Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
Conflicts of Interest
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
Conflicts of Interest
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5419-5433. doi:10.7150/thno.27876 This issue Cite
Research Paper
Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking
Hui Xu1,2*, Min Ling3*, Junchao Xue1,2*, Xiangyu Dai1,2*, Qian Sun1,2, Chao Chen1,2, Yi Liu1,2, Liang Zhou4, Jianping Liu4, Fei Luo5, Qian Bian3, Qizhan Liu1,2 ![]()
1. Institute of Toxicology, School of Public Health, Nanjing Medical University, Nanjing 211166, Jiangsu, People's Republic of China.
2. The Key Laboratory of Modern Toxicology, Ministry of Education, School of Public Health, Nanjing Medical University, Nanjing 211166, Jiangsu, People's Republic of China.
3. Jiangsu Provincial Center for Disease Control and Prevention, Nanjing 210009, Jiangsu, People's Republic of China.
4. Liyang Center for Disease Control and Prevention, Liyang 213300, Jiangsu, People's Republic of China.
5. Faculty of Public Health, School of Medicine, Shanghai Jiaotong University, Shanghai 200025, People's Republic of China.
* Theses authors contributed equally to this work.
Received 2018-6-14; Accepted 2018-10-4; Published 2018-10-29
Citation:
Xu H, Ling M, Xue J, Dai X, Sun Q, Chen C, Liu Y, Zhou L, Liu J, Luo F, Bian Q, Liu Q. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics 2018; 8(19):5419-5433. doi:10.7150/thno.27876. https://www.thno.org/v08p5419.htm
Other stylesAbstract

Rationale: Aberrant bronchial epithelium-fibroblast communication is essential for the airway remodeling that contributes to chronic obstructive pulmonary disease (COPD). Exosomes have emerged as novel mediators of intercellular communication, but their role in cigarette smoke (CS)-induced COPD is unknown. Here, we investigated the role of exosomal miR-21 in the dysfunctional epithelium-fibroblast cross-talk caused by CS.
Methods: Normal or CS extract (CSE)-treated human bronchial epithelial (HBE) cells were co-cultured with bronchial fibroblasts (MRC-5 cells). Exosomes were obtained from culture media or serum by use of commercial kits. The size distribution and concentration of exosomes were analyzed by nanoparticle tracking analysis using a ZetaView particle tracker from ParticleMetrix. Inhibition of miR-21 levels by tail vein injection of antagomir-21 into mice exposed to CS was used to demonstrate the role of miR-21 in airway remodeling leading to COPD in animals.
Results: For MRC-5 cells, co-culture with CSE-treated HBE cells or with exosomes derived from CSE-treated HBE cells resulted in the myofibroblast differentiation phenotype. Exosomal miR-21 was responsible for myofibroblast differentiation through hypoxia-inducible factor 1α (HIF-1α) signaling by targeting the von Hippel-Lindau protein (pVHL); HIF-1α transcriptionally regulated the α-SMA gene. For mice, downregulation of miR-21 prevented CS-induced airway remodeling. The levels of exosomal miR-21 were high in sera of smokers and COPD patients and inversely correlated with FEV1/FVC.
Conclusion: We demonstrate that CS triggers the modification of exosome components and identify miR-21 derived from bronchial epithelial cells as a mediator of myofibroblast differentiation through the pVHL/HIF-1α signaling pathway, which has potential value for diagnosis and treatment of COPD.
Keywords: cigarette smoking, COPD, airway remodeling, exosome, microRNA
Introduction
Chronic obstructive pulmonary disease (COPD), caused mainly by cigarette smoking, is characterized by chronic inflammation and defective tissue repair, leading to irreversible airflow limitation as a result of narrowing of the small airways and lung emphysema[1]. Airway remodeling due to exposure of cigarette smoke (CS), leads to narrowing of the small airways; it is associated with differentiation of bronchial fibroblasts to myofibroblasts and results in an inflammatory response[2, 3]. Myofibroblasts are characterized by expression of α-smooth muscle actin (α-SMA), an enhanced capacity to migrate, and production of extracellular matrix (ECM, e.g. collagen) [4]. However, the molecular mechanisms underlying the airway fibrosis response to CS are not fully understood.
Aberrant communication between the bronchial epithelium and fibroblasts is involved in the pathogenesis of COPD[5]. Bronchial epithelial cells, a first-line contact with harmful substances, are a source of various cytokines, chemokines, and factors that modulate mesenchymal fibroblasts and smooth muscle cells and recruit immune cells in response to CS[6, 7]. Exosomes, membrane-bound vesicles 50-150 nm in diameter, are modulators of cell-cell communication[8]. Exosomes exert their effects via delivering cargo, which may include proteins, DNA, messenger RNA (mRNA), and microRNAs (miRNAs) [9]. Environmental exposure can modify the composition of exosomes and cause them to participate in pathogenesis of diseases[10, 11].
MicroRNA-21 (miR-21), an oncomiR, is involved in regulation of a variety of respiratory diseases, including asthma, fibrosis, and cancer[12-14]. Our group has demonstrated that, for bronchial epithelial cells, CS extract (CSE) induces upregulation of exosomal miR-21 levels, which are involved in lung angiogenesis[15]. Although miR-21 is associated with the pathogenic process of COPD and has diagnostic potential[16, 17], its role in mediating the dysfunction of bronchial fibroblasts in the context of CS exposure is undefined.
For the present study, we hypothesized that up-regulation of exosomal miR-21 in bronchial epithelial cells modified by exposure to CS disturbs the communication between bronchial epithelial cells and fibroblasts and promotes myofibroblast differentiation. Therefore, we evaluated the expression of miR-21 in exosomes derived from bronchial epithelial cells exposed to CSE and investigated, in cell cultures and in animals, the mechanism for exosomal miR-21 in airway remodeling.
Methods
Subjects and specimens
This cross-sectional study was approved by Institutional Review Board of Nanjing Medical University. We enrolled three groups of subjects from Liyang in Jiangsu province, including 26 non-smokers without COPD, 24 smokers without COPD, and 29 smokers with COPD. COPD patients were defined as those who had dyspnea, chronic cough, and/or sputum production and a history of exposure to risk factors for the disease along with a postbronchodilator forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC) ratio (representing the percent of the lung size that can be exhaled in one second) of < 70%, according to the guidelines of the Global Initiative for Chronic Obstructive Lung Disease[18]. Patients with comorbidities, including asthma, interstitial lung diseases, heart failure, and/or neuromuscular disease, were excluded. Smoking history, defined as in our previous studies, was determined from the mean number of pack-years of cigarette consumption[19]. Venous blood samples (5 mL) were collected from individual subjects, and, after coagulation at room temperature for 30 min, the samples were centrifuged at 1,000 g for 15 min. The sera were collected in 1.5-mL Eppendorf tubes and stored at -80 °C for analysis.
Cigarette smoke (CS) exposure
Male BALB/c mice at 6-8 weeks of age were purchased from Shanghai SLRC Laboratory Animal Co. Ltd. (China) and housed in animal facilities at the Jiangsu Provincial Center for Disease Control and Prevention. Animals were treated humanely and with regard for alleviation of suffering according to a protocol approved by the Jiangsu Provincial Center for Disease Control and Prevention Institutional Animal Care and Use Committee.
Mice were exposed to smoke from 3R4F Research Cigarettes (University of Kentucky, USA) as described previously[20]. Briefly, mice were exposed to CS (500 mg/m3 total particulate matter, TPM) in a whole-body exposure system (Beijing Huironghe Technology CO., Ltd., China) for 60 min twice daily, 4 h apart, 5 days a week for a total of 8 weeks. Humidity, temperature, and O2 level in the chamber were measured continuously during the exposure period. In the first week, there was increasing exposure, as follows: mice were placed in the chamber and exposed for 20 min on the first day, 30 min on the second day, and 60 min on the third day, twice per day and until the end. Age-matched mice kept in a similar environment without exposure to CS served as controls. For inhibition of miR-21, male BALB/c mice (age 6-8 weeks) were divided into four groups (normal control, CS, CS plus antagomir-21, and CS plus antagomir negative control). Mice in the antagomir-21 and antagomir negative control (50 nmol, RiBoBio, China) groups were dosed by tail vein injection every week. CS exposure was performed as described above. Experiments were accomplished with n = 6 randomized animals per group.
Lung function measurement
For mice, airway hyper-responsiveness (AHR) was measured as the change in airway function by use of whole-body plethysmography (Buxco Electronics Ltd., USA) as previously reported[21]. Briefly, mice were placed unrestrained in a chamber connecting a sensitive pressure transducer to measure pressure changes inside the chamber. After acclimation, increasing concentrations of methacholine (0, 12.5, 25, and 50 mg/mL) were nebulized for 2 min and enhanced pause (Penh) was recorded during the response period using FinePoint software (Buxco Electronics Ltd., USA). Penh, a dimensionless unit, correlates with pulmonary resistance. Values were averaged and expressed as absolute Penh values.
