Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results and Discussion
Conclusion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results and Discussion
Conclusion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(1):265-278. doi:10.7150/thno.27246 This issue Cite
Research Paper
Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery
Hongyue Jin1,2, Yang He1,2, Pengfei Zhao1, Ying Hu3 ![]() , Jin Tao3, Jiang Chen4, Yongzhuo Huang1,2
, Jin Tao3, Jiang Chen4, Yongzhuo Huang1,2 ![]()
1. State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China,
2. University of Chinese Academy of Sciences, Beijing 100049, China
3. Zhejiang Pharmaceutical College, Ningbo 315100, China
4. Fudan University School of Pharmacy, Shanghai 201203, China
Received 2018-5-14; Accepted 2018-9-17; Published 2019-1-1
Citation:
Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J, Huang Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019; 9(1):265-278. doi:10.7150/thno.27246. https://www.thno.org/v09p0265.htm
Other stylesAbstract

Epithelial-mesenchymal transition (EMT) is closely associated with the development of drug resistance. Lipid metabolism plays an important role in EMT. This work was to study the cholesterol-lowering drug simvastatin for reversing EMT-associated resistance to chemotherapy via lipid metabolism.
Methods: The combination of simvastatin and paclitaxel was used to overcome the EMT-associated drug resistance. For dual-action on both cancer cells and tumor-associated macrophages (TAM), the tumor microenvironment-activatable multifunctional liposomes were developed for drug codelivery. The liposomes were modified with a hairpin-structured, activatable cell-penetrating peptide that is specifically responsive to the tumor-associated protease legumain.
Results: It was revealed simvastatin can disrupt lipid rafts (cholesterol-rich domains) and suppress integrin-β3 and focal adhesion formation, thus inhibiting FAK signaling pathway and re-sensitizing the drug-resistant cancer cells to paclitaxel. Furthermore, simvastatin was able to re-polarize tumor-associated macrophages (TAM), promoting M2-to-M1 phenotype switch via cholesterol-associated LXR/ABCA1 regulation. The repolarization increased TNF-α, but attenuated TGF-β, which, in turn, remodeled the tumor microenvironment and suppressed EMT. The liposomal formulation achieved enhanced treatment efficacy.
Conclusion: This study provides a promising simvastatin-based nanomedicine strategy targeting cholesterol metabolism to reverse EMT and repolarize TAM to treat drug-resistant cancer. The elucidation of the molecular pathways (cholesterol/lipid raft/integrin β3/FAK and cholesterol-associated LXR/ABCA1 regulation) for anti-EMT and the new application of simvastatin should be of clinical significance.
Keywords: Epithelial-mesenchymal transition, cholesterol metabolism, tumor-associated macrophages, simvastatin, drug resistance, legumain
Introduction
Epithelial-mesenchymal transition (EMT) is a biological process of phenotypic conversion, characterized by enhanced migratory capacity and invasiveness [1] and is therefore generally considered to be a major driver of metastasis [2]. EMT-induced drug resistance has been gradually realized and drawn much attention [3, 4]. Although it is a common perception that EMT-associated drug resistance depends on alteration of tumor cell behavior and function, such as slow growth rate, enhanced drug efflux, and dysfunction of apoptotic signal transduction [5], recent findings suggest that there are close links between EMT-associated drug resistance and lipid metabolism (e.g., fatty acids) [6].
Lipids (e.g., cholesteriol) play a critical role not only in maintaining membrane homeostasis but also in transferring signal transduction, and there has been increasing evidence that lipid metabolism is an important modulator for cancer progression and EMT as well [7],[8]. For example, lipid rafts (i.e., cholesterol-dependent microdomains) serve as an important platform for protein/lipid interaction and transportation by anchoring various proteins and modulating multiple signaling pathways (e.g., extracellular matrix (ECM)/integrin/FAK by focal adhesion formation) [9]. The activation of some lipid raft-associated pathways leads to drug resistance [10]. Therefore, lipid rafts have been a promising therapeutic target, and lipid metabolism-modulating agents (e.g., cholesterol-lowering drugs) can act on lipid rafts and interfere with tumor biological process [11]. Specifically, cholesterol-rich lipid rafts are required for TGF-β-directed epithelial plasticity, and cholesterol depletion inhibits TGF-β-induced EMT [12]. Yet, little is known about the therapeutic value of cholesterol metabolism on EMT and drug resistance. Moreover, cholesterol metabolism is involved in regulating macrophage polarization [13]. Tumor-associated macrophages (TAM) secrete high amounts of TGF-β into the tumor microenvironment (TME) and potently promote EMT [14]. Repolarization of TAM was shown to be able to reverse the EMT state of cancer cells [15]. Therefore, we hypothesized that targeting cholesterol metabolism could modulate EMT in cancer cells and repolarization of TAM simultaneously, thereby overcoming EMT-associated drug resistance.
Simvastatin (SV) is a cardiovascular drug that regulates cholesterol metabolism. Epidemiologic evidence reveals an association between simvastatin use and reduced cancer mortality [16]. But, the molecular association between cholesterol regulation and antitumor activity of SV still awaits elucidation.
In this study, we proposed a novel SV-based nanomedicine strategy for reversing EMT and overcoming the related drug resistance. We developed a TME-activatable penetration strategy using a hairpin-structured peptide that can be cleaved by the TME-associated protease legumain to modify a multifunctional liposome for codelivery of SV and paclitaxel (PTX). The hairpin peptide remained inert in the blood stream and normal tissues, but once it was cleaved by legumain in the TME, the exposed penetrating sequence mediated efficient intratumoral and intracellular penetration and achieved enhanced and specific drug delivery.
Results and Discussion
Reversal of EMT-associated drug resistance in A549T by SV
A549T is a drug-resistant variant of the parental A549 non-small-cell lung cancer (NSCLC) cell line. A549T cells were highly resistant to PTX (Figure S1A). It was found that the A549T cells exhibited EMT phenotype, characterized by low E-cadherin and high vimentin (Figure 1A-B). This suggested that EMT is an important mechanism for drug resistance in A549T cells.
To further demonstrate the association between EMT and drug resistance, an induced EMT cell model was established in the sensitive A549 cells by treatment with TGF-β (Figure 1A), and the acquired resistance to PTX was consequently developed in the TGF-β-induced mesenchymal-type A549 cells (Figure 1C). However, the drug resistance in both the A549T and TGF-β-induced A549 cells was reversed by SV at a dose of 2 µg/mL and they were re-sensitized to PTX (Figure 1D and Table S1). The attenuated drug resistance was associated with reversal of EMT. With SV treatment, the innate EMT in A549T cells was reversed, displaying an epithelial phenotype (Figure 1E and Figure S1B). A similar phenomenon of EMT reversal was observed in the TGF-β-induced A549 cells, which was in accordance with a previous report [17].
Effect of SV on lipid raft/integrin/FAK pathway
SV interrupts lipid raft formation by blocking cholesterol synthesis [18]. Our results showed that the lipid rafts on the A549T cells were reduced after SV treatment (Figure 2A and Figure S1C). Lipid rafts are an important platform for integrin recycling and recruitment [19, 20], and affect focal adhesion formation via ECM/integrin/FAK interaction [9]. Our results showed that the SV-induced lipid raft destruction was accompanied by decreased integrin β3 on the A549T cell membrane (Figure 2B and Figure S1D). Integrin β3 is a structural basis of focal adhesion, and closely associates with lipid raft-mediated focal adhesion formation [21]. As a result of integrin β3 downregulation and lipid raft disruption after SV treatment, focal adhesion formation in A549T cells was inhibited (Figure 2C). Therefore, integrin β3 could play a key role in drug resistance.
