the metastasis of KPA cancer cells. In addition to cancer cells, we also detected a high Src activity in the tumor stroma (Figure 5C). It is possible that PDGF released from cancer cells also induces Src activation in PSCs to promote desmoplasia formation. Therefore, dasatinib might target cancer and stromal cells simultaneously to suppress pancreatic cancer progression.
One important translational impact of this study is the validation of the prognostic value of PDGF/Src signaling in human pancreatic cancer patients. Many pancreatic cancer patients harbor both p53 mutation (mainly a missense mutation) and p53 loss, which is caused by gene deletion, truncation mutation, and promoter methylation. Because the p53 protein forms a tetramer to activate gene transcription, mutant p53 may interfere with the activity of wild-type p53 and act as a dominant-negative regulator in cancer cells [27, 28]. It is expected that the biological outcome elicited by p53 mutation and p53 loss is different. A recent study showed that genetically engineered mice harboring oncogenic K-ras and a mutant p53 allele developed more metastatic tumors than did identical mice harboring a p53-null allele [29]. The authors found that mutant p53 upregulated PDGFR-ß expression by relieving p73-mediated inhibition of NY-F transcriptional activity in cancer cells, and that depletion of mutant p53 or PDGFR-β significantly decreased tumor metastasis. In addition, the multiple kinase inhibitor imatinib reduced the occurrence of metastasis in the study. This raises the possibility that imatinib might be useful as a targeted therapy for pancreatic cancer. Unfortunately, various phase II clinical trials have failed to show the benefit of imatinib in pancreatic cancer patients [30, 31]. We also found that the KPC cancer cells harboring the K-ras mutation and heterozygous p53 were not highly invasive. Only 25% of mice injected with KPC cancer cells developed metastases, whereas 80% of mice injected with KPA cancer cells showed extensive tumor spread (Figure S9). The results of this study demonstrated, for the first time, that the occurrence of a third genetic hit (in addition to K-ras and p53 mutations) in the WNT/β-catenin signaling pathway upregulates PDGF expression in pancreatic cancer cells, and that constitutive production of this growth factor simultaneously stimulates cancer cells and PSCs to accelerate tumorigenesis. Higher expression of PDGFR-β has been shown to be associated with poor clinical outcome in pancreatic cancer patients [32]. Thus, pancreatic patients with high PDGF/p-Src expression might need a more aggressive treatment.
In conclusion, we showed that β-catenin activation, coupled with K-ras mutation and p53 loss, promotes pancreatic cancer progression via upregulation of PDGF/Src signaling. These data suggest that serum PDGF level might serve as a biomarker for the selection of pancreatic cancer patients who might benefit from Src inhibition.
Abbreviations
APC: adenomatous polyposis coli; PDGF: platelet-derived growth factor; KPA: Pdx1-CreKrasG12Dp53L/+APCL/+; KPC: Pdx1-CreKrasG12Dp53L/+; MMP: matrix metalloproteinase; ZNRF3: zinc and ring finger 3; RNF43: ring finger protein 43.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by the following grants: 104-2320-B-400-010-MY3 to WCH and 105-2321-B-400-010, 105-2314-B-400-003 and 106-2321-B-400-007 to LTC from the Ministry of Science and Technology, Republic of China. This study was also supported by the grants from the Ministry of Health and Welfare (CA-106-PP-15) and the Yen-Hsu Education Foundation to WCH.
Contributions
KHC, LTC, WCH designed the study and analyzed the data. TLK performed the experiments and analyzed the data. YSS, LTC contributed to the collection of tumor cohort and analyzed the clinical data. KHC, LTC, WCH wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol. 2015;12:319-34
2. Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D. et al. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 2011;8:27-33
3. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399-405
4. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
5. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature communications. 2015;6:6744
6. Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D. et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379-89
7. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656-70
8. Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C. et al. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol. 1993;142:1534-43
9. Samuel N, Hudson TJ. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;9:77-87
10. Morris JPt, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683-95
11. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M. et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488:665-9
12. Jiang X, Hao HX, Growney JD, Woolfenden S, Bottiglio C, Ng N. et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110:12649-54
13. Kuo TL, Weng CC, Kuo KK, Chen CY, Wu DC, Hung WC. et al. APC haploinsufficiency coupled with p53 loss sufficiently induces mucinous cystic neoplasms and invasive pancreatic carcinoma in mice. Oncogene. 2016;35:2223-34
14. Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ. et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS genetics. 2006;2:e146
15. Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nature genetics. 2001;29:418-25
16. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R. et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development. 2001;15:3243-8
17. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
18. Huch M, Bonfanti P, Boj SF, Sato T, Loomans CJ, van de Wetering M. et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013;32:2708-21
19. Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O. et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 2014;20:769-77
20. Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA. et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382-94
21. Luttenberger T, Schmid-Kotsas A, Menke A, Siech M, Beger H, Adler G. et al. Platelet-derived growth factors stimulate proliferation and extracellular matrix synthesis of pancreatic stellate cells: implications in pathogenesis of pancreas fibrosis. Lab Invest. 2000;80:47-55
22. Kordes C, Brookmann S, Haussinger D, Klonowski-Stumpe H. Differential and synergistic effects of platelet-derived growth factor-BB and transforming growth factor-beta1 on activated pancreatic stellate cells. Pancreas. 2005;31:156-67
23. Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S. et al. Endothelial Wnt/beta-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J Exp Med. 2012;209:1611-27
24. Gnoni A, Marech I, Silvestris N, Vacca A, Lorusso V. Dasatinib: an anti-tumour agent via Src inhibition. Curr Drug Targets. 2011;12:563-78
25. Chee CE, Krishnamurthi S, Nock CJ, Meropol NJ, Gibbons J, Fu P. et al. Phase II study of dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. Oncologist. 2013;18:1091-2
26. Evans TRJ, Van Cutsem E, Moore MJ, Bazin IS, Rosemurgy A, Bodoky G. et al. Phase 2 placebo-controlled, double-blind trial of dasatinib added to gemcitabine for patients with locally-advanced pancreatic cancer. Ann Oncol. 2017;28:354-61
27. Miller CW, Chumakov A, Said J, Chen DL, Aslo A, Koeffler HP. Mutant p53 proteins have diverse intracellular abilities to oligomerize and activate transcription. Oncogene. 1993;8:1815-24
28. Blagosklonny MV. p53 from complexity to simplicity: mutant p53 stabilization, gain-of-function, and dominant-negative effect. FASEB J. 2000;14:1901-7
29. Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B. et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246-51
30. Gharibo M, Patrick-Miller L, Zheng L, Guensch L, Juvidian P, Poplin E. A phase II trial of imatinib mesylate in patients with metastatic pancreatic cancer. Pancreas. 2008;36:341-5
31. Moss RA, Moore D, Mulcahy MF, Nahum K, Saraiya B, Eddy S. et al. A Multi-institutional Phase 2 Study of Imatinib Mesylate and Gemcitabine for First-Line Treatment of Advanced Pancreatic Cancer. Gastrointest Cancer Res. 2012;5:77-83
32. Yuzawa S, Kano MR, Einama T, Nishihara H. PDGFRbeta expression in tumor stroma of pancreatic adenocarcinoma as a reliable prognostic marker. Med Oncol. 2012;29:2824-30
Author contact
![]() Corresponding author: Dr. Wen-Chun Hung, National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan. Tel: 886-6-7000123 Fax: 886-6-2083427 E-mail: hung1228org.tw
Corresponding author: Dr. Wen-Chun Hung, National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan. Tel: 886-6-7000123 Fax: 886-6-2083427 E-mail: hung1228org.tw