Collection of bronchoalveolar lavage fluid (BALF) and cell counting
In an anesthetized animal, a transversal incision between the beginning of the rib cage and the head was made; afterwards, muscle was moved to expose the trachea. Then a cannula was inserted into the trachea down to the carina and fixed to perform a lavage (3×) with a syringe containing an isotonic saline solution (at 37 °C) in a 1:15 (volume: body weight) relation. The recovered solution was centrifuged at 2,000 rpm for 5 min, and the cell pellet was suspended in a final volume of 0.5 ml to perform cell counting with a Countess™ II Automated Cell Counter (Thermo Fisher Scientific, USA). For differential counting of mononuclear cells and neutrophils, the cells were subjected to Wright-Giemsa staining (Solarbio Life Science, China).
Masson trichrome staining
The lungs of mice were fixed with 4% paraformaldehyde and embedded in paraffin. For histological analysis and detection of collagen deposition, successive 5-µm lung sections were placed on slides and subjected to staining with trichrome stain (Masson) kits (Sigma-Aldrich, Germany), according to the manufacturer's instructions. Collagen content was determined by the ratio of collagen surface area (blue) to total surface area (red). Image J software was used to evaluate collagen deposition.
Preparation of cigarette smoke extract (CSE)
CSE was prepared as previously reported[22, 23]. Briefly, the smoke of a 3R4F Research Cigarette (University of Kentucky, USA) was bubbled into a flask containing 10 mL of warm (37 °C) minimum essential medium (MEM) by use of a vacuum pump at a constant speed (each cigarette was smoked for 5 min). The CSE solution was adjusted to pH 7.4 and then sterilized by filtration through a 0.22-µm pore filter (Schleicher & Schuell GmbH, Dassel, Germany). For quality control, the solution was standardized by monitoring the absorbance at 320 nm (A320) and 540 nm (A540). CSE quality was accepted if ΔOD (A320-A540) was between 0.9 and 1.2. The resultant CSE solution was regarded as 100% CSE and was diluted with medium to use in experiments within 1 h.
Cell culture and treatment
Primary human bronchial epithelial cells were obtained from Chi Scientific (Jiangsu, China). Human bronchial epithelial (HBE) cells, a SV40-transformed, normal HBE cell line, and fetal lung fibroblasts (MRC-5) cells, ATCC® CCL-171™ , obtained from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences, were maintained in minimum essential medium (MEM) containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific, USA), 100 mg/mL streptomycin, and 100 U/mL penicillin (Thermo Fisher Scientific, USA) under 5% CO2 at 37 °C. Cells were passaged at a ratio 1:3 every 2 days. After reaching 70-80% confluence, HBE cells were washed with PBS and then grown in MEM supplemented with 10% FBS (depleted of exosomes by ultracentrifugation at 100,000 g overnight) and exposed to 2.5% CSE for 12 h.
In a co-culture model, normal or CSE-treated HBE cells were plated on 0.4-μm pore, 24-mm hanging cell culture inserts (Millipore, Merck, Germany), and MRC-5 cells were seeded on 6-well plates. After the HBE cells were confluent, treated with 2.5% CSE, and left for 12 h in MEM media with 10% exosome-depleted FBS, the inserts were washed twice with PBS before they were placed in co-culture with MRC-5 cells.
Exosome isolation
Exosomes were isolated from the culture medium of normal or CSE-treated HBE cells as previously described[15]. The culture medium was first centrifuged at 3,000 g for 15 min, and the supernatant was filtered through a 0.22-µm PVDF filter (Millipore). Exoquick-TC exosome precipitation solution (System Biosciences) was added to the filtered culture medium at a ratio of 1:5. After mixing and refrigeration for at least 12 h, the mixture was centrifuged at 1,500 g for 30 min. For exosome isolation from serum, 63 μL of ExoQuick exosome precipitation solution (System Biosciences) was added into 250 μL of serum. After refrigeration overnight, the mixture was centrifuged at 1,500 g for 30 min. Exosome pellets were suspended in 1×PBS. The size distribution and concentration of exosomes were analyzed by nanoparticle tracking analysis using a ZetaView particle tracker from ParticleMetrix (Germany).
Exosome uptake
Exosomes derived from HBE cells were labeled with PKH67 Green Fluorescent Cell Linker Kits (Sigma-Aldrich) according to the manufacturer's protocol with minor modifications. Briefly, exosomes were suspended in 250 μL of PBS and mixed with 750 μL of 1:50 diluted PKH67 (for labeling of cell membranes, in Diluent C) and incubated at room temperature. After 5 min, 2 mL of 1% BSA was added to terminate the labeling reaction. The labeled exosomes were isolated as described above, suspended in 1×PBS, and used for uptake experiments.
For such experiments, labeled exosomes (10 μg) were incubated with MRC-5 cells seeded on 24-well plates at 37 °C for 3 h. The cells were washed twice with PBS and fixed with 4 % paraformaldehyde in PBS for 30 min at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma). Images were acquired on a Nikon Eclipse Ti-S inverted microscope (Nikon, Japan) and NIS-Elements microscope imaging software.
Transmission electron microscopy (TEM)
TEM assays were performed as previously reported[24]. Briefly, exosome pellets from cells or sera were suspended in and fixed with 4 % paraformaldehyde and 4 % glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at incubation temperature and kept at 4°C until analysis. A drop of each exosome sample was placed on a carbon-coated copper grid and immersed in 2% phosphotungstic acid solution (pH 7.0) for 30 s. The preparations were examined with a transmission electron microscope (JEM-1200EX, JEOL Ltd., Japan) at an acceleration voltage of 80 kV.
Treatment of CSE-treated HBE cells with the exosome biogenesis inhibitor, GW4869
After being exposed to 2.5% CSE for 12 h, HBE cells were treated with 2.5 mM GW4869 (Sigma-Aldrich) or 0.005% dimethyl sulfoxide (vehicle) for 3 h. Cells were used for co-culture experiments. Media were collected, and exosomes were isolated with ExoQuick.
Quantitative real-time PCR (qRT-PCR) analysis
Total RNA from cultured cells and lung tissues of mice was isolated by use of Trizol reagent (Thermo Fisher Scientific, USA) according to the manufacturer's recommendations. The pellets were suspended in 50 μL of RNase-free water and stored at -80 °C until analysis. Extraction of miRNA from exosomes was accomplished using a commercial miRNeasy Serum/Plasma kit (QIAGEN, #217184) according to the manufacturer's protocol. Synthetic C. elegans miRNA cel-miR-39 (50 fmol, RiBoBio, China) was added to the samples. The purified RNA was eluted with 25 μL of RNase-free water and stored at -80 °C until analysis. Bulge-Loop™ miRNA qRT-PCR Starter Kits (RiboBio, China) and Bulge-loopTM miRNA qRT-PCR Primer Sets (one RT primer and a pair of qPCR primers for each set) specific for miR-21, U6 snRNA, and cel-miR-39 (RiboBio, China) were used to measure the levels of miRNAs. The U6 snRNA and cel-miR-39 were used as endogenous and exogenous controls. Real-time PCR was performed by use of SYBR Green (Fermentas, USA) with a LightCycler 96 instrument (Roche, Swiss). For lung tissues and exosome samples, the formula 2-ΔCt (ΔCt = Ct miRNA - Ct control) was used to express the results of qRT-PCR. To equalize variance prior to statistical analysis, the normalized expression values were transformed to log10 values. To analyze the qRT-PCR results for cellular experiments, the 2-ΔΔCt method was used.
Western blots
The lysis buffer used for Western blotting was non-reducing buffer (Beyotime, China); the sample buffer was reducing buffer (Beyotime, China). Proteins extracted from cultured cells, lung tissues of mice, or exosomes were quantified with BCA protein assay kits (Beyotime, China). Equal amounts (80 μg) of protein were separated on 10% SDS-PAGE and transferred to PVDF membranes (Millipore, USA). Membranes were then incubated overnight at 4ºC with a primary antibody for collagen I (1:2,000, ab138492, Abcam), α-SMA (1:2,000, ab7817, Abcam), hypoxia inducible factor-1 alpha (HIF-1α) (1:1,000, #36169, Cell Signaling Technology), von Hippel Lindau protein (pVHL) (1:1,000, sc-17780, Santa Cruz Biotechnology), tubulin (1:1,000, AF0001, Beyotime), CD9 (1:2,000, ab92726, Abcam), CD63 (1:1,000, ab68418, Abcam), CD81 (1:1,000, ab109201, Abcam), or heat shock protein 90k Da beta 1 (HSP90B1) (1:1,000, #2104, Cell Signaling Technology). The membranes were then incubated with a 1:2,000 dilution of horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibodies for 1 h at room temperature and detected by ECL reagents (BIO-RAD, USA). Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed using Magna ChIP kits (Millipore, USA) according to the manufacturer's recommendations. Briefly, normal or CHBE-Exo-treated MRC-5 cells were fixed with 1% formaldehyde for 10 min. After cell lysis and nuclear lysis, the isolated chromatin was sheared by sonication to lengths mostly between 200 bp and 500 bp. Of the extracts, 10 μL was used as inputs; the remainder was incubated with antibody against HIF-1α or isotype IgG and protein A/G magnetic beads at 4ºC overnight. After reverse cross-linking of the protein/DNA complexes, the DNA was purified by use of spin columns. The primer sequences to amplify a 150-bp region spanning the putative HIF-1 response element within the promoter of the α-SMA gene (ACTA2) were as follows: 5'-ACCTTATCTCTAATACACA-3' (sense) and 5'-GGTGAAAAGAAAATAATTACACA-3' (antisense).