Accordingly, integrin β3 expression in A549T cells was much higher than that of native A549 cells (Figure S2), while other integrin variants remained at a low level in A549T cells. Integrin is a connection between focal adhesion and ECM. Our results showed that A549T cells adhered to fibronectin (FN, an integrin β3-binding ECM) much faster than the parent A549 cells, but there was no difference in cationic poly-L-lysine (PLL, an integrin β3-independent adhesion matrix) (Figure 2D). SV suppressed the interaction of integrin β3 and FN in the A549T cells (Figure 2E), and thus re-sensitized the cells to PTX (Figure 2F). By contrast, SV was not able to reverse PTX resistance in A549T cells cultured in PLL (Figure 2D). Moreover, SV had little effect on the native A549 cells cultured either in FN or PLL (Figure 2H-I) due to the low level of integrin β3 expression. This result suggested the importance of integrin β3 in drug resistance.
FAK is another major component in focal adhesions and mediates interaction with ECM. Activated FAK signaling is an integrin β3-dependent effect [22]. Therefore, it was logical to hypothesize that FAK could be a downstream pathway of integrin β3-mediated drug resistance in A549T. We took a further look at the phosphorylated FAK (pFAK) level in A549T cells. It was found that pFAK signaling and its downstream pERK/pAKT were upregulated (Figure S1E), along with overexpression of integrin β3 (Figure S2). In addition, because FAK activation is required for TGF-β-induced EMT [23], increased pFAK was also found in TGF-β-induced EMT A549 cells but not in native A549 cells without TGF-β treatment (Figure S1E). This result demonstrated a close link between pFAK and EMT.
SV treatment was able to suppress pFAK in both A549T and TGF-β-induced A549 cells; accordingly, phosphorylated ERK (pERK) was also downregulated (Figure 2J). Compared to native A549 cells, A549T and TGF-β-induced A549 with high p-FAK level were more sensitive to SV, and SV treatment significantly arrested their proliferation (Figure 2K). This result suggested that FAK signaling is an important mechanism for SV reversal of EMT-associated drug resistance. To further demonstrate the association between FAK and EMT-associated drug resistance, a FAK inhibitor defactinib was applied to the A549T cells. It was found that the A549T cells were consequently re-sensitized to PTX, but defactinib displayed a very minor effect on the native A549 cells with low pFAK level (Figure 2L). These results revealed the ability of SV-based therapy to overcome EMT-associated drug resistance, and the possible mechanism was related to the suppression of the lipid raft/integrin β3/FAK pathway.
Figure 1
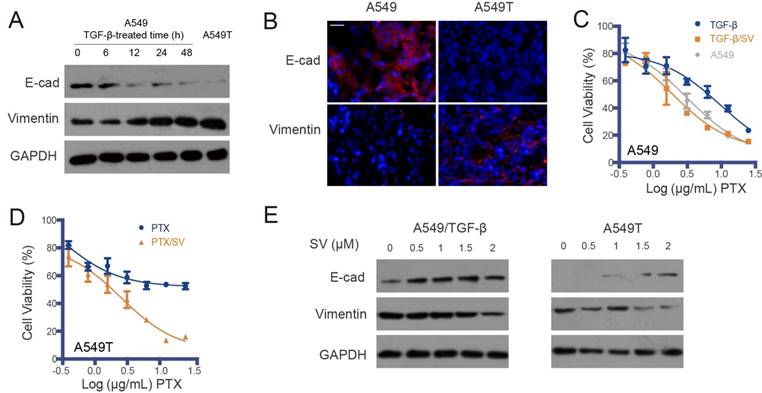
EMT-associated drug resistance and re-sensitization of A549T cells by SV. (A) Representative Western blot of EMT markers (E-cad and vimentin) in A549 cells induced by TGF-β with varying exposure duration, with the mesenchymal-type A549T cells as a control (the right lane). EMT was induced in the A549 cells exposed to TGF-β, as characterized by low E-cadherin and high vimentin. (B) Representative photomicrographs of immunofluorescence of E-cadherin and vimentin, indicated by red in the A549 and A549T xenografts (DAPI staining is blue). Scale bar, 20 μm. (C) A549 cells exposed to TGF-β showed resistance to PTX, but the TGF-β-induced drug resistance was reversed by SV. (D) SV sensitized A549T cells to PTX. (E) SV reversed EMT in both the TGF-β-treated A549 cells and A549T cells.
Figure 2
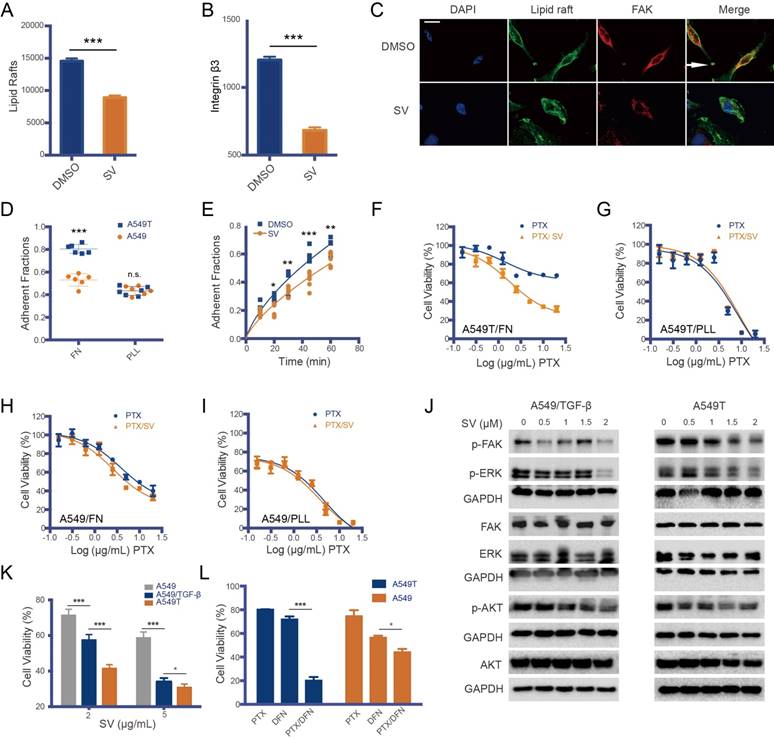
Drug resistance and the lipid raft/integrin β/FAK pathway. Lipid raft formation (A) and integrin β3 level (B) in A549T cells were reduced by SV, as measured by flow cytometry. (C) Representative photomicrograph of focal adhesion formation (white arrow) in A549T cells; the formation was inhibited by SV treatment. Scale bar, 10 μm. (D) A549T cells adhered to fibronectin (FN, an integrin β3-binding ECM) much faster than the parent A549 cells. But, there was no difference in cationic poly-L-lysine (PLL, an integrin β3-independent adhesion matrix) between A549T and A549 cells. (E) A549T cell adhesion to FN was suppressed by SV. (F) SV can re-sensitize A549T cells cultured in FN matrix to PTX due to SV-induced inhibition of integrin β3/FN interaction. (G) SV cannot re-sensitize A549T cells cultured in PLL matrix. SV showed little effect on the viability of low integrin β3-expressing A549 cells in either (H) FN or (I) PLL matrix. (J) SV inhibited FAK and its downstream signaling (ERK/AKT) in both TGF-β-induced A549 and A549T cells in a dose-dependent manner, as represented by the downregulation of the phosphorylated proteins. (K) SV inhibited the growth of the mesenchymal-type TGF-β-induced A549 and A549T cells but yielded a minor effect on A549 cells. (L) FAK inhibitor defactinib re-sensitized A549T cells to PTX, and the enhanced effect was much greater than that on the A549 cells (low pFAK level), suggesting FAK signaling mediated EMT-associated drug resistance.
