Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results and Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results and Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(1):90-103. doi:10.7150/thno.30259 This issue Cite
Research Paper
Redox Dual-Responsive and O2‑Evolving Theranostic Nanosystem for Highly Selective Chemotherapy against Hypoxic Tumors
Huachao Chen1*, Fei Li2*, Yongrong Yao1, Zhe Wang1, Zhihao Zhang1 ![]() , Ninghua Tan1,2
, Ninghua Tan1,2 ![]()
1. State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 211198, China.
2. State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, Yunnan, China.
* Contributed equally to this work.
Received 2018-9-28; Accepted 2018-11-21; Published 2019-1-1
Citation:
Chen H, Li F, Yao Y, Wang Z, Zhang Z, Tan N. Redox Dual-Responsive and O2‑Evolving Theranostic Nanosystem for Highly Selective Chemotherapy against Hypoxic Tumors. Theranostics 2019; 9(1):90-103. doi:10.7150/thno.30259. https://www.thno.org/v09p0090.htm
Other stylesAbstract

Activatable theranostic agents, which combine fluorescent reporters with masked chemotherapeutic agents that are activated by tumor-associated stimuli, would be attractive candidates to improve the tumor selectivity of chemotherapy. This work reports a ROS/GSH dual-activatable and O2‑evolving theranostic nanosystem (RA-S-S-Cy@PLGA NPs) for highly selective therapy against hypoxic tumors and in situ fluorescence-tracking of cancer chemotherapy.
Methods: In this system, the newly designed theranostic agent (RA-S-S-Cy) is composed of a disulfide bond as a cleavable linker, a near infrared (NIR) active fluorophore as a fluorescent tracker, and a natural cyclopeptide RA-V as the active anti-cancer agent. Upon reaction with the high level of intracellular glutathione (GSH), disulfide cleavage occurs, resulting in concomitant active drug RA-V release and significant NIR fluorescence increase. To further improve the tumor targeting of RA-S-S-Cy and achieve redox dual-responsiveness, RA-S-S-Cy was incorporated into the c(RGDfK)-targeted PLGA nanoparticles together with an O2-generating agent (catalase) to produce RA-S-S-Cy@PLGA NPs.
Results: The cell-specific and redox dual-activatable release of RA-V lead to enhanced therapeutic outcomes in vivo and in vitro. More significantly, the RA-S-S-Cy@PLGA NPs were successfully applied for monitoring of drug release and chemotherapeutic efficacy in situ by “turn-on” NIR fluorescence.
Conclusions: RA-S-S-Cy@PLGA NPs would be efficient theranostic nanosystems for more precise therapy against hypoxic tumors and provides a potential tool for deeper understanding of drug release mechanisms.
Keywords: cancer, theranostic system, hypoxic tumor, ROS/GSH dual-activatable, cyclopeptide RA-V, fluorescence imaging
Introduction
Chemotherapy involving the use of cytotoxic drugs is a dominant treatment modality for various cancers because of its high efficiency [1]. Although various anti-cancer drugs have been utilized extensively in clinical practice, traditional chemotherapeutics have major limitations such as poor bioavailability, nonspecific selectivity, multidrug resistance, and high systemic toxicity [2]. Therefore, the search for new chemotherapy agents has received broad attention as a promising approach to improve therapeutic outcome [3]. RA-V (deoxybouvardin), a unique homodicyclohexapeptide [4], was isolated by us from several Rubia plants [5-8], including R. yunnanensis, R. schumanniana, R. cordifolia and R. podantha. Our previous studies have shown that RA-V exhibits antitumour activities in vitro and in vivo, and suppresses inflammation, angiogenesis, and protective tumor cell autophagy and induces apoptosis [9-13]. Nevertheless, RA-V has been limited in its application to cancer therapy in vivo because of its poor solubility in physiological conditions. To improve its solubility, several drug delivery systems had been constructed by us, such as pH-responsive RA-V/squaraine-loaded micelles, and multi-organelle-targeted nanoparticles [14-15]. However, their low tumor selectivity against human cancer is still a limitation for clinical applications of RA-V.
To address these problems, an activatable theranostic agent that combines fluorescent reporters with masked chemotherapeutic agents that are activated by tumor-associated stimuli would be an attractive candidate [16-18]. Typically in such prodrug systems, the chemotherapeutic agent is linked to a fluorophore with a cleavable linker, allowing conversion to the cytotoxic drug in the cells [19]. The activatable theranostic agent can specifically kill diseased cells that differ from normal cells by inclusion of functional groups that are responsive to tumor markers [20]. Meanwhile, the cleavage of the linker also produces an easy-to-monitor fluorescence change [21]. The activatable fluorescence can realize real-time monitoring of drug release specifically in the tumor [22]. The cleavable linker in activatable theranostic agents is typically chosen to respond to tumor microenvironment-specific features including hypoxia [23], acidity [24], interstitial hypertension [25], and chronic inflammatory response [26]. Among these tumor microenvironment activatable agents, redox-sensitive drug delivery systems are of particular interest because of the higher glutathione (GSH) concentrations present in cancer cells [27]. In addition to the high level of GSH, a high level of ROS (such as H2O2) is generated by cancer cells compared to normal cells, which is primarily attributed to chronic inflammation, cell proliferation or DNA alterations [28]. Since high levels of ROS and GSH are recognized as characteristic features of cancer, many studies have focused on the construction of ROS- or GSH-activatable theranostic agents [29]. However, there are limited examples of theranostic agents specifically responsive to both ROS and GSH stimuli.
We herein reported a redox dual-activatable and O2-evolving theranostic nanosystem for highly selective chemotherapy against hypoxic tumors and in situ fluorescence-tracking of drug release (Scheme 1). In this study, the theranostic agent (designated RA-S-S-Cy) was developed with a unique natural cyclopeptide RA-V as the activatable anticancer agent, and Cy5.5 was included as a near-infrared (NIR) fluorophore to monitor activation of RA-V. The fluorescent dye Cy5.5 was linked to the disulfide linker by an amide group, and the disulfide linker could be cleaved by intracellular GSH (Scheme 1A). When Cy5.5 and RA-V were linked covalently through the disulfide linker, the fluorescence of Cy5.5 was quenched due to efficient photoinduced electron transfer (PET) [30] from the RA-V moiety (electron donor) to the singlet excited state of the Cy5.5 moiety (electron acceptor fluorophore). The presence of an electron-donating methoxyl group in RA-V lead to the effective PET process. Once the disulfide linker was cleaved by GSH, the PET efficiency decreased, and the fluorescence of Cy5.5 was “switched on”. As the fluorescence of Cy5.5 and cytotoxicity of RA-V were quenched when linked, cleavage of the disulfide bond realized an efficient increase in anticancer activity, as well as enabled monitoring of drug release in a noninvasive manner. To construct the redox dual-responsive theranostic nanoparticles (designated RA-S-S-Cy@PLGA NPs), RA-S-S-Cy was loaded into (D,L-lactic-co-glycolic acid) (PLGA) nanoparticles modified with a tumor-targeting cyclic pentapeptide c(RGDfK) [31], and catalase was incorporated into the aqueous core of RA-S-S-Cy@PLGA NPs as an O2-generating agent (Scheme 1B). H2O2 easily penetrated the lipophilic shells into the aqueous cores of RA-S-S-Cy@PLGA NPs [32] then it was catalyzed by catalase to quickly generate O2 gas, causing rupture of the PLGA shell to accelerate degradation of RA-S-S-Cy@PLGA NPs and subsequent release of RA-S-S-Cy. As a result, the released RA-S-S-Cy was selectively activated by intracellular GSH, resulting in cellular apoptosis and enhanced NIR fluorescence. More importantly, the O2 produced by RA-S-S-Cy@PLGA NPs could overcome the hypoxia problem in tumor tissue to improve chemotherapy efficiency, as hypoxia poses an obstacle for cancer therapy.
Methods
Materials
Poly(D, L -lactic-co-glycolic acid) (PLGA, with a lactide/glycolide molar ratio of 75:25 and an inherent viscosity of 0.17 dL g-1) was obtained from Daigang BIO Engineer Ltd. Co. (Shandong, China). Catalase, H2O2, GSH, 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimidehydrochloride (EDC), N-hydroxysuccinimide (NHS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl- tetrazolium bromide (MTT), 9,10-phenan-threnequinone, poly(vinyl alcohol) (PVA, MW=57-66 kDa), 3,3'-dioctadecyloxacarbocya-nineperchlorate (DiO) and other chemical reagents were all obtained from Sigma-Aldrich (St. Louis, MO, USA). c(RGDfK) was obtained from Sangon Biotech Ltd. Co (Shanghai, China). Hoechst 33342, LysoTracker Red, MitoTracker Red, ER Tracker Red were obtained from Invitrogen (Carlsbad, CA, USA). RA-V was isolated from Rubia yunnanensis as described earlier [6]. Ultrapure water was prepared using a Millipore Simplicity System (Millipore, Bedford, USA).