Luciferase reporter assays
The luciferase activity was assessed as previously reported[25]. To investigate the effect of miR-21 on the 3'UTR of pVHL (pVHL-3'UTR), the 3'UTR sequence of pVHL, which was predicted to harbor the miR-21 seed region (CAAGCACA), or a mutant sequence (CTTCGTGA), was inserted into the XhoI and NotI sites of the psiCHECK-2 promoter vector (GENEray, China). These were named psiCH-pVHL-wt and psiCH-pVHL-mut, respectively. Sub-confluent (70-80%) MRC-5 cells were co-transfected with wild-type or mutant pVHL 3'UTR luciferase plasmids and an miR-21 mimic or an miR-NC mimic using Lipofectamine 2000 (Thermo Fisher Scientific, USA). After 48 h of transfection, the cells were harvested for detection using the Dual Luciferase Reporter Assay system (Promega, USA) with an Infinite M200 PRO Multimode microplate reader (TECAN, Swiss). Renilla luciferase activities were used to normalize the transfection efficiency. For exosome treatment, wild-type or mutant pVHL 3'UTR luciferase plasmid-transfected MRC-5 cells were treated with exosomes (50 μg/mL) isolated from normal or CSE-treated HBE cells, and the luciferase activity was assessed as described above.
Statistical analyses
All experiments were performed in triplicate. Derived values are presented as means ± SD. Comparison of means among multiple groups was accomplished by one-way analysis of variance (ANOVA). The expression levels of miR-21in lung tissues and sera were log-transformed. ANOVA and post hoc analysis (Tukey) were performed. A multiple-range least significant difference (LSD) was used for inter-group comparisons. P values < 0.05 were considered statistically significant. All statistical analyses were performed with SPSS 17.0.
Results
MiR-21 is increased in the presence of CS-induced airway obstruction in mice
We first assessed the miR-21 levels in a mouse model of COPD. After 8 weeks of exposure to CS, the mice developed an airway remodeling phenotype, showing augmented AHR, airway thickening, and collagen deposition, as determined by methacholine challenge tests and Masson staining (Fig. 1A-C). An increase in total cell number in the BALF was found for mice exposed to CS, compared with controls. The inflammatory infiltrate, characterized by an increase in mononuclear cells and neutrophils, was also observed for CS-exposed mice (Fig. S1A-C). Increased differentiation of airway fibroblasts to myofibroblasts, a characteristic of enhanced α-SMA and collagen type I (collagen I), are phenotypic features associated with pathological airway remodeling[2, 26]. Western blots revealed that the protein levels of α-SMA and collagen I were upregulated in the lungs of mice exposed to CS (Fig. 1D and 1E). qRT-PCR analysis demonstrated that, in CS-exposed lungs, miR-21 was up-regulated relative to the control (Fig. 1F). These results indicated that, for mice, CS induces airway obstruction and up-regulation of miR-21.
Epithelial cell-derived miR-21 is responsible for the myofibroblast differentiation phenotype of MRC-5 cells
To determine whether miR-21 is involved in the dysfunctional cross-talk of the bronchial epithelium and fibroblasts, a co-culture cell model was developed. When normal or HBE cells treated with CSE and MRC-5 cells were placed in co-culture, miR-21 levels were increased in CSE-treated HBE cells and in MRC-5 cells co-cultured with CSE-treated HBE cells, but not in MRC-5 cells co-cultured with normal HBE cells (Fig. 2A-B). Similarly, the levels of α-SMA and collagen I were upregulated in MRC-5 cells co-cultured with CSE-treated HBE cells (Fig. 2C-D). Then, we investigated the role of miR-21 derived from HBE cells in the myofibroblast differentiation phenotype of MRC-5 cells. Inhibition of miR-21 in HBE cells caused a decrease in α-SMA and collagen I expression in MRC-5 cells co-cultured with HBE cells treated with CSE (Fig. 2E-F). For validation, a miR-21 mimic was used to overexpress miR-21 in HBE cells or MRC-5 cells. Western blotting showed that, in the MRC-5 cells co-cultured with miR-21 mimic-transfected HBE cells or directly incubated with the miR-21 mimic, the miR-21 mimic up-regulated the levels of α-SMA and collagen I expression (Fig. S2A-D).
Figure 1
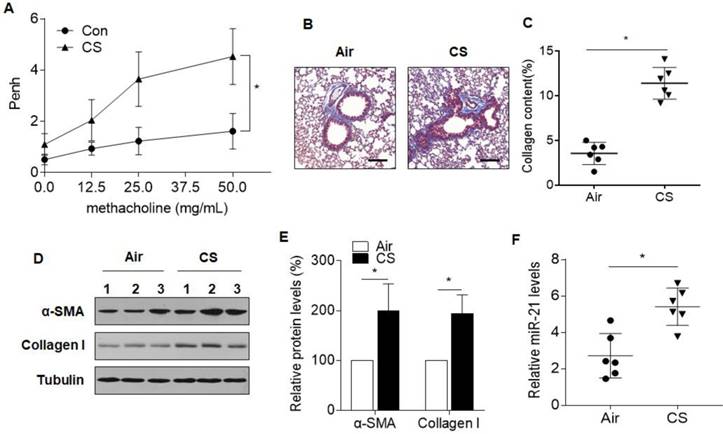
CS induces airway obstruction and increases miR-21 levels in lungs of mice. Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls. Male BALB/c mice at 6-8 weeks of age were exposed to 500 mg/m3 TPM CS for 8 weeks. (A) AHR is represented as Penh in air-exposed mice (n=6) and CS-exposed mice (n=6) response to 0, 12.5, 25, or 50 μg/mL methacholine. (B) Lung morphology was assessed by Masson staining of lung sections. Scale bars, 100 μm. Collagen: blue; nuclei: black; cytoplasm/epithelia: red. (C) Quantification of Masson staining for collagen content from air-exposed mice (n=6) and CS-exposed mice (n=6) (D) Western blots were performed, and (E) relative protein levels (n = 3) of α-SMA and collagen I in lung were determined. (F) The levels of miR-21 in the lung tissues from air-exposed mice (n=6) and CS-exposed mice (n=6) were determined by quantitative RT-PCR. All data are shown as means ± SD. *P<0.05.
Exosomes derived from CSE-treated HBE cells transfer miR-21 to MRC-5 cells and induce the myofibroblast differentiation phenotype
Exosomes, which are mediators of cell-cell communication through the transfer of their contents, are involved in lung homeostasis[27]. Thus, we predicted that miR-21 is transferred from CSE-treated HBE cells into MRC-5 cells via exosomes. Exosomes from normal and CSE-treated HBE cells were isolated, characterized, and quantified by immunoblotting and TEM. Exosomal markers, including CD9, CD63, and CD81, were evident; HSP90B1, an endoplasmic reticulum marker, was minimally expressed in exosomes isolated from normal and CSE-treated HBE cells (Fig. 3A). Furthermore, nanovesicles with diameters of ≤ 200 nm were present in the exosome fraction isolated from normal and CSE-treated HBE cells, consistent with the characteristic size range of exosomes (Fig. 3B). The particle sizes and counts of the exosomes showed no significant differences between exosomes derived from normal and CSE-treated HBE cells, as determined by a ZetaView® nanoparticle tracker (Fig. 3C). Exosomes from normal and CSE-treated HBE cells were added to MRC-5 cells. After 3 h, these cells exhibited uptake of exosomes, indicating the internalization of PKH76-labeled exosomes (Fig. 3D). qRT-PCR analyses showed that the levels of exosomal miR-21 from CSE-treated HBE cells were higher than those for normal HBE cells (Fig. 3E). Levels of miR-21 were higher in MRC-5 cells exposed to exosomes derived from CSE-treated HBE cells (Fig. 3F), and there were greater levels of α-SMA and collagen I in these cells exposed to exosomes of CSE-treated HBE cells (Fig. 3G-H). In addition, we also measured the levels of miR-21 in human primary bronchial epithelial cells and their exosomes exposed to CSE. After CSE exposure of cells, the levels of miR-21 increased in primary bronchial epithelial cells and their exosomes (Fig. S3A and 3B).