TAM repolarization by SV via LXR/ABCA1 mechanism
TAM is a major component in the TME and is highly plastic, polarizing to two major phenotypes: antitumor M1 (TAM1) and protumor M2 (TAM2). TAM constitutes up to 50% of the tumor mass and the majority is M2-like [24]. Repolarization of TAM2 to TAM1 has been demonstrated to be a promising method for cancer therapy [24]. The murine bone marrow-derived macrophage (BMDM)-induced M2 to M1 phenotype (termed M2Φ and M1Φ, respectively) was used in the cellular study (Figure S3). We found that SV was able to repolarize M2 to M1 phenotype, as evidenced by the downregulation of an M2 marker CD206 and a protumor cytokine TGF-β (Figure 3A-E). The repolarization was further confirmed by the qPCR results, displaying downregulation of M2-associated markers (e.g., IL-10, CCL-22, CCL-17, and Arg-1) and upregulation of M1-associated cytokines (e.g., IL-1β, TNF-α, IFN-γ, and IL-1α) (Figure 3F).
Figure 3
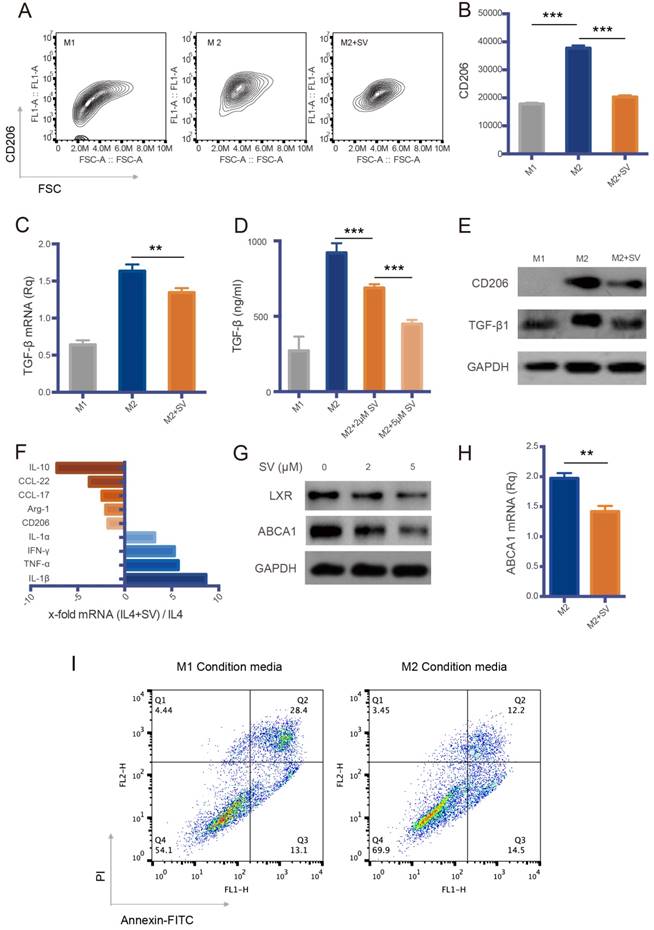
Macrophage repolarization via cholesterol metabolism modulation by SV. (A) Contour plots of flow cytometry data. SV repolarized M2Φ to M1Φ, indicated by the reduced CD206 expression. (B) Quantitative analysis of CD206 expression. (C) SV downregulated TGF-β mRNA level in M2Φ, as measured by RT-PCR. (D) SV suppressed TGF-β expression in M2Φ, as measured by ELISA. (E) SV induced repolarization from M2 to M1, indicated by the reduced expression of CD206 and TGF-β. (F) Increased levels of M1 markers (blue-colored bars) and decreased levels of M2 markers (orange-colored bars) in SV-treated M2Φ measured by RT-PCR. (G) SV suppressed ABCA1 and LXR expression in M2Φ. (H) ABCA1 level was reduced by SV treatment in M2Φ, as measured by RT-PCR. (I) M1Φ enhanced the efficacy of PTX in A549 cells, whereas M2 induced drug resistance, as reflected by the reduced apoptosis rate.
We further investigated the M2→M1 repolarization mechanism of SV. Liver X receptors (LXR)/ATP-binding cassette transporter A1 (ABCA1) is an important mechanism for regulating cholesterol homeostasis in macrophages [25]. Essentially, active LXR is required for M2 polarization [26]. The levels of LXR and its downstream gene ABCA1 were decreased by SV treatment (Figure 3G-H). This could account for the cholesterol-depletion effect of SV that led to a feedback downregulation of LXR, because cholesterol is a natural agonist of LXR, and excessive cholesterol activates LXR expression [27], but statins treatment can reduce LXR/ABCA1 expression [28].
TAM repolarization could synergistically work with SV-modulated EMT because the M2→M1 switch resulted in inhibition of TGF-β secretion and thereby reversed TGF-β-induced EMT. Accordingly, it was shown that M2Φ induced drug resistance in A549 cells, which, by contrast, were sensitized to PTX by M1Φ (Figure 3I).
Characterization of legumain-activatable liposomes (aLip)
Asparaginyl endopeptidase legumain has been demonstrated to be overexpressed on TAM, and it thus has been explored as a TAM marker and a potential drug target for cancer therapy [29, 30]. Moreover, legumain can be secreted into the TME with functions for remodeling the ECM, thus serving as an ideal target for stimuli-responsive drug delivery specific to tumors and their microenvironment [31].
We previously developed a legumain-activatable, hairpin-structured cell-penetrating peptide KC26 (Ke5Ne4GPTN2R9C) based on molecular-dynamics-simulation-driven design [32]. The liposomes were modified with KC26-PEG-DSPE and co-loaded with PTX and SV (termed aLip). The cell penetration ability was activated by legumain, which cleaved the substrate sequence (PTN) leading to detachment of the anionic segment (Ke5Ne4) and exposure of the cell-penetrating sequence (R9). Liposomes modified with PEG-DSPE were used as a control (termed PEG/Lip).