Scheme 1
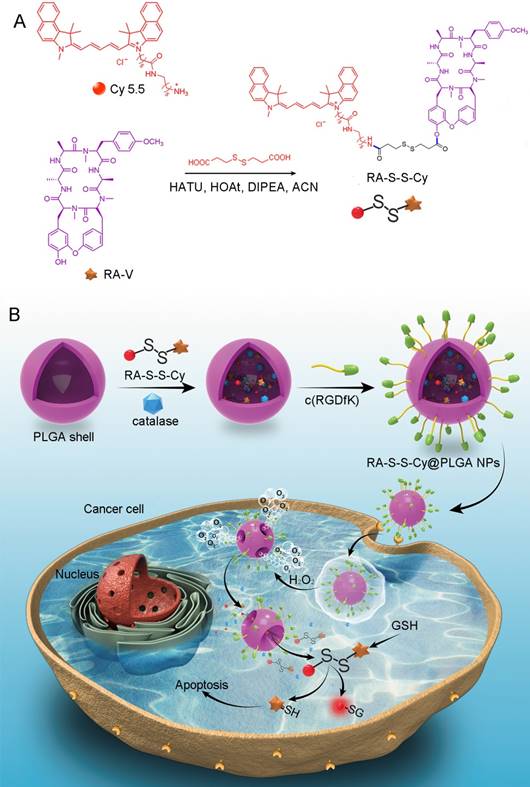
(A) Syntheses of activatable RA-S-S-Cy prodrug. (B) Schematic illustration of the RA-S-S-Cy@PLGA NPs structure and the mechanism of cytotoxicity and fluorescence enhancement produced by RA-S-S-Cy@PLGA NPs upon exposure to GSH.
Synthesis and characterization of RA-S-S-Cy
RA-V (11 mg, 1.0 eq) and 3,3'-dithiodipropionic acid (7.6 mg, 2.5 eq) were dissolved in dry ACN (1 mL) under nitrogen atmosphere. HATU (13.8 mg, 2.5 eq) and HOAt (4.9 mg, 2.5 eq) were added at 0 °C after DIPEA (6.3 μL, 2.5 eq) was added and stirred for 1 h at 0 °C. Then the reaction was maintained at 45 °C for 36 h after the reaction mixture was filtrated and concentrated in vacuo. The residue was purified by Prep-HPLC: 45% ACN/H2O to give 8.3 mg compound 2 as a white solid in 60% yield. UPLC-MS: [M+H]+: 949.40, [M+Na]+: 971.41.
Compound 2 (5.5 mg, 1.0 eq) and Cyanine 5.5 amine (4.3 mg, 1.0 eq) were dissolved in dry ACN (1 mL) under nitrogen atmosphere. HATU (6.6 mg, 3 eq) and HOAt (2.4 mg, 3 eq) were added at 0 °C after DIPEA (3 μL, 3 eq) was added and stirred for 1 h at 0 °C. Then the reaction was maintained at 45 °C for 24 h after the reaction mixture was filtrated and concentrated in vacuo. The residue was purified by silica gel flash chromatography (Chloroform: MeOH = 30:1- 10:1) to give 4.0 mg of compound 3 (HCl salt) as a dark blue powder in 43 % yield. 1H NMR (600 MHz, C5D5N, mixture of two rotamers): δ 9.92 (1H, d, J = 7.9 Hz), 8.67-8.61 (3H, overlap), 8.57 (1H, s), 8.43 (1H, m), 8.17 (2H, d, J = 7.7 Hz), 8.10-8.03 (9H, overlap), 7.67-7.61 (8H, overlap), 7.56-7.52 (2H, m), 7.44 (2H, d, J = 8.6 Hz), 7.39 (1H, d, J = 7.8 Hz), 7.35 (1H, s), 7.28-7.27 (5H, overlap), 7.08-7.01 (3H, overlap), 6.95 (1H, d, J = 8.2 Hz), 6.91-6.82 (3H, overlap), 6.59 (1H, d, J = 13.6 Hz), 6.41 (1H, d, J = 13.6 Hz), 5.91-5.84 (2H, m), 5.73 (1H, d, J = 11.3 Hz), 5.53-5.46 (3H, overlap), 5.36 (2H, brs), 5.04-5.02 (1H, overlap), 4.95 (1H, d, J = 11.8 Hz), 4.89-4.85 (2H, m), 4.64 (1H, s), 4.23-4.19 (3H, overlap), 4.10-4.08 (1H, m), 3.93 (1H, t, J = 12.7 Hz), 3.81 (1H, dd, J = 13.3, 3.4 Hz), 3.71 (3H, s), 3.67-3.59 (6H, overlap), 3.50 (1H, s), 3.44-3.43 (5H, overlap), 3.30-3.24 (5H, overlap), 3.21-3.15 (5H, overlap), 3.08 (1H, s), 2.99-2.97 (7H, overlap), 2.88 (2H, t, J = 6.8 Hz), 2.85 (2H, t, J = 7.4 Hz), 2.61 (1H, d, J = 11.0 Hz), 2.54 (1H, d, J = 7.7 Hz), 2.47-2.45 (5H, overlap), 2.13-2.11 (4H, overlap), 2.05- 2.03 (4H, overlap), 1.90-1.88 (9H, overlap), 1.86-1.85 (5H, overlap), 1.57-1.56 (6H, overlap), 1.47-1.46 (6H, overlap), 0.89-0.87 (9H, overlap). 13C NMR (150 MHz, C5D5N, mixture of two rotamers): δ 175.9 and 175.2, 175.1 and 174.8, 173.8 and 173.7, 173.5 and 173.0, 172.9 and 172.2, 172.4 and 172.1, 171.3, 171.1, 170.6, 170.1, 169.2, 159.2 and 158.5, 155.8 and 153.8, 153.2 and 153.1, 148.7, 148.6, 148.2, 141.1 and 140.4, 140.0 and 139.8, 139.4 and 139.4, 136.8, 134.5, 134.2 and 133.6, 132.7 and 132.7, 131.2 and 130.9, 130.6 and 130.5, 128.8, 128.7, 128.5, 128.5, 126.7 and 126.5, 126.0, 125.9 and 125.8, 125.6, 125.4, 125.1, 123.0, 120.1, 115.6 and 114.9, 114.9 and 114.8, 112.0 and 111.8, 104.2, 103.9, 80.2 and 79.9, 69.0 and 67.5, 67.1 and 65.0, 58.1 and 57.7, 55.5 and 55.4, 54.9, 51.7, 51.6, 48.4 and 47.2, 45.0 and 44.6, 40.3, 39.8, 39.7, 36.7, 35.6 and 35.5, 35.0 and 34.6, 34.4 and 34.4, 32.9, 32.5 and 31.8, 31.1 and 30.7, 30.4 and 30.3, 30.3, 30.1, 30.0, 29.8, 29.6, 27.9, 27.5, 27.4, 27.3, 27.1 and 26.6, 26.0 and 24.1, 23.3 and 14.7. HRESIMS calcd for C92H111N10O12S2: [M]+ 1611.7806, found 1611.7824.
HPLC analysis
Analysis was performed on a Waters Acquity HPLC with 2998 PDA system (Waters, UK). The samples were separated on a 4.6 mm × 50 mm, 2.7 μm CORTECS C18 column (Waters, UK). A gradient of 0.1% formic acid in water (A) and acetonitrile (B) was used as follows: a linear gradient of 10-100% B over 0-30 min. The flow rate was 0.5 mL min-1. The chromatographic column and autosampler were maintained at 45 °C and 4 °C, respectively. Every 10 µL sample solution was injected.
LC/MS analysis
Analysis was performed on a Waters Acquity UPLC with XEVO-TQD-MS system (Waters, UK). The samples were separated on a 2.1 mm × 50 mm, 1.7 μm BEH C18 column (Waters, UK). A gradient of 0.1% formic acid in water (A) and acetonitrile (B) was used as follows: a linear gradient of 10-100% B over 0-25 min. The flow rate was 0.3 mL min-1. The chromategraphic column and autosampler were maintained at 45 and 4°C, respectively. Every 2 µL sample solution was injected.