Figure 2
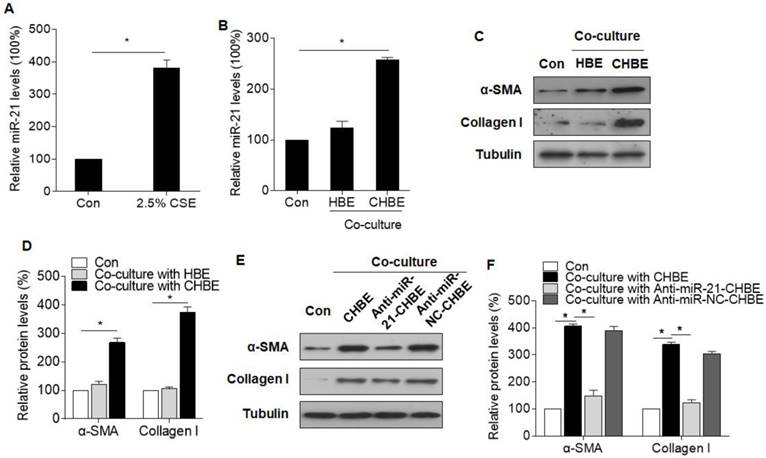
miR-21 derived from CSE-treated HBE cells is involved in the myofibroblast differentiation phenotype of MRC-5 cells. HBE, normal HBE cells; CHBE, HBE cells treated with CSE; Anti-miR-21-CHBE, HBE cells transfected with a miR-21 inhibitor and then treated with CSE; Anti-miR-NC-CHBE, HBE cells transfected with miR-NC inhibitor and then treated with CSE. Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls. HBE cells were treated with 0 or 2.5% CSE for 12 h. (A) The levels of miR-21 in HBE cells were determined by qRT-PCR. MRC-5 cells were co-cultured with normal or CSE-treated HBE cells for 24 h. (B) The levels of miR-21 in MRC-5 cells were determined by qRT-PCR. (C) Western blots were performed, and (D) relative protein levels of α-SMA and collagen I were determined for MRC-5 cells. MRC-5 cells were co-cultured with CSE-treated HBE cells transfected with a miR-21 inhibitor or miR-NC inhibitor for 24 h. (E) Western blots were performed, and (F) relative protein levels of α-SMA and collagen I were determined for MRC-5 cells. Data are means ± SD, n=3 cultures. *P<0.05.
Figure 3
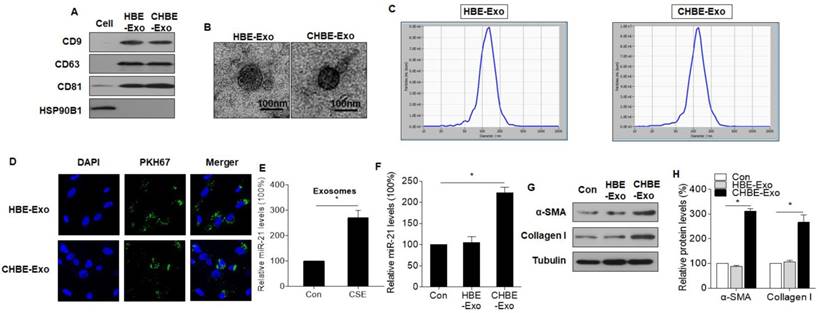
Exosomes derived from CSE-treated HBE cells transfer miR-21 to MRC-5 cells, which promotes the myofibroblast differentiation phenotype. HBE-Exo, exosomes derived from normal HBE cells; CHBE-Exo, exosomes derived from HBE cells treated with CSE. Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls. Exosomes from normal or CSE-treated HBE cells were fractionated by ExoQuick-TC. (A) Western blots of CD9, CD63, CD81, and HSP90B1 in exosomes. (B) Electron microscopic images of exosomes (bars = 100 nm). (C) Particle number and size analysis of HBE-exo or CHBE-exo were determined by dynamic light scattering using a ZetaView® nanoparticle tracker (ParticleMetrix, Germany). (D) Microscopic images of MRC-5 cells after incubation with PKH67-labeled (green) exosomes; nuclei were stained with DAPI (blue). (E) The levels of miR-21 in exosomes derived from normal or CSE-treated HBE cells were determined by qRT-PCR. MRC-5 cells were treated exosomes (50 μg/mL) derived from normal or CSE-treated HBE cells. (F) The levels of miR-21 in MRC-5 cells were determined by qRT-PCR. (G) Western blots were performed, and (H) relative protein levels of α-SMA and collagen I were determined for MRC-5 cells. Data are means ± SD, n=3 cultures. *P<0.05.
Exosomes derived from epithelial cells promote the myofibroblast differentiation phenotype of MRC-5 cells through cargo miR-21
We hypothesized that the exosomal miR-21 derived from CSE-treated HBE cells was involved in myofibroblast differentiation of MRC-5 cells. To confirm this, MRC-5 cells and CSE-treated HBE cells were placed in co-culture with dimethyl sulfoxide (vehicle) or GW4869, an inhibitor of exosome generation.[24] There were fewer exosomes released from CHBE cells treated with GW4869 (Fig. 4A). The elevated levels of miR-21 in MRC-5 cells co-cultured with CSE-treated HBE cells were prevented by GW4869 (Fig. 4B). In addition, the increases of α-SMA and collagen I expression in MRC-5 cells were blocked by GW4869 (Fig. 4C and D). Inhibition of miR-21 lowered the miR-21 levels in CSE-treated HBE cells and in their exosomes (Fig. 4E and F), which caused lower miR-21 levels in MRC-5 cells (Fig. 4G). For MRC-5 cells, down-regulation of exosomal miR-21 blocked the exosome-induced myofibroblast differentiation phenotype (Fig. 4H-I). In addition, to exclude the possibility that exosomes derived from HBE cells deliver the α-SMA and collagen I into MRC-5 cells, we analyzed the expression levels of α-SMA and collagen I in exosomes after CSE exposure or miR-21 inhibitor transfection. The results showed that there was no expression of α-SMA or collagen I in the exosomes, which confirmed that exosomal miR-21 was involved in the myofibroblast differentiation phenotype (Fig. S4).
Figure 4
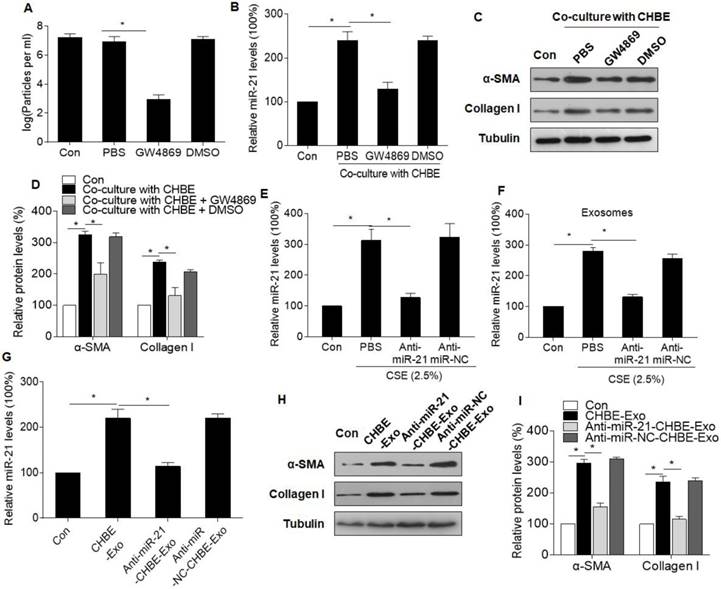
Exosomal miR-21 derived from CSE-treated HBE cells promotes the myofibroblast differentiation phenotype of MRC-5 cells. CHBE, HBE cells treated with CSE; CHBE-Exo, exosomes derived from HBE cells treated with CSE; Anti-miR-21-CHBE, HBE cells transfected with a miR-21 inhibitor and then treated with CSE; Anti-miR-NC-CHBE, HBE cells transfected with a miR-NC inhibitor and then treated with CSE. Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls. MRC-5 cells were co-cultured with CSE-treated HBE cells after incubation with DMSO or GW4869 (an inhibitor of exosome generation). (A) Nanoparticle tracking analysis was accomplished for exosome samples isolated using ExoQuick and normalized to the cell count. (B) The levels of miR-21 in MRC-5 cells were determined by qRT-PCR. (C) Western blots were performed, and (D) protein levels of α-SMA and collagen I were determined. MRC-5 cells were incubated with exosomes (50 μg/mL) derived from CSE-treated HBE cells transfected with a miR-21 inhibitor or miR-NC inhibitor. (E) The levels of miR-21 in normal HBE cells or CSE-treated HBE cells transfected with a miR-21 inhibitor or miR-NC inhibitor were determined by qRT-PCR. (F) The levels of miR-21 in exosomes derived from normal HBE cells or CSE-treated HBE cells transfected with a miR-21 inhibitor or miR-NC inhibitor were determined by qRT-PCR. (G) The levels of miR-21 in MRC-5 cells treated with exosomes derived from normal HBE cells or CSE-treated HBE cells transfected with a miR-21 inhibitor or miR-NC inhibitor were determined by qRT-PCR. (H) Western blots were performed, and (I) protein levels of α-SMA and collagen I were determined. All data are shown as means ± SD, n=3 cultures. *P<0.05.