Figure 4
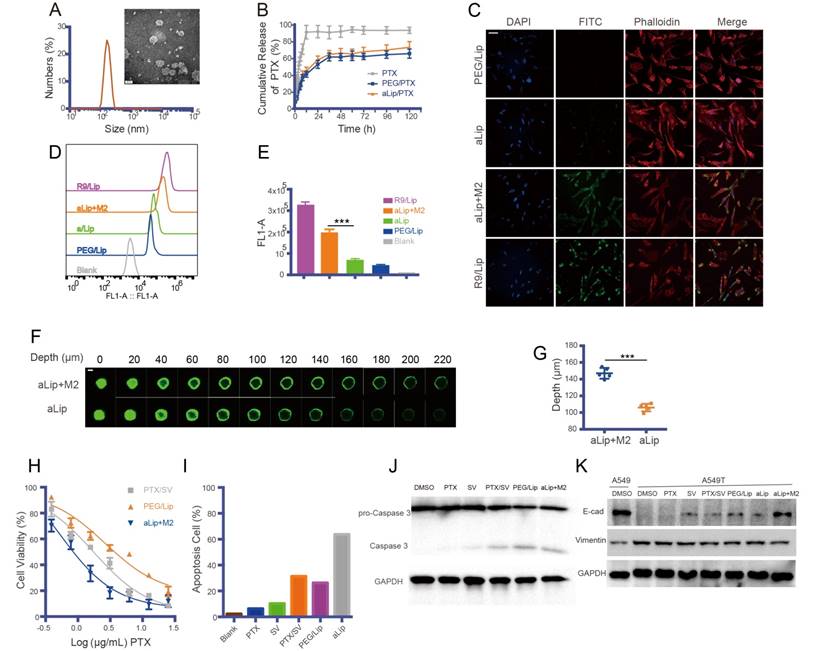
Characterization of legumain-activatable liposomes (aLip). (A) Size and TEM of aLip. (B) In vitro release of PTX. (C) Representative confocal imaging (scale bar, 20 μm) and (D) flow cytometry analysis of aLip uptake in A549T cells with or without pretreatment with M2Φ. The cell-penetrating R9/Lip was used as a positive control, while PEG/Lip was the negative control. (E) Quantitative analysis of the uptake efficiency in A549T cells. (F) Penetration of the pre-cleaved (top panel) or uncleaved (bottom panel) aLip in the cultured tumor spheroids. The aLip pretreated with legumain-containing M2 lysis would have activated cell-penetrating peptide-assisted intratumor penetration. Z-axis continuous top-down scanning layers by confocal microscopy of the spheroids. Scale bar, 200 μm. (G) Analysis of intratumor penetration depth in tumor spheroids. (H) MTT assay of free PTX/SV, PEG/Lip, and pre-cleaved aLip by M2Φ in A549T cells. (I) Cell apoptosis assay of free drug and aLip in A549T cells. (J) Western blot analysis of caspase 3 in A549T cells. aLip enhanced the level of activated caspase 3. (K) Western blot analysis of EMT reversal after drug treatment.
The aLip were spherical with a particle size of about 140 nm (Figure 4A), with a neutral zeta potential due to the PEG chain mask. The loading efficiencies of PTX and SV in aLip were 1.6% and 3.4%, and the encapsulation efficiencies were 73.2% and 81.8%, respectively. The liposomes showed a sustained PTX drug release profile (Figure 4B), with a similar profile of SV release (Figure S4A). It was confirmed that legumain was overexpressed in M2Φ (Figure S4B), with high enzymatic activity detected by the Z-AAN-AMC substrate probe (Figure S4C). The legumain-activatable function of the liposomes was demonstrated by fluorescence assay (Figure S4D-E).
The cellular uptake of aLip was investigated. PEG/Lip and liposomes modified with the cell-penetrating peptide polyR (R9) (termed R9/Lip) were used as negative and positive controls, respectively. The cell penetration ability of aLip was activated by pre-incubation with the legumain-containing M2Φ lysate (Figure 4C-E). The cellular uptake of the activated aLip was significantly higher than that of the non-pretreated aLip. The non-pretreated aLip, due to the non-exposure of R9, showed poor cellular uptake, similar to that of PEG/Lip. In addition, aLip activated by TAM2 displayed much deeper penetration in the cultured A549T spheroid than the non-activated aLip (Figure 4F-G). Moreover, the uptake of aLip in M2Φ was much higher than that in M1Φ (Figure S5), which rendered aLip to preferentially act on the legumain-overexpressed TAM2. It should be mentioned that M2Φ displayed considerable tolerance to both the blank aLip and the drug-loaded aLip (Figure S6A-B).
The TAM2-activated, PTX/SV-encapsulated aLip showed dose-dependent antitumor activity in A549T cells (Figure 4H). aLip displayed a low IC50 of 0.51 μg/mL while that of PEG/Lip was more than five-fold higher. A flow cytometric assay further confirmed the efficacy of aLip, of which the apoptotic rate was 63.7%, compared with 26.3% for PEG/Lip and 31.4% for free drugs SV/PTX (Figure 4I and Figure S6C). Accordingly, the cleaved caspase-3 level was upregulated in the aLip group (Figure 4J).
In addition, the mechanisms of SV-mediated reversal of EMT-associated drug resistance were further demonstrated in another cancer cell line (i.e., PC9). The result confirmed that SV reversed TGF-β-induced EMT in PC9 cells, and suppressed the activation of FAK and its downstream pathways (ERK/AKT), thus effectively re-sensitizing the cells to PTX (Figure S7A-B). Furthermore, we also found that the TGF-β-induced mesenchymal-type PC9 and A549 cells were resistant to gefitinib, and the resistance could also be reversed by SV (Fig S7C-D).
Tumor-targeted delivery and in vivo imaging studies
The tumor targeting effect was investigated via i.v. injection of aLip labeled with DIR dye in a A549T xenograft mouse model. Due to overexpression of legumain in the tumor, aLip was efficiently activated and achieved effective tumor penetration and cell uptake, thereby exhibiting higher tumor accumulation than the control PEG/Lip (Figure 5A). At the experimental endpoint, the major organs were dissected for ex vivo imaging. The highest signal in the tumor was seen in the aLip group (Figure 5B-C), further demonstrating the activatable cell penetration ability of aLip. Free DIR significantly accumulated in the lung due to its hydrophobicity, but the liposomal formulation reduced this unwanted lung exposure.
Legumain expression was very low in both A549T and A549 cells, but high levels of legumain were found in their xenografts due to the presence of TAM2 in the TME (Figure 5D). The dissected A549T tumors were processed by cryosectioning and immunofluorescence staining. The result showed that aLip colocalized with TAM2 (Figure 5E). Furthermore, efficient uptake of aLip by TAM2 in the tumor tissues was observed by flow cytometry (Figure 5F), which demonstrated activation of aLip by TAM2-associated legumain.
The biodistribution of aLip was further investigated by measuring the PTX concentration in the organs. The tumor drug accumulation was significantly increased in the aLip group (Figure S8), in accordance with the in vivo imaging results.
In vivo therapy
The antitumor efficacy of aLip was investigated in a subcutaneous A549T tumor mouse model. Because of the drug resistance of A549T cells, free PTX hardly arrested tumor growth. However, the combination of PTX/SV displayed considerable efficacy, suggesting that SV was able to reverse drug resistance. The efficacy of the combination therapy was enhanced by the liposomal formulations (Figure 6A-C), mainly due to the improved tumor accumulation. aLip exhibited significantly higher efficacy than PEG/Lip. This result was in line with the imaging observation of higher tumor accumulation of aLip due to its activatable cell penetration ability. Tumor growth in the aLip group was fully inhibited.