Fabrication and characterization of RA-S-S-Cy@PLGA NPs
RA-S-S-Cy@PLGA NPs were prepared using a water-in-oil-in-water (W/O/W) double-emulsion, solvent-diffusion-evaporation approach method [33]. A 2.5 mg catalase powder was added to 1 mL PVA aqueous solution (10 mg mL-1) and then mixed thoroughly for 15 min. Subsequently, the mixture was emulsified with 2 mL of PLGA solution (5 mg mL-1 in CH2Cl2) containing DiO (1 mg mL-1) and RA-S-S-Cy (1 mg mL-1) to obtain the primary W/O emulsion. The primary emulsification was carried out by using an ultrasonicator for 1 min in an ice bath. The primary emulsion was then added to 6 mL of PVA aqueous (20 mg mL-1) and emulsified by using an ultrasonicator for 1 min in an ice bath to form the W/O/W double-emulsion. To evaporate CH2Cl2 and solidify the particles, the resultant double-emulsion was transferred into 20 mL of ultrapure water and stirred overnight at room temperature. After washing three times with ultrapure water, the PLGA NPs were collected by centrifugation. The reaction processes for conjugation of NH2-containing c(RGDfK) with carboxyl-containing PLGA nanoparticles via covalent amide bond involves EDC/NHS activation of carboxylic acids and the following amidation reaction [34]. A NP suspension (150 μL, 10 mg mL-1) was treated with 400 mM EDC and 100 mM NHS in water for 15 min at room temperature with agitation to give the corresponding PLGA-NHS ester. The activated NPs were washed twice using the Amicon centrifugal filter units to remove unreacted EDC and NHS and then c(RGDfK) was added into the PLGA NPs suspension at a mass ratio of 10% to PLGA, agitating for 2 h at room temperature. The resulting NPs were washed three times with water with Amicon centrifugal units and re-suspended in water for storage. The surface morphology of the as-prepared RA-S-S-Cy@PLGA NPs was investigated by scanning electron microscopy (SEM, Hitachi s-4800 high resolution). The transmission electron microscopy (TEM, JEM-2100) measurement of RA-S-S-Cy@PLGA NPs was prepared by dropping the solution onto a carbon-coated copper grid following negative staining with 2.0% (w/v) phosphotungstic acid. The particle size of RA-S-S-Cy@PLGA NPs were measured by dynamic light scattering (DLS) (a Mastersizer 2000 particle size analyzer) with a fixed scattering angle of 90°. Zeta potential measurement was performed at 25 °C on a Malvern Zeta sizer-Nano Z instrument. An ultrasound imaging system with a 7 MHz linear-array transducer (Toshiba Nemio 30, Japan) was utilized to visualize the O2 bubbles generated by RA-S-S-Cy@PLGA NPs in a test tube containing H2O2 at 25 °C.
Cell culture and confocal fluorescence imaging
Human colon cancer cell line (HCT-116 cells) and human breast cancer cell line (MCF-7 cells) were seeded at a density of 1×106 cells mL-1 in RPMI 1640 supplemented with 10% FBS, NaHCO3 (2 g L-1) and 1% antibiotics (penicillin/streptomycin, 100 U mL-1). The cells were maintained in a humidified incubator at 37 °C, in 5% CO2/95% air. One day before imaging, cells were passed and plated on 18-mm glass bottom dishes. Cell imaging was carried out after washing cells with PBS three times. Confocal fluorescence microscopy images of the cells were obtained using a ZEISS Laser Scanning Microscope: LSM 710.
Cytotoxicity assay
The cytotoxicities of empty NPs, RA-S-S-Cy@PLGA NPs (without catalase), free RA-V, RA-S-S-Cy, and RA-S-S-Cy@PLGA NPs were investigated by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Cells (106 cells mL-1) were dispersed within replicate 96-well microtiter plates to a total volume of 200 μL/well. The empty NPs, RA-S-S-Cy@PLGA NPs (without catalase), free RA-V, RA-S-S-Cy, and RA-S-S-Cy@PLGA NPs at specific concentrations were added to the culture medium. Cells were incubated for an additional 48 h and then 20 μL of 5 mg mL-1 MTT solution in pH 7.4 PBS was added to each well. After incubation for 4 h, the culture plates were centrifuged and the medium was removed. DMSO (150 μL) was added to each well and the absorbance of the dissolved formazan was measured at 570 nm using an ELISA plate reader. Calculation of the half lethal dose (IC50) values was done according to Huber and Koella [35].
In vivo imaging study
Tumor models were established in BALB/c nude mice by injecting 1×106 HCT-116 cells subcutaneously into the selected positions. All animal operations were performed in accordance with institutional animal use and care regulations approved by the Model Animal Research Center of China Pharmaceutical University. Time-dependent in vivo fluorescence images of subcutaneous HCT-116 tumor-bearing mice after i.v. injection of 10 mg kg-1 RA-S-S-Cy@PLGA NPs (~30 μg RA-S-S-Cy per 1 mg RA-S-S-Cy@PLGA NPs), Cy NPs, or RA-S-S-Cy@PLGA NPs (without catalase). At 8 h and 24 h, the mice were placed in the chamber of an in vivo imaging system (CRi, MA, USA). After the 24 h scanning, the mice were euthanized and the tumor, heart, liver, spleen, lung, kidney and intestine were excised for ex vivo imaging.
In vivo antitumor efficacy on subcutaneous tumor model
In vivo targeted treatment was performed using HCT-116 tumor-bearing mice. The tumor-bearing mice were randomly divided into different groups and treated i.v. with saline, empty NPs, RA-S-S-Cy@PLGA NPs (without catalase), RA-V cNPs, RA-S-S-Cy, and RA-S-S-Cy@PLGA NPs at a RA-V dosage of 1 mg kg-1, respectively. Each group contained six mice. From day 0, the mice were intravenously injected with sample every two days for 12 days, and tumors were measured using a caliper every two days. The therapeutic effects were evaluated by monitoring the tumor volumes and by H&E staining.
Results and Discussion
Characterization of RA-S-S-Cy and RA-S-S-Cy@PLGA NPs
RA-S-S-Cy is comprised of RA-V as an antitumor agent, Cy5.5 as an imaging agent, and a disulfide linker that is readily cleaved by GSH, which is relatively abundant in cancer cells. RA-S-S-Cy was prepared according to the synthetic route outlined in Scheme S1. The structure of RA-S-S-Cy was characterized with 1H NMR, 13C-NMR and HRMS (Figures S1-S3). The chromatograms from HPLC and LCMS (Figure S4 and Figure S5) were used to assess the purity of the conjugate.
The loading capacity and the encapsulation efficiency of RA-V in RA-S-S-Cy@PLGA NPs were determined with HPLC to be about 3±0.1% and 65±2.5%, respectively. Transmission electron microscopy (TEM) indicated that RA-S-S-Cy@PLGA NPs were spherical with a relatively uniform size (Figure 1A). The average hydrodynamic diameter of RA-S-S-Cy@PLGA NPs was approximately 220 nm (PDI=0.11) as measured by dynamic light scattering (DLS) (Figure 1B). The zeta potential of the NPs was -20 mV, suggesting stability in aqueous medium. After conjugation with c(RGDfK), the zeta potential of RA-S-S-Cy@PLGA NPs changed to -16 mV due to the positive charge from arginine in c(RGDfK), confirming the presence of c(RGDfK) at the surface. The long-term stability of RA-S-S-Cy@PLGA NPs, in terms of size and fluorescence, was investigated by DLS and fluorescence spectroscopy, respectively. The hydrodynamic diameter and fluorescence of RA-S-S-Cy@PLGA NPs did not change for at least one week in two commonly used biological media (Figure S6 and Figure S7), indicating that RA-S-S-Cy@PLGA NPs have long-term stability in physiological environments. The morphological changes of RA-S-S-Cy@PLGA NPs in the presence of H2O2 were investigated by SEM. When incubated with 100 µM H2O2, small pores were observed on the shell of RA-S-S-Cy@PLGA NPs after 4 h and the pore size increased over time. After 24 h incubation with H2O2, the shells of RA-S-S-Cy@PLGA NPs were completely ruptured. In contrast, the RA-S-S-Cy@PLGA NPs without H2O2 incubation and RA-S-S-Cy@PLGA NPs (without catalase) in the presence of H2O2 had a smooth surface and a dense morphology over 24 h (Figure 1C). Ultrasound imaging demonstrated that once H2O2 infiltrated the PLGA shell wall and was catalysed by catalase in the aqueous core, O2 bubbles were generated instantly to disrupt RA-S-S-Cy@PLGA NPs (Figure 1D).