Exosomal miR-21 derived from CSE-treated HBE cells targets pVHL in MRC-5 cells
HIF-1α is a factor in the development of fibrosis[28]. To determine if HIF-1α is involved in myofibroblast differentiation of bronchial fibroblasts, Western blots were performed. The results showed that the levels of HIF-1α were up-regulated in MRC-5 cells after exposure to exosomes derived from CSE-treated HBE cells (Fig. 5A-B). By use of three independent online databases, including Targetscan (targetscan.org), miRTarBase (mirtarbase.mbc.nctu.edu.tw), and Miranda (microrna.org) (Table S1), we predicted that targets of miR-21 contribute to HIF-1α activity and focused on pVHL, a factor related to HIF-1α stabilization. Western blots showed that pVHL levels were lower in MRC-5 cells exposed to exosomes derived from CSE-treated HBE cells (Fig. 5A-B). Consistently, the changes of pVHL and HIF-1α levels were evident in lung tissues of mice exposed to CS (Fig. S5A-B).
Figure 5
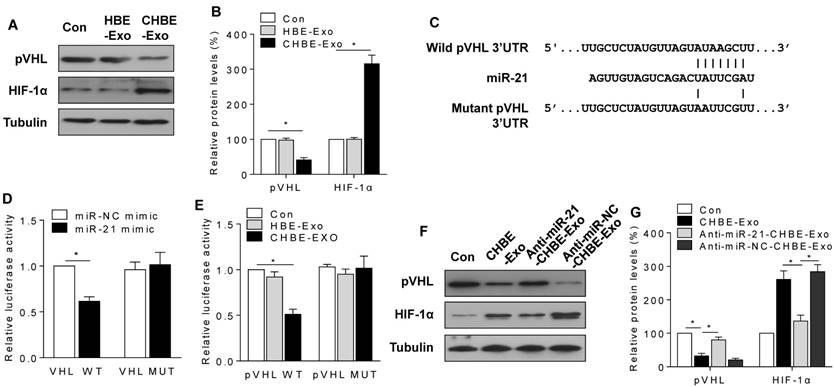
Exosomal miR-21 regulates HIF-1α stabilization by targeting pVHL in MRC-5 cells. HBE-Exo, exosomes derived from normal HBE cells. CHBE-Exo, exosomes derived from HBE cells treated with CSE; Anti-miR-21-CHBE-Exo, exosomes derived from HBE cells transfected with a miR-21 inhibitor and then treated with CSE; Anti-miR-NC-CHBE-Exo, exosomes derived from HBE cells transfected with a miR-NC inhibitor and then treated with CSE. Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls. MRC-5 cells were treated with exosomes (50 μg/mL) derived from normal or CSE-treated HBE cells for 24 h. (A) Western blots were performed, and (B) protein levels of pVHL and HIF-1α were determined. (C) Schematic of miR-21 putative target sites in the 3'UTR of pVHL and the sequences of mutant UTRs. (D) Luciferase reporter assay was performed on MRC-5 cells after co-transfected with the miR-21 mimic or miR-NC mimic (100 nM) and the pVHL-wt plasmid or pVHL-mut plasmid (1 μg). (E) Luciferase readout from wild-type or mutant pVHL 3'UTR reporter transfected into MRC-5 cells, which were then incubated with exosomes (50 μg/mL) derived from normal or CSE-treated HBE cells. MRC-5 cells were treated with exosomes (50 μg/mL) derived from CSE-treated HBE cells transfected with a miR-21 inhibitor or miR-NC inhibitor for 24 h. (F) Western blots were performed, and (G) protein levels of pVHL and HIF-1α were determined. All data are shown as means ± SD, n=3 cultures. *P<0.05.
Next, luciferase reporter assays were conducted by use of plasmids containing wild-type or mutant pVHL 3'UTR with miR-21 binding sites (Fig. 5C). Following co-transfection of MRC-5 cells with a plasmid containing wild-type pVHL 3'UTR and an miR-21 mimic, there was low luciferase activity; however, co-transfection with a miR-21 mimic with mutated forms of pVHL 3'UTR resulted in no appreciable change in luciferase activity (Fig. 5D). Consistently, exosomes derived from CSE-treated HBE cells also decreased the luciferase activity of pVHL 3'UTR, but not that for mutant pVHL 3'UTR (Fig. 5E). Furthermore, down-regulation of exosomal miR-21 blocked the changes of pVHL and HIF-1α levels caused by exosomes derived from CSE-treated HBE cells (Fig. 5F-G). After incubation of cells with the miR-21 mimic for 48 h, a decrease of pVHL levels and an increase of HIF-1α levels were evident (Fig. S6A-B). In addition, to exclude the possibility that the exosomes deliver pVHL and HIF-1α from HBE cells to MRC-5 cells, we analyzed the expression levels of pVHL and HIF-1α in exosomes after the cells were exposed to CSE or transfected with an miR-21 inhibitor. The results showed that there was no expression of pVHL or HIF-1α in the exosomes (Fig. S4).
Exosomal miR-21 derived from CSE-treated HBE cells promotes the myofibroblast differentiation phenotype of MRC-5 cells through pVHL/HIF-1α signaling
As shown by Western blots, down-regulation of HIF-1α in MRC-5 cells by use of an HIF-1α siRNA blocked the increases of protein levels of α-SMA and collagen I that were induced by exosomes derived from CSE-treated HBE cells (Fig. 6A-B). To determine the mechanism by which HIF-1α regulates myofibroblast differentiation, we searched the promoter regions of profibrotic genes and found a putative HIF-1α response element (A/GCGTG) within the promoter of the α-SMA gene (ACTA2), at 363 bp upstream of the transcriptional starting site (Fig. 6C). Moreover, ChIP assays confirmed that HIF-1α bound to this region after MRC-5 cells were exposed to exosomes derived from CSE-treated HBE cells (Fig. 6D).
To determine whether exosomal miR-21 derived from CSE-treated HBE cells promotes myofibroblast differentiation through regulation of pVHL, control or pVHL siRNA-transduced MRC-5 cells were exposed to exosomes derived from CSE-treated HBE cells with or without transfection of a miR-21 inhibitor, and the protein levels of pVHL, HIF-1α, α-SMA, and collagen I were determined. Western blotting results showed that downregulation of exosomal miR-21 blocked the changes of pVHL, HIF-1α, α-SMA, and collagen I expression in MRC-5 cells caused by exosomes derived from CSE-treated HBE cells, but knockdown of pVHL attenuated the effect of the miR-21 inhibitor (Fig. 6E-F).