Figure 5
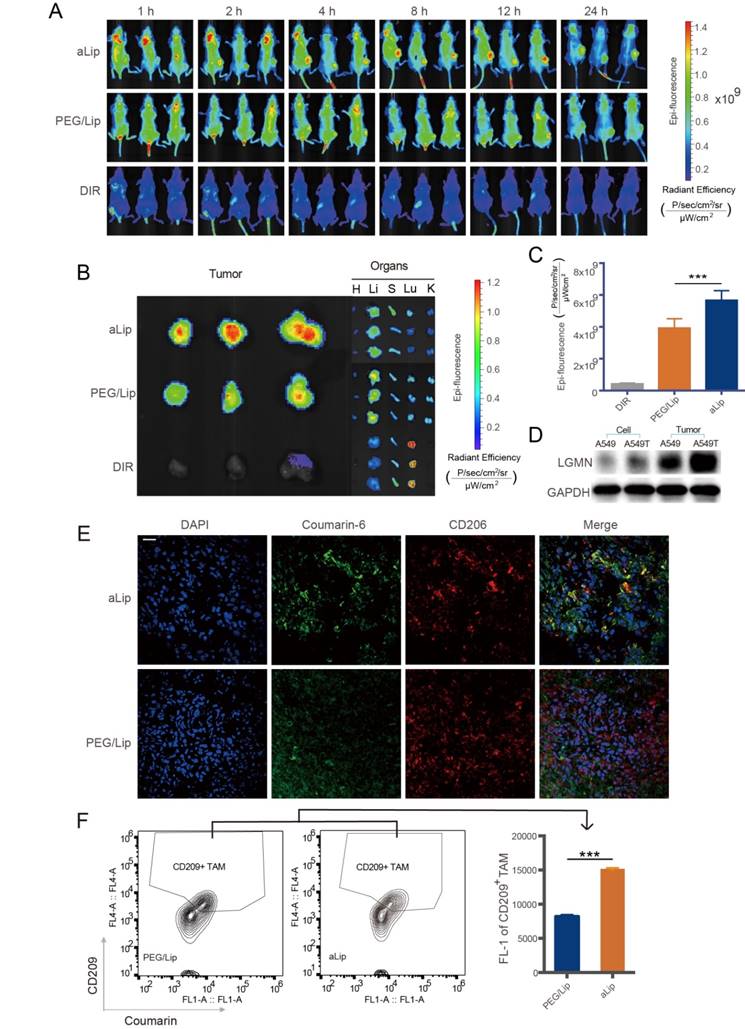
In vivo imaging and distribution of aLip. (A) Distribution of aLip, PEG/Lip, and free DIR in A549T tumor-bearing mice (n = 3). (B) Representative photomicrographs of ex vivo imaging of the tumors and major organs. aLip showed enhanced accumulation in the tumors. (C) Ex vivo radiant efficiency in the tumors. (D) Legumain expression in A549 and A549T cells and their xenografts detected by Western blotting. (E) Representative photomicrographs of the co-localization of aLip and CD206 by immunofluorescence staining. Scale bar, 20 μm. (F) The uptake efficiency of aLip measured in TAM2 (labeling with a M2 marker CD209) sorted from the tumor tissues.
Figure 6
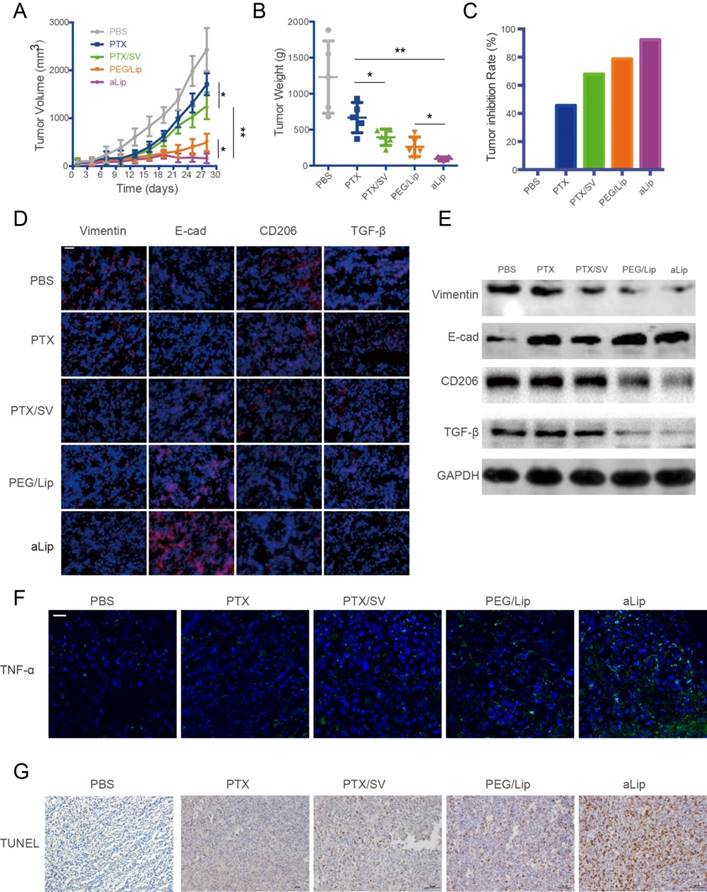
Treatment efficacy of aLip in subcutaneous A549T-xenograft model. (A) Tumor growth curve. (B) Tumor weight. (C) Tumor growth inhibition rate. (D) Representative immunofluorescence of markers of EMT (vimentin and E-cad) and TMA2 (CD206 and TGF-β), indicated by red, in the tumor tissues after treatment (DAPI staining is blue). Scale bar, 20 μm. (E) Western blots of vimentin, E-cadherin, CD206, and TGF-β expression in the A549T xenografts after treatment. (F) Representative immunofluorescence of a M1-associated marker (TNF-α) in the tumors. (G) TUNEL staining of the A549T xenografts. Scale bar, 20 μm.
EMT was reversed after treatment with aLip, as evidenced by upregulation of E-cadherin and downregulation of vimentin in the tumor (Figure 6D-E). The in vivo results further confirmed that EMT reversal is an important mechanism for overcoming drug resistance. The TAM2 markers, CD206 and TGF-β, were greatly downregulated in the aLip group compared to other groups (Figure 6D-E). Repolarization of TAM2 could be a mechanism for suppression of EMT because TGF-β is an essential driving factor of EMT. Moreover, the M2→M1 repolarization also resulted in increased level of the M1-associated cytokine TNF-α (Figure 6F). TUNEL staining revealed that there were a large number of apoptotic cells in the aLip group (Figure 6G).
The safety of the treatment was also evaluated. Both body weight changes and histological examination demonstrated the good biocompatibility of aLip, and no obvious toxicities were found in the major organs by histological examination (Figure S9).
Conclusion
In summary, we proposed a dual-targeting nano codelivery system for modulating lipid metabolism as a novel strategy to reverse drug resistance in cancer therapy. SV has been reported to be able to inhibit tumorigenesis and cancer EMT progress,[33] but little is known about its mechanisms. We revealed that modulation of cholesterol metabolism by SV is a major mechanism for reversing EMT-associated drug resistance via two pathways. One was to act on the cancer cells to downregulate lipid rafts and the focal adhesion formation, thus leading to suppression of integrin/FAK/ERK signaling and EMT. Another was to act on TAMs and repolarize the protumor M2 to the antitumor M1 phenotype via inhibition of LXR/ABCA1 that is responsible for cholesterol homeostasis and M2 polarization. The benefit of this dual-action system was that it not only reversed EMT of the cancer cells but also repolarized TAM2. As a result, TAM2 reprogramming led to a reduction of TGF-β secretion, which thus suppressed TGF-β-driven EMT and yielded a combination effect.