Figure 1
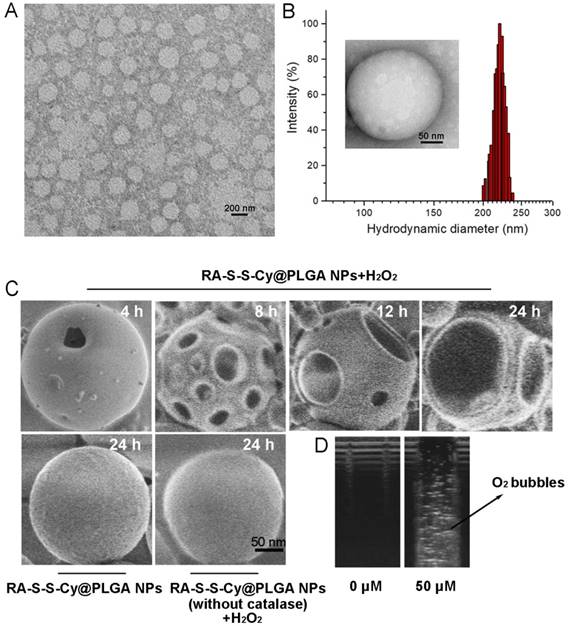
(A) TEM micrographs of RA-S-S-Cy@PLGA NPs. Scale bar: 200 nm. (B) Size distribution of RA-S-S-Cy@PLGA NPs characterized by DLS at 25 °C. Insets: TEM micrograph of RA-S-S-Cy NP. Scale bars: 50 nm. (C) SEM micrographs of RA-S-S-Cy@PLGA NPs incubated with and without 100 µM H2O2, or RA-S-S-Cy@PLGA NPs (without catalase) incubated with 100 µM H2O2 for 24 h. Scale bars: 50 nm. (D) Ultrasound images of RA-S-S-Cy@PLGA NPs suspended in media with H2O2 at different concentrations.
Fluorescence response of RA-S-S-Cy and in vitro release of RA-V from RA-S-S-Cy
The responsiveness of RA-S-S-Cy to GSH was investigated by its fluorescence intensity response. As shown in Figure 2A, a 10-fold fluorescence enhancement was observed with increasing amount of GSH due to cleavage of the disulfide linker. There was good linearity (R=0.995) between fluorescence intensity at 710 nm and the amount of GSH (0-24 equivalence). The fluorescence intensity of RA-S-S-Cy in the absence and presence of GSH at different pH values was also investigated. As shown in Figure 2B, RA-S-S-Cy was stable between pH 4.5 and 10.5, but when exposed to 1 mM GSH, significant fluorescence enhancement was observed over a pH range of 4.5-7.5, which can be explained by the pH-dependent conformational behavior of GSH [36]. The results indicated that RA-S-S-Cy could undergo thiol-induced cleavage within the physiological pH range. Interference of other intracellular substances was tested to study the selectivity of RA-S-S-Cy. Figure S8 shows that RA-S-S-Cy exhibited significant fluorescence enhancement when treated with GSH, whereas its response toward other substances was negligible, indicating the specificity of RA-S-S-Cy to GSH. The GSH-triggered drug release from RA-S-S-Cy was monitored by high performance liquid chromatography (HPLC) (Figure S9). In the presence of 1 mM GSH, cumulative release of RA-V reached ~100% within 24 h. Fluorometric time-dependent analysis showed that the amount of RA-V released was correlated with the increase in fluorescence intensity at 710 nm in the presence of GSH (Figure 2C). Furthermore, the drug release and fluorescence responsiveness of RA-S-S-Cy@PLGA NPs were both investigated in the presence or absence of H2O2 and GSH (Figure S10 and Figure 2D). The results showed that RA-S-S-Cy@PLGA NPs could achieve precisely triggered smart release in response to H2O2/GSH-dual stimuli, thereby enhancing therapy efficiency.
Figure 2
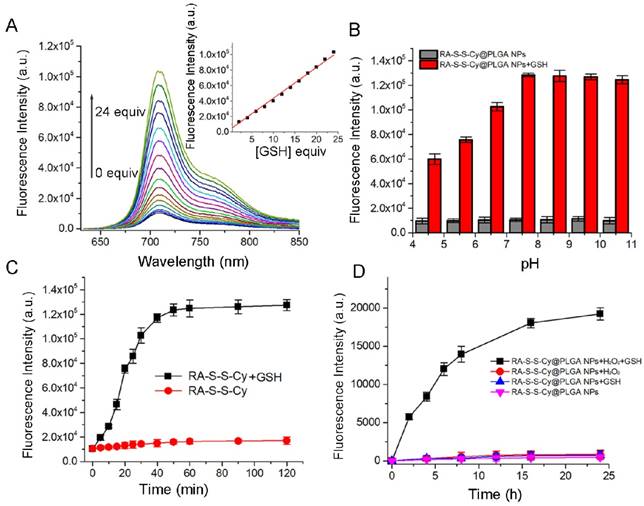
(A) Fluorescence spectra of RA-S-S-Cy (5 μM) recorded in the presence of different concentrations of GSH in PBS buffer. (B) Fluorescence intensity at 710 nm determined in the absence and presence of GSH (1 mM) at different pH values. All measurements were made at 37 °C using an excitation wavelength of 630 nm. (C) Fluorescence response of RA-S-S-Cy (5 μM) as a function of time in the presence and absence of GSH. Excitation was at 630 nm. (D) Fluorescence response of RA-S-S-Cy@PLGA NPs as a function of time in the presence and absence of GSH (1 mM) and H2O2 (50 μM). Excitation was at 630 nm.
Cellular selectivity and localization of RA-S-S-Cy@PLGA NPs
In order to visualize and localize RA-S-S-Cy@PLGA NPs in cancer cells, a fluorescent tracker, 3,3'-dioctadecyloxacarbocyanineperchlorate (DiO), was inserted into the shell of RA-S-S-Cy@PLGA NPs. After incubating with αvβ3 integrin-rich HCT-116 cells for 2 h, green fluorescence of DiO was observed in the cytoplasm, indicating efficient internalization of RA-S-S-Cy@PLGA NPs into HCT-116 cells. In contrast, MCF-7 cells, which lack significant expression of αvβ3 [37], showed very weak fluorescence signal after incubation with RA-S-S-Cy@PLGA NPs for 2 h (Figure 3). To further confirm that the specific internalization of RA-S-S-Cy@PLGA NPs was due to selective recognition of c(RGDfK), immunoglobulin G antibody [38] was used to replace the c(RGDfK) on RA-S-S-Cy@PLGA NPs as a negative control. The resulting RA-S-S-Cy@PLGA NPs (modified with IgG) were not efficiently uptaken by HCT-116 cells after 2 h incubation (Figure 3). The results suggested that RA-S-S-Cy@PLGA NPs are specifically recognized by αvβ3 integrin-rich tumor cells by virtue of c(RGDfK).
To further confirm that cRGDfk plays an important role in the selective internalization of RA-S-S-Cy@PLGA NPs into αvβ3 integrin-rich tumor cells, the cellular uptake of RA-S-S-Cy@PLGA NPs (without cRGD) was also investigated. HCT-116 cells showed negligible fluorescence signal after being incubated with RA-S-S-Cy@PLGA NPs (without cRGD) for 2 h. As an additional control, excess cRGDfk was added for 30 min before RA-S-S-Cy@PLGA NPs incubation; internalization of RA-S-S-Cy@PLGA NPs into HCT-116 cells was obviously blocked (Figure 3), which indicated that the targeted delivery of RA-S-S-Cy@PLGA NPs was aided by the specific interaction of c(RGDfK) with αvβ3 integrin.
To investigate the subcellular localization of RA-S-S-Cy@PLGA NPs, intracellular co-localization assays were carried out with ER Tracker Red, LysoTracker Red, Mito Tracker Red and Hoechst 33342. According to Figure 4, the co-localization staining clearly showed that the fluorescence appears mainly around the lysosome (colocalization efficient= 96%), whereas negligible fluorescence was observed in mitochondria (colocalization efficient=3.2%), endoplasmic reticulum (colocalization efficient=4.1%) or nucleus (colocalization efficient=0.9%). This observation suggested that RA-S-S-Cy@PLGA NPs was internalized into lysosomes of αvβ3 integrin-rich cells via receptor-mediated endocytosis.