Figure 6
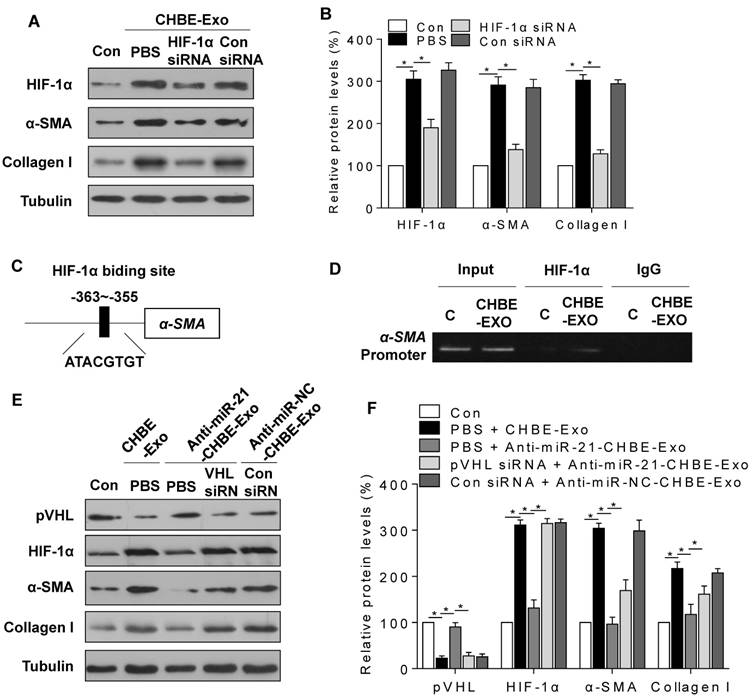
Exosomal miR-21 promotes the myofibroblast differentiation phenotype of MRC-5 cells by regulating pVHL/HIF-1α signaling. CHBE-Exo, exosomes derived from HBE cells treated with CSE; Anti-miR-21-CHBE-Exo, exosomes derived from HBE cells transfected with a miR-21 inhibitor and then treated with CSE; Anti-miR-NC-CHBE-Exo, exosomes derived from HBE cells transfected with a miR-NC inhibitor and then treated with CSE. Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls. MRC-5 cells were transfected with HIF-1α siRNA or control siRNA (20 nM) and then incubated with exosomes (50 μg/mL) derived from CSE-treated HBE cells. (A) Western blots were performed, and (B) relative protein levels of HIF-1α, α-SMA, and collagen I were determined. (C) Schematic of HIF-1α putative binding sites in the promoter region of the α-SMA gene (ACTA2). (D) ChIP assays were performed after the chromatin was immunoprecipitated with antibodies against HIF-1α. MRC-5 cells were transfected with pVHL siRNA or control siRNA (20 nM) and then incubated with exosomes (50 μg/mL) derived from CSE-treated HBE cells transfected with a miR-21 inhibitor or miR-NC inhibitor. (E) Western blots were performed, and (F) relative protein levels of pVHL, HIF-1α, α-SMA, and collagen I were determined. All data are shown as means ± SD, n=3 cultures. *P<0.05.
Downregulation of miR-21 prevents CS-induced airway injury in mice
To confirm the role of miR-21 in CS-induced COPD in mice, antagomir-21 or an antagomir negative control was injected via the tail vein weekly, simultaneous with the first CS exposure (Fig. 7A). Lungs of mice receiving antagomir-21 had a decrease of miR-21 levels, showing that miR-21 was down-regulated (Fig. 7B). Results of AHR assays, BALF cells counts, Masson staining, and Western blots showed that, in CS-exposed mice, down-expression of miR-21 prevented changes in pulmonary function and attenuated inflammation and airway obstruction (Fig. 7C-G, S7A-C). Moreover, the expression of pVHL was restored in mice treated with antagomir-21, whereas HIF-1α levels were decreased (Fig. 7F, G).
Figure 7
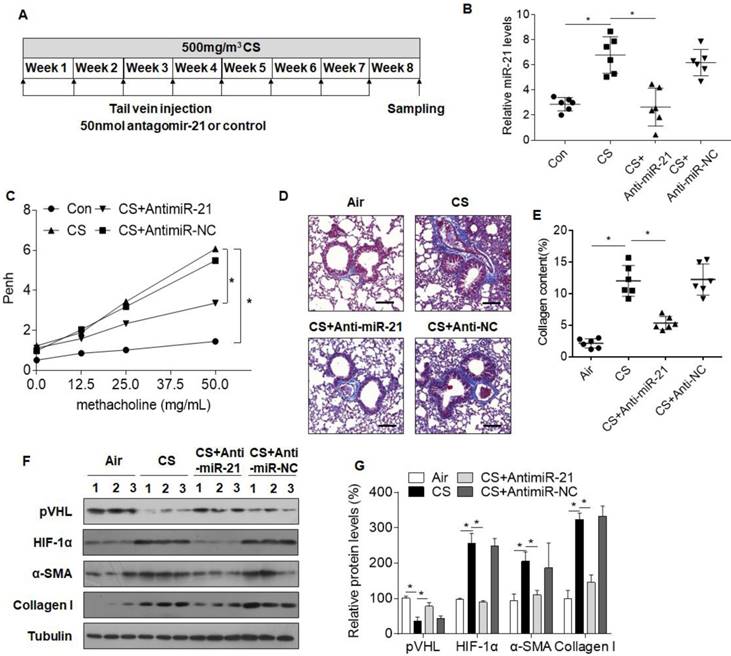
Inhibition of miR-21 prevents CS-induced airway obstruction in mice. Densities of bands were quantified by Image J software. Tubulin levels, measured in parallel, served as controls. Male BALB/c mice (age 6-8 weeks) were divided into four groups: normal control (n=6), CS (n=6), CS plus antagomir-21 (n=6), and CS plus antagomir-NC (n=6). Antagomir-21 and an antagomir negative control were administered by tail vein injections every week for 8 weeks. The mice were exposed to air or to 500 mg/m3 TPM CS every 5 days/week for 8 weeks. (A) A schematic diagram illustrating the experimental design. (B) The levels of miR-21 in lungs of mice determined by quantitative RT-PCR. Data are means ± SD, n=6 for each group. (C) AHR is represented as Penh in mice responding to 0, 12.5, 25, or 50 μg/mL methacholine. Data are means ± SD, n=6 for each group. *P < 0.05. (D) Lung morphology was assessed by Masson staining of lung sections from mice. Scale bars, 100 μm. Collagen: blue; nuclei: purple; cytoplasm/epithelia: pink. (E) Quantification of Masson staining for collagen content. Data are means ± SD, n=6 for each group. (F) Western blots were performed, and (G) relative protein levels of pVHL, HIF-1α, α-SMA, and collagen I in lungs were determined. Data are means ± SD, n=3 for each group. *P < 0.05.
Cigarette smoking induces aberrant levels of miR-21 in serum exosomes
Exosomes contain specific proteins, lipids, and nucleic acids that can serve as disease biomarkers[9, 29]. To determine whether miR-21 is a mediator for CS-induced COPD, the exosomal miR-21 expression levels in non-smokers (n=26), smokers (n=24), and smokers with COPD (n=29) were compared (Table 1). Exosomes from the sera of these three groups were isolated and characterized. Western blotting showed the presence of exosome markers (Fig. 8A). Electron microscopy demonstrated that the exosomes had a size distribution consistent with exosome vesicles (Fig. 8B). The levels of exosomal miR-21 from smokers with COPD were at higher levels than those for non-smokers and smokers without airflow obstruction (Fig. 8C). Similarly, there were higher expressions of exosomal miR-21 in the sera of the smoker group compared with those in the non-smoker group (Fig. 8C). Furthermore, the levels of serum exosomal miR-21 inversely correlated with FEV1/FVC% (Fig. 8D). We additionally measured the serum miR-21 levels in the three groups. In accordance with the exosomal miR-21 results, the levels of serum miR-21 from smokers with COPD were higher than those for non-smokers and for smokers without airflow obstruction, and there were higher expressions of miR-21 in the sera of the smoker group compared with those in the non-smoker group. Furthermore, the levels of serum miR-21 inversely correlated with FEV1/FVC% (Fig. S8).
Table 1
Characteristics of subjects in the study
| Non-smokers | Smokers | COPD smokers | |
|---|---|---|---|
| Number | 26 | 24 | 29 |
| Male, n (%) | 24 (92.3%) | 20 (83.3%) | 27 (93.1%) |
| Age, years | 54±7 | 55±6 | 59±7** |
| BMI, kg/m2 | 25±2 | 24±3 | 25±3 |
| Smoking history, pack-years | 0 | 38±18** | 42±24** |
| FEV1/FVC, % | 88.4±2.3 | 75.6±2.7** | 55.3±8.3**## |
FEV1: the forced expiratory volume 1.
FVC: forced vital capacity.
Data are presented as means ± SD, unless otherwise stated.
**p<0.01 versus non-smokers, ##p<0.01 versus smokers.
Discussion
This work revealed an exosome-mediated intercellular communication mechanism between bronchial epithelial cells and fibroblasts that promotes the development of CS-induced airway remodeling. We elucidated the mechanism of differentiation of bronchial fibroblasts to myofibroblasts attributed to the CSE-induced, HBE cells-derived exosomal miR-21 regulation of pVHL/HIF-1α signaling.