This study revealed that cholesterol metabolism is a promising drug target and SV-based nanomedicine is a potential therapeutic strategy for overcoming EMT-induced drug resistance. The activatable cell-penetrating liposomal codelivery system was able to enhance treatment efficacy of drug-resistant lung cancer, offering a promising druggable formulation for targeting EMT and related drug resistance in cancer therapy. The elucidation of the molecular mechanisms of an old drug (SV) for a new application of overcoming EMT-associated MDR will be helpful for promoting clinical translation. It would also benefit cancer patients with MDR because of the ready availability of the low-cost SV with well-established PK, safety, and tolerance.
Methods
Materials
PTX, defactinib, and SV were purchased from Melone Pharmaceutical Company (Dalian, China). SPC, cholesterol, DSPE-PEG2000 and DSPE-PEG2000-mal were obtained from A.V.T. Pharmaceutical company (Shanghai, China). E-CAD, vimentin, ERK (p42/44), P-ERK (Thr202/Tyr204), AKT, P-AKT (Ser473), and caspase-3 antibodies were obtained from CST (Boston, USA). P-FAK (Tyr397), FAK, CD206, LXR, and TGF-β antibodies were purchased from Abcam (Cambridge, UK). CD86, F4/80, CD61(Integrin β3-APC) antibodies, and CTB-488 were purchased from Thermo Fisher Scientific (Waltham, USA). ABCA1 antibodies were purchased from R&D Systems (Minneapolis, USA). TGF-β1 ELISA kit was obtained from DAKEWEI Biotech (Shenzhen, China). Human TGF-β, IL-4, IFN-γ, and M-CSF cytokines were purchased from PeproTech (Quebec, Canada). KC26 and R9 peptides were synthesized by Bankpeptide Biotechnology (Hefei, China). Mouse legumain was purchased from SinoBiological (Beijing, China). The culture medium was purchased from Thermo Fisher.
Cells
A549 and A549T NSCLC cells and PC9 cells were incubated in Roswell Park Memorial Institute medium (RPMI 1640) containing 10% FBS and antibiotics (100 μg/mL of streptomycin and 100 U/mL of penicillin, Amresco, USA) at 37 °C and 5% CO2. Induced EMT models of A549 and PC9 cells were developed by TGF-β treatment (5 ng/mL, 48 h) [34].
Bone marrow-derived macrophages (BMDM) were collected from BALB/c mice (male, 4-5 weeks, SPF) using a standard procedure. Briefly, the bone marrow was flushed with DMEM medium, and the cells were incubation in DMEM with 10% FBS and 20 ng/mL M-CSF for 5 days. The adherent macrophages were exposed to LPS (100 ng/mL)/IFN-γ (20 ng/mL) or IL4 (40 ng/mL) for 24 h to induce M1 or M2 phenotype, respectively. M1 Φ and M2Φ were characterized by immunofluorescence staining with the antibodies of F4/80-488 (macrophage marker), CD86-PE (M1 marker), or CD206-FITC (M2 marker), and measured by FACS. SV (5 µM) was added with IL-4 (40 ng/mL) to the BMDM to investigate its reversal of M2 polarization. After incubation for 48 h, the cells were digested and collected for RT-PCR test. The RT-PCR primers are listed in Table S2.
DMSO was used as a solvent for the free drugs in cell culture study at a safe concentration of 0.2% that was biocompatible to the cells (Figure S10).
Animals
BALB/c nude mice (male, 4-5 weeks) were supplied by Shanghai Laboratory Animal Center (SLAC) Co., Ltd. (Shanghai, China), and housed at the SPF care facility. All the animal experimental procedures were approved by Institutional Animal Care and Use Committee (IACUC), Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences. (Note: SIMM is an institution with AAALAC International accreditation).
Cellular mechanism studies
TGF-β-induced EMT cells (A549 and PC9 cells) were established by treatment with TGF-β (5 ng/mL) for 24 h. EMT reversal effect in A549T cells was investigated by exposure to SV (dissolved in 0.2% DMSO), while the A549 and PC9 cells were co-treated with SV (2 µM) and TGF-β (5 ng/mL) for 24 h. The total proteins were extracted using a lysate buffer containing protease inhibitor cocktail and quantified by BCA assay. The procedures for Western blotting assay are described as follows. Samples with the same protein concentration were loaded onto 12% SDS-PAGE gels and run for 30 min at 90 V and then 90 min at 120 V. Semi-dry transfer to a membrane was conducted at 9 V for 120 min. The membranes were blocked with TBST buffer containing 5% BSA. Primary antibody incubation was performed at 4 °C overnight at a dilution of 1:1000 with TBST/5% BSA buffer for the antibodies to the targeted proteins and 1:20000 for the housekeeping protein. After washing 3 times with TBST (each for 10 min). The membranes were incubated with anti-rabbit IgG, HRP-linked antibody (1:1000 dilution, Beyotime, China) for 45 min at room temperature. After thorough washing and a standard staining process, the membranes were exposed to X-ray film or a digital imager. Quantification of protein bands was conducted using densitometry with ImageJ and the results are shown in Table S3-5.
Cell viability of the A549T, TGF-β-induced A549, and PC9 cells was tested by a standard MTT assay after treatment with SV and PTX for 48 h.
Measurement of lipid raft and focal adhesion formation
A549T cells were seeded in 6-well plates and SV was added 24 h later. After incubation for another 24 h, the lipid rafts of the cells were labeled with CTB-488 following a previously reported method.[35] The cells were then measured by FACS. For FAK and lipid raft co-localization assay, the cells were pretreated with SV for 24 h, and then collected and seeded at 5×104 per well with FN-coated coverslips. After 2 h incubation, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, blocked with 5% BSA, and then incubated with the FAK primary antibody for 2 h. After labeling by the secondary antibody and DAPI, the cells on the coverslip were subjected to confocal microscopy.
Measurement of cell surface integrin
After the designated treatments, the cells were fixed and then incubated with anti-integrin β3-APC (Thermo Fisher Scientific, USA) for a standard flow cytometry assay.
Cell adherent assay
A549 and A549T cells were labeled with Calcein AM (Thermo Fisher Scientific, USA) and measured according to the manufacturer's protocol. The labeled cells were suspended in RIMI 1640 medium with 10% FBS and added to FN (5 μg/cm2) or PLL (4 μg/cm2) pre-coated 96-well plates. The 1640 medium was removed after 30 min incubation, and the adherent cells were washed thoroughly with PBS. The fluorescence of the adherent cells was measured by a microplate reader (PE, USA).
For the adherence assay of SV-treated cells, A549T cells were seeded in 6-well plates and incubated for 24 h and then exposed to medium containing 2 μM SV or DMSO (0.2%, control) for another 24 h. The cells were harvested and labeled with Calcein AM. The labeled A549T cells were seeded onto FN or PLL pre-coated 96-well plates for 30 min. After washing, the fluorescence of the adherent cells was measured by a microplate reader (PE, USA). The viability of A549T cells in FN or PLL matrix was also conducted using a standard MTT test procedure.
Synthesis of DSPE-PEG2000-KC26
KC26 peptide was dissolved in methanol, and DSPE-PEG2000-MAL was dissolved in chloroform. The KC26 solution was added to the DSPE-PEG2000-MAL solution dropwise at a molar ratio of 1.3:1 and stirred at room temperature for 6 h. The organic solvent was then removed by rotary evaporation. The residue was dissolved in chloroform and filtered. The solution was evaporated and the final product stored under -20 °C.