Figure 3
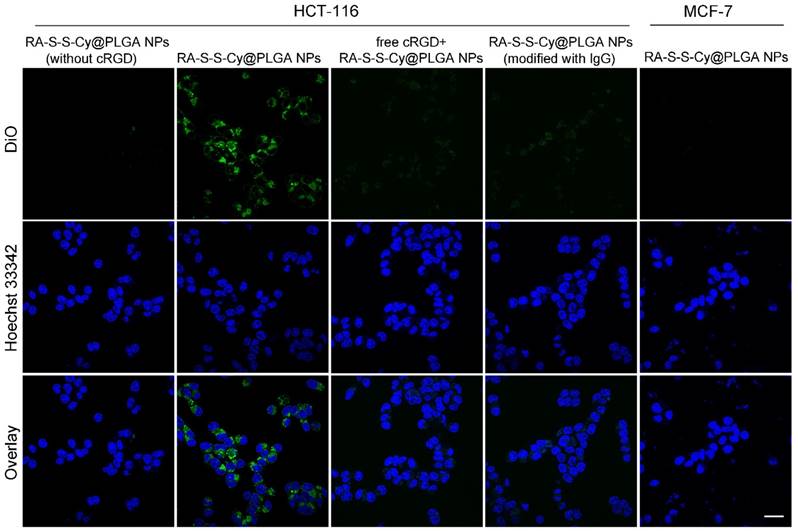
Confocal fluorescence imaging of HCT-116 cells and MCF-7 cells. HCT-116 cells were incubated with 50 μg mL-1 RA-S-S-Cy@PLGA NPs (without cRGD), RA-S-S-Cy@PLGA NPs (modified with IgG) or 50 μg mL-1 RA-S-S-Cy@PLGA NPs for 2 h. For a competitive inhibition assay, HCT-116 cells were pretreated with excess free cRGD followed by incubation with 50 μg mL-1 RA-S-S-Cy@PLGA NPs for 2 h. MCF-7 cells were incubated with 50 μg mL-1 RA-S-S-Cy@PLGA NPs for 2 h. Scale bars: 20 μm.
Real-time monitoring of intracellular release of RA-V
The unique redox dual-responsiveness and the activatable fluorescence of RA-S-S-Cy@PLGA NPs made them suitable for selective imaging of cancer cells and monitoring of drug release in situ. To assess this capability, the intracellular delivery of RA-S-S-Cy@PLGA NPs and the redox-triggered release of RA-V were tracked in real-time by confocal fluorescence imaging. Phorbol myristate acetate (PMA), through the activation of NADPH oxidase and the formation of superoxide, is converted to other reactive oxygen species such as H2O2 [39]. Thus, HCT-116 cells were stimulated by PMA to induce H2O2 generation [40]. The PMA-stimulated HCT-116 cells displayed strong green fluorescence in lysosomes after a 2 h incubation of RA-S-S-Cy@PLGA NPs (Figure 5), showing that RA-S-S-Cy@PLGA NPs were efficiently uptaken by cancer cells via receptor-mediated endocytosis. The PMA-stimulated HCT-116 cells were then incubated with fresh culture medium for an additional 4 h; the green fluorescence of DiO in lysosomes had no obvious change, whereas the red fluorescence of Cy5.5 in cell cytoplasm was increased significantly (Figure 5). The results demonstrate the specific H2O2-mediated release and subsequent GSH-induced activation of RA-S-S-Cy. The redox-activatable fluorescence imaging capability of RA-S-S-Cy@PLGA NPs allowed for more precise theranostics at the subcellular level, providing a convenient method for real-time monitoring of drug delivery and therapeutic efficacy in a noninvasive manner.
Evaluation of antitumor activity of RA-S-S-Cy@PLGA NPs in vitro
The in vitro cytotoxicity of empty NPs, RA-S-S-Cy@PLGA NPs (without catalase), free RA-V, RA-S-S-Cy, and RA-S-S-Cy@PLGA NPs was investigated by the MTT assay. The empty NPs showed no cytotoxicity to HCT-116 cells and NCM460 cells (Figure S11). RA-S-S-Cy showed stronger cytotoxicity than free RA-V to HCT-116 cells. The enhancement was attributed to the GSH-triggered rapid release of RA-V in the cells, which led to a buildup of intracellular local drug and thus induced cell apoptosis. Moreover, the positive charge of the NH3+ group on the Cy5.5 of RA-S-S-Cy enhanced selective cellular uptake [41]. RA-S-S-Cy@PLGA NPs showed significant cytotoxicity to HCT-116 cells, which was higher than that of free RA-V (Figure 6), indicating the enhancement of antitumor activity by incorporating RA-V into the redox-responsive nanosystem. To investigate whether the c(RGDfK) and H2O2-responsive features of RA-S-S-Cy@PLGA NPs played a key role in improving the antitumor activity of RA-V, RA-S-S-Cy@PLGA NPs (without catalase) and RA-S-S-Cy were also used for comparison in MTT assay. As shown in Figure 6, RA-S-S-Cy@PLGA NPs (without catalase) and RA-S-S-Cy exhibited lower cytotoxicity at the same concentration compared to RA-S-S-Cy@PLGA NPs. The MTT assay results revealed that the cytotoxicity of RA-V in RA-S-S-Cy@PLGA NPs was improved by exploiting H2O2-responsive payload release and GSH-activated disulfide bond breaking, which increase the local drug concentration to effectively induce tumor cells apoptosis. After being uptaken by HCT-116 cells via receptor-mediated endocytosis into the lysosome (Figure 4) and triggered by the intracellular stimulus, RA-S-S-Cy@PLGA NPs rapidly released their drug payload, leading to a buildup of an intracellular local drug concentration that was higher than the cell-killing threshold and thus led to cell death. These observations suggested that the role of the RGD-targeted and redox-responsive nanosystem was analogous to that of a “Trojan Horse” which hides and transports soldiers (anticancer drugs) across the walls of the castle (cell membranes) [42].
Figure 4
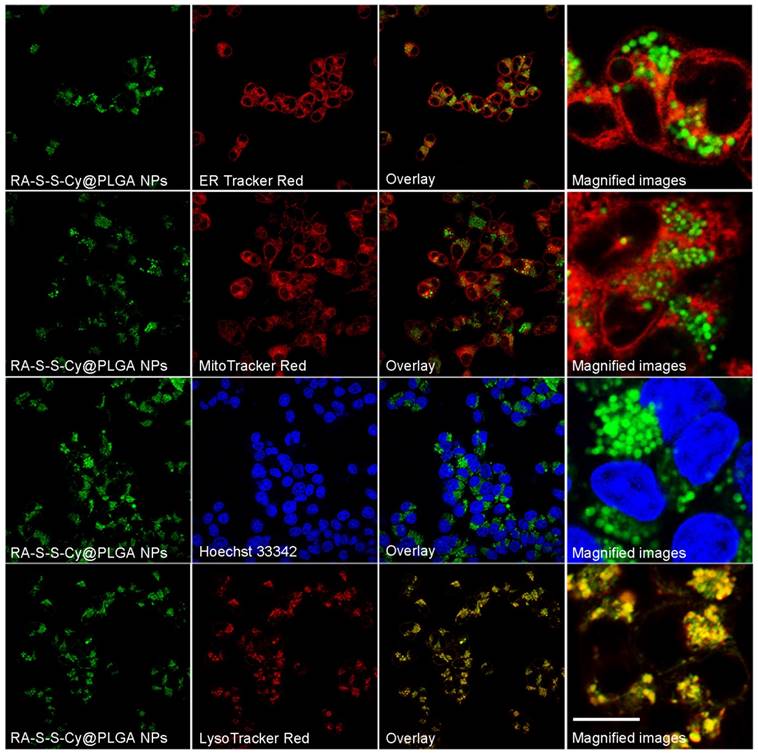
Co-localization images of RA-S-S-Cy@PLGA NPs in HCT-116 cells. HCT-116 cells were incubated with RA-S-S-Cy@PLGA NPs at 37 °C for 2 h, and then incubated with 100 nM LysoTracker Red, ER Tracker Red, Hoechst 33342 and MitoTracker Red 10 min. Scale bar: 20 μm.