Figure 8
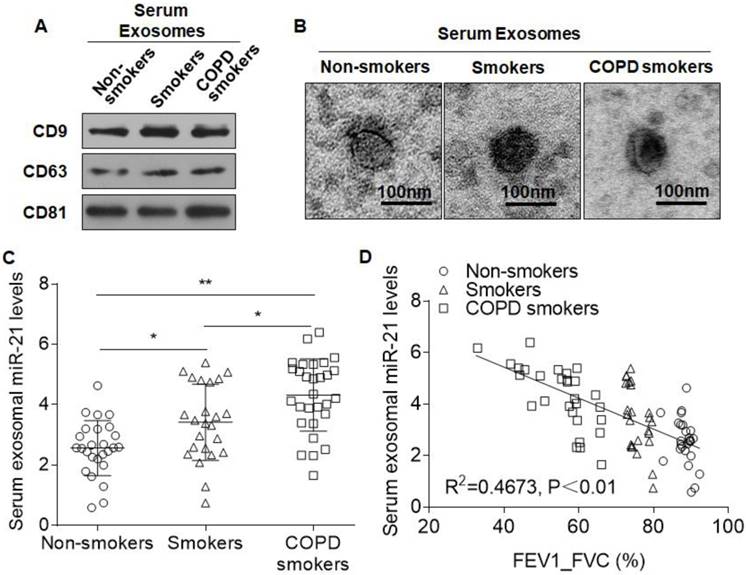
Levels of serum exosomal miR-21 are over-expressed in smokers and COPD patients and are negatively related to lung function. Exosomes from sera of non-smokers (n=26), smokers (n=24), and smokers with COPD (n=29) were isolated by ExoQuick. (A) Western blots of CD9, CD63, and CD81 in serum exosomes. (B) Representative electron micrograph of serum exosomes (bars = 200 nm). (C) The levels of serum exosomal miR-21 were determined by quantitative RT-PCR. Data are means ± SD. *P<0.05. (D) The relationship between the levels of serum exosomal miR-21 and FEV1/FVC (%). Data shown are individual values of the mean levels from three separate experiments. *p <0.05, **p <0.01.
COPD, caused mainly by exposure to CS, is characterized by abnormal activation of bronchial epithelial cells, which secrete mucus and various mediators of immune cell activation and cause changes in the interaction between epithelial and mesenchymal cell types, such as fibroblasts and airway smooth muscle cells[27]. A defining feature of airway remodeling in COPD pathogenesis is the presence and activation of bronchial myofibroblasts[26]. Exosomes are mediators of intercellular communication through the transfer of their contents, which can be modified by environmental stimuli[30]. An analysis of the pathophysiological functions of lung epithelial cell-derived exosomes shows that CSE induces exosomes enriched in CCN1 (CYR61/CTGF/NOV family 1), which is involved in tissue remodeling and serves as an extracellular matrix protein[31]. Analysis of the RNA profile in exosomes derived from bronchial epithelial cells shows that they contain small RNAs, including miRNAs[3]. Furthermore, CSE induces upregulation of miR-210 expression in HBE cell-derived extracellular vesicles, and, in COPD pathogenesis, promotes myofibroblast differentiation by suppression of autophagy[3]. Here, we showed that CSE induced upregulation of miR-21 levels in exosomes derived from HBE cells and, in fibroblasts, promoted myofibroblast differentiation through increased HIF-1α transcriptional activity. These observations indicate that exposure of bronchial epithelial cells to CS damages the epithelial barrier and changes the composition of exosomes, which, in turn, disrupts the balance between epithelial cells and fibroblasts and thereby induces airway remodeling.
HIF-1α is a transcriptional factor activated in response to reduced oxygen levels and environmental changes[32]. Under normoxic conditions, HIF-1α is subject to hydroxylation by prolyl hydroxylase domain proteins (PHDs). The von Hippel-Lindau protein (pVHL) recognizes hydroxylated HIF-1α for subsequent degradation through the proteasome[33]. In hypoxia or in response to environmental cues, the hydroxylation reactions are diminished, resulting in HIF-1α accumulation, dimerization with HIF-1β, and binding to and activation of target genes[33]. Increased HIF-1α activity is likely involved in the pathogenesis of fibrosis through transcriptional regulation of profibrotic genes and/or through other regulation[34]. Here, we found that, in COPD pathogenesis, HIF-1α was required for the differentiation of airway fibroblasts to myofibroblasts, which was accomplished through regulation of the profibrotic gene, α-SMA, by binding to the HIF-1α response element within the promoter.
MiRNAs are involved in regulation of HIF-1α activity. For example, miR-543 increases HIF-1α expression via targeting the 3′-UTR of protein arginine methyltransferase 9 (PRMT9), which promotes proliferation of osteosarcoma cells[35]. Some miRNAs, such as miR-199a and miR-186, suppress expression of HIF-1α downstream genes by targeting the 3′UTR of HIF-1α[36, 37]. In the present study, we demonstrated that miR-21 was a regulator for stabilization of HIF-1α by targeting pVHL and that it promoted profibrotic phenotypes. In glioblastomas, pVHL is a target of miR-21[38]. Although pVHL is a component of a ubiquitin protein ligase complex targeting HIFs to the proteasome for degradation, it also regulates the fibronectin and collagen matrix[34]. The role of the pVHL/HIF-1α pathway in profibrotic pathogenesis may be complex. The present results showed that the protein levels of α-SMA and collagen I were only partly modulated by miR-21/pVHL, which may due to the regulation of airway fibroblast differentiation by multiple pathways. For example, the TGF-β1/Wnt pathway is involved in fibroblast differentiation in COPD[39]. The PI3K/AKT signaling pathway also contributes to up-regulation of collagen I/III in COPD[40]. In the present study, we showed that HIF-1α regulation of α-SMA by binding to the response element within the promoter is an important pathway in the differentiation of airway fibroblasts to myofibroblasts and COPD.
MiR-21 is associated with myofibroblast differentiation via an enhanced TGF-β signaling pathway[13]. Although TGF-β1 is a mediator of fibrogenesis, other factors, including HIF-1α, could contribute to the differentiation of myofibroblasts, a process that involves various transcriptional and epigenetic mechanisms[28]. Indeed, we determined here a role for miR-21 in a CS-induced animal model of COPD. Consistent with data derived with cell cultures, miR-21 was up-regulated in lungs of mice exposed to CS, and, with these mice, pVHL downregulation and enhanced HIF-1α signaling were evident. In mice, inhibition of miR-21 attenuated CS-induced inflammation and airway remodeling. Airway remodeling is a complex process, associated with various types of cellular dysfunction and various stages of COPD pathogenesis, including metaplasia of goblet cells, proliferation of airway smooth muscle cells, and airway fibroblast differentiation into myofibroblasts[2]. Therefore, the role of exosomes in the dysfunctional cross-talk of the types of airway cells needs to be clarified.
Circulating exosomes are potentially useful as disease biomarkers because they carry specific proteins and nucleic acids and, because of their phospholipid bilayer, are stable in body fluids[16, 27]. As described in the present study, we isolated and identified exosomes from sera of smoking COPD patients, smokers without airflow obstruction, and healthy, never-smoking volunteers. The results showed that miR-21 was over-expressed in the exosomes of smokers and smoking COPD patients and that there was a positive correlation between miR-21 levels and FEV1/FVC. For asymptomatic heavy smokers, the levels of serum miR-21 and miR-181a and their ratio have potential biomarker utility for predicting the development of COPD[17]. Due to differential changes in miRNAs in plasma, measurement of exosomal miRNAs is not equivalent to measurement of miRNAs[41]. Our data demonstrate that miR-21 levels of serum and serum exosomes are similar. We found that the coefficient of determination (R2) between serum exosomal miR-21 and FEV1/FVC% was higher than that between serum miR-21 and FEV1/FVC%, suggesting a reliable correlation between serum exosomal miR-21 and FEV1/FVC%. The results suggest that exosomal miRNAs have the potential to be biomarkers of diseases, including COPD, although technical and scientific obstacles must be overcome prior to clinical application.
In conclusion, for bronchial epithelial cells of lung, CS exposure induces increases of miR-21 levels, which are transfered from bronchial epithelial cells into bronchial fibroblast cells by exosomes. In bronchial fibroblast cells, miR-21 causes increases of HIF-1α levels by targeting pVHL, which elevates the levels of α-SMA and collagen I, inducing the myofibroblast differentiation phenotype. These results have potential value for diagnosis and treatment of COPD.
Abbreviations
AHR: airway hyper-responsiveness; α-SMA: α-smooth muscle actin; BALF: bronchoalveolar lavage fluid; ChIP: chromatin immunoprecipitation; COPD: chronic obstructive pulmonary disease; CS: cigarette smoke; CSE: cigarette smoke extract; FEV1: forced expiratory volume in 1 second; FVC: forced vital capacity; HIF-1α: hypoxia-inducible factor 1α; PHDs: prolyl hydroxylase domain proteins; pVHL: von Hippel-Lindau protein; TPM: total particulate matter.
Supplementary Material
Supplementary table S1.
Acknowledgements
This work was supported by the Natural Science Foundations of China (81473011), the Postgraduate Innovation Project of Jiangsu province (KYLX16_1128), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (2014). The authors thank Donald L. Hill (University of Alabama at Birmingham, USA), an experienced, English-speaking scientific editor, for editing.
Conflicts of Interest
The authors have declared that no competing interest exists.