Preparation and characterization of liposomes
The liposomes were prepared by a thin film hydration method. Briefly, SPC, cholesterol, DSPE-PEG2000-KC26, and DSPE-PEG2000 at a molar ratio of 59:33:6:2 were dissolved in methanol/chloroform (1:1), and this lipid solution was then mixed with a PTX/SV methanol solution. The organic solvent was removed by rotary evaporation in a 42 °C water bath. The lipid film was hydrated with 5% glucose solution followed by sonication. Free PTX and SV were removed by a Sephadex G-50 column. The particle size was measured with a dynamic light scattering analyzer (Nano-ZS90, Malvern, UK). The nanostructure was observed using TEM (JEOL JEM-100C×2, Japan) with an acceleration voltage of 180 kV. The quantitation of PTX and SV was determined by HPLC. The drug loading efficiency (DL%) and encapsulation efficiency (EE%) in the liposomes were calculated by the equations:
DL% = WDE / WL × 100%
EE% = WDE / WT × 100%
where WDE is the drug amount encapsulated in the liposomes, WL is the total weight of the liposomes, and WT is the total amount of drug.
In vitro drug release
Release of PTX or SV from the liposomes was studied using a dialysis method. Briefly, liposomes in a dialysis bag (MWCO 10-12 kD) were incubated in 20 mL of release medium (pH 7.2, PBS containing 0.5% w/v SDS). The experiment was conducted at 37 °C with continuous stirring. A small portion of release medium (0.5 mL) was withdrawn at each experimental time point, and an equivalent volume of fresh medium was replenished. The PTX or SV concentration was measured by HPLC.
Activity assay of legumain from macrophages
M1Φ and M2Φ were induced from the primary murine bone marrow-derived macrophages as described above. The macrophage lysates were prepared in citric acid buffer (pH 5.8). The legumain probe AMC-AAN was used to evaluate enzymatic activity. AMC-AAN solution was incubated with various macrophage lysates or commercial legumain for 30 min at 37 °C, followed by fluorescence measurement at an excitation wavelength of 352 nm.
Legumain cleavage assay
To investigate activation of aLip in the presence of legumain, the liposomes were labeled with DSPE-PEG2000-FITC and KC26-Cy3 (CR9N2TPGE4NE5K-Cy3). The M2Φ lysate was prepared by suspending the cells in the lysis buffer (citric acid buffer, pH 5.8) and incubating on ice for 10 min, following by three cycles of freezing and thawing. The cells were centrifuged at 21,000 rcf for 20 min at 4 °C. The supernatant was collected for further use. The macrophage lysates and legumain were incubated with the liposomes in citric acid buffer at 37 °C for 30 min. The liposomes were then purified with a G-50 column and examined by turbidity measurement at 400 nm. The cleavage efficiency of legumain was evaluated by the ratio of fluorescence intensities of Cy3 (λ 570 nm) and FITC (λ 520 nm).
In vitro uptake studies
A549T cells were seeded in 12-well plates at a density of 5×104/well and incubated for 24 h. Liposomes were treated with TAM2 lysate in citric acid buffer for 30 min before being added to the cells. The cells were incubated with the liposomes for 2 h and washed with PBS, followed by FACS assay. Confocal microscope experiment was conducted as the same procedure, with additional steps of fixation by 4% paraformaldehyde and staining with DAPI and phalloidin.
Penetration of A549T tumor spheroid
A549T cells were seeded at a density of 1000 cells per well in 96-well plates coated with 50 μL of 1% (w/v) agarose. The cells were incubated for 5 days to form tumor spheroids. The tumor spheroids were incubated with coumarin-6-loaded liposomes (at a dose equal to 100 ng/mL of coumarin-6) for 8 h and measured by confocal microscopy after washing with PBS. The fluorescence signal in the tumor spheroid was measured in a top-down mode (Z-axis) at a spatial distance of 10 µm for each scan layer. The penetration depth was measured by the Olympus FV1000 Fluoview workstation.
In vitro cytotoxicity studies
Cytotoxicity was measured by MTT assay. Briefly, A549, A549T, and PC9 cells were seeded in 96-well plates at a density of 5000 per well for 24 h. PTX, SV and defactinib were dissolved in DMSO. The final DMSO concentration was 0.2%. After pretreatment with TAM2 lysate, liposomes co-loaded with PTX and SV (1:2 w/w) were added to the medium at different concentrations. MTT assay was measured after 48 h. For apoptosis assay, A549T cells were seeded in 6-well plates and treated with liposomes or free drugs at a dose of 1 μg/mL PTX and 2 μg/mL SV. The cells were harvested after 24 h incubation. After washing with PBS, the cells were collected and stained with FITC Annexin V Apoptosis Kit (BD USA). The cells were subjected to apoptosis analysis using FACS. Caspase-3 was detected by Western blot after 24 h drug treatment.
The gefitinib treatment efficacy studies on the TGF-β-induced A549 and PC9 cells were conducted using the same procedures as above.
In vivo imaging and bio-distribution analysis
An A549T xenograft tumor model was developed by subcutaneous injection of A549T cells (108 cells per mouse) at the right hind legs in BALB/c nude mice. For the in vivo imaging studies, after the xenografted tumors reached 80 mm3 in size, the mice were injected via the tail vein with DIR-loaded liposomes. Fluorescence imaging was conducted at pre-determined time points using an IVIS instrument (Caliper Life Science, USA). After in vivo imaging at 24 h, the mice were sacrificed and the major organs collected for ex vivo fluorescence imaging.
For FACS analysis, the mice were injected with coumarin-6-loaded liposomes and sacrificed 4 h later. The tumors were collected and digested by collagenase and measured by FACS for determining the uptake efficiency of the liposomes in TAM2, which were labeled with anti-CD209-APC (Thermo Fisher Scientific, USA) against the M2 marker CD209.
For bio-distribution analysis, when the tumors reached a volume of 150-200 mm3, the mice were randomly assigned to three groups and intravenously injected with PTX dissolved in Cremophor EL, PEG/Lip, and aLip at a dose of 10 mg/kg. At predetermined time points after administration, mice were sacrificed (n = 4 for each time point) and the tumor and major organs were collected. PTX was extracted by methanol and quantified by HPLC analysis.
In vivo antitumor efficacy and systemic toxicity
BALB/c nude mice bearing A549T subcutaneous xenograft tumors were used for the study. After the tumors reached ~80 mm3, the mice were treated with saline or SV/PTX-loaded liposomes (equal to PTX: 4 mg/kg, SV: 8 mg/kg). The tumor volume (V) was calculated as V = (L × W2)/2. At the end of the experiment, the tumors, livers, lungs, hearts, spleens, and kidneys were collected for immunofluorescence and histological examination as well as Western blot assay using standard procedures.
Statistical analyses
Statistical analyses were performed using GraphPad Software. Data are expressed as mean ± SD (n>3). Statistical significance was determined using Student's t-test. Statistically significant difference was defined as *P<0.05, **P<0.01, and ***P<0.001.