Figure 5
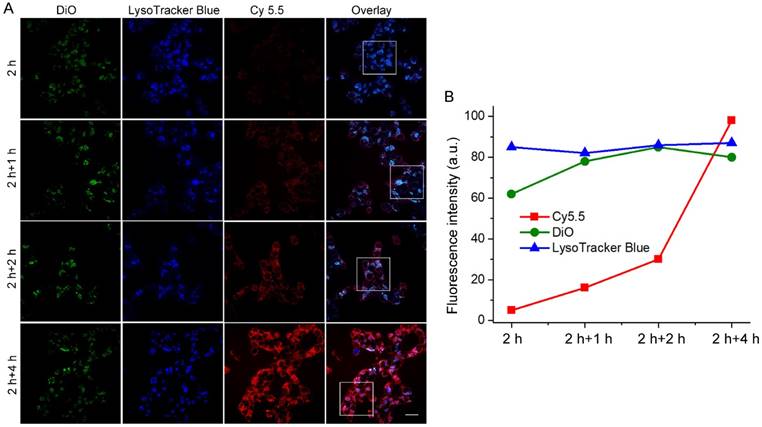
(A) Real-time confocal fluorescence imaging and (B) quantitative fluorescence intensities of PMA-stimulated HCT-116 cells incubated with RA-S-S-Cy@PLGA NPs at 37 °C for 2 h and further incubated with fresh culture medium for an additional 4 h. RA-S-S-Cy@PLGA NPs were tracked using DiO inserted into the PLGA shells in the green channel with excitation at 488 nm. Release of Cy 5.5 was recorded in the red channel with excitation at 633 nm. Scale bars: 20 μm.
Figure 6
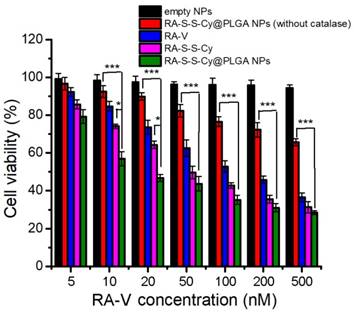
MTT assay of HCT-116 cells in the presence of different concentrations of empty NPs, RA-S-S-Cy@PLGA NPs (without catalase), free RA-V, RA-S-S-Cy, and RA-S-S-Cy @PLGA NPs. Data are mean ± SD (n=3). *P < 0.05, **P < 0.01, ***P < 0.001.
In vivo targeted imaging and in situ tracking of drug release in subcutaneous tumor-bearing mice
Owing to the intrinsic redox-activatable fluorescence of RA-S-S-Cy@PLGA NPs, in situ monitoring of drug release specifically in tumors could be realized by non-invasive near-infrared fluorescence imaging. To assess this, time-dependent in vivo imaging was performed in subcutaneous HCT-116 tumor-bearing BALB/c nude mice. After injection of 10 mg kg-1 RA-S-S-Cy@PLGA NPs, the fluorescence in the tumor region increased significantly and could be distinguished from the normal tissues within 24 h post injection (Figure 7A). The ex vivo fluorescence images showed that only the tumor tissue displayed strong fluorescence signal, while other organs (liver, lung, etc.) displayed negligible fluorescence signal, indicating tumor-specific activation of the RA-S-S-Cy@PLGA NPs (Figures 7B-C). To further confirm that the highly specific tumor imaging was attributed to H2O2-triggered payload release and GSH-activatable fluorescence of RA-S-S-Cy@PLGA NPs, Cy NPs (RA-S-S-Cy replaced with Cy5.5) and RA-S-S-Cy@PLGA NPs (without catalase) were used for in vivo imaging as controls. At 24 h post injection of Cy NPs, the fluorescence in liver and kidney were higher than that of RA-S-S-Cy@PLGA NPs-treated mice, which suggested that the redox dual-responsiveness played a key role in tumor-specific fluorescence activation. The fluorescence in the tumor after treatment with RA-S-S-Cy@PLGA NPs (without catalase) was very weak because the fluorescence of Cy5.5 in RA-S-S-Cy was quenched when linked to RA-V. Due to the lack of H2O2-triggered drug release feature, RA-S-S-Cy@PLGA NPs (without catalase) could not release RA-S-S-Cy and activate the fluorescence of Cy5.5. To demonstrate the role of c(RGDfK) moiety in enhancing selective celluar uptake of RA-S-S-Cy@PLGA NPs, a RA-S-S-Cy@PLGA NPs treatment group and a free cRGD peptide blocking group were also used for in vivo imaging. The tumor-targeted fluorescence activation of RA-S-S-Cy@PLGA NPs (without cRGD) was lower than that of RA-S-S-Cy@PLGA NPs, and the fluorescence activation of RA-S-S-Cy@PLGA NPs was blocked by excess free c(RGDfK) (Figure S12). These results confirm that RA-S-S-Cy@PLGA NPs could achieve in vivo targeted tumor imaging and in situ tracking of drug release, which provides significant advancement in cancer therapeutic monitoring and deeper exploration of theranostic drug-delivery systems.
Evaluation of the antitumor activity of RA-S-S-Cy@PLGA NPs in vivo
The in vivo targeted antitumor activity of RA-S-S-Cy@PLGA NPs was investigated using a xenograft mouse model of HCT-116 cells. To assess tumor growth inhibition, tumor-bearing mice were intravenously injected with saline, empty NPs, RA-S-S-Cy@PLGA NPs (without catalase), RA-V cNPs, RA-S-S-Cy, and RA-S-S-Cy @PLGA NPs, and the tumor volume was measured after treatment (Figure 8A). Tumor growth was significantly suppressed after administration of RA-S-S-Cy@PLGA NPs and the body weights of the mice showed no significant changes (Figure 8D), indicating the high in vivo antitumor efficiency of RA-S-S-Cy@PLGA NPs. Also, the tumor volume was decreased in vivo with increasing RA-S-S-Cy@PLGA NPs dose (Figure S13). Its therapeutic efficacy was comparable to the same dose of RA-V cNPs, which are RA-V-loaded control NPs formed from a widely applied commercial polymer Pluronic F-127. Since the PEO-PPO-PEO block copolymer has no biological responsiveness, it was used as a control polymer to demonstrate the role of redox dual-responsive vesicle in improving the antitumor activity of RA-V. The mice treated with RA-V cNPs exhibited lower tumor growth inhibition rates compared with those treated with RA-S-S-Cy@PLGA NPs, validating the enhancement in RA-V efficiency by its incorporation inside the redox dual-responsive vesicles. Similarly, the mice treated with RA-S-S-Cy@PLGA NPs (without catalase) or RA-S-S-Cy exhibited lower antitumor effect than those treated with RA-S-S-Cy@PLGA NPs. The results suggested that the ROS/GSH dual-responsive feature led to precisely triggered release of the payload, which enhanced the antitumor activity of RA-V.