References
1. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389:1931-40
2. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L. et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645-53
3. Fujita Y, Araya J, Ito S, Kobayashi K, Kosaka N, Yoshioka Y. et al. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J Extracell Vesicles. 2015;4:28388
4. Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J. et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340-55
5. Osei ET, Florez-Sampedro L, Tasena H, Faiz A, Noordhoek JA, Timens W. et al. miR-146a-5p plays an essential role in the aberrant epithelial-fibroblast cross-talk in COPD. Eur Respir J. 2017:49
6. Nyunoya T, Mebratu Y, Contreras A, Delgado M, Chand HS, Tesfaigzi Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am J Respir Cell Mol Biol. 2014;50:471-82
7. Gohy ST, Hupin C, Pilette C, Ladjemi MZ. Chronic inflammatory airway diseases: the central role of the epithelium revisited. Clin Exp Allergy. 2016;46:529-42
8. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255-89
9. Fujita Y, Kosaka N, Araya J, Kuwano K, Ochiya T. Extracellular vesicles in lung microenvironment and pathogenesis. Trends Mol Med. 2015;21:533-42
10. Harischandra DS, Ghaisas S, Rokad D, Kanthasamy AG. Exosomes in Toxicology: Relevance to Chemical Exposure and Pathogenesis of Environmentally Linked Diseases. Toxicol Sci. 2017
11. Benedikter BJ, Wouters EFM, Savelkoul PHM, Rohde GGU, Stassen FRM. Extracellular vesicles released in response to respiratory exposures: implications for chronic disease. J Toxicol Environ Health B Crit Rev. 2018;21:142-60
12. Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E. et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. 2010;18:282-93
13. Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ. et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589-97
14. Kim RY, Horvat JC, Pinkerton JW, Starkey MR, Essilfie AT, Mayall JR. et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J Allergy Clin Immunol. 2017;139:519-32
15. Liu Y, Luo F, Wang B, Li H, Xu Y, Liu X. et al. STAT3-regulated exosomal miR-21 promotes angiogenesis and is involved in neoplastic processes of transformed human bronchial epithelial cells. Cancer Lett. 2016;370:125-35
16. Pattarayan D, Thimmulappa RK, Ravikumar V, Rajasekaran S. Diagnostic Potential of Extracellular MicroRNA in Respiratory Diseases. Clin Rev Allergy Immunol. 2016
17. Xie L, Wu M, Lin H, Liu C, Yang H, Zhan J. et al. An increased ratio of serum miR-21 to miR-181a levels is associated with the early pathogenic process of chronic obstructive pulmonary disease in asymptomatic heavy smokers. Mol Biosyst. 2014;10:1072-81
18. Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A. et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347-65
19. Lu L, Xu H, Luo F, Liu X, Lu X, Yang Q. et al. Epigenetic silencing of miR-218 by the lncRNA CCAT1, acting via BMI1, promotes an altered cell cycle transition in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2016;304:30-41
20. Jobse BN, Rhem RG, Wang IQ, Counter WB, Stampfli MR, Labiris NR. Detection of lung dysfunction using ventilation and perfusion SPECT in a mouse model of chronic cigarette smoke exposure. J Nucl Med. 2013;54:616-23
21. Rajavelu P, Chen G, Xu Y, Kitzmiller JA, Korfhagen TR, Whitsett JA. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J Clin Invest. 2015;125:2021-31
22. Milara J, Serrano A, Peiro T, Artigues E, Gavalda A, Miralpeix M. et al. Aclidinium inhibits cigarette smoke-induced lung fibroblast-to-myofibroblast transition. Eur Respir J. 2013;41:1264-74
23. Benedikter BJ, Volgers C, van Eijck PH, Wouters EFM, Savelkoul PHM, Reynaert NL. et al. Cigarette smoke extract induced exosome release is mediated by depletion of exofacial thiols and can be inhibited by thiol-antioxidants. Free Radic Biol Med. 2017;108:334-44
24. Chen C, Luo F, Liu X, Lu L, Xu H, Yang Q. et al. NF-kB-regulated exosomal miR-155 promotes the inflammation associated with arsenite carcinogenesis. Cancer Lett. 2017;388:21-33
25. Xu H, Sun Q, Lu L, Luo F, Zhou L, Liu J. et al. MicroRNA-218 acts by repressing TNFR1-mediated activation of NF-kappaB, which is involved in MUC5AC hyper-production and inflammation in smoking-induced bronchiolitis of COPD. Toxicol Lett. 2017;280:171-80
26. Salazar LM, Herrera AM. Fibrotic response of tissue remodeling in COPD. Lung. 2011;189:101-9
27. Kadota T, Fujita Y, Yoshioka Y, Araya J, Kuwano K, Ochiya T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. Int J Mol Sci. 2016;17:E1801
28. Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM. et al. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am J Respir Crit Care Med. 2015;192:1462-74
29. Barile L, Vassalli G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol Ther. 2017;174:63-78
30. Beninson LA, Fleshner M. Exosomes: an emerging factor in stress-induced immunomodulation. Semin Immunol. 2014;26:394-401
31. Moon HG, Kim SH, Gao J, Quan T, Qin Z, Osorio JC. et al. CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 2014;307:L326-37
32. Li Q, Chen H, Huang X, Costa M. Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1alpha) and HIF-regulated genes. Toxicol Appl Pharmacol. 2006;213:245-55
33. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399-408
34. Haase VH. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006;69:1302-7
35. Zhang H, Guo X, Feng X, Wang T, Hu Z, Que X. et al. MiRNA-543 promotes osteosarcoma cell proliferation and glycolysis by partially suppressing PRMT9 and stabilizing HIF-1alpha protein. Oncotarget. 2017;8:2342-55
36. Liu L, Wang Y, Bai R, Yang K, Tian Z. MiR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1alpha regulation. Oncogenesis. 2016;5:e224
37. Li B, He L, Zuo D, He W, Wang Y, Zhang Y. et al. Mutual Regulation of MiR-199a-5p and HIF-1alpha Modulates the Warburg Effect in Hepatocellular Carcinoma. J Cancer. 2017;8:940-9
38. Zhang KL, Han L, Chen LY, Shi ZD, Yang M, Ren Y. et al. Blockage of a miR-21/EGFR regulatory feedback loop augments anti-EGFR therapy in glioblastomas. Cancer Lett. 2014;342:139-49
39. Spanjer AI, Baarsma HA, Oostenbrink LM, Jansen SR, Kuipers CC, Lindner M. et al. TGF-beta-induced profibrotic signaling is regulated in part by the WNT receptor Frizzled-8. FASEB J. 2016;30:1823-35
40. Wang Z, Li R, Zhong R. Extracellular matrix promotes proliferation, migration and adhesion of airway smooth muscle cells in a rat model of chronic obstructive pulmonary disease via upregulation of the PI3K/AKT signaling pathway. Mol Med Rep. 2018;18:3143-52
41. Xie JX, Fan X, Drummond CA, Majumder R, Xie Y, Chen T. et al. MicroRNA profiling in kidney disease: Plasma versus plasma-derived exosomes. Gene. 2017;627:1-8
Author contact
![]() Corresponding author: Qizhan Liu, Institute of Toxicology, School of Public Health, Nanjing Medical University, Nanjing 211166, Jiangsu, People's Republic of China. Telephone: +86-25-8686-8424, Fax: +86-25-8686-8499, E-mail: drqzliucom
Corresponding author: Qizhan Liu, Institute of Toxicology, School of Public Health, Nanjing Medical University, Nanjing 211166, Jiangsu, People's Republic of China. Telephone: +86-25-8686-8424, Fax: +86-25-8686-8499, E-mail: drqzliucom
Citation styles
APA
Xu, H., Ling, M., Xue, J., Dai, X., Sun, Q., Chen, C., Liu, Y., Zhou, L., Liu, J., Luo, F., Bian, Q., Liu, Q. (2018). Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics, 8(19), 5419-5433. https://doi.org/10.7150/thno.27876.
ACS
Xu, H.; Ling, M.; Xue, J.; Dai, X.; Sun, Q.; Chen, C.; Liu, Y.; Zhou, L.; Liu, J.; Luo, F.; Bian, Q.; Liu, Q. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics 2018, 8 (19), 5419-5433. DOI: 10.7150/thno.27876.
NLM
Xu H, Ling M, Xue J, Dai X, Sun Q, Chen C, Liu Y, Zhou L, Liu J, Luo F, Bian Q, Liu Q. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics 2018; 8(19):5419-5433. doi:10.7150/thno.27876. https://www.thno.org/v08p5419.htm
CSE
Xu H, Ling M, Xue J, Dai X, Sun Q, Chen C, Liu Y, Zhou L, Liu J, Luo F, Bian Q, Liu Q. 2018. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics. 8(19):5419-5433.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.