Abbreviations
ABCA1: ATP-binding cassette transporter A1; aLip: legumain-activatable liposomes; BMDM: bone marrow-derived macrophages; DMSO: dimethyl sulfoxide; E-CAD: E-cadherin; ECM: extracellular matrix; EMT: epithelial-mesenchymal transitio; FACS: fluorescence activated cell sorting; FN: fibronectin; HPLC: high-performance liquid chromatography; LXR: liver X receptors; MDR: multidrug resistance; MWCO: molecular weight cut-off; NSCLC: non-small-cell lung cancer; PLL: poly-L-lysine; PTX: paclitaxel; RT-PCR: reverse transcription polymerase chain reaction; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; SV: simvastatin; TAM: tumor-associated macrophages; TEM: transmission electron microscopy; TME: tumor microenvironment; TBST: tris-buffered saline containing 0.05% Tween 20.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We are thankful for the support of the 973 Program, China (2014CB931900), NFSC (81673382, 81422048, and 81521005), Strategic Priority Research Program of CAS (XDA12050307), National Special Project for Significant New Drugs Development (2018ZX09711002-010-002), CAS Scientific Research and Equipment Development Project (YZ201437), and Fudan-SIMM Joint Research Fund (FU-SIMM20174009). We also thank the National Center for Protein Science Shanghai, CAS, and the Department of Molecular Imaging, SIMM, for TEM and fluorescence imaging technical assistance.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Morandi A, Taddei ML, Chiarugi P, Giannoni E. Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front Oncol. 2017;7:40
2. Nurwidya F, Takahashi F, Murakami A, Takahashi K. Epithelial mesenchymal transition in drug resistance and metastasis of lung cancer. Cancer Res Treat. 2012;44:151-6
3. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525-30
4. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472-6
5. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611-29
6. Yang L, Zhang F, Wang X, Tsai Y, Chuang KH, Keng PC. et al. A FASN-TGF-beta1-FASN regulatory loop contributes to high EMT/metastatic potential of cisplatin-resistant non-small cell lung cancer. Oncotarget. 2016;7:55543-54
7. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189
8. Marro M, Nieva C, Sanz-Pamplona R, Sierra A. Molecular monitoring of epithelial-to-mesenchymal transition in breast cancer cells by means of Raman spectroscopy. Biochim Biophys Acta. 2014;1843:1785-95
9. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56-68
10. Goldman A, Majumder B, Dhawan A, Ravi S, Goldman D, Kohandel M. et al. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat Commun. 2015;6:6139
11. Hryniewicz-Jankowska A, Augoff K, Biernatowska A, Podkalicka J, Sikorski AF. Membrane rafts as a novel target in cancer therapy. Biochim Biophys Acta. 2014;1845:155-65
12. Zuo W, Chen YG. Specific activation of mitogen-activated protein kinase by transforming growth factor-beta receptors in lipid rafts is required for epithelial cell plasticity. Mol Biol Cell. 2009;20:1020-9
13. Medbury MJ, Williams H, Li S, Fletcher JP. The bidirectional relationship between cholesterol and macrophage polarization. J Clin Cell Immunol. 2015;6:303
14. Chockley PJ, Keshamouni VG. Immunological consequences of epithelial-mesenchymal transition in tumor progression. J Immunol. 2016;197:691-8
15. Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T. et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS One. 2015;10:e0141392
16. Cardwell CR, Mc Menamin U, Hughes CM, Murray LJ. Statin use and survival from lung cancer: a population-based cohort study. Cancer Epidemiol Biomarkers Prev. 2015;24:833-41
17. Yang T, Chen M, Sun T. Simvastatin attenuates TGF-beta1-induced epithelial-mesenchymal transition in human alveolar epithelial cells. Cell Physiol Biochem. 2013;31:863-74
18. Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115:959-68
19. Runz S, Mierke CT, Joumaa S, Behrens J, Fabry B, Altevogt P. CD24 induces localization of beta1 integrin to lipid raft domains. Biochem Biophys Res Commun. 2008;365:35-41
20. Wang C, Yoo Y, Fan H, Kim E, Guan KL, Guan JL. Regulation of Integrin beta 1 recycling to lipid rafts by Rab1a to promote cell migration. J Biol Chem. 2010;285:29398-405
21. Wang R, Bi J, Ampah KK, Ba X, Liu W, Zeng X. Lipid rafts control human melanoma cell migration by regulating focal adhesion disassembly. Biochim Biophys Acta. 2013;1833:3195-205
22. Shah PP, Fong MY, Kakar SS. PTTG induces EMT through integrin alphaVbeta3-focal adhesion kinase signaling in lung cancer cells. Oncogene. 2012;31:3124-35
23. Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A. et al. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314:143-52
24. Zheng X, Turkowski K, Mora J, Brune B, Seeger W, Weigert A. et al. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget. 2017;8:48436-52
25. Pennings M, Meurs I, Ye D, Out R, Hoekstra M, Van Berkel TJ. et al. Regulation of cholesterol homeostasis in macrophages and consequences for atherosclerotic lesion development. FEBS Lett. 2006;580:5588-96
26. Kimura T, Nada S, Takegahara N, Okuno T, Nojima S, Kang S. et al. Polarization of M2 macrophages requires Lamtor1 that integrates cytokine and amino-acid signals. Nat Commun. 2016;7:13130
27. Zhao C, Dahlman-Wright K. Liver X receptor in cholesterol metabolism. J Endocrinol. 2010;204:233-40
28. Qiu G, Hill JS. Atorvastatin inhibits ABCA1 expression and cholesterol efflux in THP-1 macrophages by an LXR-dependent pathway. J Cardiovasc Pharmacol. 2008;51:388-95
29. Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C. et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132-41
30. Lewen S, Zhou H, Hu HD, Cheng T, Markowitz D, Reisfeld RA. et al. A Legumain-based minigene vaccine targets the tumor stroma and suppresses breast cancer growth and angiogenesis. Cancer Immunol Immunother. 2008;57:507-15
31. Liu Z, Xiong M, Gong J, Zhang Y, Bai N, Luo Y. et al. Legumain protease-activated TAT-liposome cargo for targeting tumours and their microenvironment. Nat Commun. 2014;5:4280
32. Jiang Y, Lu J, Wang Y, Zeng F, Wang H, Peng H. et al. Molecular-dynamics-simulation-driven design of a protease-responsive probe for in-vivo tumor imaging. Adv Mater. 2014;26:8174-8
33. Kidera Y, Tsubaki M, Yamazoe Y, Shoji K, Nakamura H, Ogaki M. et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J Exp Clin Cancer Res. 2010;29:127
34. Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. 2005;6:56
35. Huang Q, Shen HM, Shui G, Wenk MR, Ong CN. Emodin inhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer Res. 2006;66:5807-15
Author contact
![]() Corresponding author: Yongzhuo Huang, Ph.D., Professor, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 501 Haike Rd, Shanghai 201203, China. Tel/Fax: +86-21-2023-1981 Email: yzhuangac.cn Ying Hu, PhD. Professor Zhejiang Pharmaceutical College, Ningbo 315100, China. pharmhawkcom
Corresponding author: Yongzhuo Huang, Ph.D., Professor, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 501 Haike Rd, Shanghai 201203, China. Tel/Fax: +86-21-2023-1981 Email: yzhuangac.cn Ying Hu, PhD. Professor Zhejiang Pharmaceutical College, Ningbo 315100, China. pharmhawkcom
Citation styles
APA
Jin, H., He, Y., Zhao, P., Hu, Y., Tao, J., Chen, J., Huang, Y. (2019). Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics, 9(1), 265-278. https://doi.org/10.7150/thno.27246.
ACS
Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019, 9 (1), 265-278. DOI: 10.7150/thno.27246.
NLM
Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J, Huang Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019; 9(1):265-278. doi:10.7150/thno.27246. https://www.thno.org/v09p0265.htm
CSE
Jin H, He Y, Zhao P, Hu Y, Tao J, Chen J, Huang Y. 2019. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics. 9(1):265-278.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.