Figure 7
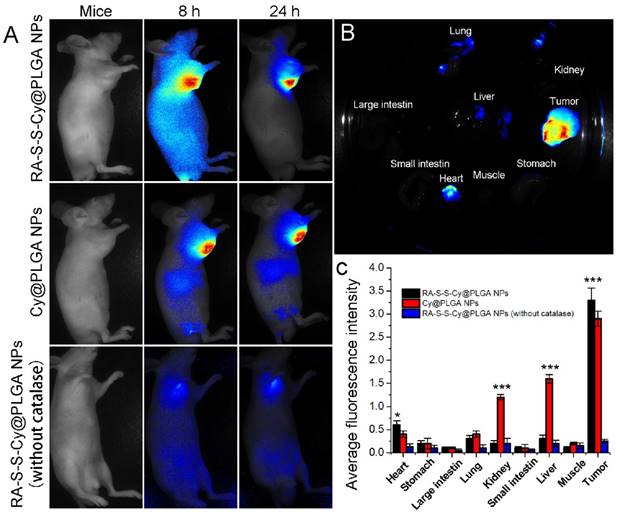
(A) Time-dependent in vivo fluorescence images of subcutaneous HCT-116 tumor-bearing mice after i.v. injection of 10 mg kg-1 RA-S-S-Cy@PLGA NPs, Cy@PLGA NPs, or RA-S-S-Cy@PLGA NPs (without catalase). The fluorescence images were acquired using an IVIS Spectrum instrument equipped with 675/30 nm excitation and 720/20 nm emission filters. (B) Ex vivo fluorescence imaging of tumor and normal tissues harvested from the euthanized tumor-bearing nude mice at 24 h post injection of RA-S-S-Cy@PLGA NPs. (C) Ex vivo fluorescence intensity of the major organs and tumor tissues of nude mice 24 h after receiving RA-S-S-Cy@PLGA NPs, Cy@PLGA NPs, or RA-S-S-Cy@PLGA NPs (without catalase) via tail vein injection. Data are mean ± SD (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 8
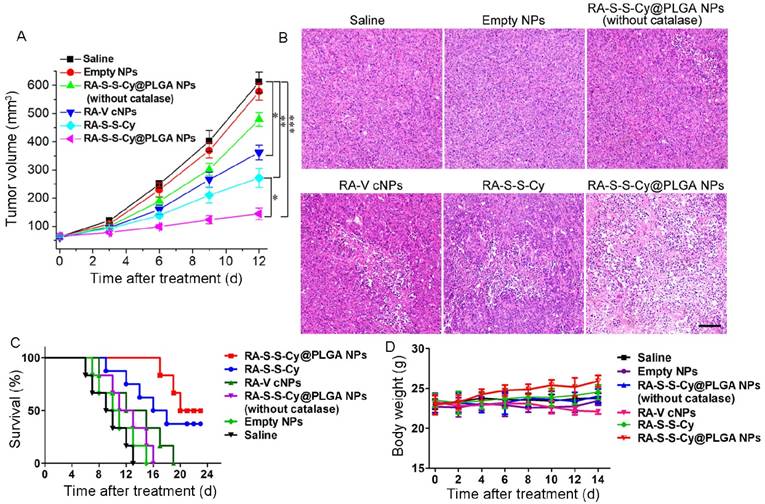
(A) Change in tumor volume with various treatments. Data are mean ± SD (n=6), *P < 0.05, **P < 0.01, ***P < 0.001. (B) Histological observation of the tumor tissues from various treatment groups stained with H&E. Scale bar: 100 µm. (C) Survival rates of tumor-bearing mice with various treatments (n = 6). (D) Body weight variation of tumour-bearing mice during treatment.
Hematoxylin and eosin (H&E) staining of tissue sections from the different treatment groups at day 7 post-treatment was performed to further assess the in vivo chemotherapeutic effect of RA-S-S-Cy@PLGA NPs (Figure 8B). Large numbers of apoptotic and necrotic tumor cells were observed after treatment with RA-S-S-Cy@PLGA NPs, while fewer apoptotic cells were observed in the tumors of control mice after other treatments. More importantly, the survival rates were significantly improved in the mice treated with RA-S-S-Cy@PLGA NPs compared to other groups (Figure 8C). The results indicated that RA-S-S-Cy@PLGA NPs exhibited high in vivo anti-tumor efficiency owing to the targeted ROS/GSH dual-activatable cytotoxicity in the tumor. Moreover, H&E-stained images of tissue sections from different organs of mice after RA-S-S-Cy@PLGA NPs treatment and age-matched healthy mice without treatment showed that RA-S-S-Cy@PLGA NPs selectively treated the tumors and no obvious pathological abnormalities were observed on major normal organs including heart, liver, spleen, lung and kidney of mice. (Figure S14).
Overcoming tumor hypoxia by RA-S-S-Cy@PLGA NPs
First, generation of O2 from RA-S-S-Cy@PLGA NPs was investigated by ultrasound imaging, which showed that a large number of O2 bubbles were produced when RA-S-S-Cy@PLGA NPs were incubited with a low concentration of H2O2 (Figure 1D). Furthermore, to confirm whether RA-S-S-Cy@PLGA NPs could overcome hypoxia in vivo, hypoxia-inducible factor (HIF)-1α staining assay [43] was carried out. As shown in Figure S15, the tumor tissues of the untreated group and the RA-S-S-Cy@PLGA NPs (without catalase) treated-group were stained dark brown with hypoxic characteristics, indicating accumulation of HIF-1α under hypoxic conditions. In contrast, for the RA-S-S-Cy@PLGA NPs treated-group, few cells were stained dark brown, suggesting that RA-S-S-Cy@PLGA NPs could overcome hypoxia in the tumor owing to generation of O2.
Conclusion
In summary, we have successfully constructed a ROS/GSH dual-activatable and O2‑evolving theranostic nanosystem (RA-S-S-Cy@PLGA NPs) for highly efficient therapy against hypoxic tumors and for real-time monitoring of drug release. Modification with c(RGDfK) on the PLGA shell targeted RA-S-S-Cy@PLGA NPs to tumor cells via αvβ3 integrin-mediated uptake. After being selectively taken up by αvβ3 integrin-rich cancer cells, intracellular H2O2 penetrated into the RA-S-S-Cy@PLGA NPs, leading to release of the theranostic molecular probe (RA-S-S-Cy) accompanied by production of gaseous O2. Subsequently, the fluorescence and cytotoxicity of RA-S-S-Cy were activated upon specific GSH-induced cleavage of the disulfide bond. This ROS/GSH dual-responsive feature enables controllable release of the anticancer agent RA-V, which significantly improved its chemotherapeutic efficiency in the tumor. More importantly, the activatable NIR fluorescence made it possible to realize in vivo and in situ dynamic monitoring of drug release, making significant advances toward investigations of further mechanisms of RA-V internalization and cytotoxicity in tumor cells. Overall, RA-S-S-Cy@PLGA NPs would be efficient theranostic nanosystems that could achieve more precise therapy in hypoxic tumors by monitoring prodrug activation by NIR fluorescence imaging.
Abbreviations
MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl- tetrazolium bromide; NIR: near infrared; PET: photoinduced electron transfer; PLGA: D,L-lactic-co- glycolic acid; SEM: scanning electron microscopy; TEM: transmission electron microscopy; DLS: dynamic light scattering; HPLC: high performance liquid chromatography; DiO: 3,3'-dioctadecyloxacarbocyanineperchlorate; PMA: Phorbol myristate acetate.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank the National Natural Science Foundation of China (21602255 and 31470428), the Natural Science Foundation of Jiangsu Province (BK20160745), the National New Drug Innovation Major Project of Ministry of Science and Technology of China (2017ZX09309027), the Program of Innovative Research Team of Jiangsu Province, the Fund of Chinese Academy of Sciences (XDA09030301-4), the Fund for Introduction of High-level Talents from China Pharmaceutical University, and “111” Project (B16046) from the Ministry of Education of China and the State Administration of Foreign Experts Affairs of China for financial support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang X, Guo Z. Targeting and delivery of platinum-based anticancer drugs. Chem Soc Rev. 2013;42:202-224
2. Bhattarai P, Hameed S, Dai Z. Recent advances in anti-angiogenic nanomedicines for cancer therapy. Nanoscale. 2018;10:5393-5423
3. Liang C, Xu L, Song G. et al. Emerging nanomedicine approaches fighting tumor metastasis: animal models, metastasis-targeted drug delivery, phototherapy, and immunotherapy. Chem Soc Rev. 2016;45:6250-6269
4. Tan N, Zhou J. Plant cyclopeptides. Chem Rev. 2006;106:840-895
5. Chen X, Zhao S, Wang Z. et al. Rubicordins A-C, new cyclopeptides from Rubia cordifolia, with cytotoxicity and inhibiting NF-κB signaling pathway. Tetrahedron. 2015;71:9673-9678
6. Fan J, Su J, Peng Y. et al. Rubiyunnanins C-H, cytotoxic cyclic hexapeptides from Rubia yunnanensis inhibiting nitric oxide production and NF-κB activation. Bioorg Med Chem. 2010;18:8226-8234
7. Wang Z, Zhao S, Zhao L. et al. Rubipodanin A, the first natural N-desmonomethyl rubiaceae-type cyclopeptide from Rubia podantha, indicating an important role of the N9-methyl group in the conformation and bioactivity. Plos One. 2015;10:e0144950
8. Huang M, Zhao S, Zeng G. et al. Rubischumanins A-C, new cytotoxic cyclopeptides from Rubia schumanniana. Tetrahedron. 2014;70:7627-7631
9. Yue G, Fan J, Lee J. et al. Cyclopeptide RA-V inhibits angiogenesis by down-regulating ERK1/2 phosphorylation in HUVEC and HMEC-1 endothelial cells. Br J Pharm. 2011;164:1883-1898
10. Leung H, Wang Z, Yue G. et al. Cyclopeptide RA-V inhibits cell adhesion and invasion in both estrogen receptor positive and negative breast cancer cells via PI3K/AKT and NF-κB signaling pathways. Biochim Biophys Acta. 2015;1853:1827-1840
11. Wang Z, Zhao S, Song L. et al. Natural cyclopeptide RA-V inhibits the NF-κB signaling pathway by targeting TAK1. Cell Death Dis. 2018;9:715-731
12. Fang X, Chen W, Fan J. et al. Plant cyclopeptide RA-V kills human breast cancer cells by inducing mitochondria-mediated apoptosis through blocking PDK1-AKT interaction. Toxicol Appl Pharmacol. 2013;267:95-103
13. Yang J, Yang T, Yan W. et al. TAK1 inhibition by natural cyclopeptide RA-V promotes apoptosis and inhibits protective autophagy in Kras-dependent non-small-cell lung carcinoma cells. RSC Adv. 2018;8:23451-23458
14. Qiao Z, Zhang D, Hou C. et al. A pH-responsive natural cyclopeptide RA-V drug formulation for improved breast cancer therapy. J Mater Chem B. 2015;3:4514-4523
15. Chen H, Wang Y, Yao Y. et al. Sequential delivery of cyclopeptide RA-V and doxorubicin for combination therapy on resistant tumor and in situ monitoring of cytochrome c release. Theranostics. 2017;7:3781-3793
16. Santra S, Kaittanis C, Santiesteban OJ. et al. Cell-specific, activatable and theranostic prodrug for dual targeted cancer imaging and therapy. J Am Chem Soc. 2011;133:16680-16688
17. Kim EJ, Bhuniya S, Lee H. et al. An activatable prodrug for the treatment of metastatic tumors. J Am Chem Soc. 2014;136:13888-13894
18. Wu X, Sun X, Guo Z. et al. In vivo and in situ tracking cancer chemotherapy by highly photostable nir fluorescent theranostic prodrug. J Am Chem Soc. 2014;136:3579-3588
19. Lee MH, Kim EJ, Lee H. et al. Liposomal texaphyrin theranostics for metastatic liver cancer. J Am Chem Soc. 2016;138:16380-16387
20. Kang Y, Ju X, Ding L. et al. Reactive oxygen species and glutathione dual redox-responsive supramolecular assemblies with controllable release capability. ACS Appl Mater Interfaces. 2017;9:4475-4484
21. Chen H, Jia H, Tham HP. et al. Theranostic prodrug vesicles for imaging guided codelivery of camptothecin and siRNA in synergetic cancer therapy. ACS Appl Mater Interfaces. 2017;9:23536-23543
22. Dai Y, Xu C, Sun X. et al. Nanoparticle design strategies for enhanced anticancer therapy by exploiting the tumour microenvironment. Chem Soc Rev. 2017;46:3830-3852
23. Chen H, Bi Q, Yao Y. et al. Dimeric BODIPY-loaded liposomes for dual hypoxia marker imaging and activatable photodynamic therapy against tumors. J Mater Chem B. 2018;6:4351-4362
24. Tian J, Zhou J, Zhen S. et al. A pH-activatable and aniline-substituted photosensitizer for near-infrared cancer theranostics. Chem Sci. 2015;6:5969-5977
25. Chung M, Chen K, Liang H. et al. A liposomal system capable of generating CO2 bubbles to induce transient cavitation, lysosomal rupturing, and cell necrosis. Angew Chem Int Edit. 2012;124:10236-10240
26. Wen A, Steinmetz NF. Design of virus-based nanomaterials for medicine, biotechnology, and energy. Chem Soc Rev. 2016;45:4074-4126
27. Dai L, Cai R, Li M. et al. Dual-targeted cascade-responsive prodrug micelle system for tumor therapy in vivo. Chem Mater. 2017;29:6976-6992
28. Chen H, He W, Guo Z. An H2O2-responsive nanocarrier for dual-release of platinum anticancer drugs and O2: controlled release and enhanced cytotoxicity against cisplatin resistant cancer cells. Chem Commun. 2014;50:9714-9717
29. Chen H, Tian J, He W. et al. H2O2-activatable and O2-evolving nanoparticles for highly efficient and selective photodynamic therapy against hypoxic tumor cells. J Am Chem Soc. 2015;137:1539-1547
30. Abo M, Urano Y, Hanaoka K. et al. Development of a highly sensitive fluorescence probe for hydrogen peroxide. J Am Chem Soc. 2011;133:10629-10637
31. Wang J, Li W, Lu Z. et al. The use of RGD-engineered exosomes for enhanced targeting ability and synergistic therapy toward angiogenesis. Nanoscale. 2017;9:15598-15605
32. Kim G, Lee YEK, Xu H. et al. Nanoencapsulation method for high selectivity sensing of hydrogen peroxide inside live cells. Anal Chem. 2010;82:2165-2169
33. Tu F, Lee D. Controlling the stability and size of double-emulsion-templated poly(lactic-co-glycolic) acid microcapsules. Langmuir. 2012;28:9944-9952
34. Wang C, Yan Q, Liu H. et al. Different EDC/NHS activation mechanisms between PAA and PMAA brushes and the following amidation reactions. Langmuir. 2011;27:12058-12068
35. Huber W, Koella JC. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993;55:257-261
36. Vila-Viçosa D, Teixeira VH, Santos HAF. Conformational study of GSH and GSSG using constant-pH molecular dynamics simulations. J Phys Chem B. 2013;117:7507-7517
37. Yuan Y, Kwok RTK, Tang B. et al. Targeted theranostic platinum(IV) prodrug with a built-in aggregation-induced emission light-up apoptosis sensor for noninvasive early evaluation of its therapeutic responses in situ. J Am Chem Soc. 2014;136:2546-2554
38. Bilgiçer B, Thomas III SW, Shaw BF. et al. A non-chromatographic method for the purification of a bivalently active monoclonal IgG antibody from biological fluids. J Am Chem Soc. 2009;131:9361-9367
39. de Gracia Lux C, Joshi-Barr S, Nguyen T. et al. Biocompatible polymeric nanoparticles degrade and release cargo in response to biologically relevant levels of hydrogen peroxide. J Am Chem Soc. 2012;134:15758-15764
40. Qu L, Li D, Qin L. et al. Selective and sensitive detection of intracellular O2(·-) using Au NPs/cytochrome c as SERS nanosensors. Anal Chem. 2013;85:9549-9555
41. Yu B, Zhang Y, Zheng W. et al. Positive surface charge enhances selective cellular uptake and anticancer efficacy of selenium nanoparticles. Inorg Chem. 2012;51:8956-8963
42. Cai Y, Shen H, Zhan J. et al. Supramolecular “ Trojan Horse ” for nuclear delivery of dual anticancer drugs. J Am Chem Soc. 2017;139:2876-2879
43. Jin CS, Lovell JF, Chen J. et al. Ablation of hypoxic tumors with dose-equivalent photothermal, but not photodynamic, therapy using a nanostructured porphyrin assembly. ACS Nano. 2013;7:2541-2450
Author contact
![]() Corresponding authors: Ninghua Tan, Email: nhtanedu.cn; Zhihao Zhang, zzh-198518com
Corresponding authors: Ninghua Tan, Email: nhtanedu.cn; Zhihao Zhang, zzh-198518com
Citation styles
APA
Chen, H., Li, F., Yao, Y., Wang, Z., Zhang, Z., Tan, N. (2019). Redox Dual-Responsive and O2‑Evolving Theranostic Nanosystem for Highly Selective Chemotherapy against Hypoxic Tumors. Theranostics, 9(1), 90-103. https://doi.org/10.7150/thno.30259.
ACS
Chen, H.; Li, F.; Yao, Y.; Wang, Z.; Zhang, Z.; Tan, N. Redox Dual-Responsive and O2‑Evolving Theranostic Nanosystem for Highly Selective Chemotherapy against Hypoxic Tumors. Theranostics 2019, 9 (1), 90-103. DOI: 10.7150/thno.30259.
NLM
Chen H, Li F, Yao Y, Wang Z, Zhang Z, Tan N. Redox Dual-Responsive and O2‑Evolving Theranostic Nanosystem for Highly Selective Chemotherapy against Hypoxic Tumors. Theranostics 2019; 9(1):90-103. doi:10.7150/thno.30259. https://www.thno.org/v09p0090.htm
CSE
Chen H, Li F, Yao Y, Wang Z, Zhang Z, Tan N. 2019. Redox Dual-Responsive and O2‑Evolving Theranostic Nanosystem for Highly Selective Chemotherapy against Hypoxic Tumors. Theranostics. 9(1):90-103.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.